Detoxification- and Immune-Related Transcriptomic Analysis of Gills from Bay Scallops (Argopecten irradians) in Response to Algal Toxin Okadaic Acid

 ,
,  ,
,
Abstract
1. Introduction
2. Results
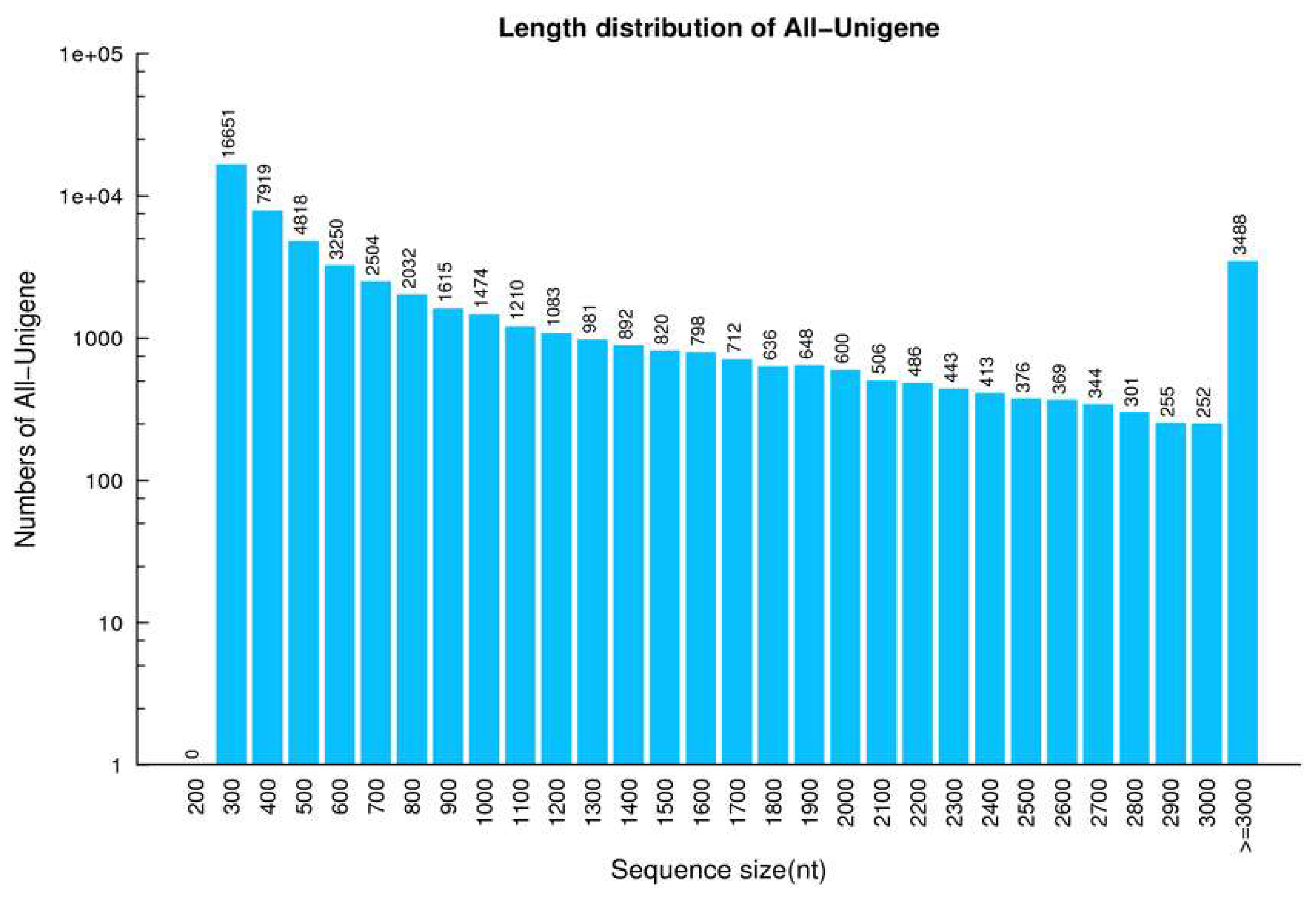
2.1. Analysis of DGE Libraries
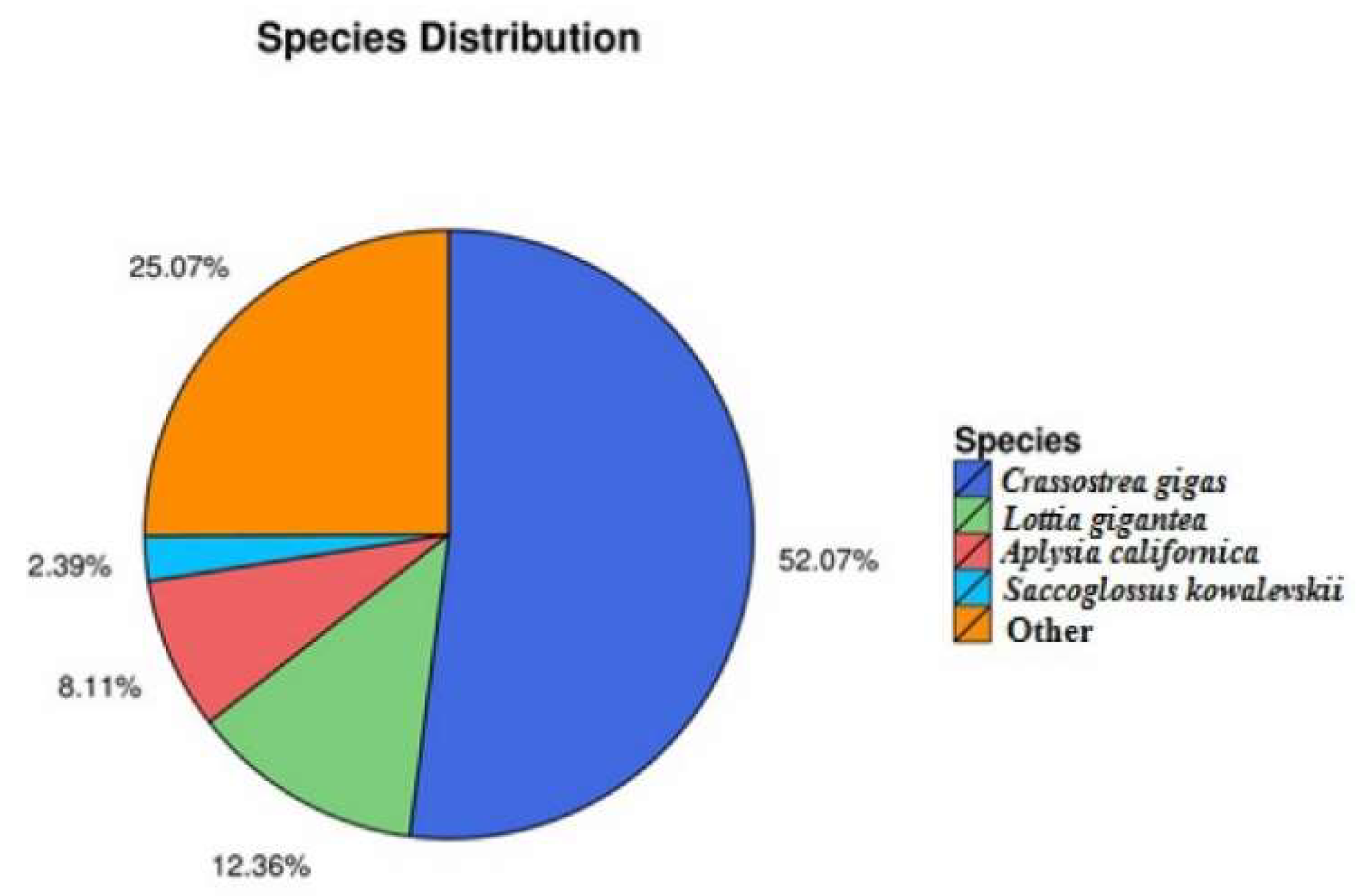
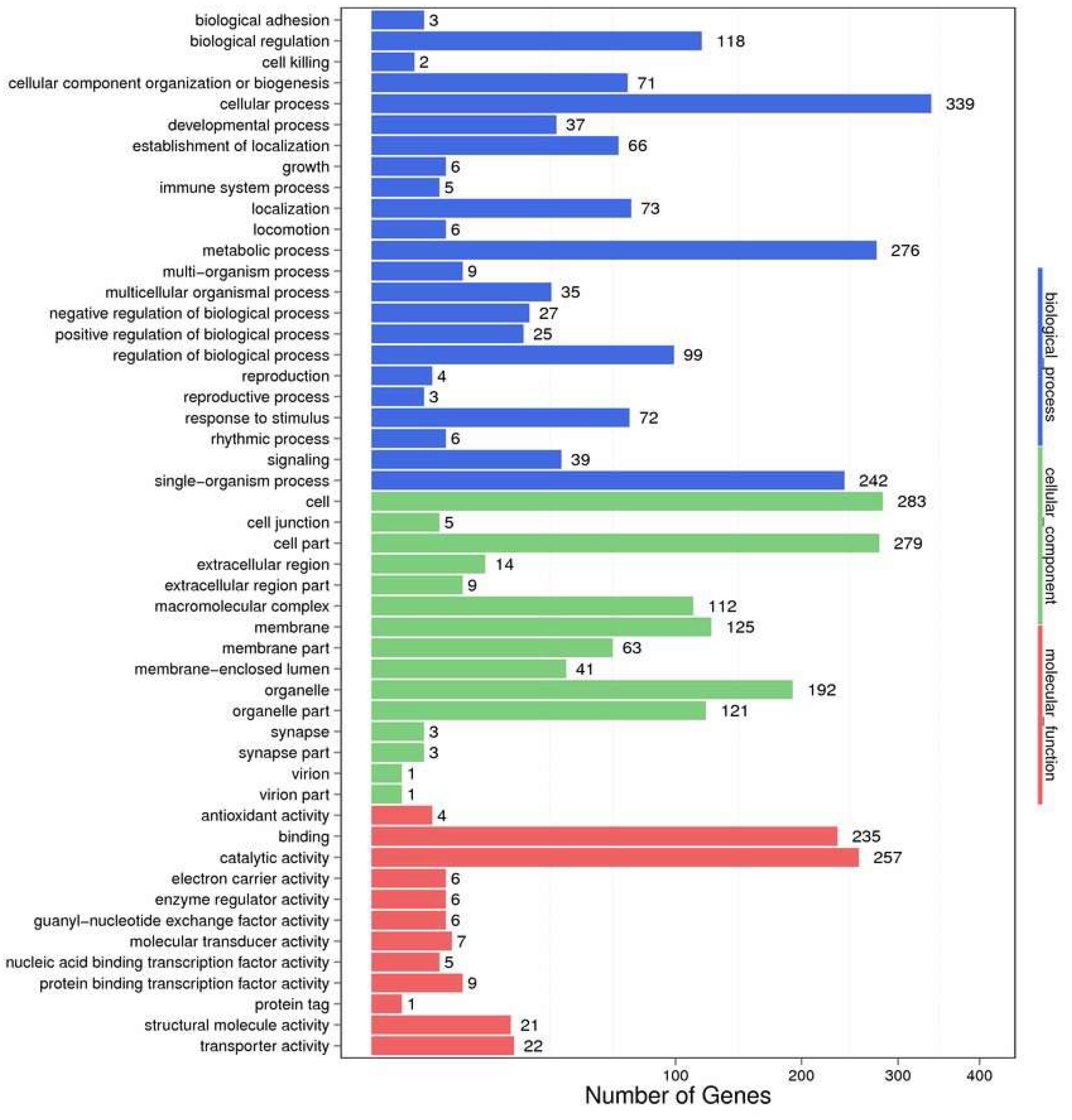
2.2. Functional Annotation and Species Distribution
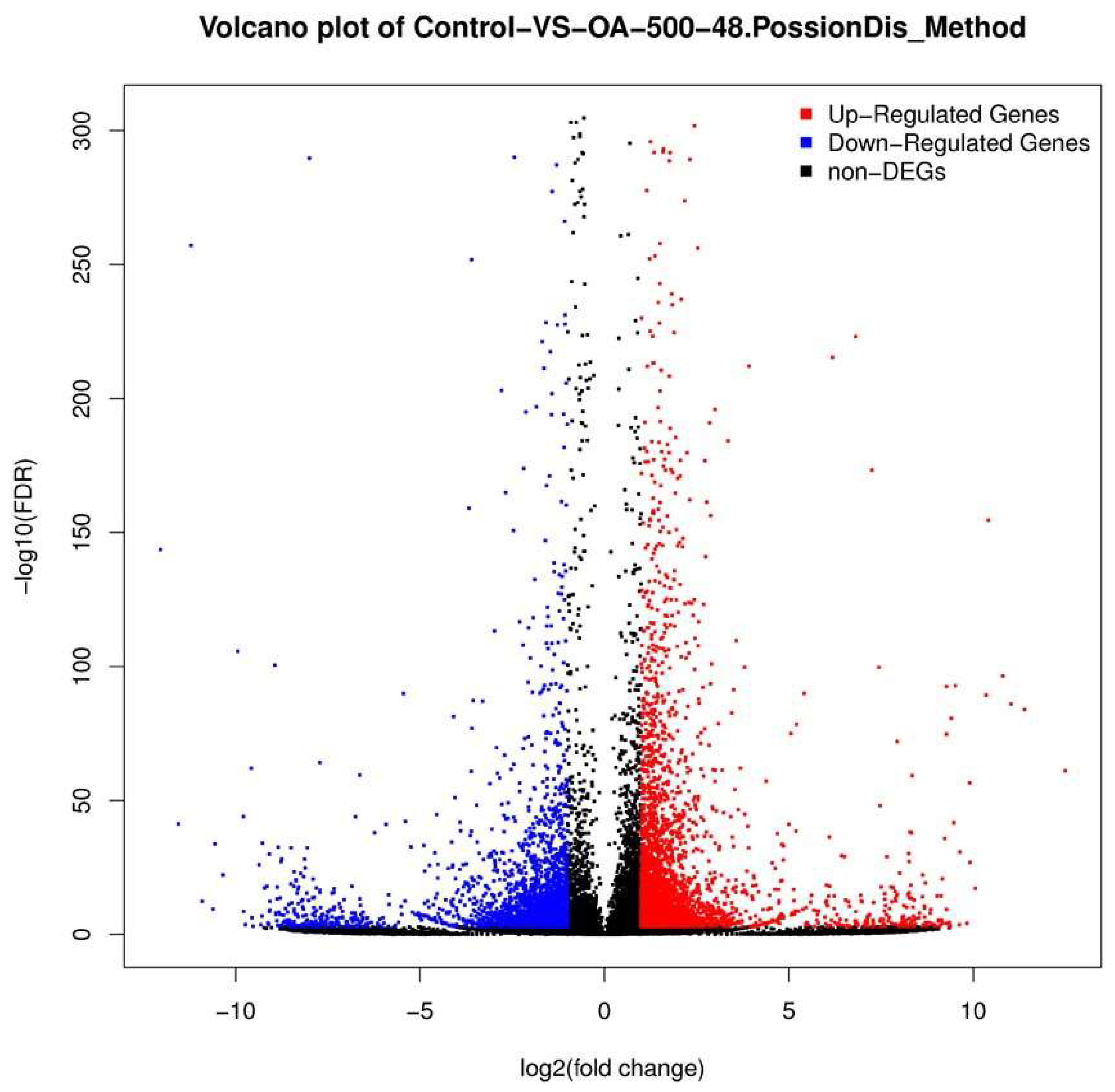
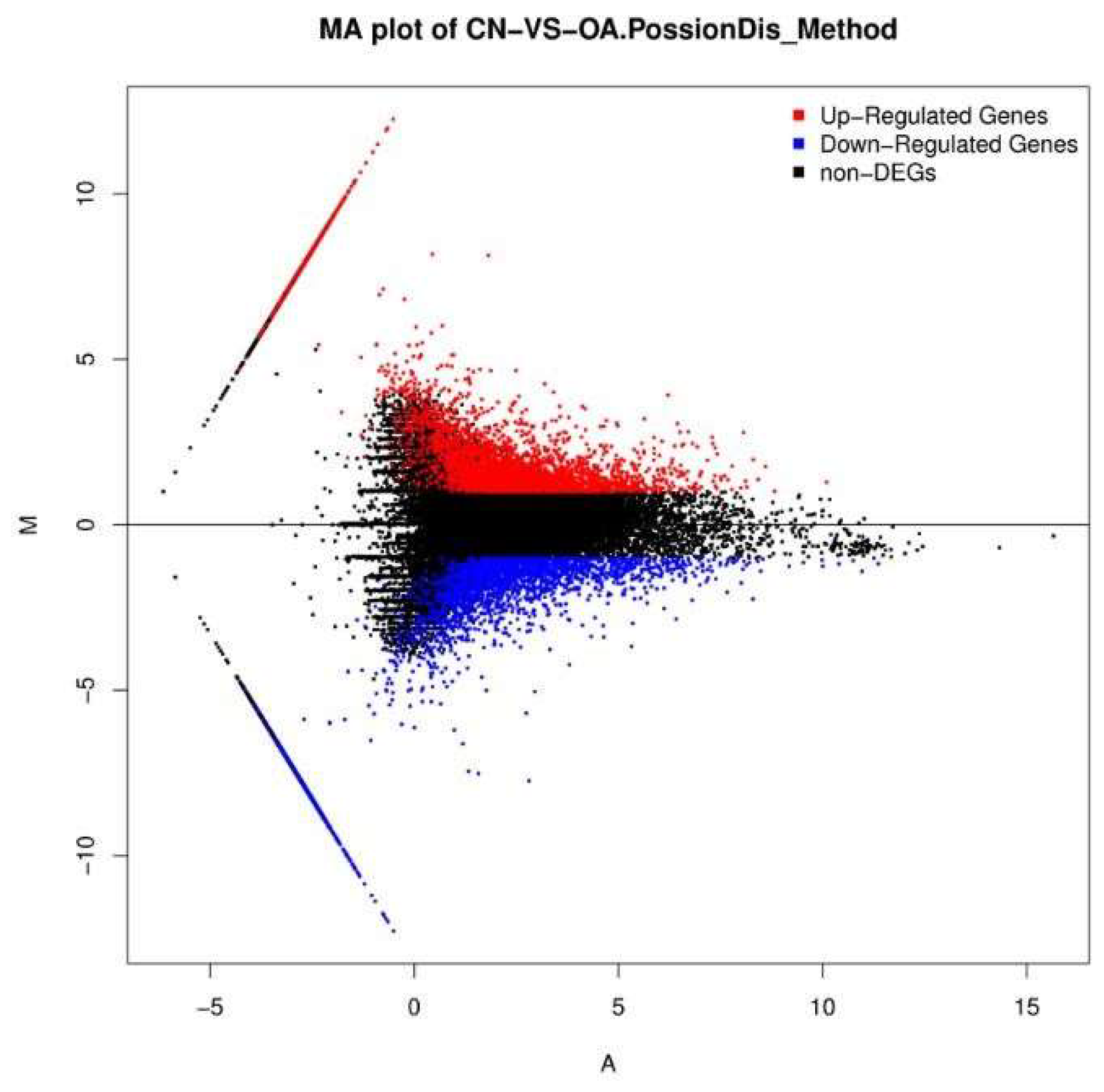
2.3. Differential Gene Expression Analysis
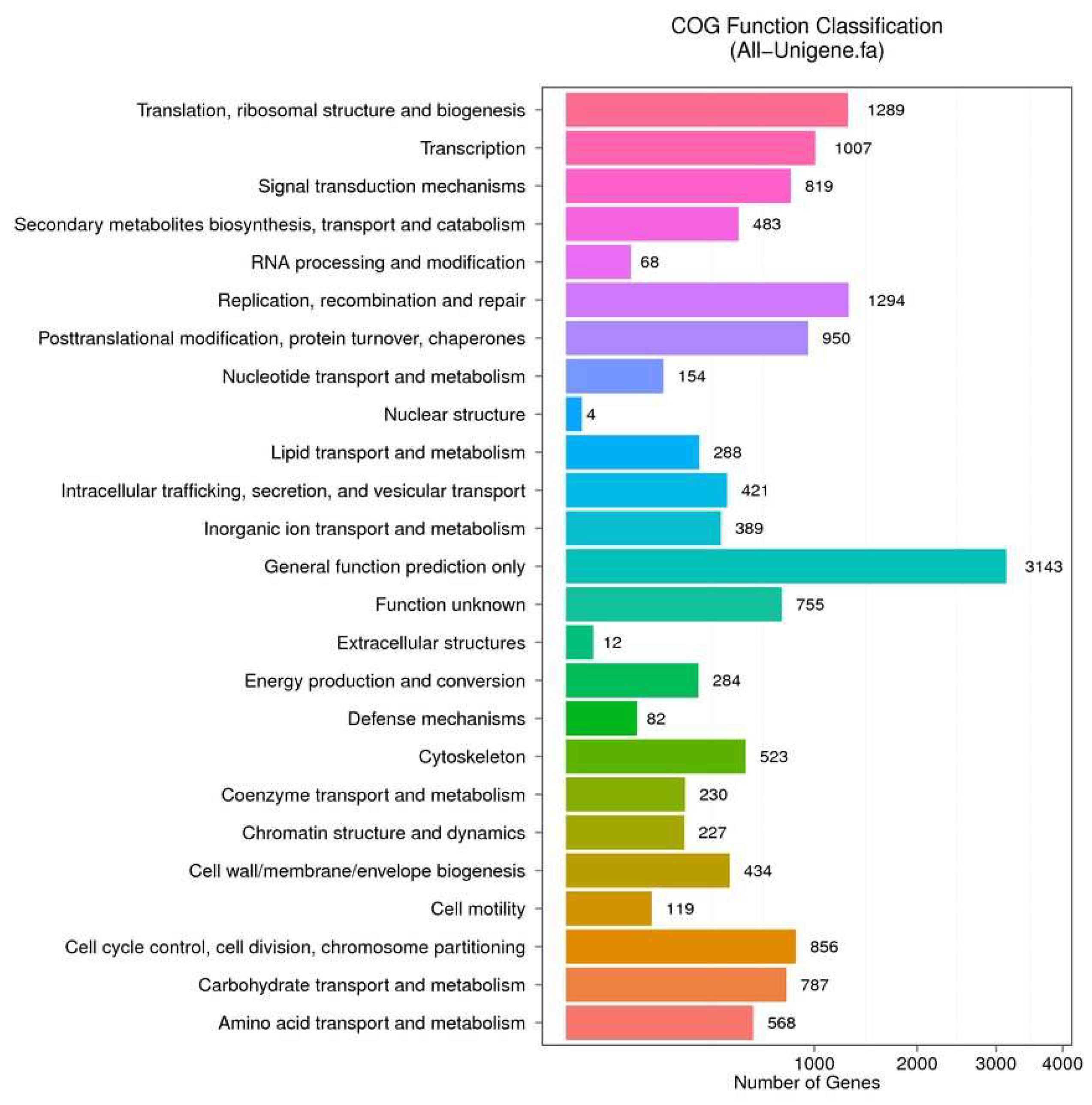
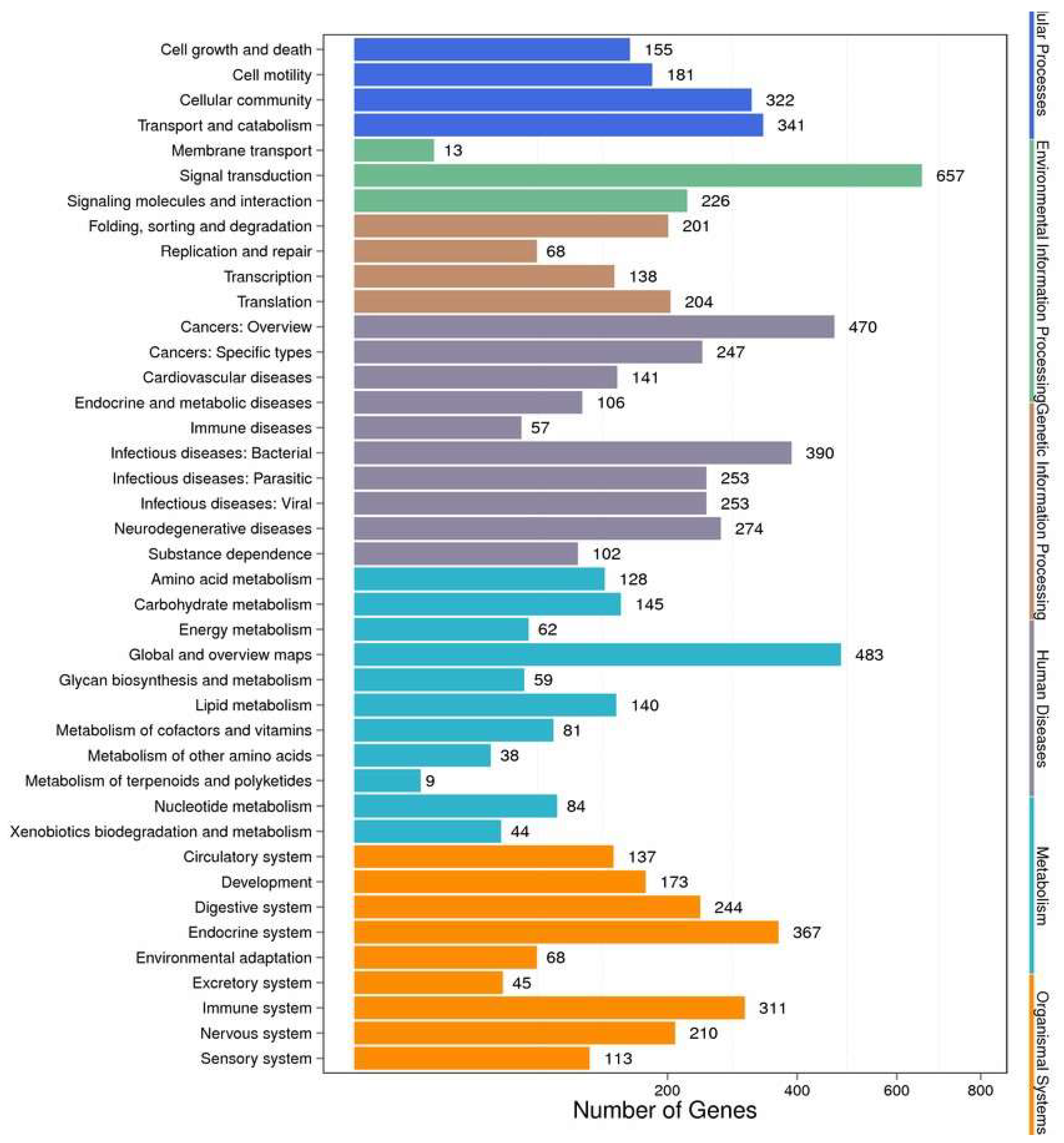
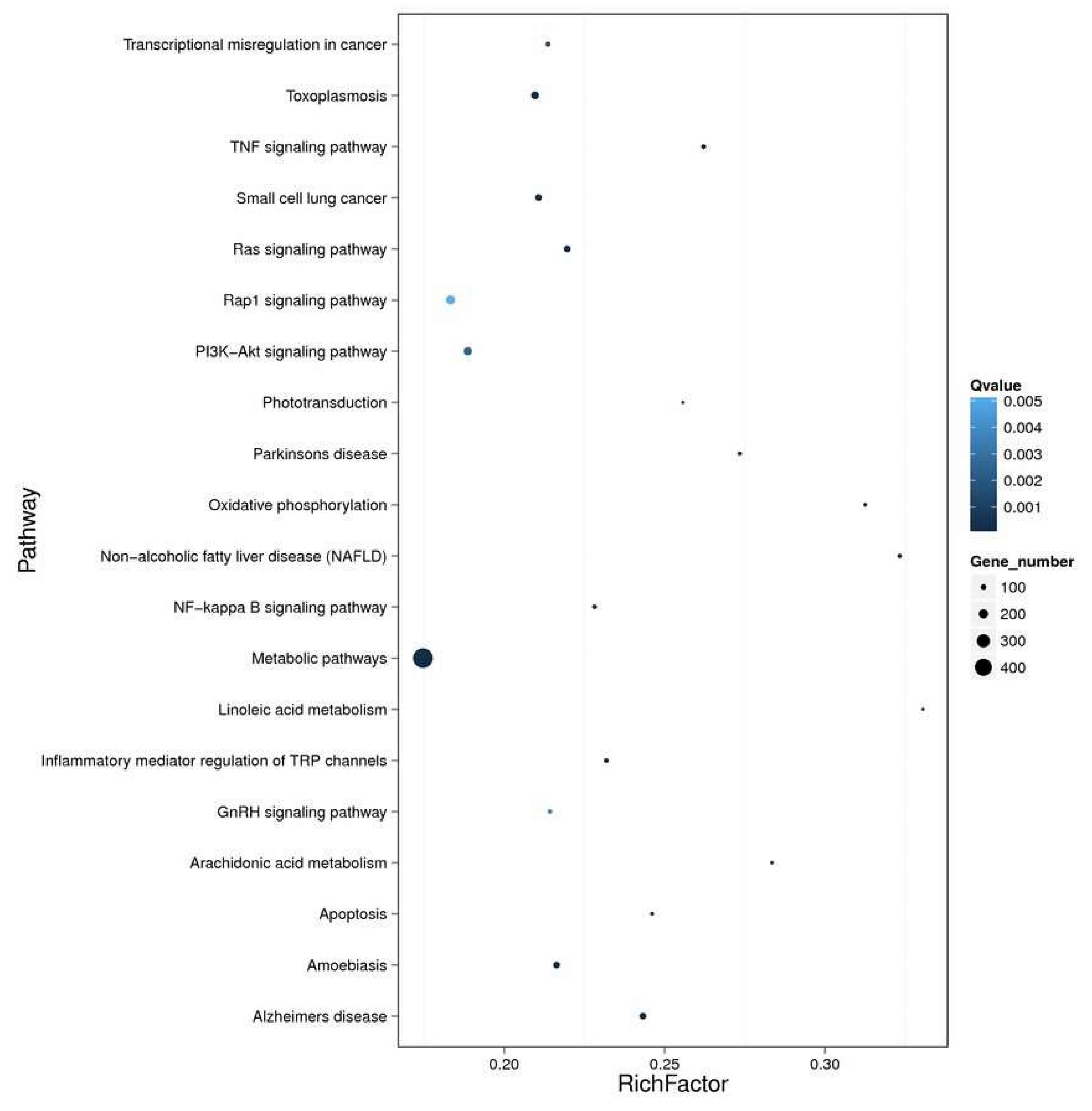
2.4. Enrichment and Pathway Analysis
2.5. Identification of Genes Related to OA-Induced Stress Response
3. Discussion
4. Materials and Methods
4.1. Maintenance of Scallops
4.2. Okadaic Acid Exposure and RNA Extraction
4.3. Library Preparation and Illumina Sequencing
4.4. De Novo Transcriptome Assembly
4.5. Gene Annotation and Analysis
4.6. Differential Expression Analysis
4.7. GO and KEGG Enrichment Analysis of Differentially Expressed Genes
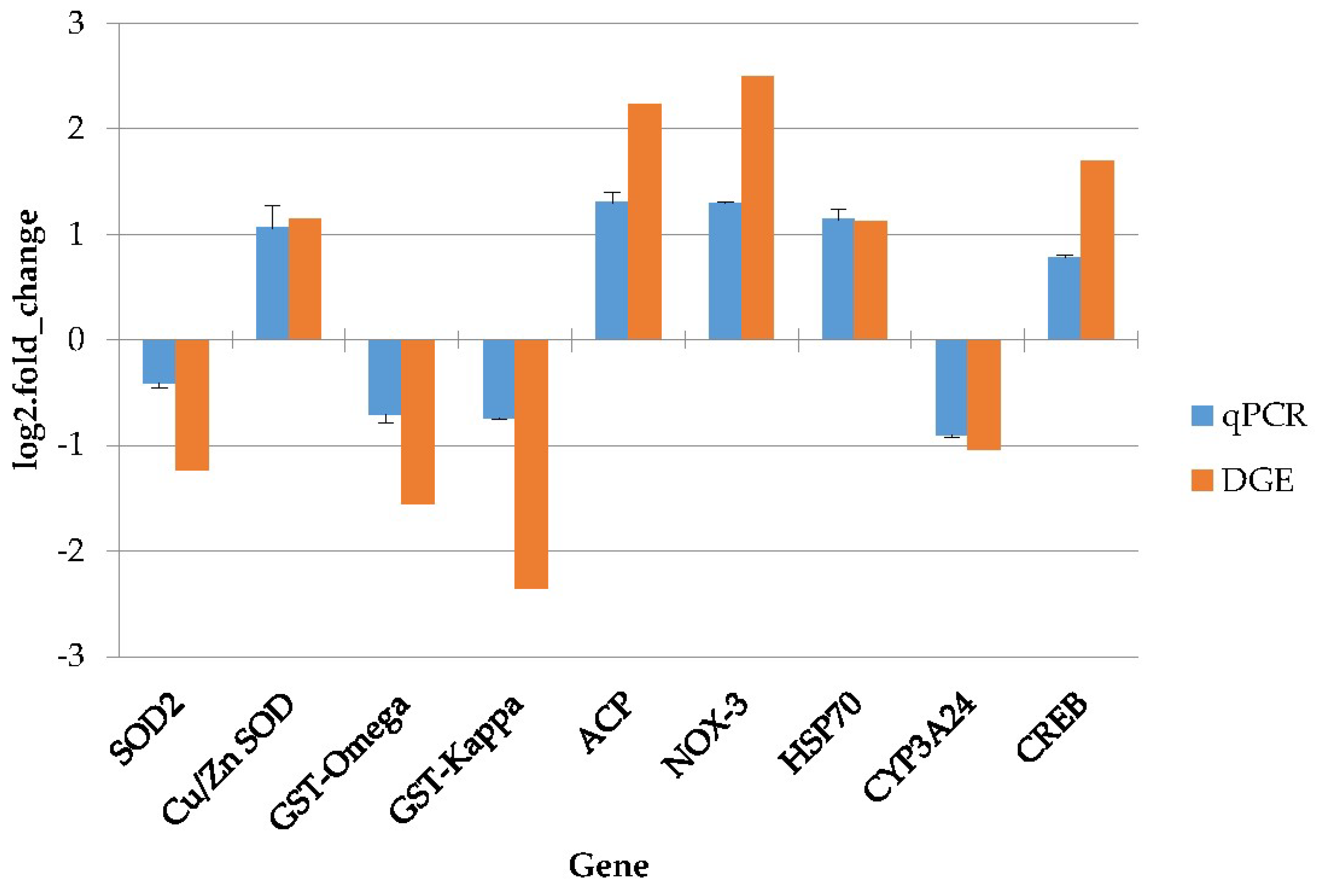
4.8. Quantitative Real-Time PCR Validation
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cai, Y.; Pan, L.; Hu, F.; Jin, Q.; Liu, T. Deep sequencing-based transcriptome profiling analysis of Chlamys farreri exposed to benzo [a] pyrene. Gene 2014, 551, 261–270. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, E.D.; Bowen, V.T.; Farrington, J.W.; Harvey, G.; Martin, J.H.; Parker, P.L.; Risebrough, R.W.; Robertson, W.; Schneider, E.; Gamble, E. The mussel watch. Environ. Conserv. 1978, 5, 101–125. [Google Scholar] [CrossRef]
- Hu, F.; Pan, L.; Cai, Y.; Liu, T.; Jin, Q. Deep sequencing of the scallop Chlamys farreri transcriptome response to tetrabromobisphenol A (TBBPA) stress. Mar. Genom. 2015, 19, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Pan, L.; Wang, J.; Yang, H.; Liu, D. Application of the biomarker responses in scallop (Chlamys farreri) to assess metals and PAHs pollution in Jiaozhou Bay, China. Mar. Environ. Res. 2012, 80, 38–45. [Google Scholar] [CrossRef] [PubMed]
- De Jesús Romero-Geraldo, R.; García-Lagunas, N.; Hernández-Saavedra, N.Y. Crassostrea gigas exposure to the dinoflagellate Prorocentrum lima: Histological and gene expression effects on the digestive gland. Mar. Environ. Res. 2016, 120, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Zingone, A.; Enevoldsen, H.O. The diversity of harmful algal blooms: A challenge for science and management. Ocean. Coast. Manag. 2000, 43, 725–748. [Google Scholar] [CrossRef]
- Anderson, D.M. Approaches to monitoring, control and management of harmful algal blooms (HABs). Ocean. Coast. Manag. 2009, 52, 342–347. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.; Giri, S.S.; Jun, J.W.; Kim, H.J.; Kim, S.W.; Yun, S.; Park, S.C. Effects of algal toxin okadaic acid on the non-specific immune and antioxidant response of bay scallop (Argopecten irradians). Fish. Shellfish Immunol. 2017, 65, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Glibert, P.M.; Anderson, D.M.; Gentien, P.; Granéli, E.; Sellner, K.G. The global, complex phenomena of harmful algal blooms. Oceanography 2005, 18, 136–147. [Google Scholar] [CrossRef]
- Basti, L.; Hégaret, H.; Shumway, S.E. Harmful algal blooms and shellfish. In Harmful Algal Blooms: A Compendium Desk Reference; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2018; pp. 135–190. [Google Scholar]
- Huang, L.; Zou, Y.; Weng, H.W.; Li, H.Y.; Liu, J.S.; Yang, W.D. Proteomic profile in Perna viridis after exposed to Prorocentrum lima, a dinoflagellate producing DSP toxins. Environ. Pollut. 2015, 196, 350–357. [Google Scholar] [CrossRef] [PubMed]
- Chi, C.; Giri, S.S.; Jun, J.W.; Kim, H.J.; Yun, S.; Kim, S.G.; Park, S.C. Marine Toxin Okadaic Acid Affects the Immune Function of Bay Scallop (Argopecten irradians). Molecules 2016, 21, 1108. [Google Scholar] [CrossRef] [PubMed]
- Espiña, B.; Louzao, M.; Cagide, E.; Alfonso, A.; Vieytes, M.R.; Yasumoto, T.; Botana, L.M. The methyl ester of okadaic acid is more potent than okadaic acid in disrupting the actin cytoskeleton and metabolism of primary cultured hepatocytes. Br. J. Pharmacol. 2010, 159, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Ulloa, V.; Fernandez-Tajes, J.; Aguiar-Pulido, V.; Prego-Faraldo, M.V.; Florez-Barros, F.; Sexto-Iglesias, A.; Mendez, J.; Eirin-Lopez, J.M. Unbiased high-throughput characterization of mussel transcriptomic responses to sublethal concentrations of the biotoxin okadaic acid. PeerJ 2015, 3, e1429. [Google Scholar] [CrossRef] [PubMed]
- Mouratidou, T.; Kaniou-Grigoriadou, I.; Samara, C.; Kouimtzis, T. Detection of the marine toxin okadaic acid in mussels during a diarrhetic shellfish poisoning (DSP) episode in Thermaikos Gulf, Greece, using biological, chemical and immunological methods. Sci. Total Environ. 2006, 366, 894–904. [Google Scholar] [CrossRef] [PubMed]
- Stonik, V.A.; Stonik, I.V. Toxins Produced by Marine Microorganisms: A Short Review. Mar. Freshw. Toxins 2016, 3–21. [Google Scholar] [CrossRef]
- Prego-Faraldo, M.V.; Valdiglesias, V.; Laffon, B.; Eirín-López, J.M.; Méndez, J. In vitro analysis of early genotoxic and cytotoxic effects of okadaic acid in different cell types of the mussel Mytilus galloprovincialis. J. Toxicol. Environ. Health Part A 2015, 78, 814–824. [Google Scholar] [CrossRef] [PubMed]
- Mello, D.F.; Proença, L.A. d. O.; Barracco, M.A. Comparative study of various immuneparameters in three bivalve species during a natural bloom of Dinophysis acuminata in Santa Catarina Island, Brazil. Toxins 2010, 2, 1166–1178. [Google Scholar] [CrossRef] [PubMed]
- Burgos-Aceves, M.A.; Faggio, C. An approach to the study of the immunity functions of bivalve haemocytes: Physiology and molecular aspects. Fish Shellfish Immunol. 2017, 67, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Prado-Alvarez, M.; Flórez-Barrós, F.; Méndez, J.; Fernandez-Tajes, J. Effect of okadaic acid on carpet shell clam (Ruditapes decussatus) haemocytes by in vitro exposure and harmful algal bloom simulation assays. Cell. Biol. Toxicol. 2013, 29, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Fabioux, C.; Sulistiyani, Y.; Haberkorn, H.; Hégaret, H.; Amzil, Z.; Soudant, P. Exposure to toxic Alexandrium minutum activates the detoxifying and antioxidant systems in gills of the oyster Crassostrea gigas. Harmful Algae 2015, 48, 55–62. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.S.; Lee, S.G.; Lee, C.K.; Kim, H.G.; Jung, J. Reactive oxygen species as causative agents in the ichthyotoxicity of the red tide dinoflagellate Cochlodinium polykrikoides. J. Plankton Res. 1999, 21, 2105–2115. [Google Scholar] [CrossRef]
- Flores, H.S.; Wikfors, G.H.; Dam, H.G. Reactive oxygen species are linked to the toxicity of the dinoflagellate Alexandrium spp. to protists. Aquat. Microb. Ecol. 2012, 66, 199–209. [Google Scholar] [CrossRef]
- Reuter, J.A.; Spacek, D.V.; Snyder, M.P. High-throughput sequencing technologies. Mol. Cell 2015, 58, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Huang, Q.; Dong, S.; Fang, C.; Wu, X.; Ye, T.; Lin, Y. Deep sequencing-based transcriptome profiling analysis of Oryzias melastigma exposed to PFOS. Aquat. Toxicol. 2012, 120, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Yu, H.; Kong, L.; Li, Q. Transcriptomic responses to salinity stress in the Pacific. oyster Crassostrea gigas. PLoS ONE 2012, 7, e46244. [Google Scholar] [CrossRef]
- Fu, X.; Sun, Y.; Wang, J.; Xing, Q.; Zou, J.; Li, R.; Wang, Z.; Wang, S.; Hu, X.; Zhang, L. Sequencing-based gene network analysis provides a core set of gene resource for understanding thermal adaptation in Zhikong scallop Chlamys farreri. Mol. Ecol. Resour. 2014, 14, 184–198. [Google Scholar] [CrossRef] [PubMed]
- Philipp, E.E.; Kraemer, L.; Melzner, F.; Poustka, A.J.; Thieme, S.; Findeisen, U.; Schreiber, S.; Rosenstiel, P. Massively parallel RNA sequencing identifies a complex immune gene repertoire in the lophotrochozoan Mytilus edulis. PLoS ONE 2012, 7, e33091. [Google Scholar] [CrossRef] [PubMed]
- Svensson, S.; Särngren, A.; Förlin, L. Mussel blood cells, resistant to the cytotoxic effects of okadaic acid, do not express cell membrane p-glycoprotein activity (multixenobiotic resistance). Aquat. Toxicol. 2003, 65, 27–37. [Google Scholar] [CrossRef]
- Ravindran, J.; Gupta, N.; Agrawal, M.; Bhaskar, A.B.; Rao, P.L. Modulation of ROS/MAPK signaling pathways by okadaic acid leads to cell death via, mitochondrial mediated caspase-dependent mechanism. Apoptosis 2011, 16, 145–161. [Google Scholar] [CrossRef] [PubMed]
- Von Zezschwitz, C.; Vorwerk, H.; Tergau, F.; Steinfelder, H.J. Apoptosis induction by inhibitors of Ser/Thr phosphatases 1 and 2A is associated with transglutaminase activation in two different human epithelial tumour lines. FEBS Lett. 1997, 413, 147–151. [Google Scholar] [CrossRef]
- Soliño, L.; Sureda, F.X.; Diogène, J. Evaluation of okadaic acid, dinophysistoxin-1 and dinophysistoxin-2 toxicity on Neuro-2a, NG108-15 and MCF-7 cell lines. Toxicol. Vitro 2015, 29, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Marioni, J.C.; Mason, C.E.; Mane, S.M.; Stephens, M.; Gilad, Y. RNA-seq: An assessment of technical reproducibility and comparison with gene expression arrays. Genome Res. 2008, 18, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Simon, P. Q-Gene: Processing quantitative real-time RT–PCR data. Bioinformatics 2003, 19, 1439–1440. [Google Scholar] [CrossRef] [PubMed]
- Clayton, M.E.; Steinmann, R.; Fent, K. Different expression patterns of heat shock proteins hsp 60 and hsp 70 in zebra mussels (Dreissena polymorpha) exposed to copper and tributyltin. Aquat. Toxicol. 2000, 47, 213–226. [Google Scholar] [CrossRef]
- Boutet, I.; Tanguy, A.; Moraga, D. Response of the Pacific oyster Crassostrea gigas to hydrocarbon contamination under experimental conditions. Gene 2004, 329, 147–157. [Google Scholar] [CrossRef] [PubMed]
- Faggio, C.; Pagano, M.; Alampi, R.; Vazzana, I.; Felice, M.R. Cytotoxicity, haemolymphatic parameters, and oxidative stress following exposure to sub-lethal concentrations of quaternium-15 in Mytilus galloprovincialis. Aquat. Toxicol. 2016, 180, 258–265. [Google Scholar] [CrossRef] [PubMed]
- Piano, A.; Valbonesi, P.; Fabbri, E. Expression of cytoprotective proteins, heat shock protein 70 and metallothioneins, in tissues of Ostrea edulis exposed to heat andheavy metals. Cell. Stress Chaperones 2004, 9, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Anzenbacher, P.; Anzenbacherová, E. Cytochromes P450 and metabolism of xenobiotics. Cell. Mol. Life Sci. 2001, 58, 737–747. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Pan, L.; Cai, Y.; Li, Z.; Miao, J.J. Response of detoxification gene mRNA expression and selection of molecular biomarkers in the clam Ruditapes philippinarum exposed to benzo[a]pyrene. Environ. Pollut. 2014, 189, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.; An, T.; Rein, K.S. The algal hepatoxoxin okadaic acid is a substrate for human cytochromes CYP3A4 and CYP3A5. Toxicon 2010, 55, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Wang, J.; Chen, W.C.; Li, H.Y.; Liu, J.S.; Jiang, T.; Yang, W.D. P-glycoprotein expression in Perna viridis after exposure to Prorocentrum lima, a dinoflagellate producing DSP toxins. Fish Shellfish Immunol. 2014, 39, 254–262. [Google Scholar] [CrossRef] [PubMed]
- Donaghy, L.; Hong, H.K.; Jauzein, C.; Choi, K.S. The known and unknown sources of reactive oxygen and nitrogen species in haemocytes of marine bivalve molluscs. Fish Shellfish Immunol. 2015, 42, 91–97. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Pan, L.; Li, Z.; Cai, Y.; Miao, J. Metabolites analysis, metabolic enzyme activities and bioaccumulation in the clam Ruditapes philippinarum exposed to benzo [a] pyrene. Ecotoxicol. Environ. Saf. 2014, 107, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Prego-Faraldo, M.; Vieira, L.; Eirin-Lopez, J.; Méndez, J.; Guilhermino, L. Transcriptional and biochemical analysis of antioxidant enzymes in the mussel Mytilus galloprovincialis during experimental exposures to the toxic dinoflagellate Prorocentrum lima. Mar. Environ. Res. 2017, 129, 304–315. [Google Scholar] [CrossRef] [PubMed]
- Jin, Q.; Pan, L.; Liu, T.; Hu, F. RNA-seq based on transcriptome reveals differ genetic expressing in Chlamys farreri exposed to carcinogen PAHs. Environ. Toxicol. Pharmacol. 2015, 39, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Regoli, F.; Benedetti, M.; Giuliani, M.E. Antioxidant defenses and acquisition of tolerance to chemical stress. Toler. Environ. Contam. 2011, 153–173. [Google Scholar]
- Regoli, F.; Giuliani, M.E.; Benedetti, M.; Arukwe, A. Molecular and biochemical biomarkers in environmental monitoring: A comparison of biotransformation and antioxidant defense systems in multiple tissues. Aquat. Toxicol. 2011, 105, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Manfrin, C.; Dreos, R.; Battistella, S.; Beran, A.; Gerdol, M.; Varotto, L.; Lanfranchi, G.; Venier, P.; Pallavicini, A. Mediterranean mussel gene expression profile induced by okadaic acid exposure. Environ. Sci. Technol. 2010, 44, 8276–8283. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.Y.; Yang, H.S.; Delaporte, M.; Zhao, S.J.; Xing, K. Immune responses of the scallop Chlamys farreri after air exposure to different temperatures. J. Exp. Mar. Biol. Ecol. 2007, 345, 52–60. [Google Scholar] [CrossRef]
- Zhou, J.; Xiong, Q.; Chen, H.; Yang, C.; Fan, Y. Identification of the spinal expression profile of non-coding RNAs involved in neuropathic pain following spared nerve injury by sequence analysis. Front. Mol. Neurosci. 2017, 10, 91. [Google Scholar] [CrossRef] [PubMed]
- Cock, P.J.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S.; García-Gómez, J.M.; Terol, J.; Talón, M.; Robles, M. Blast2GO: A universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 2005, 21, 3674–3676. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [PubMed]
- Audic, S.; Claverie, J.-M. The significance of digital gene expression profiles. Genome Res. 1997, 7, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT–PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef] [PubMed]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Total Number | Total Length | Mean Length | N50 | N70 | N90 | GC(%) |
|---|---|---|---|---|---|---|---|
| Control | 51,465 | 41,105,722 | 798 | 1411 | 704 | 302 | 39.48 |
| OA-treated | 49,453 | 43,129,157 | 872 | 1646 | 803 | 318 | 39.63 |
| All-unigene | 55,876 | 53,465,429 | 956 | 1840 | 960 | 345 | 39.42 |
| Function | Transcript | Log2 (Fold Change) (RNAseq) | Regulation |
|---|---|---|---|
| Immune system | C-type lectin superfamily 17 member A | −4.255 | Down |
| C-type lectin domain family 4 member E | −3.507 | Down | |
| Complement C1q tumor necrosis factor-related protein 2 | −4.791 | Down | |
| Fibrinogen C domain-containing protein 1 | −2.100 | Down | |
| Toll-like receptor 4 | 2.880 | Up | |
| Toll-like receptor 13 | 1.347 | Up | |
| Acid phosphatase | 2.238 | Up | |
| NADPH oxidase 3 | 2.493 | Up | |
| Detoxification | ATP-binding cassette, subfamily C, member 1 | 1.773 | Up |
| ATP-binding cassette sub-family B member 10 | 1.165 | Up | |
| ATP-binding cassette, sub-family C member 5 | 1.280 | Up | |
| Cyclic AMP-responsive element-binding protein | 1.953 | Up | |
| Nuclear factor erythroid 2-related factor 2 | 1.231 | Up | |
| NADPH2:quinone reductase | 1.677 | Up | |
| Cytochrome P450 3A80 | 1.207 | Up | |
| Cytochrome P450 3A64 | 1.783 | Up | |
| Cytochrome P450 1A5 | −1.686 | Down | |
| Cytochrome P450 3A24 | −2.315 | Down | |
| Superoxide dismutase Cu-Zn family | 1.139 | Up | |
| Superoxide dismutase 2 | −1.126 | Down | |
| Glutathione S-transferase 1 | −1.552 | Down | |
| Glutathione S-transferase 2 | −2.511 | Down | |
| Glutathione S-transferase omega | −1.775 | Down | |
| Glutathione S-transferase theta-1 | −1.254 | Down | |
| Glutathione S-transferase A | −1.218 | Down | |
| Glutathione S-transferase kappa | −2.356 | Down |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chi, C.; Giri, S.S.; Jun, J.W.; Kim, S.W.; Kim, H.J.; Kang, J.W.; Park, S.C. Detoxification- and Immune-Related Transcriptomic Analysis of Gills from Bay Scallops (Argopecten irradians) in Response to Algal Toxin Okadaic Acid. Toxins 2018, 10, 308. https://doi.org/10.3390/toxins10080308
Chi C, Giri SS, Jun JW, Kim SW, Kim HJ, Kang JW, Park SC. Detoxification- and Immune-Related Transcriptomic Analysis of Gills from Bay Scallops (Argopecten irradians) in Response to Algal Toxin Okadaic Acid. Toxins. 2018; 10(8):308. https://doi.org/10.3390/toxins10080308
Chicago/Turabian StyleChi, Cheng, Sib Sankar Giri, Jin Woo Jun, Sang Wha Kim, Hyoun Joong Kim, Jeong Woo Kang, and Se Chang Park. 2018. "Detoxification- and Immune-Related Transcriptomic Analysis of Gills from Bay Scallops (Argopecten irradians) in Response to Algal Toxin Okadaic Acid" Toxins 10, no. 8: 308. https://doi.org/10.3390/toxins10080308
APA StyleChi, C., Giri, S. S., Jun, J. W., Kim, S. W., Kim, H. J., Kang, J. W., & Park, S. C. (2018). Detoxification- and Immune-Related Transcriptomic Analysis of Gills from Bay Scallops (Argopecten irradians) in Response to Algal Toxin Okadaic Acid. Toxins, 10(8), 308. https://doi.org/10.3390/toxins10080308