Impact of Altered Intestinal Microbiota on Chronic Kidney Disease Progression


 ,
,  and
and 
Abstract
1. CKD and CKD Progression
2. The Intestinal Microbiota in CKD
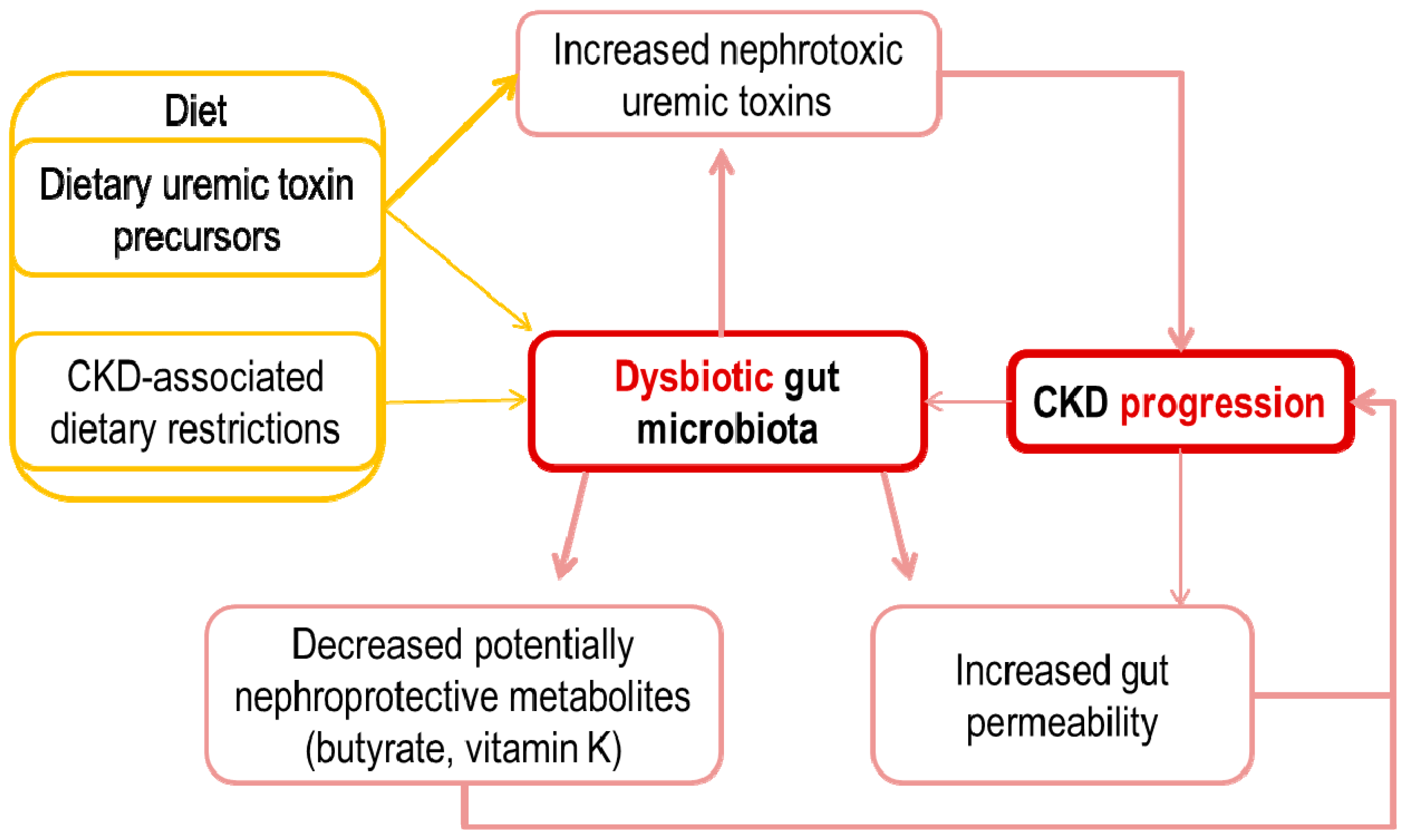
3. Potential Pathogenic Links between an Altered Intestinal Microbiota and CKD Progression
4. Increased Gut Permeability and Inflammation
5. Metabolite Overload: Microbiota-Generated Nephrotoxins
5.1. pCS and pCG
5.2. IS and IAA
5.3. TMAO
6. The Missing Ones: Microbiota-Generated Nephroprotective Factors
6.1. Vitamin K
6.2. Butyrate
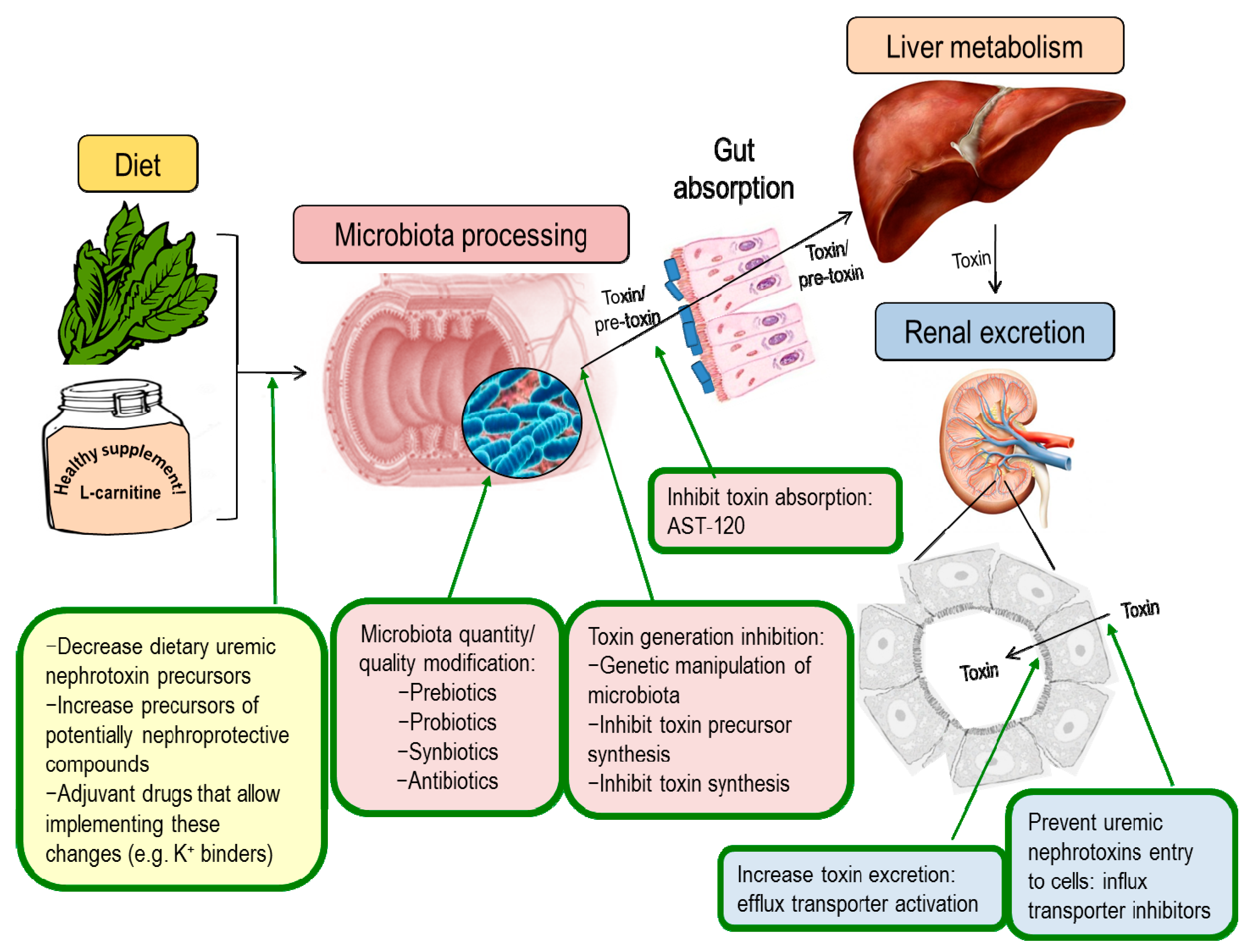
7. Nephroprotection Interventional Studies Targeting the Intestinal Microbiota or Microbiota-Derived Toxins
7.1. Modulation of Uremic Toxins Synthesis
7.2. Modulation of Renal Transporters
7.3. Adsorbents
7.4. Modulation of Microbiota Composition
8. The Way Forward
Author Contributions
Funding
Conflicts of Interest
References
- Sanchez-Niño, M.D.; Sanz, A.B.; Ramos, A.M.; Ruiz-Ortega, M.; Ortiz, A. Translational science in chronic kidney disease. Clin. Sci. 2017, 131, 1617–1629. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, A.; Covic, A.; Fliser, D.; Fouque, D.; Goldsmith, D.; Kanbay, M.; Mallamaci, F.; Massy, Z.A.; Rossignol, P.; Vanholder, R.; et al. Epidemiology, contributors to and clinical trials of mortality risk in chronic kidney failure. Lancet 2014, 383, 1831–1843. [Google Scholar] [CrossRef]
- Vanholder, R.; Fouque, D.; Glorieux, G.; Heine, G.H.; Kanbay, M.; Mallamaci, F.; Massy, Z.A.; Ortiz, A.; Rossignol, P.; Wiecek, A.; et al. Clinical management of the uraemic syndrome in chronic kidney disease. Lancet Diabetes Endocrinol. 2016, 4, 360–373. [Google Scholar] [CrossRef]
- Fernandez-Fernandez, B.; Izquierdo, M.C.; Valiño-Rivas, L.; Nastou, D.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Albumin downregulates Klotho in tubular cells. Nephrol. Dial. Transplant. 2018. [Google Scholar] [CrossRef] [PubMed]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A. European uremic toxin work group normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Andres, O.; Sanchez-Niño, M.D.; Moreno, J.A.; Ruiz-Ortega, M.; Ramos, A.M.; Sanz, A.B.; Ortiz, A. Downregulation of kidney protective factors by inflammation: Role of transcription factors and epigenetic mechanisms. Am. J. Physiol. Renal Physiol. 2016, 311, F1329–F1340. [Google Scholar] [CrossRef] [PubMed]
- Zoccali, C.; Vanholder, R.; Massy, Z.A.; Ortiz, A.; Sarafidis, P.; Dekker, F.W.; Fliser, D.; Fouque, D.; Heine, G.H.; Jager, K.J.; et al. The systemic nature of CKD. Nat. Rev. Nephrol. 2017, 13, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Glorieux, G. The intestine and the kidneys: A bad marriage can be hazardous. Clin. Kidney J. 2015, 8, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, A.; Massy, Z.A.; Meijers, B.; Evenepoel, P.; Vanholder, R.; Raj, D.S. Role of the gut microbiome in uremia: A potential therapeutic target. Am. J. Kidney Dis. 2016, 67, 483–498. [Google Scholar] [CrossRef] [PubMed]
- Nallu, A.; Sharma, S.; Ramezani, A.; Muralidharan, J.; Raj, D. Gut microbiome in chronic kidney disease: Challenges and opportunities. Transl. Res. 2017, 179, 24–37. [Google Scholar] [CrossRef] [PubMed]
- Packham, D.K.; Rasmussen, H.S.; Lavin, P.T.; El-Shahawy, M.A.; Roger, S.D.; Block, G.; Qunibi, W.; Pergola, P.; Singh, B. Sodium zirconium cyclosilicate in hyperkalemia. N. Engl. J. Med. 2015, 372, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Weir, M.R.; Bakris, G.L.; Bushinsky, D.A.; Mayo, M.R.; Garza, D.; Stasiv, Y.; Wittes, J.; Christ-Schmidt, H.; Berman, L.; Pitt, B.; et al. Patiromer in patients with kidney disease and hyperkalemia receiving RAAS inhibitors. N. Engl. J. Med. 2015, 372, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Prado, R.; Esteras, R.; Perez-Gomez, M.V.; Gracia-Iguacel, C.; Gonzalez-Parra, E.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Nutrients turned into toxins: Microbiota modulation of nutrient properties in chronic kidney disease. Nutrients 2017, 9, 489. [Google Scholar] [CrossRef] [PubMed]
- Meert, N.; Schepers, E.; Glorieux, G.; Van Landschoot, M.; Goeman, J.L.; Waterloos, M.-A.; Dhondt, A.; Van der Eycken, J.; Vanholder, R. Novel method for simultaneous determination of p-cresylsulphate and p-cresylglucuronide: Clinical data and pathophysiological implications. Nephrol. Dial. Transplant. 2012, 27, 2388–2396. [Google Scholar] [CrossRef] [PubMed]
- Poesen, R.; Evenepoel, P.; de Loor, H.; Kuypers, D.; Augustijns, P.; Meijers, B. Metabolism, protein binding and renal clearance of microbiota-derived p-cresol in patients with CKD. Clin. J. Am. Soc. Nephrol. 2016, 11, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Wu, I.-W.; Hsu, K.-H.; Lee, C.-C.; Sun, C.-Y.; Hsu, H.-J.; Tsai, C.-J.; Tzen, C.-Y.; Wang, Y.-C.; Lin, C.-Y.; Wu, M.-S. p-Cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrol. Dial. Transplant. 2011, 26, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-J.; Wu, V.; Wu, P.-C.; Wu, C.-J. Meta-analysis of the associations of p-cresyl sulfate (PCS) and indoxyl sulfate (IS) with cardiovascular events and all-cause mortality in patients with chronic renal failure. PLoS ONE 2015, 10, e0132589. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: a systematic review. J. Am. Soc. Nephrol. 2014, 25, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Poveda, J.; Sanchez-Niño, M.D.; Glorieux, G.; Sanz, A.B.; Egido, J.; Vanholder, R.; Ortiz, A. p-Cresyl sulphate has pro-inflammatory and cytotoxic actions on human proximal tubular epithelial cells. Nephrol. Dial. Transplant. 2014, 29, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Niño, M.D.; Fernandez-Fernandez, B.; Perez-Gomez, M.V.; Poveda, J.; Sanz, A.B.; Cannata-Ortiz, P.; Ruiz-Ortega, M.; Egido, J.; Selgas, R.; Ortiz, A. Albumin-induced apoptosis of tubular cells is modulated by BASP1. Cell Death Dis. 2015, 6, e1644. [Google Scholar] [CrossRef] [PubMed]
- Bolati, D.; Shimizu, H.; Higashiyama, Y.; Nishijima, F.; Niwa, T. Indoxyl sulfate induces epithelial-to-mesenchymal transition in rat kidneys and human proximal tubular cells. Am. J. Nephrol. 2011, 34, 318–323. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-Y.; Hsu, H.-H.; Wu, M.-S. p-Cresol sulfate and indoxyl sulfate induce similar cellular inflammatory gene expressions in cultured proximal renal tubular cells. Nephrol. Dial. Transplant. 2013, 28, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, A.; Takeda, M.; Tojo, A.; Sekine, T.; Cha, S.H.; Khamdang, S.; Takayama, F.; Aoyama, I.; Nakamura, S.; Endou, H.; et al. Role of organic anion transporters in the tubular transport of indoxyl sulfate and the induction of its nephrotoxicity. J. Am. Soc. Nephrol. 2002, 13, 1711–1720. [Google Scholar] [CrossRef] [PubMed]
- Niwa, T.; Ise, M. Indoxyl sulfate, a circulating uremic toxin, stimulates the progression of glomerular sclerosis. J. Lab. Clin. Med. 1994, 124, 96–104. [Google Scholar] [PubMed]
- Niwa, T.; Ise, M.; Miyazaki, T. Progression of glomerular sclerosis in experimental uremic rats by administration of indole, a precursor of indoxyl sulfate. Am. J. Nephrol. 1994, 14, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, T.; Ise, M.; Seo, H.; Niwa, T. Indoxyl sulfate increases the gene expressions of TGF-β1, TIMP-1 and pro-α1(I) collagen in uremic rat kidneys. Kidney Int. Suppl. 1997, 62, S15–S22. [Google Scholar] [PubMed]
- Edamatsu, T.; Fujieda, A.; Ezawa, A.; Itoh, Y. Classification of five uremic solutes according to their effects on renal tubular cells. Int. J. Nephrol. 2014, 2014, 512178. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Hayashi, H.; Watanabe, M.; Ueda, K.; Yamato, H.; Yoshioka, T.; Motojima, M. Uremic toxins overload accelerates renal damage in a rat model of chronic renal failure. Nephron. Exp. Nephrol. 2003, 95, e111–e118. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.B.; Morse, B.L.; Djurdjev, O.; Tang, M.; Muirhead, N.; Barrett, B.; Holmes, D.T.; Madore, F.; Clase, C.M.; Rigatto, C.; et al. Advanced chronic kidney disease populations have elevated trimethylamine N-oxide levels associated with increased cardiovascular events. Kidney Int. 2016, 89, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Muir, J.G.; Lu, Z.X.; Young, G.P.; Cameron-Smith, D.; Collier, G.R.; O’Dea, K. Resistant starch in the diet increases breath hydrogen and serum acetate in human subjects. Am. J. Clin. Nutr. 1995, 61, 792–799. [Google Scholar] [CrossRef] [PubMed]
- Wolever, T.M.; Josse, R.G.; Leiter, L.A.; Chiasson, J.L. Time of day and glucose tolerance status affect serum short-chain fatty acid concentrations in humans. Metabolism. 1997, 46, 805–811. [Google Scholar] [CrossRef]
- Pryde, S.E.; Duncan, S.H.; Hold, G.L.; Stewart, C.S.; Flint, H.J. The microbiology of butyrate formation in the human colon. FEMS Microbiol. Lett. 2002, 217, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Guo, H.-L.; Deng, X.; Zhu, T.-T.; Xiong, J.-F.; Xu, Y.-H.; Xu, Y. Short-chain fatty acids inhibit oxidative stress and inflammation in mesangial cells induced by high glucose and lipopolysaccharide. Exp. Clin. Endocrinol. Diabetes 2017, 125, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, M.; Mikami, D.; Kimura, H.; Kamiyama, K.; Morikawa, Y.; Yokoi, S.; Kasuno, K.; Takahashi, N.; Taniguchi, T.; Iwano, M. Short-chain fatty acids, GPR41 and GPR43 ligands, inhibit TNF-α-induced MCP-1 expression by modulating p38 and JNK signaling pathways in human renal cortical epithelial cells. Biochem. Biophys. Res. Commun. 2017, 486, 499–505. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Zhang, B.; Hong, X.; Zhang, X.; Kong, X. Histone deacetylase inhibitor, sodium butyrate, attenuates gentamicin-induced nephrotoxicity by increasing prohibitin protein expression in rats. Eur. J. Pharmacol. 2013, 707, 147–154. [Google Scholar] [CrossRef] [PubMed]
- Machado, R.A.; de Constantino, L.S.; Tomasi, C.D.; Rojas, H.A.; Vuolo, F.S.; Vitto, M.F.; Cesconetto, P.A.; de Souza, C.T.; Ritter, C.; Dal-Pizzol, F. Sodium butyrate decreases the activation of NF-κB reducing inflammation and oxidative damage in the kidney of rats subjected to contrast-induced nephropathy. Nephrol. Dial. Transplant. 2012, 27, 3136–3140. [Google Scholar] [CrossRef] [PubMed]
- Perez-Gomez, M.V.; Sanchez-Niño, M.D.; Sanz, A.B.; Zheng, B.; Martín-Cleary, C.; Ruiz-Ortega, M.; Ortiz, A.; Fernandez-Fernandez, B. Targeting inflammation in diabetic kidney disease: early clinical trials. Expert Opin. Inv. Drugs 2016, 25, 1045–1058. [Google Scholar] [CrossRef] [PubMed]
- Kooman, J.P.; Dekker, M.J.; Usvyat, L.A.; Kotanko, P.; van der Sande, F.M.; Schalkwijk, C.G.; Shiels, P.G.; Stenvinkel, P. Inflammation and premature aging in advanced chronic kidney disease. Am. J. Physiol. Renal Physiol. 2017, 313, F938–F950. [Google Scholar] [CrossRef] [PubMed]
- Castillo-Rodríguez, E.; Pizarro-Sánchez, S.; Sanz, A.B.; Ramos, A.M.; Sanchez-Niño, M.D.; Martin-Cleary, C.; Fernandez-Fernandez, B.; Ortiz, A. Inflammatory cytokines as uremic toxins: “Ni son todos los que estan, ni estan todos los que son”. Toxins 2017, 9, 114. [Google Scholar] [CrossRef] [PubMed]
- Mafra, D.; Fouque, D. Gut microbiota and inflammation in chronic kidney disease patients. Clin. Kidney J. 2015, 8, 332–334. [Google Scholar] [CrossRef] [PubMed]
- Lau, W.L.; Kalantar-Zadeh, K.; Vaziri, N.D. The gut as a source of inflammation in chronic kidney disease. Nephron 2015, 130, 92–98. [Google Scholar] [CrossRef] [PubMed]
- Sabatino, A.; Regolisti, G.; Brusasco, I.; Cabassi, A.; Morabito, S.; Fiaccadori, E. Alterations of intestinal barrier and microbiota in chronic kidney disease. Nephrol. Dial. Transplant. 2015, 30, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Anders, H.-J.; Andersen, K.; Stecher, B. The intestinal microbiota, a leaky gut and abnormal immunity in kidney disease. Kidney Int. 2013, 83, 1010–1016. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Jiang, H.; Shi, K.; Ren, Y.; Zhang, P.; Cheng, S. Gut bacterial translocation is associated with microinflammation in end-stage renal disease patients. Nephrol. 2012, 17, 733–738. [Google Scholar] [CrossRef] [PubMed]
- Shi, K.; Wang, F.; Jiang, H.; Liu, H.; Wei, M.; Wang, Z.; Xie, L. Gut bacterial translocation may aggravate microinflammation in hemodialysis patients. Dig. Dis. Sci. 2014, 59, 2109–2117. [Google Scholar] [CrossRef] [PubMed]
- Stenvinkel, P. Inflammation in end-stage renal disease—A fire that burns within. Contrib. Nephrol. 2005, 149, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Feroze, U.; Kalantar-Zadeh, K.; Sterling, K.A.; Molnar, M.Z.; Noori, N.; Benner, D.; Shah, V.; Dwivedi, R.; Becker, K.; Kovesdy, C.P.; et al. Examining associations of circulating endotoxin with nutritional status, inflammation and mortality in hemodialysis patients. J. Renal Nutr. 2012, 22, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Szeto, C.-C.; Kwan, B.C.-H.; Chow, K.-M.; Lai, K.-B.; Chung, K.-Y.; Leung, C.-B.; Li, P.K.-T. Endotoxemia is related to systemic inflammation and atherosclerosis in peritoneal dialysis patients. Clin. J. Am. Soc. Nephrol. 2008, 3, 431–436. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, C.W.; Harrison, L.E.A.; Eldehni, M.T.; Jefferies, H.J.; Szeto, C.-C.; John, S.G.; Sigrist, M.K.; Burton, J.O.; Hothi, D.; Korsheed, S.; et al. Circulating endotoxemia: A novel factor in systemic inflammation and cardiovascular disease in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 133–141. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Goshtasbi, N.; Yuan, J.; Jellbauer, S.; Moradi, H.; Raffatellu, M.; Kalantar-Zadeh, K. Uremic plasma impairs barrier function and depletes the tight junction protein constituents of intestinal epithelium. Am. J. Nephrol. 2012, 36, 438–443. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Yuan, J.; Norris, K. Role of urea in intestinal barrier dysfunction and disruption of epithelial tight junction in chronic kidney disease. Am. J. Nephrol. 2013, 37, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zambetti, L.P.; Mortellaro, A. NLRPs, microbiota and gut homeostasis: Unravelling the connection. J. Pathol. 2014, 233, 321–330. [Google Scholar] [CrossRef] [PubMed]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. p-Cresyl sulfate. Toxins 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Miyamoto, Y.; Honda, D.; Tanaka, H.; Wu, Q.; Endo, M.; Noguchi, T.; Kadowaki, D.; Ishima, Y.; Kotani, S.; et al. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 2013, 83, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Mutsaers, H.A.M.; Caetano-Pinto, P.; Seegers, A.E.M.; Dankers, A.C.A.; van den Broek, P.H.H.; Wetzels, J.F.M.; van den Brand, J.A.J.G.; van den Heuvel, L.P.; Hoenderop, J.G.; Wilmer, M.J.G.; et al. Proximal tubular efflux transporters involved in renal excretion of p-cresyl sulfate and p-cresyl glucuronide: Implications for chronic kidney disease pathophysiology. Toxicol. Vitr. 2015, 29, 1868–1877. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.-Y.; Chang, S.-C.; Wu, M.-S. Uremic toxins induce kidney fibrosis by activating intrarenal renin-angiotensin-aldosterone system associated epithelial-to-mesenchymal transition. PLoS ONE 2012, 7, e34026. [Google Scholar] [CrossRef] [PubMed]
- Gulyassy, P.F.; Jarrard, E.; Stanfel, L.A. Contributions of hippurate, indoxyl sulfate and o-hydroxyhippurate to impaired ligand binding by plasma in azotemic humans. Biochem. Pharmacol. 1987, 36, 4215–4220. [Google Scholar] [CrossRef]
- Ortiz, A.; Justo, P.; Sanz, A.; Melero, R.; Caramelo, C.; Guerrero, M.F.; Strutz, F.; Müller, G.; Barat, A.; Egido, J. Tubular cell apoptosis and cidofovir-induced acute renal failure. Antivir. Ther. 2005, 10, 185–190. [Google Scholar] [PubMed]
- Humanes, B.; Lazaro, A.; Camano, S.; Moreno-Gordaliza, E.; Lazaro, J.A.; Blanco-Codesido, M.; Lara, J.M.; Ortiz, A.; Gomez-Gomez, M.M.; Martín-Vasallo, P.; et al. Cilastatin protects against cisplatin-induced nephrotoxicity without compromising its anticancer efficiency in rats. Kidney Int. 2012, 82, 652–663. [Google Scholar] [CrossRef] [PubMed]
- Ichii, O.; Otsuka-Kanazawa, S.; Nakamura, T.; Ueno, M.; Kon, Y.; Chen, W.; Rosenberg, A.Z.; Kopp, J.B. Podocyte injury caused by indoxyl sulfate, a uremic toxin and aryl-hydrocarbon receptor ligand. PLoS ONE 2014, 9, e108448. [Google Scholar] [CrossRef] [PubMed]
- Motojima, M.; Hosokawa, A.; Yamato, H.; Muraki, T.; Yoshioka, T. Uraemic toxins induce proximal tubular injury via organic anion transporter 1-mediated uptake. Br. J. Pharmacol. 2002, 135, 555–563. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Sallée, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J. Am. Soc. Nephrol. 2015, 26, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallée, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Chitalia, V.C.; Shivanna, S.; Martorell, J.; Balcells, M.; Bosch, I.; Kolandaivelu, K.; Edelman, E.R. Uremic serum and solutes increase post-vascular interventional thrombotic risk through altered stability of smooth muscle cell tissue factor. Circulation 2013, 127, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Shivanna, S.; Kolandaivelu, K.; Shashar, M.; Belghasim, M.; Al-Rabadi, L.; Balcells, M.; Zhang, A.; Weinberg, J.; Francis, J.; Pollastri, M.P.; et al. The aryl hydrocarbon receptor is a critical regulator of tissue factor stability and an antithrombotic target in uremia. J. Am. Soc. Nephrol. 2016, 27, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Ng, H.-Y.; Yisireyili, M.; Saito, S.; Lee, C.-T.; Adelibieke, Y.; Nishijima, F.; Niwa, T. Indoxyl sulfate downregulates expression of Mas receptor via OAT3/AhR/Stat3 pathway in proximal tubular cells. PLoS ONE 2014, 9, e91517. [Google Scholar] [CrossRef] [PubMed]
- Moraes, C.; Fouque, D.; Amaral, A.C.F.; Mafra, D. Trimethylamine N-oxide from gut microbiota in chronic kidney disease patients: Focus on diet. J. Renal Nutr. 2015, 25, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Schugar, R.C.; Willard, B.; Wang, Z.; Brown, J.M. Postprandial gut microbiota-driven choline metabolism links dietary cues to adipose tissue dysfunction. Adipocyte 2018, 7, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Bennett, B.J.; de Aguiar Vallim, T.Q.; Wang, Z.; Shih, D.M.; Meng, Y.; Gregory, J.; Allayee, H.; Lee, R.; Graham, M.; Crooke, R.; et al. Trimethylamine-N-oxide, a metabolite associated with atherosclerosis, exhibits complex genetic and dietary regulation. Cell Metab. 2013, 17, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Miyake, T.; Mizuno, T.; Mochizuki, T.; Kimura, M.; Matsuki, S.; Irie, S.; Ieiri, I.; Maeda, K.; Kusuhara, H. Involvement of organic cation transporters in the kinetics of trimethylamine N-oxide. J. Pharm. Sci. 2017, 106, 2542–2550. [Google Scholar] [CrossRef] [PubMed]
- Teft, W.A.; Morse, B.L.; Leake, B.F.; Wilson, A.; Mansell, S.E.; Hegele, R.A.; Ho, R.H.; Kim, R.B. Identification and characterization of trimethylamine-N-oxide uptake and efflux transporters. Mol. Pharm. 2017, 14, 310–318. [Google Scholar] [CrossRef] [PubMed]
- Gessner, A.; König, J.; Fromm, M.F. Contribution of multidrug and toxin extrusion protein 1 (MATE1) to renal secretion of trimethylamine-N-oxide (TMAO). Sci. Rep. 2018, 8, 6659. [Google Scholar] [CrossRef] [PubMed]
- Hoyles, L.; Jiménez-Pranteda, M.L.; Chilloux, J.; Brial, F.; Myridakis, A.; Aranias, T.; Magnan, C.; Gibson, G.R.; Sanderson, J.D.; Nicholson, J.K.; et al. Metabolic retroconversion of trimethylamine N-oxide and the gut microbiota. Microbiome 2018, 6, 73. [Google Scholar] [CrossRef] [PubMed]
- Bain, M.A.; Faull, R.; Fornasini, G.; Milne, R.W.; Evans, A.M. Accumulation of trimethylamine and trimethylamine-N-oxide in end-stage renal disease patients undergoing haemodialysis. Nephrol. Dial. Transplant. 2006, 21, 1300–1304. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J.A.P.; Wheeler, D.C. The role of trimethylamine N-oxide as a mediator of cardiovascular complications in chronic kidney disease. Kidney Int. 2017, 92, 809–815. [Google Scholar] [CrossRef] [PubMed]
- Yancey, P.H. Organic osmolytes as compatible, metabolic and counteracting cytoprotectants in high osmolarity and other stresses. J. Exp. Biol. 2005, 208, 2819–2830. [Google Scholar] [CrossRef] [PubMed]
- Missailidis, C.; Hällqvist, J.; Qureshi, A.R.; Barany, P.; Heimbürger, O.; Lindholm, B.; Stenvinkel, P.; Bergman, P. Serum Trimethylamine-N-oxide is strongly related to renal function and predicts outcome in chronic kidney disease. PLoS ONE 2016, 11, e0141738. [Google Scholar] [CrossRef] [PubMed]
- Gruppen, E.G.; Garcia, E.; Connelly, M.A.; Jeyarajah, E.J.; Otvos, J.D.; Bakker, S.J.L.; Dullaart, R.P.F. TMAO is associated with mortality: Impact of modestly impaired renal function. Sci. Rep. 2017, 7, 13781. [Google Scholar] [CrossRef] [PubMed]
- Rhee, E.P.; Clish, C.B.; Ghorbani, A.; Larson, M.G.; Elmariah, S.; McCabe, E.; Yang, Q.; Cheng, S.; Pierce, K.; Deik, A.; et al. A combined epidemiologic and metabolomic approach improves CKD prediction. J. Am. Soc. Nephrol. 2013, 24, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Yin, Z.; Liu, N.; Bian, X.; Yu, R.; Su, X.; Zhang, B.; Wang, Y. Gut microbial metabolite TMAO contributes to renal dysfunction in a mouse model of diet-induced obesity. Biochem. Biophys. Res. Commun. 2017, 493, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.-K.; Stafford, D.W. Functional study of the vitamin K cycle enzymes in live cells. Methods Enzymol. 2017, 584, 349–394. [Google Scholar] [CrossRef] [PubMed]
- Harshman, S.G.; Shea, M.K. The role of vitamin K in chronic aging diseases: Inflammation, cardiovascular disease and osteoarthritis. Curr. Nutr. Rep. 2016, 5, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Beulens, J.W.J.; Booth, S.L.; van den Heuvel, E.G.H.M.; Stoecklin, E.; Baka, A.; Vermeer, C. The role of menaquinones (vitamin K2) in human health. Br. J. Nutr. 2013, 110, 1357–1368. [Google Scholar] [CrossRef] [PubMed]
- Holden, R.M.; Morton, A.R.; Garland, J.S.; Pavlov, A.; Day, A.G.; Booth, S.L. Vitamins K and D status in stages 3–5 chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Booth, S.L.; Broe, K.E.; Peterson, J.W.; Cheng, D.M.; Dawson-Hughes, B.; Gundberg, C.M.; Cupples, L.A.; Wilson, P.W.F.; Kiel, D.P. Associations between vitamin K biochemical measures and bone mineral density in men and women. J. Clin. Endocrinol. Metab. 2004, 89, 4904–4909. [Google Scholar] [CrossRef] [PubMed]
- Fusaro, M.; Plebani, M.; Iervasi, G.; Gallieni, M. Vitamin K deficiency in chronic kidney disease: Evidence is building up. Am. J. Nephrol. 2017, 45, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.-F.; Drummen, N.E.A.; Thijs, L.; Jacobs, L.; Herfs, M.; Van’t Hoofd, C.; Vermeer, C.; Staessen, J.A. Vitamin-K-dependent protection of the renal microvasculature: Histopathological studies in normal and diseased kidneys. Pulse 2016, 4, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Van Ballegooijen, A.J.; Beulens, J.W. The role of vitamin K status in cardiovascular health: Evidence from observational and clinical studies. Curr. Nutr. Rep. 2017, 6, 197–205. [Google Scholar] [CrossRef] [PubMed]
- Thamratnopkoon, S.; Susantitaphong, P.; Tumkosit, M.; Katavetin, P.; Tiranathanagul, K.; Praditpornsilpa, K.; Eiam-Ong, S. Correlations of plasma desphosphorylated uncarboxylated matrix GLA protein with vascular calcification and vascular stiffness in chronic kidney disease. Nephron 2017, 135, 167–172. [Google Scholar] [CrossRef] [PubMed]
- Kaesler, N.; Magdeleyns, E.; Herfs, M.; Schettgen, T.; Brandenburg, V.; Fliser, D.; Vermeer, C.; Floege, J.; Schlieper, G.; Krüger, T. Impaired vitamin K recycling in uremia is rescued by vitamin K supplementation. Kidney Int. 2014, 86, 286–293. [Google Scholar] [CrossRef] [PubMed]
- O’Seaghdha, C.M.; Hwang, S.-J.; Holden, R.; Booth, S.L.; Fox, C.S. Phylloquinone and vitamin D status: associations with incident chronic kidney disease in the Framingham Offspring cohort. Am. J. Nephrol. 2012, 36, 68–77. [Google Scholar] [CrossRef] [PubMed]
- Nigwekar, S.U.; Thadhani, R.; Brandenburg, V.M. Calciphylaxis. N. Engl. J. Med. 2018, 378, 1704–1714. [Google Scholar] [CrossRef] [PubMed]
- Caluwé, R.; Pyfferoen, L.; De Boeck, K.; De Vriese, A.S. The effects of vitamin K supplementation and vitamin K antagonists on progression of vascular calcification: ongoing randomized controlled trials. Clin. Kidney J. 2016, 9, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Brodsky, S.V.; Satoskar, A.; Chen, J.; Nadasdy, G.; Eagen, J.W.; Hamirani, M.; Hebert, L.; Calomeni, E.; Nadasdy, T. Acute kidney injury during warfarin therapy associated with obstructive tubular red blood cell casts: A report of 9 cases. Am. J. kidney Dis. 2009, 54, 1121–1126. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Martín-Cleary, C.; Gutiérrez, E.; Toldos, O.; Blanco-Colio, L.M.; Praga, M.; Ortiz, A.; Egido, J. AKI associated with macroscopic glomerular hematuria: Clinical and pathophysiologic consequences. Clin. J. Am. Soc. Nephrol. 2012, 7, 175–184. [Google Scholar] [CrossRef] [PubMed]
- Ware, K.; Brodsky, P.; Satoskar, A.A.; Nadasdy, T.; Nadasdy, G.; Wu, H.; Rovin, B.H.; Bhatt, U.; Von Visger, J.; Hebert, L.A.; et al. Warfarin-related nephropathy modeled by nephron reduction and excessive anticoagulation. J. Am. Soc. Nephrol. 2011, 22, 1856–1862. [Google Scholar] [CrossRef] [PubMed]
- Yao, X.; Tangri, N.; Gersh, B.J.; Sangaralingham, L.R.; Shah, N.D.; Nath, K.A.; Noseworthy, P.A. Renal outcomes in anticoagulated patients with atrial fibrillation. J. Am. Coll. Cardiol. 2017, 70, 2621–2632. [Google Scholar] [CrossRef] [PubMed]
- Golbin, L.; Vigneau, C.; Touchard, G.; Thervet, E.; Halimi, J.-M.; Sawadogo, T.; Lagoutte, N.; Siohan, P.; Zagdoun, E.; Hertig, A.; et al. Warfarin-related nephropathy induced by three different vitamin K antagonists: Analysis of 13 biopsy-proven cases. Clin. Kidney J. 2017, 10, 381–388. [Google Scholar] [CrossRef] [PubMed]
- Moeckel, G.W.; Luciano, R.L.; Brewster, U.C. Warfarin-related nephropathy in a patient with mild IgA nephropathy on dabigatran and aspirin. Clin. Kidney J. 2013, 6, 507–509. [Google Scholar] [CrossRef] [PubMed]
- Wächtershäuser, A.; Stein, J. Rationale for the luminal provision of butyrate in intestinal diseases. Eur. J. Nutr. 2000, 39, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Xie, S.; Lv, D.; Wang, P.; He, H.; Zhang, T.; Zhou, Y.; Lin, Q.; Zhou, H.; Jiang, J.; et al. Alteration of the gut microbiota in Chinese population with chronic kidney disease. Sci. Rep. 2017, 7, 2870. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Xie, S.; Lv, D.; Zhang, Y.; Deng, J.; Zeng, L.; Chen, Y. A reduction in the butyrate producing species Roseburia spp. and Faecalibacterium prausnitzii is associated with chronic kidney disease progression. Antonie Van Leeuwenhoek 2016, 109, 1389–1396. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Piceno, Y.M.; DeSantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Gao, Z.; Yin, J.; Zhang, J.; Ward, R.E.; Martin, R.J.; Lefevre, M.; Cefalu, W.T.; Ye, J. Butyrate improves insulin sensitivity and increases energy expenditure in mice. Diabetes 2009, 58, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Maslowski, K.M.; Vieira, A.T.; Ng, A.; Kranich, J.; Sierro, F.; Yu, D.; Schilter, H.C.; Rolph, M.S.; Mackay, F.; Artis, D.; et al. Regulation of inflammatory responses by gut microbiota and chemoattractant receptor GPR43. Nature 2009, 461, 1282–1286. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Chow, M.Z.Y.; Wang, Z.; Zhang, L.; Liu, B.; Liu, X.; Zhou, Z. Histone H4 lysine 16 hypoacetylation is associated with defective DNA repair and premature senescence in Zmpste24-deficient mice. Proc. Natl. Acad. Sci. USA 2011, 108, 12325–12330. [Google Scholar] [CrossRef] [PubMed]
- Walsh, M.E.; Bhattacharya, A.; Sataranatarajan, K.; Qaisar, R.; Sloane, L.; Rahman, M.M.; Kinter, M.; Van Remmen, H. The histone deacetylase inhibitor butyrate improves metabolism and reduces muscle atrophy during aging. Aging Cell 2015, 14, 957–970. [Google Scholar] [CrossRef] [PubMed]
- Fontecha-Barriuso, M.; Martin-Sanchez, D.; Ruiz-Andres, O.; Poveda, J.; Sanchez-Niño, M.D.; Valiño-Rivas, L.; Ruiz-Ortega, M.; Ortiz, A.; Sanz, A.B. Targeting epigenetic DNA and histone modifications to treat kidney disease. Nephrol. Dial. Transplant. 2018, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Izquierdo, M.C.; Sanchez-Niño, M.D.; Suárez-Alvarez, B.; Lopez-Larrea, C.; Jakubowski, A.; Blanco, J.; Ramirez, R.; Selgas, R.; Ruiz-Ortega, M.; et al. The inflammatory cytokines TWEAK and TNFα reduce renal klotho expression through NFκB. J. Am. Soc. Nephrol. 2011, 22, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Andres, O.; Sanchez-Niño, M.D.; Cannata-Ortiz, P.; Ruiz-Ortega, M.; Egido, J.; Ortiz, A.; Sanz, A.B. Histone lysine crotonylation during acute kidney injury in mice. Dis. Model. Mech. 2016, 9, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.J.; Goldsworthy, S.M.; Barnes, A.A.; Eilert, M.M.; Tcheang, L.; Daniels, D.; Muir, A.I.; Wigglesworth, M.J.; Kinghorn, I.; Fraser, N.J.; et al. The orphan G protein-coupled receptors GPR41 and GPR43 are activated by propionate and other short chain carboxylic acids. J. Biol. Chem. 2003, 278, 11312–13319. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Niño, M.D.; Ortiz, A. Differential effects of oral and intravenous l-carnitine on serum lipids: Is the microbiota the answer? Clin. Kidney J. 2014, 7, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Devlin, A.S.; Marcobal, A.; Dodd, D.; Nayfach, S.; Plummer, N.; Meyer, T.; Pollard, K.S.; Sonnenburg, J.L.; Fischbach, M.A. Modulation of a circulating uremic solute via rational genetic manipulation of the gut microbiota. Cell Host Microbe 2016, 20, 709–715. [Google Scholar] [CrossRef] [PubMed]
- Kusumoto, M.; Kamobayashi, H.; Sato, D.; Komori, M.; Yoshimura, M.; Hamada, A.; Kohda, Y.; Tomita, K.; Saito, H. Alleviation of cisplatin-induced acute kidney injury using phytochemical polyphenols is accompanied by reduced accumulation of indoxyl sulfate in rats. Clin. Exp. Nephrol. 2011, 15, 820–830. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Yoshimura, M.; Saigo, C.; Komori, M.; Nomura, Y.; Yamamoto, Y.; Sagata, M.; Wakida, A.; Chuman, E.; Nishi, K.; et al. Hepatic sulfotransferase as a nephropreventing target by suppression of the uremic toxin indoxyl sulfate accumulation in ischemic acute kidney injury. Toxicol. Sci. 2014, 141, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Saigo, C.; Nomura, Y.; Yamamoto, Y.; Sagata, M.; Matsunaga, R.; Jono, H.; Nishi, K.; Saito, H. Meclofenamate elicits a nephropreventing effect in a rat model of ischemic acute kidney injury by suppressing indoxyl sulfate production and restoring renal organic anion transporters. Drug Des. Dev. Ther. 2014, 8, 1073–1082. [Google Scholar] [CrossRef]
- Suzuki, T.; Toyohara, T.; Akiyama, Y.; Takeuchi, Y.; Mishima, E.; Suzuki, C.; Ito, S.; Soga, T.; Abe, T. Transcriptional regulation of organic anion transporting polypeptide SLCO4C1 as a new therapeutic modality to prevent chronic kidney disease. J. Pharm. Sci. 2011, 100, 3696–3707. [Google Scholar] [CrossRef] [PubMed]
- Toyohara, T.; Suzuki, T.; Morimoto, R.; Akiyama, Y.; Souma, T.; Shiwaku, H.O.; Takeuchi, Y.; Mishima, E.; Abe, M.; Tanemoto, M.; et al. SLCO4C1 transporter eliminates uremic toxins and attenuates hypertension and renal inflammation. J. Am. Soc. Nephrol. 2009, 20, 2546–2555. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, Y.; Kikuchi, K.; Saigusa, D.; Suzuki, T.; Takeuchi, Y.; Mishima, E.; Yamamoto, Y.; Ishida, A.; Sugawara, D.; Jinno, D.; et al. Indoxyl sulfate down-regulates SLCO4C1 transporter through up-regulation of GATA3. PLoS ONE 2013, 8, e66518. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, J.; Tanaka, T.; Inagi, R. Effect of AST-120 in chronic kidney disease treatment: Still a controversy? Nephron 2017, 135, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Saigusa, D.; Mishima, E.; Uchida, T.; Miura, D.; Morikawa-Ichinose, T.; Kisu, K.; Sekimoto, A.; Saito, R.; Oe, Y.; et al. Impact of the oral adsorbent AST-120 on organ-specific accumulation of uremic toxins: LC-MS/MS and MS imaging techniques. Toxins 2017, 10, 19. [Google Scholar] [CrossRef] [PubMed]
- Schulman, G.; Berl, T.; Beck, G.J.; Remuzzi, G.; Ritz, E.; Arita, K.; Kato, A.; Shimizu, M. Randomized placebo-controlled EPPIC trials of AST-120 in CKD. J. Am. Soc. Nephrol. 2015, 26, 1732–1746. [Google Scholar] [CrossRef] [PubMed]
- Schulman, G.; Berl, T.; Beck, G.J.; Remuzzi, G.; Ritz, E.; Shimizu, M.; Kikuchi, M.; Shobu, Y. Risk factors for progression of chronic kidney disease in the EPPIC trials and the effect of AST-120. Clin. Exp. Nephrol. 2018, 22, 299–308. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Cupisti, A.; Bolasco, P. Keto-analogues and essential aminoacids and other supplements in the conservative management of chronic kidney disease. Panminerva Med. 2017, 59, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Koh, G.Y.; Rowling, M.J. Resistant starch as a novel dietary strategy to maintain kidney health in diabetes mellitus. Nutr. Rev. 2017, 75, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Normand, G.; Lemoine, S.; Villien, M.; Le Bars, D.; Merida, I.; Irace, Z.; Troalen, T.; Costes, N.; Juillard, L. AGE content of a protein load is responsible for renal performances: A pilot study. Diabetes Care 2018, 41, 1292–1294. [Google Scholar] [CrossRef] [PubMed]
- Yacoub, R.; Nugent, M.; Cai, W.; Nadkarni, G.N.; Chaves, L.D.; Abyad, S.; Honan, A.M.; Thomas, S.A.; Zheng, W.; Valiyaparambil, S.A.; et al. Advanced glycation end products dietary restriction effects on bacterial gut microbiota in peritoneal dialysis patients; a randomized open label controlled trial. PLoS ONE 2017, 12, e0184789. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, T.; Cai, W.; Peppa, M.; Dardaine, V.; Baliga, B.S.; Uribarri, J.; Vlassara, H. Advanced glycoxidation end products in commonly consumed foods. J. Am. Diet. Assoc. 2004, 104, 1287–1291. [Google Scholar] [CrossRef] [PubMed]
- Poesen, R.; Evenepoel, P.; de Loor, H.; Delcour, J.A.; Courtin, C.M.; Kuypers, D.; Augustijns, P.; Verbeke, K.; Meijers, B. The influence of prebiotic arabinoxylan oligosaccharides on microbiota derived uremic retention solutes in patients with chronic kidney disease: A randomized controlled trial. PLoS ONE 2016, 11, e0153893. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Hazen, S.L. The contributory role of gut microbiota in cardiovascular disease. J. Clin. Invest. 2014, 124, 4204–4211. [Google Scholar] [CrossRef] [PubMed]
- Effectiveness of Probiotics to Treat End Stage Renal Disease. Available online: https://www.clinicaltrials.gov/ct2/show/NCT02929225 (accessed on 18 July 2018).
- Van der Wal, W.M.; Noordzij, M.; Dekker, F.W.; Boeschoten, E.W.; Krediet, R.T.; Korevaar, J.C.; Geskus, R.B. Full loss of residual renal function causes higher mortality in dialysis patients; findings from a marginal structural model. Nephrol. Dial. Transplant. 2011, 26, 2978–2983. [Google Scholar] [CrossRef] [PubMed]
- Marrón, B.; Remón, C.; Pérez-Fontán, M.; Quirós, P.; Ortíz, A. Benefits of preserving residual renal function in peritoneal dialysis. Kidney Int. Suppl. 2008, 73, S42–S51. [Google Scholar] [CrossRef] [PubMed]
- Glorieux, G.; Tattersall, J. Uraemic toxins and new methods to control their accumulation: game changers for the concept of dialysis adequacy. Clin. Kidney J. 2015, 8, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.C.; Eloot, S.; Glorieux, G.L.R.L. Future avenues to decrease uremic toxin concentration. Am. J. Kidney Dis. 2016, 67, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Koppe, L.; Fouque, D.; Soulage, C.O. The role of gut microbiota and diet on uremic retention solutes production in the context of chronic kidney disease. Toxins 2018, 10, 155. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Compound | Total Plasma Concentration in CKD G4/G5 Non-Dialysis (µM) | Lowest Concentration Active on Cultured Renal Cells (µM) | Effects on Cultured Renal Cells | Effects on Kidneys In Vivo | References |
|---|---|---|---|---|---|
| pCS | Median 50 Maximum 500 | 100 | Decreased viability, increased oxidative stress, increased inflammatory and profibrotic responses, decreased expression of nephroprotective factors and viability, decreased function of (MRP4/ABCC4 and BCRP/ABCG2) in tubular cells. | Progression of CKD, kidney fibrosis Promote epithelial-to-mesenchymal transition Activate the renal-angiotensin system | [5,14,15,16,17,18,19,20] |
| pCG | Median 0.22 Maximum 8 | 25 | No actions observed on human proximal tubule cell inflammatory response Decreased the function of proximal cell membrane transporters (MRP4) | Kidney fibrosis Promotes epithelial-to-mesenchymal transition | [5,14,15] |
| IS | Median 211 Maximum 1100 | 1000 | Decreased viability, increased oxidative stress, increased inflammatory and profibrotic responses, decreased expression of nephroprotective factors and viability, nuclear AhR translocation in tubular cells. Podocyte injury | Accelerated fibrosis and CKD progression Podocyte injury | [18,21,22,23,24,25,26] |
| IAA | Median 5 Maximum 50 | 250 | Reduced viability through induction of apoptosis in tubular cells | Accelerated CKD progression | [27,28] |
| TMAO | Median 25 Upper quartile > 38 | ND | No data | Kidney tubulointerstitial fibrosis | [29,30] |
| Butyrate | Not data in CKD patients 2–65 in blood 5000–20,000 in gut lumen in non-CKD | 500–1000 | Reduced inflammation | Protects from gentamicin and contrast nephrotoxicity | [31,32,33,34,35,36,37] |
| Supplement | Putative Benefits or Indications | Resulting Uremic Toxins with Nephrotoxicity Potential |
|---|---|---|
| l-tyrosine (para-tyrosine, 4-hydroxyphenylalanine) | Enhanced physical performance, enhanced cognitive performance, | pCS, pCG |
| Tryptophan | Antidepressant, anxiolytic, sleep aid | IS, IAA |
| Choline/phosphatidylcholine/lecithin | Liver health, memory, Alzheimer disease, enhanced physical performance, pregnancy | TMAO |
| l-carnitine | Enhanced physical performance, haemodialysis | TMAO |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castillo-Rodriguez, E.; Fernandez-Prado, R.; Esteras, R.; Perez-Gomez, M.V.; Gracia-Iguacel, C.; Fernandez-Fernandez, B.; Kanbay, M.; Tejedor, A.; Lazaro, A.; Ruiz-Ortega, M.; et al. Impact of Altered Intestinal Microbiota on Chronic Kidney Disease Progression. Toxins 2018, 10, 300. https://doi.org/10.3390/toxins10070300
Castillo-Rodriguez E, Fernandez-Prado R, Esteras R, Perez-Gomez MV, Gracia-Iguacel C, Fernandez-Fernandez B, Kanbay M, Tejedor A, Lazaro A, Ruiz-Ortega M, et al. Impact of Altered Intestinal Microbiota on Chronic Kidney Disease Progression. Toxins. 2018; 10(7):300. https://doi.org/10.3390/toxins10070300
Chicago/Turabian StyleCastillo-Rodriguez, Esmeralda, Raul Fernandez-Prado, Raquel Esteras, Maria Vanessa Perez-Gomez, Carolina Gracia-Iguacel, Beatriz Fernandez-Fernandez, Mehmet Kanbay, Alberto Tejedor, Alberto Lazaro, Marta Ruiz-Ortega, and et al. 2018. "Impact of Altered Intestinal Microbiota on Chronic Kidney Disease Progression" Toxins 10, no. 7: 300. https://doi.org/10.3390/toxins10070300
APA StyleCastillo-Rodriguez, E., Fernandez-Prado, R., Esteras, R., Perez-Gomez, M. V., Gracia-Iguacel, C., Fernandez-Fernandez, B., Kanbay, M., Tejedor, A., Lazaro, A., Ruiz-Ortega, M., Gonzalez-Parra, E., Sanz, A. B., Ortiz, A., & Sanchez-Niño, M. D. (2018). Impact of Altered Intestinal Microbiota on Chronic Kidney Disease Progression. Toxins, 10(7), 300. https://doi.org/10.3390/toxins10070300