Evaluating the Performance of De Novo Assembly Methods for Venom-Gland Transcriptomics

 , , , and
, , , and 
Abstract
1. Introduction
2. Results
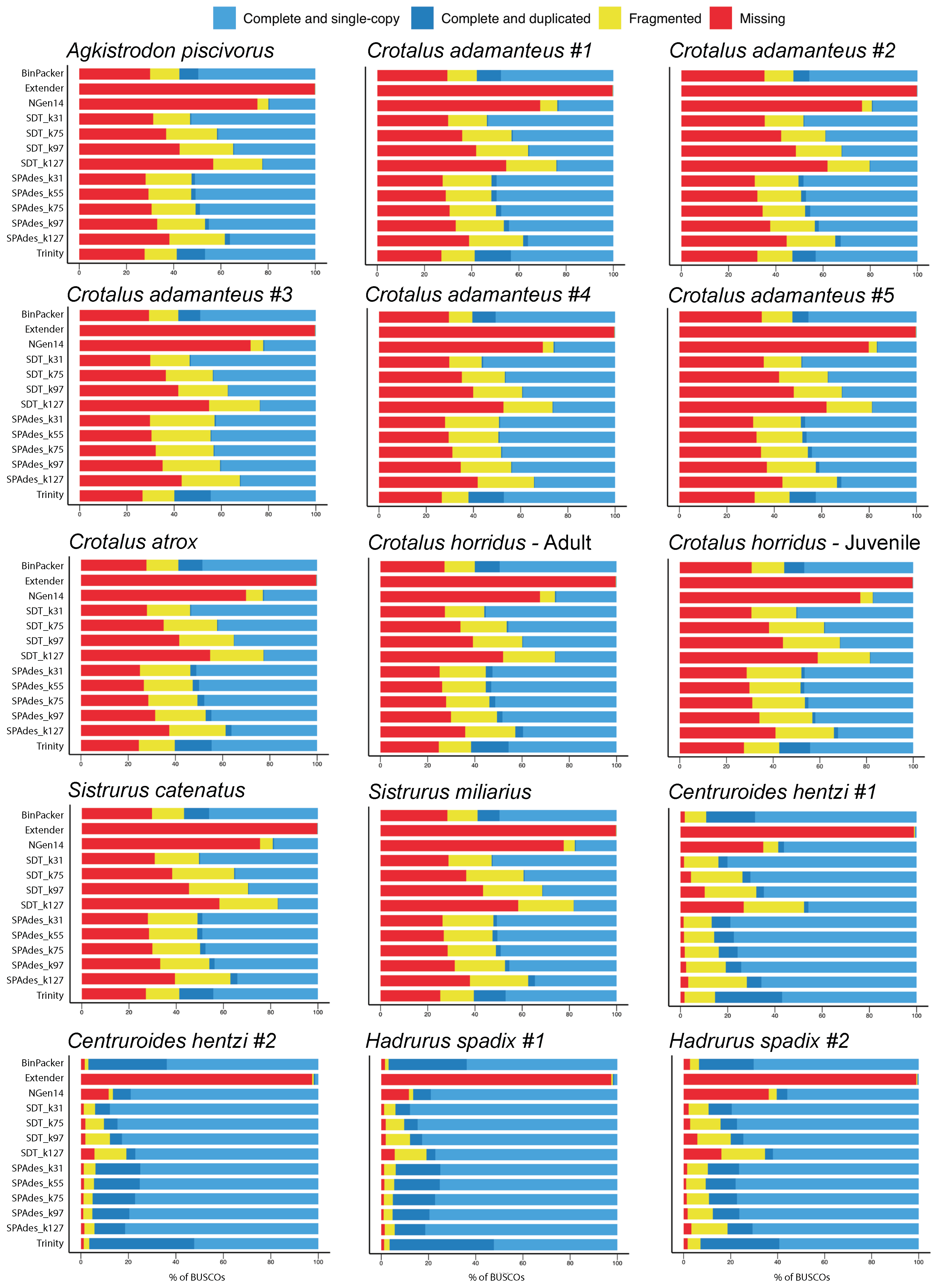
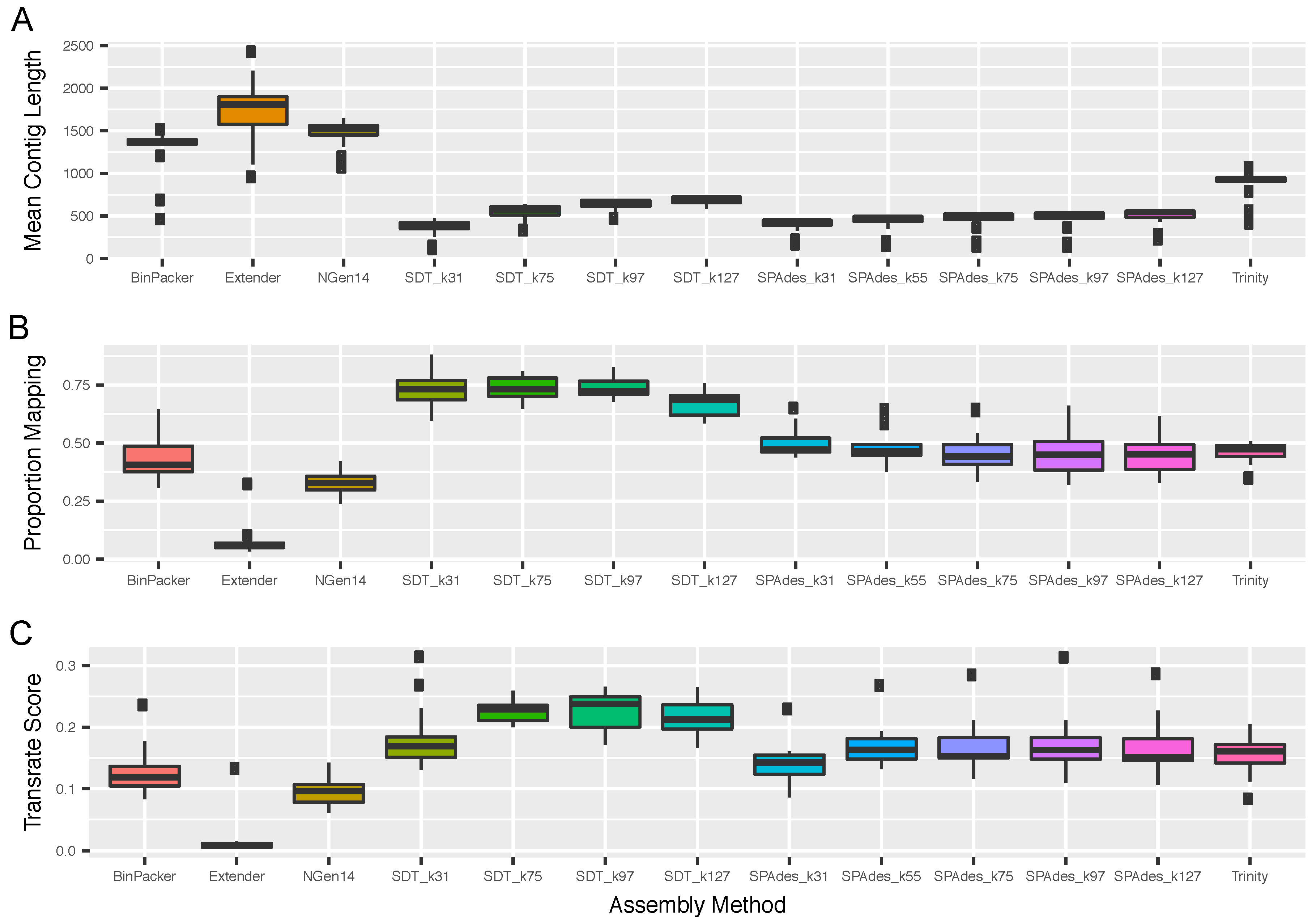
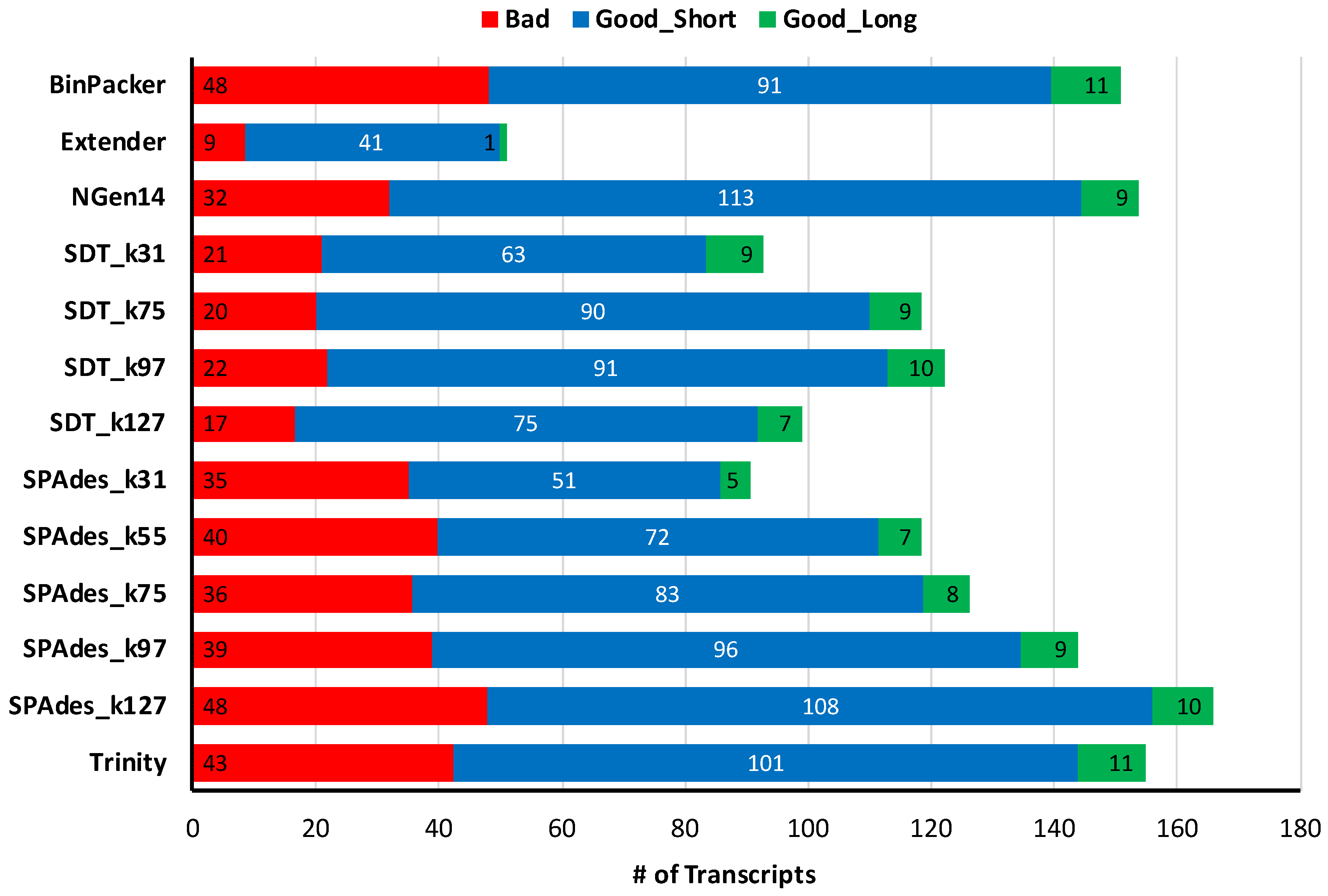
2.1. Overall Assembly Quality and Recovery of Nontoxins
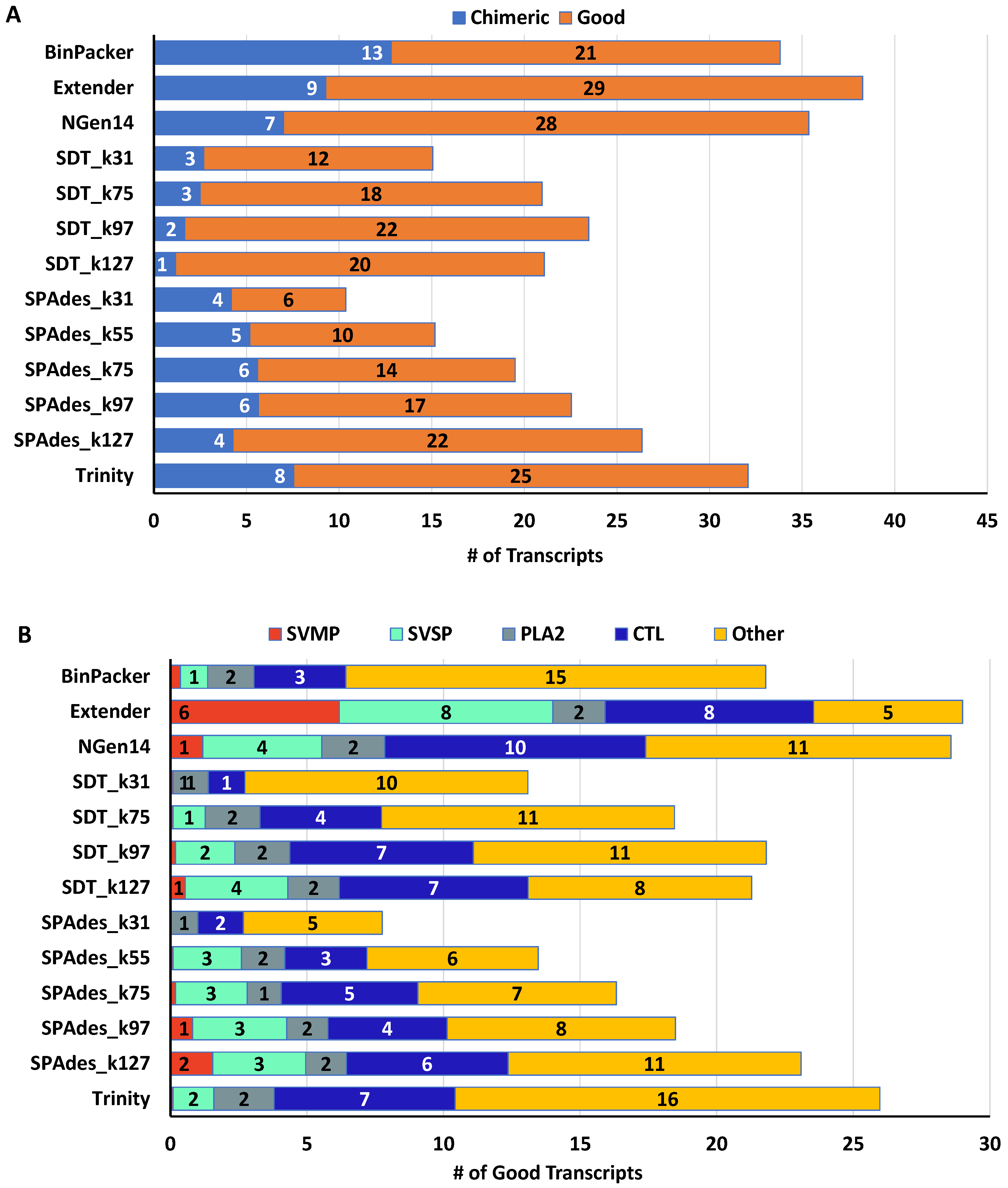
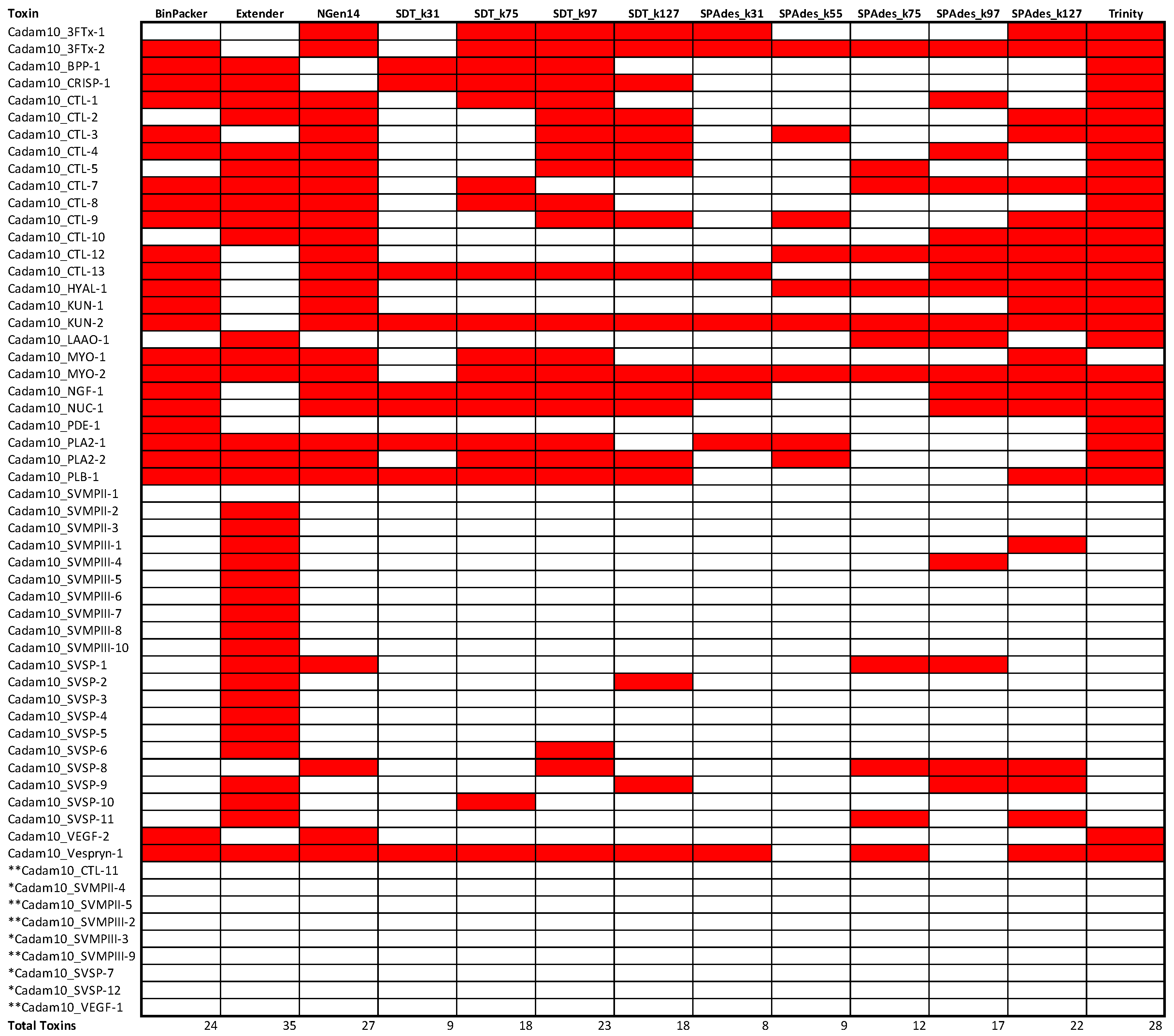
2.2. Snake Toxin Assembly Quality
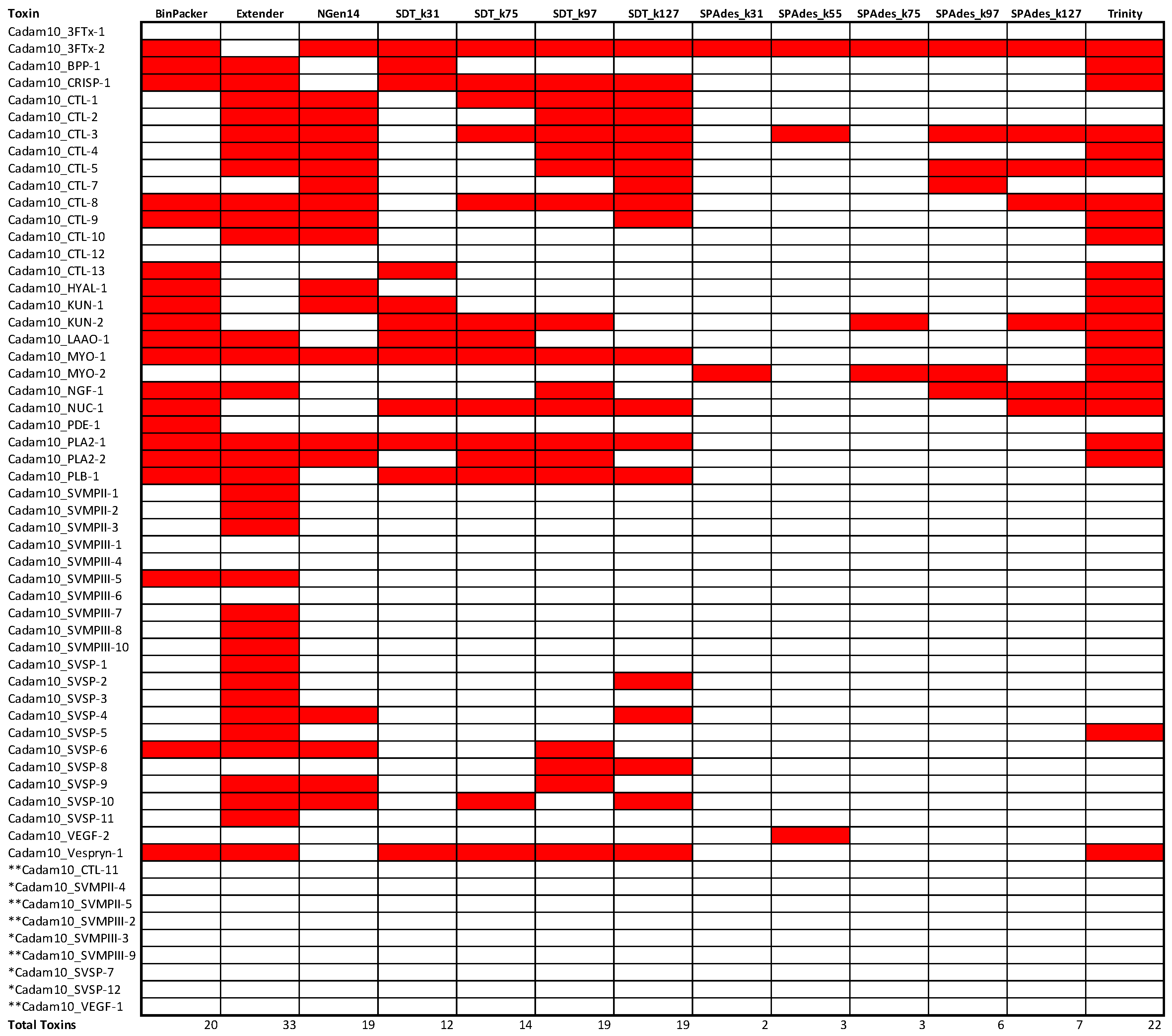
2.3. Assembly Completeness for the Crotalus adamanteus Transcriptome
2.4. Scorpion Assembly Quality
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Transcriptome Sequencing
4.2. Tissue Preparation and Sequencing
4.3. Transcriptome Assemblies
4.4. Full Assembly Quality and Recovery of Nontoxins
4.5. Evaluating Toxin Gene Assembly
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 3FTx | three-finger toxin |
| bp | base pair |
| BPP | bradykinin potentiating peptide |
| CRISP | cysteine rich secretory protein |
| CTL | C-type lectin |
| HYAL | hyaluronidase |
| KUN | Kunitz-like protein |
| LAAO | L-amino acid oxidase |
| NGen14 | SeqMan NGen v.14 |
| MYO | myotoxin |
| PDE | phosphodiesterase |
| PLA2 | phospholipase A2 |
| PLB | phospholipase B |
| SDT | SOAPdenovo-Trans |
| SPAdes | rnaSPAdes |
| SVMP | snake venom metalloproteinase |
| SVSP | snake venom serine protease |
| VESP | vespryn |
| VEGF | vascular endothelial growth factor |
References
- Brodie, E.D., 3rd; Feldman, C.R.; Hanifin, C.T.; Motychak, J.E.; Mulcahy, D.G.; Williams, B.L.; Brodie, E.D., Jr. Parallel arms races between garter snakes and newts involving tetrodotoxin as the phenotypic interface of coevolution. J. Chem. Ecol. 2005, 31, 343–356. [Google Scholar] [CrossRef] [PubMed]
- Toju, H.; Ueno, S.; Taniguchi, F.; Sota, T. Metapopulation structure of a seed-predator weevel and its host plant in arms race coevolution. Evolution 2011, 65, 1707–1722. [Google Scholar] [CrossRef] [PubMed]
- Ehrlich, P.R.; Raven, P.H. Butterflies and plants: A study in coevolution. Evolution 1964, 18, 586–608. [Google Scholar] [CrossRef]
- Margres, M.J.; Wray, K.P.; Hassinger, A.T.; Ward, M.J.; McGivern, J.J.; Lemmon, E.M.; Lemmon, A.R.; Rokyta, D.R. Quantity, not quality: Rapid adaptation in a polygenic trait proceeded exclusively through expression differentiation. Mol. Biol. Evol. 2017, 34, 3099–3110. [Google Scholar] [CrossRef] [PubMed]
- Rokyta, D.R.; Margres, M.J.; Calvin, K. Post-transcriptional mechanisms contribute little to phenotypic variation in snake venoms. G3 Genes Genomes Genet. 2015, 5, 2375–2382. [Google Scholar] [CrossRef] [PubMed]
- Calvete, J.J.; Juárez, P.; Sanz, L. Snake venomics. Strategy and applications. J. Mass Spectrom. 2007, 42, 1405–1414. [Google Scholar] [CrossRef] [PubMed]
- Mackessy, S.P.; Baxter, L.M. Bioweapons synthesis and storage: The venom gland of front-fanged snakes. Zool. Anz. J. Comp. Zool. 2006, 245, 147–159. [Google Scholar] [CrossRef]
- Margres, M.J.; Wray, K.P.; Seavy, M.; McGivern, J.J.; Herrera, N.D.; Rokyta, D.R. Expression differentiation is constrained to low-expression proteins over ecological timescales. Genetics 2016, 202, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Sunagar, K.; Morgenstern, D.; Reitzel, A.M.; Moran, Y. Ecological venomics: How genomics, transcriptomics and proteomics can shed new light on the ecology and evolution of venom. J. Proteom. 2016, 135, 62–72. [Google Scholar] [CrossRef] [PubMed]
- Calvete, J.J. Next-generation snake venomics: Protein-locus resolution through venom proteome decomplexation. Expert Rev. Proteom. 2014, 11, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.; et al. Full-length transcriptome assemby from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Wu, G.; Tang, J.; Luo, R.; Patterson, J.; Liu, S.; Huang, W.; He, G.; Gu, S.; Li, S.; et al. SOAPdenovo-Trans: De novo transcriptome assembly with short RNA-Seq reads. Bioinformatics 2014, 30, 1660–1666. [Google Scholar] [CrossRef] [PubMed]
- Schulz, M.H.; Zerbino, D.R.; Vingron, M.; Birney, E. Oases: Robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 2012, 28, 1086–1092. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Leung, H.C.M.; Yiu, S.M.; Lv, M.J.; Zhu, X.G.; Chin, F.Y.L. IDBA-tran: A more robust de novo de Bruijn graph assembler for transcriptomes with uneven expression levels. Bioinformatics 2013, 29, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Robertson, G.; Schein, J.; Chiu, R.; Corbett, R.; Field, M.; Jackman, S.D.; Mungall, K.; Lee, S.; Okada, H.M.; Qian, J.Q.; et al. De novo assembly and analysis of RNA-seq data. Nat. Methods 2010, 7, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Charuvaka, A.; Rangwala, H. Evaluation of short read metagenomic assembly. BMC Genom. 2011, 12, S8. [Google Scholar] [CrossRef] [PubMed]
- Cahais, V.; Gayral, P.; Tsagkogeorga, G.; Melo-Ferreira, J.; Ballenghien, M.; Weinert, L.; Chiari, Y.; Belkhir, K.; Ranwez, V.; Galtier, N. Reference-free transcriptome assembly in non-model animals from next-generation sequencing data. Mol. Ecol. Resour. 2012, 12, 834–845. [Google Scholar] [CrossRef] [PubMed]
- Haney, R.A.; Ayoub, N.A.; Clarke, T.H.; Hayashi, C.Y.; Garb, J.E. Dramatic expansion of the black widow toxin arsenal uncovered by multi-tissue transcriptomics and venom proteomics. BMC Genom. 2014, 15, 366. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Yu, H.; Xue, W.; Yue, Y.; Liu, S.; Xing, R.; Li, P. Jellyfish venomics and venom gland transcriptomics analysis of Stomolophus meleagris to reveal the toxins associated with sting. J. Proteom. 2014, 106, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Luna-Ramírez, K.; Quintero-Hernández, V.; Rivelino Juárez-González, V.; Possani, L.D. Whole transcriptome of the venom gland from Urodacus yaschenkoi Scorpion. PLoS ONE 2015, 10, e0127883. [Google Scholar] [CrossRef] [PubMed]
- Tan, C.H.; Tan, K.Y.; Fung, S.Y.; Tan, N.H. Venom-gland transcriptome and venom proteome of the Malaysian king cobra (Ophiophagus hannah). BMC Genom. 2015, 16, 687. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, X.; Hu, T.; Zhou, W.; Cui, Q.; Tian, J.; Zheng, Y.; Fan, Q. Discovery of toxin-encoding genes from the false viper Macropisthodon rudis, a rear-fanged snake, by transcriptome analysis of venom gland. Toxicon 2015, 106, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Santibáñez-López, C.E.; Cid-Uribe, J.I.; Batista, C.V.; Ortiz, E.; Possani, L.D. Venom gland transcriptomic and proteomic analyses of the enigmatic scorpion Superstitionia donensis (Scorpiones: Superstitioniidae), with insights on the evolution of its venom components. Toxins 2016, 8, 367. [Google Scholar] [CrossRef] [PubMed]
- Gomes De Oliveira Júnior, N.; Da, G.; Fernandes, R.; Cardoso, M.H.; Costa, F.F.; De Souza Cândido, E.; Neto, D.G.; Mortari, M.R.; Schwartz, E.F.; Franco, O.L.; et al. Venom gland transcriptome analyses of two freshwater stingrays (Myliobatiformes: Potamotrygonidae) from Brazil. Sci. Rep. 2016, 6, 21935. [Google Scholar] [CrossRef] [PubMed]
- Amorim, F.G.; Morandi-Filho, R.; Fujimura, P.T.; Ueira-Vieira, C.; Sampaio, S.V. New findings from the first transcriptome of the Bothrops moojeni snake venom gland. Toxicon 2017, 140, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Kazemi-Lomedasht, F.; Khalaj, V.; Bagheri, K.P.; Behdani, M.; Shahbazzadeh, D. The first report on transcriptome analysis of the venom gland of Iranian scorpion, Hemiscorpius lepturus. Toxicon 2017, 125, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Zhao, H.Y.; Yin, Y.; Shen, S.S.; Shan, L.L.; Chen, C.X.; Zhang, Y.X.; Gao, J.F.; Ji, X. Combined venomics, antivenomics and venom gland transcriptome analysis of the monocoled cobra (Naja kaouthia) from China. J. Proteom. 2017, 159, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Martinson, E.O.; Mrinalini; Kelkar, Y.D.; Chang, C.H.; Werren, J.H. The Evolution of Venom by Co-option of Single-Copy Genes. Curr. Biol. 2017, 27, 2007–2013. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.Y.; Tan, C.H.; Chanhome, L.; Tan, N.H. Comparative venom gland transcriptomics of Naja kaouthia (monocled cobra) from Malaysia and Thailand: Elucidating geographical venom variation and insights into sequence novelty. PeerJ 2017, 5, e3142. [Google Scholar] [CrossRef] [PubMed]
- Aird, S.; da Silva, N.; Qiu, L.; Villar-Briones, A.; Saddi, V.; Pires de Campos Telles, M.; Grau, M.; Mikheyev, A. Coralsnake Venomics: Analyses of Venom Gland Transcriptomes and Proteomes of Six Brazilian Taxa. Toxins 2017, 9, 187. [Google Scholar] [CrossRef] [PubMed]
- Cusumano, A.; Duvic, B.; Jouan, V.; Ravallec, M.; Legeai, F.; Peri, E.; Colazza, S.; Volkoff, A.N. First extensive characterization of the venom gland from an egg parasitoid: Structure, transcriptome and functional role. J. Insect Physiol. 2018, 107, 68–80. [Google Scholar] [CrossRef] [PubMed]
- Rokyta, D.R.; Lemmon, A.R.; Margres, M.J.; Aronow, K. The venom-gland transcriptome of the eastern diamondback rattlesnake (Crotalus adamanteus). BMC Genom. 2012, 13, 312. [Google Scholar] [CrossRef] [PubMed]
- Barghi, N.; Concepcion, G.P.; Olivera, B.M.; Lluisma, A.O. High conopeptide diversity in Conus tribblei revealed through analysis of venom duct transcriptome using two high-throughput sequencing platforms. Mar. Biotechnol. 2015, 17, 81–98. [Google Scholar] [CrossRef] [PubMed]
- Brinkman, D.L.; Jia, X.; Potriquet, J.; Kumar, D.; Dash, D.; Kvaskoff, D.; Mulvenna, J. Transcriptome and venom proteome of the box jellyfish Chironex fleckeri. BMC Genom. 2015, 16, 407. [Google Scholar] [CrossRef] [PubMed]
- Dhaygude, K.; Trontti, K.; Paviala, J.; Morandin, C.; Wheat, C.; Sundström, L.; Helanterä, H. Transcriptome sequencing reveals high isoform diversity in the ant Formica exsecta. PeerJ 2017, 5, e3998. [Google Scholar] [CrossRef] [PubMed]
- Honaas, L.A.; Wafula, E.K.; Wickett, N.J.; Der, J.P.; Zhang, Y.; Edger, P.P.; Altman, N.S.; Chris Pires, J.; Leebens-Mack, J.H.; DePamphilis, C.W. Selecting superior de novo transcriptome assemblies: Lessons learned by leveraging the best plant genome. PLoS ONE 2016, 11, e0146062. [Google Scholar] [CrossRef] [PubMed]
- Cabau, C.; Escudié, F.; Djari, A.; Guiguen, Y.; Bobe, J.; Klopp, C. Compacting and correcting Trinity and Oases RNA-Seq de novo assemblies. PeerJ 2017, 5, e2988. [Google Scholar] [CrossRef] [PubMed]
- Rana, S.B.; Zadlock, F.J., IV; Zhang, Z.; Murphy, W.R.; Bentivegna, C.S. Comparison of De Novo Transcriptome Assemblers and k-mer Strategies Using the Killifish, Fundulus heteroclitus. PLoS ONE 2016, 11, e0153104. [Google Scholar] [CrossRef] [PubMed]
- Macrander, J.; Broe, M.; Daly, M. Multi-copy venom genes hidden in de novo transcriptome assemblies, a cautionary tale with the snakelocks sea anemone Anemonia sulcata (Pennant, 1977). Toxicon 2015, 108, 184–188. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Zhao, S.; Chen, Y.; Cao, Q.; Wei, C.; Cheng, X.; Zhang, Y. Optimal assembly strategies of transcriptome related to ploidies of eukaryotic organisms. BMC Genom. 2015, 16, 65. [Google Scholar] [CrossRef] [PubMed]
- Brandley, M.C.; Bragg, J.G.; Singhal, S.; Chapple, D.G.; Jennings, C.K.; Lemmon, A.R.; Lemmon, E.M.; Thompson, M.B.; Moritz, C. Evaluating the performance of anchored hybrid enrichment at the tips of the tree of life: A phylogenetic analysis of Australian Eugongylus group scincid lizards. BMC Evol. Biol. 2015, 15, 62. [Google Scholar] [CrossRef] [PubMed]
- Rokyta, D.R.; Margres, M.J.; Ward, M.J.; Sanchez, E.E. The genetics of venom ontogeny in the eastern diamondback rattlesnake (Crotalus adamanteus). PeerJ 2017, 5, e3249. [Google Scholar] [CrossRef] [PubMed]
- Simão, F.A.; Waterhouse, R.M.; Ioannidis, P.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO: Assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 2015, 31, 3210–3212. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Li, G.; Chang, Z.; Yu, T.; Liu, B.; McMullen, R.; Chen, P.; Huang, X. BinPacker: Packing-based de novo transcriptome assembly from RNA-seq data. PLoS Comput. Biol. 2016, 12, e1004772. [Google Scholar] [CrossRef] [PubMed]
- Haas, B.J.; Papanicolaou, A.; Yassour, M.; Grabherr, M.; Blood, P.D.; Bowden, J.; Couger, M.B.; Eccles, D.; Li, B.; Lieber, M.; et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat. Protoc. 2013, 8, 1494–1512. [Google Scholar] [CrossRef] [PubMed]
- Smith-Unna, R.; Boursnell, C.; Patro, R.; Hibberd, J.M.; Kelly, S. TransRate: Reference-free quality assessment of de novo transcriptome assemblies. Genome Res. 2016, 26, 1134–1144. [Google Scholar] [CrossRef] [PubMed]
- Ward, M.J.; Ellsworth, S.A.; Rokyta, D.R. Venom-gland transcriptomics and venom proteomics of the Hentz striped scorpion (Centruroides hentzi; Buthidae) reveal high toxin diversity in a harmless member of a lethal family. Toxicon 2018, 142, 14–29. [Google Scholar] [CrossRef] [PubMed]
- Haney, R.A.; Clarke, T.H.; Gadgil, R.; Fitzpatrick, R.; Hayashi, C.Y.; Ayoub, N.A.; Garb, J.E. Effects of gene duplication, positive selection, and shifts in gene expression on the evolution of the venom gland transcriptome in widow spiders. Genome Biol. Evol. 2016, 8, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, R.M.; Seppey, M.; Simão, F.A.; Manni, M.; Ioannidis, P.; Klioutchnikov, G.; Kriventseva, E.V.; Zdobnov, E.M. BUSCO applications from quality assessments to gene prediction and phylogenomics. Mol. Biol. Evol. 2018, 35, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Surget-Groba, Y.; Montoya-Burgos, J.I. Optimization of de novo transcriptome assembly from next-generation sequencing data. Genome Res. 2010, 20, 1432–1440. [Google Scholar] [CrossRef] [PubMed]
- Sonal, S. De novo transcriptomic analyses for non-model organisms: An evaluation of methods across a multi-species data set. Mol. Ecol. Resour. 2013, 13, 403–416. [Google Scholar]
- Archer, J.; Whiteley, G.; Casewell, N.R.; Harrison, R.A.; Wagstaff, S.C. VTBuilder: A tool for the assembly of multi isoform transcriptomes. BMC Bioinform. 2014, 15, 389. [Google Scholar] [CrossRef] [PubMed]
- Margres, M.J.; Wray, K.P.; Seavy, M.; McGivern, J.J.; Sanader, D.; Rokyta, D.R. Phenotypic integration in the feeding system of the eastern diamondback rattlesnake (Crotalus adamanteus). Mol. Ecol. 2015, 24, 3405–3420. [Google Scholar] [CrossRef] [PubMed]
- Nakasugi, K.; Crowhurst, R.; Bally, J.; Waterhouse, P. Combining transcriptome assemblies from multiple de novo assemblers in the allo-tetraploid plant Nicotiana benthamiana. PLoS ONE 2014, 9, e91776. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- MacManes, M. Oster River Protocol For Tranascriptome Assembl. Available online: http://oyster-river-protocol.readthedocs.io/en/latest/ (accessed on 29 May 2018).
- Rokyta, D.R.; Ward, M.J. Venom-gland transcriptomics and venom proteomics of the black-back scorpion (Hadrurus spadix) reveal detectability challenges and an unexplored realm of animal toxin diversity. Toxicon 2017, 128, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Margres, M.J.; Bigelow, A.T.; Lemmon, E.M.; Lemmon, A.R.; Rokyta, D.R. Selection to increase expression, not sequence diversity, precedes gene family origin and expansion in rattlesnake venom. Genetics 2017, 206, 1569–1580. [Google Scholar] [CrossRef] [PubMed]
- Margres, M.J.; McGivern, J.J.; Seavy, M.; Wray, K.P.; Facente, J.; Rokyta, D.R. Contrasting modes and tempos of venom expression evolution in two snake species. Genetics 2015, 199, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Currier, R.B.; Calvete, J.J.; Sanz, L.; Harrison, R.A.; Rowley, P.D.; Wagstaff, S.C. Unusual stability of messenger RNA in snake venom reveals gene expression dynamics of venom replenishment. PLoS ONE 2012, 7, e41888. [Google Scholar] [CrossRef] [PubMed]
- Marçais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar]
- Krueger, F. Trim Galore! A Wrapper Tool around Cutadapt and FastQC to Consistently Apply Quality and Adapter Trimming to FastQ files. Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 27 April 2018).
- Zhang, J.; Kobert, K.; Flouri, T.; Stamatakis, A. PEAR: A fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 2014, 30, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Zdobnov, E.M.; Tegenfeldt, F.; Kuznetsov, D.; Waterhouse, R.M.; Simão, F.A.; Ioannidis, P.; Seppey, M.; Loetscher, A.; Kriventseva, E.V. OrthoDB v9.1: Cataloging evolutionary and functional annotations for animal, fungal, plant, archaeal, bacterial and viral orthologs. Nucleic Acids Res. 2017, 45, D744–D749. [Google Scholar] [CrossRef] [PubMed]
- Gertz, E.M.; Yu, Y.K.; Agarwala, R.; Schäffer, A.A.; Altschul, S.F. Composition-based statistics and translated nucleotide searches: Improving the TBLASTN module of BLAST. BMC Biol. 2006, 4, 41. [Google Scholar] [CrossRef] [PubMed]
- Mistry, J.; Finn, R.D.; Eddy, S.R.; Bateman, A.; Punta, M. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res. 2013, 41, e121. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | Sample ID | Read Pairs | Short Read Archive Accession |
|---|---|---|---|
| Pitvipers | |||
| Agkistrodon piscivorus | KW1750 | 17,165,966 | SRR6916474 |
| Crotalus adamanteus #1 | KW1942 | 23,568,901 | SRR5259496, SRR5259495 |
| Crotalus adamanteus #2 | KW2161 | 19,500,323 | SRR5259494, SRR5259493 |
| Crotalus adamanteus #3 | KW2170 | 10,758,708 | SRR5259492, SRR5259491 |
| Crotalus adamanteus #4 | KW2171 | 15,862,828 | SRR5259490, SRR5259489 |
| Crotalus adamanteus #5 | MM0127 | 13,497,975 | SRR5259486, SRR5259485 |
| Crotalus cerastes | KW1744 | 17,607,533 | SRR6768689 |
| Crotalus horridus-Juvenile | KW1369 | 12,409,168 | SRR6916473 |
| Crotalus horridus-Adult | KW1089 | 14,798,031 | SRR6916472 |
| Sistrurus catenatus | KW1090 | 13,171,023 | SRR6916476 |
| Sistrurus miliarius | KW1749 | 15,292,491 | SRR6916477 |
| Scorpions | |||
| Centruroides hentzi #1 | C0136 | 18,977,620 | SRR6041834 |
| Centruroides hentzi #2 | C0148 | 20,032,620 | SRR6041835 |
| Hadrurus spadix #1 | C0195 | 12,205,269 | SRR4069277 |
| Hadrurus spadix #2 | C0196 | 24,243,211 | SRR4069278 |
| Species/Assembly | CTL | PLA2 | SVSP | SVMP | Other Toxins | Total Toxins |
|---|---|---|---|---|---|---|
| Agkistrodon contortrix | ||||||
| BinPacker | 5 | 2 | — | — | 16 | 23 |
| Extender | 8 | 3 | 11 | 11 | 9 | 42 |
| NGen14 | 9 | 5 | 8 | 6 | 12 | 40 |
| SDT_k31 | — | 2 | — | — | 12 | 14 |
| SDT_k75 | 3 | 4 | 1 | — | 12 | 20 |
| SDT_k97 | 5 | 4 | 2 | — | 13 | 24 |
| SDT_k127 | 5 | 3 | 5 | 1 | 8 | 22 |
| SPAdes_k31 | 1 | 1 | — | — | 7 | 9 |
| SPAdes_k55 | 1 | 1 | — | 1 | 8 | 11 |
| SPAdes_k75 | 5 | 1 | — | 2 | 8 | 16 |
| SPAdes_k97 | 4 | 1 | 5 | 5 | 12 | 27 |
| SPAdes_k127 | 5 | 1 | 6 | 3 | 12 | 27 |
| Trinity | 7 | 3 | — | 1 | 17 | 28 |
| Crotalus adamanteus | ||||||
| BinPacker | 2.6 (1–5) | 1.8 (1–2) | 1.0 (1–1) | 0.2 (0–1) | 16.6 (14–19) | 21.6 (19–25) |
| Extender | 8.4 (6–11) | 1.4 (1–2) | 7.2 (2–11) | 5.0 (2–7) | 5.6 (2–9) | 27.6 (13–34) |
| NGen14 | 9.2 (5–12) | 1.8 (1–2) | 3.4 (1–5) | 0.4 (0–2) | 12.4 (8–16) | 27.2 (22–33) |
| SDT_k31 | 1.0 (1–1) | 1.0 (1–1) | — | — | 10.0 (8–13) | 11.4 (10–14) |
| SDT_k75 | 4.4 (2–6) | 1.2 (1–2) | 1.2 (1–2) | — | 11.4 (10–15) | 18.8 (15–22) |
| SDT_k97 | 6.8 (5–8) | 2.0 (1–2) | 2.0 (1–3) | — | 11.8 (9–15) | 22.2 (17–27) |
| SDT_k127 | 6.4 (6–7) | 1.3 (1–2) | 3.3 (2–5) | 0.2 (0–1) | 10.4 (8–14) | 20.6 (16–24) |
| SPAdes_k31 | 1.0 (1–1) | — | — | — | 3.6 (2–8) | 4.0 (2–9) |
| SPAdes_k55 | 2.3 (1–3) | 2.0 (2–2) | — | — | 5.0 (3–7) | 7.2 (4–12) |
| SPAdes_k75 | 2.7 (2–3) | 1.0 (1–1) | 2.3 (1–4) | — | 7.0 (5–11) | 10.2 (5–16) |
| SPAdes_k97 | 2.6 (1–6) | 1.0 (1–1) | 2.7 (1–4) | — | 7.4 (5–12) | 11.8 (6–21) |
| SPAdes_k127 | 3.0 (1–6) | 1.0 (1–1) | 2.8 (1–5) | 0.8 (0–2) | 12.2 (7–23) | 18.6 (8–33) |
| Trinity | 6.8 (3–11) | 1.8 (1–2) | 2.0 (2–2) | — | 17.4 (12–21) | 26.0 (21–34) |
| Crotalus cerastes | ||||||
| BinPacker | 5 | 1 | — | — | 16 | 23 |
| Extender | 6 | 1 | 9 | 10 | 5 | 32 |
| NGen14 | 12 | 1 | 7 | 1 | 13 | 35 |
| SDT_k31 | 3 | 1 | — | — | 11 | 16 |
| SDT_k75 | 10 | 1 | 1 | — | 13 | 26 |
| SDT_k97 | 11 | 1 | 4 | — | 11 | 27 |
| SDT_k127 | 12 | — | 4 | 1 | 8 | 26 |
| SPAdes_k31 | 4 | — | — | — | 7 | 12 |
| SPAdes_k55 | 4 | — | 2 | — | 8 | 14 |
| SPAdes_k75 | 11 | — | 4 | — | 6 | 22 |
| SPAdes_k97 | 7 | — | 3 | 1 | 8 | 20 |
| SPAdes_k127 | 10 | — | 6 | 1 | 13 | 31 |
| Trinity | 6 | 1 | 1 | — | 16 | 26 |
| Crotalus horridus—Juvenile | ||||||
| BinPacker | 4 | 1 | — | — | 15 | 21 |
| Extender | 4 | 1 | 9 | 6 | 4 | 24 |
| NGen14 | 6 | — | 6 | 1 | 11 | 25 |
| SDT_k31 | 1 | 1 | — | — | 11 | 13 |
| SDT_k75 | 2 | 1 | 2 | — | 10 | 17 |
| SDT_k97 | 4 | 1 | 3 | 1 | 10 | 21 |
| SDT_k127 | 5 | 1 | 5 | 1 | 6 | 18 |
| SPAdes_k31 | 2 | — | — | — | 5 | 7 |
| SPAdes_k55 | 2 | — | — | — | 7 | 10 |
| SPAdes_k75 | 5 | — | 2 | — | 9 | 17 |
| SPAdes_k97 | 6 | — | 3 | — | 9 | 19 |
| SPAdes_k127 | 6 | — | 1 | 3 | 8 | 19 |
| Trinity | 7 | 1 | — | — | 13 | 22 |
| Agkistrodon contortrix | ||||||
| BinPacker | 5 | 2 | — | — | 16 | 23 |
| Extender | 8 | 3 | 11 | 11 | 9 | 42 |
| NGen14 | 9 | 5 | 8 | 6 | 12 | 40 |
| SDT_k31 | — | 2 | — | — | 12 | 14 |
| SDT_k75 | 3 | 4 | 1 | — | 12 | 20 |
| SDT_k97 | 5 | 4 | 2 | — | 13 | 24 |
| SDT_k127 | 5 | 3 | 5 | 1 | 8 | 22 |
| SPAdes_k31 | 1 | 1 | — | — | 7 | 9 |
| SPAdes_k55 | 1 | 1 | — | 1 | 8 | 11 |
| SPAdes_k75 | 5 | 1 | — | 2 | 8 | 16 |
| SPAdes_k97 | 4 | 1 | 5 | 5 | 12 | 27 |
| SPAdes_k127 | 5 | 1 | 6 | 3 | 12 | 27 |
| Trinity | 7 | 3 | — | 1 | 17 | 28 |
| Crotalus adamanteus | ||||||
| BinPacker | 2.6 (1–5) | 1.8 (1–2) | 1.0 (1–1) | 0.2 (0–1) | 16.6 (14–19) | 21.6 (19–25) |
| Extender | 8.4 (6–11) | 1.4 (1–2) | 7.2 (2–11) | 5.0 (2–7) | 5.6 (2–9) | 27.6 (13–34) |
| NGen14 | 9.2 (5–12) | 1.8 (1–2) | 3.4 (1–5) | 0.4 (0–2) | 12.4 (8–16) | 27.2 (22–33) |
| SDT_k31 | 1.0 (1–1) | 1.0 (1–1) | — | — | 10.0 (8–13) | 11.4 (10–14) |
| SDT_k75 | 4.4 (2–6) | 1.2 (1–2) | 1.2 (1–2) | — | 11.4 (10–15) | 18.8 (15–22) |
| SDT_k97 | 6.8 (5–8) | 2.0 (1–2) | 2.0 (1–3) | — | 11.8 (9–15) | 22.2 (17–27) |
| SDT_k127 | 6.4 (6–7) | 1.3 (1–2) | 3.3 (2–5) | 0.2 (0–1) | 10.4 (8–14) | 20.6 (16–24) |
| SPAdes_k31 | 1.0 (1–1) | — | — | — | 3.6 (2–8) | 4.0 (2–9) |
| SPAdes_k55 | 2.3 (1–3) | 2.0 (2–2) | — | — | 5.0 (3–7) | 7.2 (4–12) |
| SPAdes_k75 | 2.7 (2–3) | 1.0 (1–1) | 2.3 (1–4) | — | 7.0 (5–11) | 10.2 (5–16) |
| SPAdes_k97 | 2.6 (1–6) | 1.0 (1–1) | 2.7 (1–4) | — | 7.4 (5–12) | 11.8 (6–21) |
| SPAdes_k127 | 3.0 (1–6) | 1.0 (1–1) | 2.8 (1–5) | 0.8 (0–2) | 12.2 (7–23) | 18.6 (8–33) |
| Trinity | 6.8 (3–11) | 1.8 (1–2) | 2.0 (2–2) | — | 17.4 (12–21) | 26.0 (21–34) |
| Crotalus cerastes | ||||||
| BinPacker | 5 | 1 | — | — | 16 | 23 |
| Extender | 6 | 1 | 9 | 10 | 5 | 32 |
| NGen14 | 12 | 1 | 7 | 1 | 13 | 35 |
| SDT_k31 | 3 | 1 | — | — | 11 | 16 |
| SDT_k75 | 10 | 1 | 1 | — | 13 | 26 |
| SDT_k97 | 11 | 1 | 4 | — | 11 | 27 |
| SDT_k127 | 12 | — | 4 | 1 | 8 | 26 |
| SPAdes_k31 | 4 | — | — | — | 7 | 12 |
| SPAdes_k55 | 4 | — | 2 | — | 8 | 14 |
| SPAdes_k75 | 11 | — | 4 | — | 6 | 22 |
| SPAdes_k97 | 7 | — | 3 | 1 | 8 | 20 |
| SPAdes_k127 | 10 | — | 6 | 1 | 13 | 31 |
| Trinity | 6 | 1 | 1 | — | 16 | 26 |
| Crotalus horridus—Juvenile | ||||||
| BinPacker | 4 | 1 | — | — | 15 | 21 |
| Extender | 4 | 1 | 9 | 6 | 4 | 24 |
| NGen14 | 6 | — | 6 | 1 | 11 | 25 |
| SDT_k31 | 1 | 1 | — | — | 11 | 13 |
| SDT_k75 | 2 | 1 | 2 | — | 10 | 17 |
| SDT_k97 | 4 | 1 | 3 | 1 | 10 | 21 |
| SDT_k127 | 5 | 1 | 5 | 1 | 6 | 18 |
| SPAdes_k31 | 2 | — | — | — | 5 | 7 |
| SPAdes_k55 | 2 | — | — | — | 7 | 10 |
| SPAdes_k75 | 5 | — | 2 | — | 9 | 17 |
| SPAdes_k97 | 6 | — | 3 | — | 9 | 19 |
| SPAdes_k127 | 6 | — | 1 | 3 | 8 | 19 |
| Trinity | 7 | 1 | — | — | 13 | 22 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Holding, M.L.; Margres, M.J.; Mason, A.J.; Parkinson, C.L.; Rokyta, D.R. Evaluating the Performance of De Novo Assembly Methods for Venom-Gland Transcriptomics. Toxins 2018, 10, 249. https://doi.org/10.3390/toxins10060249
Holding ML, Margres MJ, Mason AJ, Parkinson CL, Rokyta DR. Evaluating the Performance of De Novo Assembly Methods for Venom-Gland Transcriptomics. Toxins. 2018; 10(6):249. https://doi.org/10.3390/toxins10060249
Chicago/Turabian StyleHolding, Matthew L., Mark J. Margres, Andrew J. Mason, Christopher L. Parkinson, and Darin R. Rokyta. 2018. "Evaluating the Performance of De Novo Assembly Methods for Venom-Gland Transcriptomics" Toxins 10, no. 6: 249. https://doi.org/10.3390/toxins10060249
APA StyleHolding, M. L., Margres, M. J., Mason, A. J., Parkinson, C. L., & Rokyta, D. R. (2018). Evaluating the Performance of De Novo Assembly Methods for Venom-Gland Transcriptomics. Toxins, 10(6), 249. https://doi.org/10.3390/toxins10060249