Cell-Based Reporter Release Assay to Determine the Potency of Proteolytic Bacterial Neurotoxins

Abstract
1. Introduction
2. Results
2.1. Suitability for Testing BoNT Activity in Pharmacological Preparations
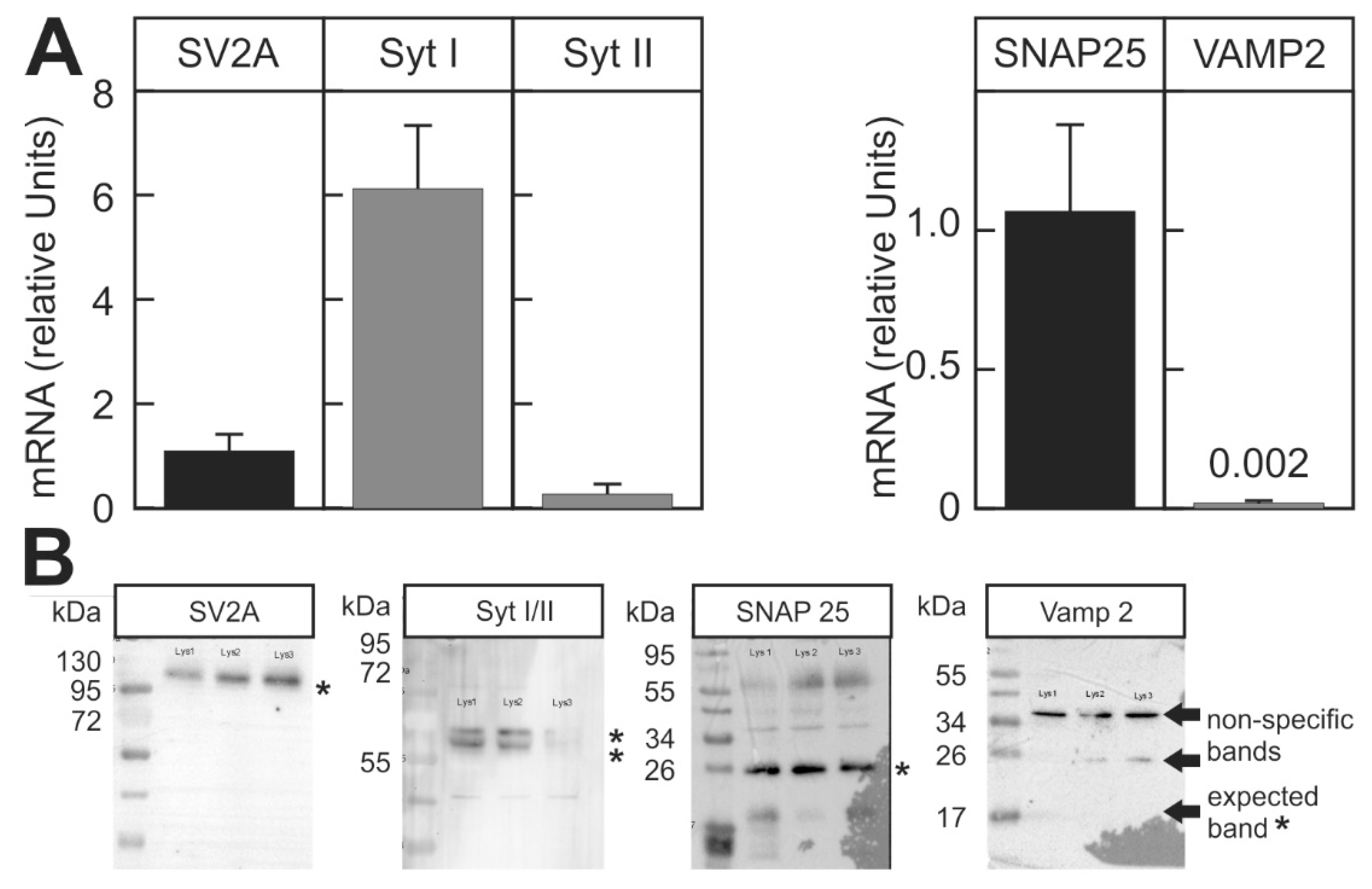
2.2. Suitability of the Assay for Different BoNT Serotypes
3. Discussion
3.1. Suitability of the Sssay as Replacement Method
3.2. Potential Reasons for the Limited Sensitivity for BoNT-B
4. Conclusions
5. Materials and Methods
5.1. Materials
5.2. Cell Culture
5.3. Luciferase Release from BoNT or TeT Treated Cells
5.4. Western Blot Analysis
5.5. Real Time RT-PCR
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hill, K.K.; Smith, T.J. Genetic diversity within Clostridium botulinum serotypes, botulinum neurotoxin gene clusters and toxin subtypes. Curr. Top. Microbiol. Immunol. 2013, 364, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Halpin, J.L.; Hill, K.; Johnson, S.L.; Bruce, D.C.; Shirey, T.B.; Dykes, J.K.; Lúquez, C. Finished Whole-Genome Sequences of Clostridium butyricum Toxin Subtype E4 and Clostridium baratii Toxin Subtype F7 Strains. Genome Announc. 2017, 5, e00375-17. [Google Scholar] [CrossRef] [PubMed]
- Peck, M.W.; Smith, T.J.; Anniballi, F.; Austin, J.W.; Bano, L.; Bradshaw, M.; Cuervo, P.; Cheng, L.W.; Derman, Y.; Dorner, B.G.; et al. Historical Perspectives and Guidelines for Botulinum Neurotoxin Subtype Nomenclature. Toxins 2017, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Jeffery, I.A.; Karim, S. StatPearls: Botulism. Treasure Island (FL); 2018. Available online: https://www.ncbi.nlm.nih.gov/books/NBK459273/ (accessed on 6 November 2017).
- Bigalke, H. Botulinum toxin: Application, safety, and limitations. Curr. Top. Microbiol. Immunol. 2013, 364, 307–317. [Google Scholar] [CrossRef] [PubMed]
- Jankovic, J. Botulinum toxin: State of the art. Mov. Disord. 2017, 32, 1131–1138. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.S.; Hong, E.S.; Kim, H.S. Botulinum Toxin in the Field of Dermatology: Novel Indications. Toxins 2017, 9, 403. [Google Scholar]
- Pellett, S.; Tepp, W.H.; Toth, S.I.; Johnson, E.A. Comparison of the primary rat spinal cord cell (RSC) assay and the mouse bioassay for botulinum neurotoxin type A potency determination. J. Pharmacol. Toxicol. Methods 2010, 61, 304–310. [Google Scholar] [CrossRef] [PubMed]
- Fujinaga, Y.; Sugawara, Y.; Matsumura, T. Uptake of botulinum neurotoxin in the intestine. Curr. Topics Microbiol. Immunol. 2013, 364, 45–59. [Google Scholar] [CrossRef]
- Rummel, A. Double receptor anchorage of botulinum neurotoxins accounts for their exquisite neurospecificity. Curr. Top. Microbiol. Immunol. 2013, 364, 61–90. [Google Scholar] [CrossRef]
- Binz, T. Clostridial neurotoxin light chains: Devices for SNARE cleavage mediated blockade of neurotransmission. Curr. Top. Microbiol. Immunol. 2013, 364, 139–157. [Google Scholar] [CrossRef] [PubMed]
- Pellett, S. Progress in cell based assays for botulinum neurotoxin detection. Curr. Topics Microbiol. Immunol. 2013, 364, 257–285. [Google Scholar] [CrossRef]
- Wild, E.; Bonifas, U.; Klimek, J.; Trösemeier, J.H.; Krämer, B.; Kegel, B.; Behrensdorf-Nicol, H.A. In vitro potency determination of botulinum neurotoxin B based on its receptor-binding and proteolytic characteristics. Toxicol. In Vitro 2016, 34, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Fernándezsalas, E.; Wang, J.; Molina, Y.; Nelson, J.B.; Jacky, B.P.S.; Aoki, K.R. Botulinum Neurotoxin Serotype a Specific Cell-Based Potency Assay to Replace the Mouse Bioassay. PLoS ONE 2012, 7, e49516. [Google Scholar] [CrossRef]
- Pellett, S.; Tepp, W.H.; Johnson, E.A.; Sesardic, D. Assessment of ELISA as endpoint in neuronal cell-based assay for BoNT detection using hiPSC derived neurons. J. Pharmacol. Toxicol. Methods 2017, 88, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Pathe-Neuschäfer-Rube, A.; Neuschäfer-Rube, F.; Genz, L.; Püschel, G.P. Botulinum neurotoxin dose-dependently inhibits release of neurosecretory vesicle-vargeted luciferase from neuronal cells. ALTEX 2015, 32, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Mander, G.; Bruenn, C.; Jatzke, C.; Eisele, K.H.; Taylor, H.V.; Pellett, S.; Johnson, E.A.; Fink, K. 134. Potency assay for botulinum neurotoxin type A based on neuronal cells as a replacement for the mouse bioassay. Toxicon 2015, 93, S41–S42. [Google Scholar] [CrossRef]
- Takakura, H.; Kojima, R.; Urano, Y.; Terai, T.; Hanaoka, K.; Nagano, T. Aminoluciferins as functional bioluminogenic substrates of firefly luciferase. Chem.—Asian J. 2011, 6, 1800–1810. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Peng, L.; Berntsson, R.P.A.; Liu, S.M.; Park, S.H.; Yu, F.; Boone, C.; Palan, S.; Beard, M.; Chabrier, P.E. Engineered botulinum neurotoxin B with improved efficacy for targeting human receptors. Nat. Commun. 2017, 8, 53. [Google Scholar] [CrossRef] [PubMed]
- Pappert, E.J.; Germanson, T. Botulinum toxin type B. vs. type A in toxin-naïve patients with cervical dystonia: Randomized, double-blind, noninferiority trial. Mov. Disord. 2008, 23, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Rust, A.; Doran, C.; Hart, R.; Binz, T.; Stickings, P.; Sesardic, D.; Peden, A.A.; Davletov, B. A Cell Line for Detection of Botulinum Neurotoxin Type B. Front. Pharmacol. 2017, 8, 15–19. [Google Scholar] [CrossRef] [PubMed]
- Schiavo, G.G.; Benfenati, F.; Poulain, B.; Rossetto, O.; Laureto, P.P.D.; Dasgupta, B.R.; Montecucco, C. Tetanus and botulinum-B neurotoxins block neurotransmitter release by proteolytic cleavage of synaptobrevin. Nature 1992, 359, 832–835. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| GAPDH | 5′-TGATGACATCAAGAAGGTGG | 5′-TTACTCCTTGGAGGCCATGT |
| SNAP25 | 5′-ACCAGTTGGCTGATGAGTCG | 5′-GTTCGTCCACTACACGAGCA |
| VAMP-2 | 5′-CCATAGAGGGAGGGTGTTGC | 5′-GTCCCCACCCTTACCTTGAG |
| SV2A | 5′-GAAGGTGGTGCATCCAGTGA | 5′-AGGCCTAGCATGCCTTTGTT |
| SytI | 5′-TCCTGACCTGCTGCTTTTGT | 5′-GGGTTTTGCCACCCAATTCC |
| SytII | 5′-CATTGGACCCGTGGACAACT | 5′-AGAACGCCCACAGTAAGCTG |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pathe-Neuschäfer-Rube, A.; Neuschäfer-Rube, F.; Haas, G.; Langoth-Fehringer, N.; Püschel, G.P. Cell-Based Reporter Release Assay to Determine the Potency of Proteolytic Bacterial Neurotoxins. Toxins 2018, 10, 360. https://doi.org/10.3390/toxins10090360
Pathe-Neuschäfer-Rube A, Neuschäfer-Rube F, Haas G, Langoth-Fehringer N, Püschel GP. Cell-Based Reporter Release Assay to Determine the Potency of Proteolytic Bacterial Neurotoxins. Toxins. 2018; 10(9):360. https://doi.org/10.3390/toxins10090360
Chicago/Turabian StylePathe-Neuschäfer-Rube, Andrea, Frank Neuschäfer-Rube, Gerald Haas, Nina Langoth-Fehringer, and Gerhard Paul Püschel. 2018. "Cell-Based Reporter Release Assay to Determine the Potency of Proteolytic Bacterial Neurotoxins" Toxins 10, no. 9: 360. https://doi.org/10.3390/toxins10090360
APA StylePathe-Neuschäfer-Rube, A., Neuschäfer-Rube, F., Haas, G., Langoth-Fehringer, N., & Püschel, G. P. (2018). Cell-Based Reporter Release Assay to Determine the Potency of Proteolytic Bacterial Neurotoxins. Toxins, 10(9), 360. https://doi.org/10.3390/toxins10090360