The Binary Toxin CDT of Clostridium difficile as a Tool for Intracellular Delivery of Bacterial Glucosyltransferase Domains

 and
and
Abstract
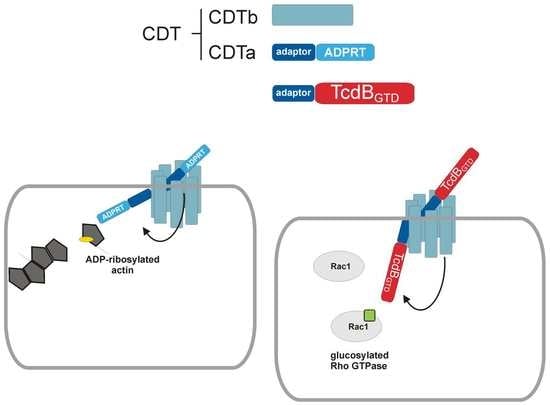
1. Introduction
2. Results
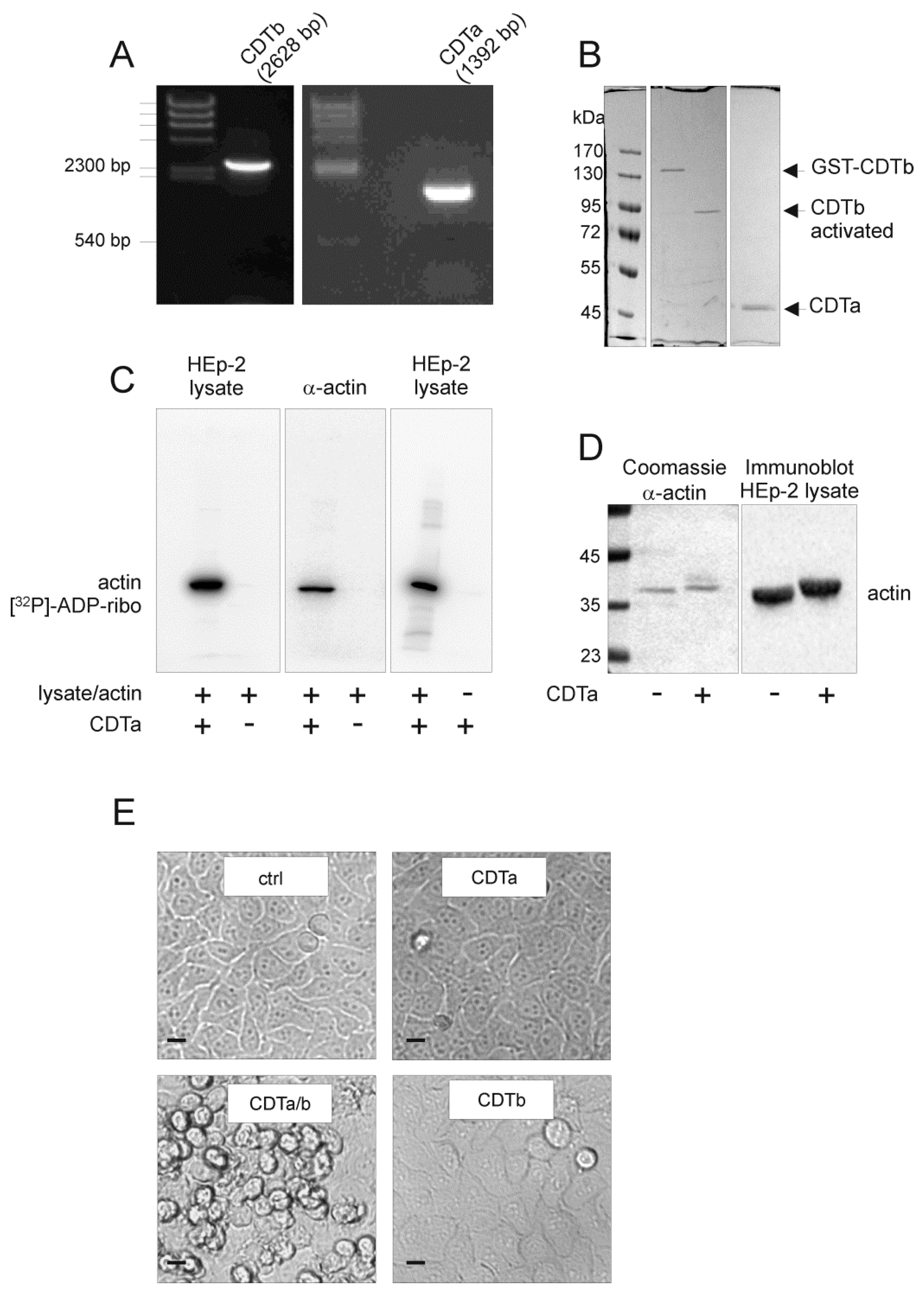
2.1. Cloning and Characterization of CDT
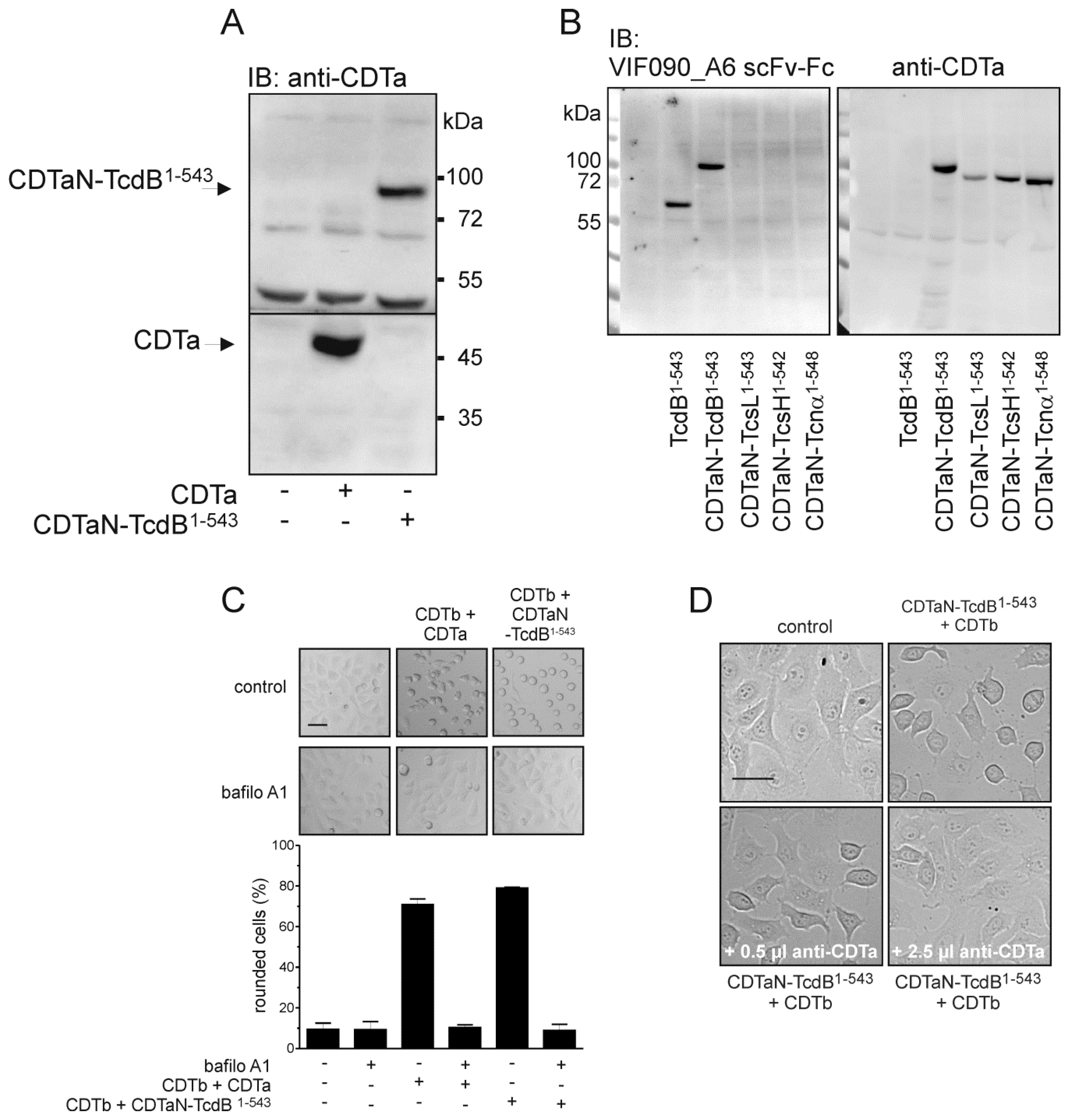
2.2. C-Terminal Fusion Proteins are Delivered into Target Cells
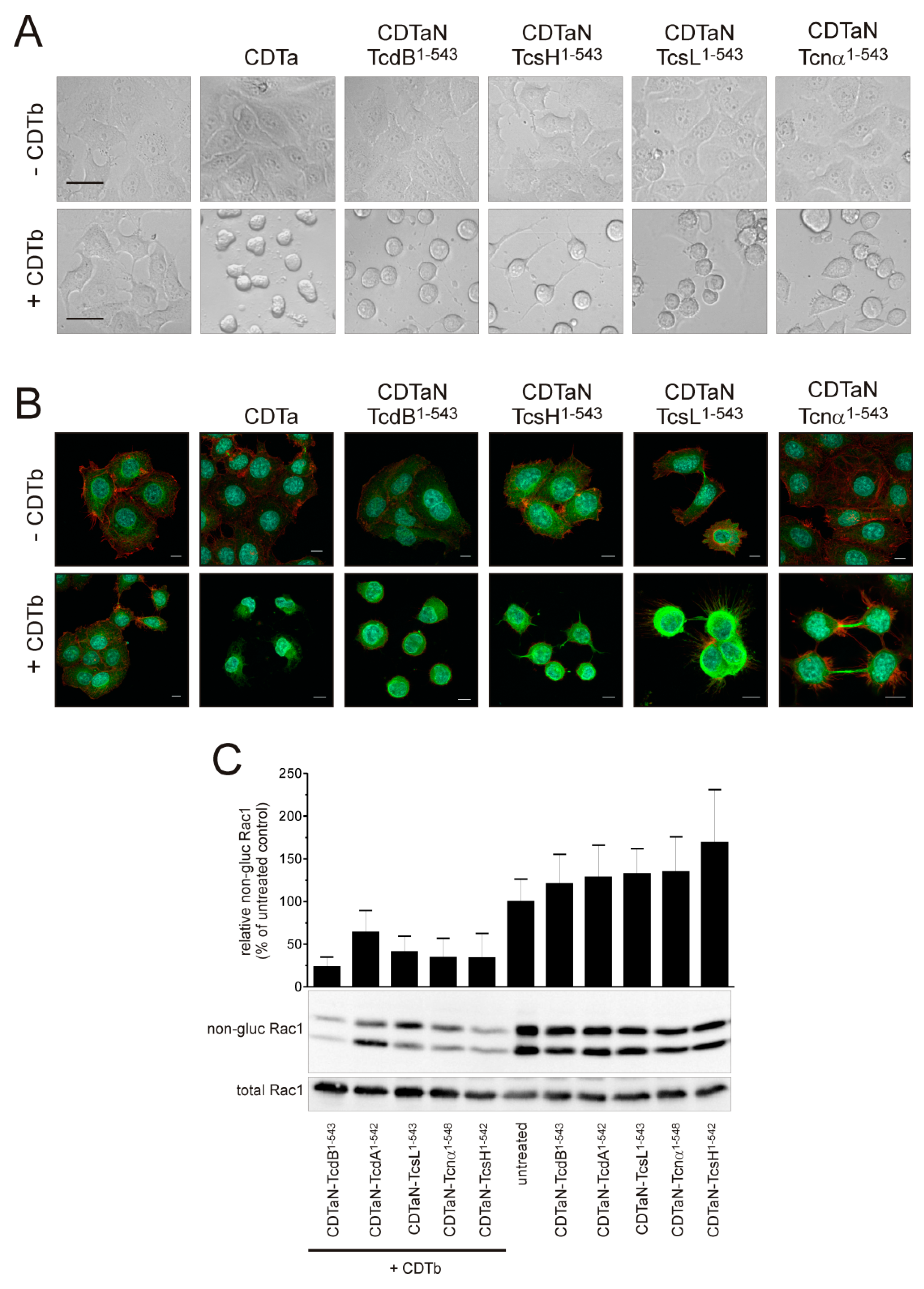
2.3. Application of the CDT Delivery System with a Variety of Glucosyltransferase Domains
2.4. Cell Surface Binding of CDTaN Fusion Proteins
3. Discussion
4. Materials and Methods
4.1. Cloning of CDTa, CDTb, and CDTaN Fusion Proteins
4.2. Purification and Activation of CDTa and CDTb
4.3. In Vitro ADP-Ribosylation and Glucosylation Assays
4.4. Cell Culture and Cell Rounding Assay
4.5. Toxin Binding Assay
4.6. Immunostaining and Fluorescence Microscopy
4.7. Generation of a Monoclonal Anti-TcdB Antibody
4.8. Polyclonal Anti-CDTa Rabbit Serum
4.9. Western Blot
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Smits, W.K.; Lyras, D.; Lacy, D.B.; Wilcox, M.H.; Kuijper, E.J. Clostridium difficile infection. Nat. Rev. Dis. Prim. 2016, 2, 16020. [Google Scholar] [CrossRef] [PubMed]
- Just, I.; Selzer, J.; Wilm, M.; von Eichel-Streiber, C.; Mann, M.; Aktories, K. Glucosylation of rho proteins by Clostridium difficile toxin b. Nature 1995, 375, 500–503. [Google Scholar] [CrossRef] [PubMed]
- Popoff, M.R.; Rubin, E.J.; Gill, D.M.; Boquet, P. Actin-specific ADP-ribosyltransferase produced by a Clostridium difficile strain. Infect. Immun. 1988, 56, 2299–2306. [Google Scholar] [PubMed]
- Bauer, M.P.; Notermans, D.W.; van Benthem, B.H.; Brazier, J.S.; Wilcox, M.H.; Rupnik, M.; Monnet, D.L.; van Dissel, J.T.; Kuijper, E.J.; Group, E.S. Clostridium difficile infection in europe: A hospital-based survey. Lancet 2011, 377, 63–73. [Google Scholar] [CrossRef]
- Smits, W.K. Hype or hypervirulence: A reflection on problematic C. difficile strains. Virulence 2013, 4, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Hunt, J.J.; Ballard, J.D. Variations in virulence and molecular biology among emerging strains of Clostridium difficile. Microbiol. Mol. Biol. Rev. 2013, 77, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Stabler, R.A.; He, M.; Dawson, L.; Martin, M.; Valiente, E.; Corton, C.; Lawley, T.D.; Sebaihia, M.; Quail, M.A.; Rose, G.; et al. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol. 2009, 10, R102. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Aktories, K.; Popoff, M.R.; Stiles, B.G. Binary bacterial toxins: Biochemistry, biology, and applications of common clostridium and bacillus proteins. Microbiol. Mol. Biol. Rev. 2004, 68, 373–402. [Google Scholar] [CrossRef] [PubMed]
- Gerding, D.N.; Johnson, S.; Rupnik, M.; Aktories, K. Clostridium difficile binary toxin CDT: Mechanism, epidemiology, and potential clinical importance. Gut Microbes 2014, 5, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Perelle, S.; Gibert, M.; Bourlioux, P.; Corthier, G.; Popoff, M.R. Production of a complete binary toxin (actin-specific ADP-ribosyltransferase) by Clostridium difficile cd196. Infect. Immun. 1997, 65, 1402–1407. [Google Scholar] [PubMed]
- Sundriyal, A.; Roberts, A.K.; Shone, C.C.; Acharya, K.R. Structural basis for substrate recognition in the enzymatic component of ADP-ribosyltransferase toxin CDTa from Clostridium difficile. J. Biol. Chem. 2009, 284, 28713–28719. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, P.; Carette, J.E.; Bell, G.W.; Schwan, C.; Guttenberg, G.; Brummelkamp, T.R.; Aktories, K. Lipolysis-stimulated lipoprotein receptor (LSR) is the host receptor for the binary toxin Clostridium difficile transferase (CDT). Proc. Natl. Acad. Sci. USA 2011, 108, 16422–16427. [Google Scholar] [CrossRef] [PubMed]
- Papatheodorou, P.; Wilczek, C.; Nolke, T.; Guttenberg, G.; Hornuss, D.; Schwan, C.; Aktories, K. Identification of the cellular receptor of Clostridium spiroforme toxin. Infect. Immun. 2012, 80, 1418–1423. [Google Scholar] [CrossRef] [PubMed]
- Aktories, K.; Papatheodorou, P.; Schwan, C. Binary Clostridium difficile toxin (CDT)—A virulence factor disturbing the cytoskeleton. Anaerobe 2018. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Blocker, D.; Aktories, K. The uptake machinery of clostridial actin ADP-ribosylating toxins—A cell delivery system for fusion proteins and polypeptide drugs. Naunyn Schmiedebergs Arch. Pharmacol. 2002, 366, 501–512. [Google Scholar] [CrossRef] [PubMed]
- Arora, N.; Klimpel, K.R.; Singh, Y.; Leppla, S.H. Fusions of anthrax toxin lethal factor to the ADP-ribosylation domain of Pseudomonas exotoxin A are potent cytotoxins which are translocated to the cytosol of mammalian cells. J. Biol. Chem. 1992, 267, 15542–15548. [Google Scholar] [PubMed]
- Spyres, L.M.; Qa’Dan, M.; Meader, A.; Tomasek, J.J.; Howard, E.W.; Ballard, J.D. Cytosolic delivery and characterization of the TcdB glucosylating domain by using a heterologous protein fusion. Infect. Immun. 2001, 69, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Hofmann, F.; Olenik, C.; Just, I.; Aktories, K. The N-terminal part of the enzyme component (C2I) of the binary Clostridium botulinum C2 toxin interacts with the binding component C2II and functions as a carrier system for a Rho ADP-ribosylating C3-like fusion toxin. Infect. Immun. 1998, 66, 1364–1369. [Google Scholar] [PubMed]
- Wilde, C.; Chhatwal, G.S.; Schmalzing, G.; Aktories, K.; Just, I. A novel C3-like ADP-ribosyltransferase from Staphylococcus aureus modifying RhoE and Rnd3. J. Biol. Chem. 2001, 276, 9537–9542. [Google Scholar] [CrossRef] [PubMed]
- Pust, S.; Hochmann, H.; Kaiser, E.; von Figura, G.; Heine, K.; Aktories, K.; Barth, H. A cell-permeable fusion toxin as a tool to study the consequences of actin-ADP-ribosylation caused by the Salmonella enterica virulence factor spvB in intact cells. J. Biol. Chem. 2007, 282, 10272–10282. [Google Scholar] [CrossRef] [PubMed]
- Fahrer, J.; Rieger, J.; van Zandbergen, G.; Barth, H. The c2-streptavidin delivery system promotes the uptake of biotinylated molecules in macrophages and t-leukemia cells. Biol. Chem. 2010, 391, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Fahrer, J.; Schweitzer, B.; Fiedler, K.; Langer, T.; Gierschik, P.; Barth, H. C2-streptavidin mediates the delivery of biotin-conjugated tumor suppressor protein p53 into tumor cells. Bioconjug. Chem. 2013, 24, 595–603. [Google Scholar] [CrossRef] [PubMed]
- Marvaud, J.C.; Stiles, B.G.; Chenal, A.; Gillet, D.; Gibert, M.; Smith, L.A.; Popoff, M.R. Clostridium perfringens Iota toxin. Mapping of the ia domain involved in docking with Ib and cellular internalization. J. Biol. Chem. 2002, 277, 43659–43666. [Google Scholar] [CrossRef] [PubMed]
- Riese, M.J.; Goehring, U.M.; Ehrmantraut, M.E.; Moss, J.; Barbieri, J.T.; Aktories, K.; Schmidt, G. Auto-ADP-ribosylation of Pseudomonas aeruginosa exoS. J. Biol. Chem. 2002, 277, 12082–12088. [Google Scholar] [CrossRef] [PubMed]
- Genth, H.; Huelsenbeck, J.; Hartmann, B.; Hofmann, F.; Just, I.; Gerhard, R. Cellular stability of Rho-GTPases glucosylated by Clostridium difficile toxin B. FEBS Lett. 2006, 580, 3565–3569. [Google Scholar] [CrossRef] [PubMed]
- Just, I.; Gerhard, R. Large clostridial cytotoxins. Rev. Physiol. Biochem. Pharmacol. 2004, 152, 23–47. [Google Scholar] [PubMed]
- Goy, S.D.; Olling, A.; Neumann, D.; Pich, A.; Gerhard, R. Human neutrophils are activated by a peptide fragment of Clostridium difficile toxin B presumably via formyl peptide receptor. Cell. Microbiol. 2015, 17, 893–909. [Google Scholar] [CrossRef] [PubMed]
- Barth, H.; Stiles, B.G. Binary actin-ADP-ribosylating toxins and their use as molecular trojan horses for drug delivery into eukaryotic cells. Curr. Med. Chem. 2008, 15, 459–469. [Google Scholar] [CrossRef] [PubMed]
- Haug, G.; Wilde, C.; Leemhuis, J.; Meyer, D.K.; Aktories, K.; Barth, H. Cellular uptake of Clostridium botulinum C2 toxin: Membrane translocation of a fusion toxin requires unfolding of its dihydrofolate reductase domain. Biochemistry 2003, 42, 15284–15291. [Google Scholar] [CrossRef] [PubMed]
- Haug, G.; Barth, H.; Aktories, K. Purification and activity of the Rho ADP-ribosylating binary C2/C3 toxin. Methods Enzymol. 2006, 406, 117–127. [Google Scholar] [PubMed]
- Fahrer, J.; Plunien, R.; Binder, U.; Langer, T.; Seliger, H.; Barth, H. Genetically engineered clostridial C2 toxin as a novel delivery system for living mammalian cells. Bioconjug. Chem. 2010, 21, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Fahrer, J.; Kuban, J.; Heine, K.; Rupps, G.; Kaiser, E.; Felder, E.; Benz, R.; Barth, H. Selective and specific internalization of clostridial C3 ADP-ribosyltransferases into macrophages and monocytes. Cell. Microbiol. 2010, 12, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Rohrbeck, A.; Schroder, A.; Hagemann, S.; Pich, A.; Holtje, M.; Ahnert-Hilger, G.; Just, I. Vimentin mediates uptake of C3 exoenzyme. PLoS ONE 2014, 9, e101071. [Google Scholar] [CrossRef] [PubMed]
- Rohrbeck, A.; von Elsner, L.; Hagemann, S.; Just, I. Uptake of Clostridium botulinum C3 exoenzyme into intact HT22 and J774a.1 cells. Toxins (Basel) 2015, 7, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Muetzelburg, M.V.; Hofmann, F.; Just, I.; Pich, A. Identification of biomarkers indicating cellular changes after treatment of neuronal cells with the C3 exoenzyme from Clostridium botulinum using the ITRAQ protocol and LC-MS/MS analysis. J. Chromatogr. B 2009, 877, 1344–1351. [Google Scholar] [CrossRef] [PubMed]
- Adolf, A.; Leondaritis, G.; Rohrbeck, A.; Eickholt, B.J.; Just, I.; Ahnert-Hilger, G.; Holtje, M. The intermediate filament protein vimentin is essential for axonotrophic effects of Clostridium botulinum C3 exoenzyme. J. Neurochem. 2016, 139, 234–244. [Google Scholar] [CrossRef] [PubMed]
- Fantini, J.; Di Scala, C.; Evans, L.S.; Williamson, P.T.; Barrantes, F.J. A mirror code for protein-cholesterol interactions in the two leaflets of biological membranes. Sci. Rep. 2016, 6, 21907. [Google Scholar] [CrossRef] [PubMed]
- Jank, T.; Bohmer, K.E.; Tzivelekidis, T.; Schwan, C.; Belyi, Y.; Aktories, K. Domain organization of legionella effector setA. Cell. Microbiol. 2012, 14, 852–868. [Google Scholar] [CrossRef] [PubMed]
- Belyi, Y.; Tartakovskaya, D.; Tais, A.; Fitzke, E.; Tzivelekidis, T.; Jank, T.; Rospert, S.; Aktories, K. Elongation factor 1a is the target of growth inhibition in yeast caused by Legionella pneumophila glucosyltransferase Lgt1. J. Biol. Chem. 2012, 287, 26029–26037. [Google Scholar] [CrossRef] [PubMed]
- Thalmann, J.; Janik, K.; May, M.; Sommer, K.; Ebeling, J.; Hofmann, F.; Genth, H.; Klos, A. Actin re-organization induced by Chlamydia trachomatis serovar d--evidence for a critical role of the effector protein CT166 targeting Rac. PLoS ONE 2010, 5, e9887. [Google Scholar] [CrossRef] [PubMed]
- Reaves, D.K.; Fagan-Solis, K.D.; Dunphy, K.; Oliver, S.D.; Scott, D.W.; Fleming, J.M. The role of lipolysis stimulated lipoprotein receptor in breast cancer and directing breast cancer cell behavior. PLoS ONE 2014, 9, e91747. [Google Scholar] [CrossRef] [PubMed]
- Hemmasi, S.; Czulkies, B.A.; Schorch, B.; Veit, A.; Aktories, K.; Papatheodorou, P. Interaction of the Clostridium difficile binary toxin CDT and its host cell receptor, lipolysis-stimulated lipoprotein receptor (LSR). J. Biol. Chem. 2015, 290, 14031–14044. [Google Scholar] [CrossRef] [PubMed]
- Fagan-Solis, K.D.; Reaves, D.K.; Rangel, M.C.; Popoff, M.R.; Stiles, B.G.; Fleming, J.M. Challenging the roles of CD44 and lipolysis stimulated lipoprotein receptor in conveying Clostridium perfringens iota toxin cytotoxicity in breast cancer. Mol. Cancer 2014, 13, 163. [Google Scholar] [CrossRef] [PubMed]
- Schorch, B.; Heni, H.; Zahaf, N.I.; Brummer, T.; Mione, M.; Schmidt, G.; Papatheodorou, P.; Aktories, K. Targeting oncogenic ras by the Clostridium perfringens toxin TpeL. Oncotarget 2018, 9, 16489–16500. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed]
- Kugler, J.; Wilke, S.; Meier, D.; Tomszak, F.; Frenzel, A.; Schirrmann, T.; Dubel, S.; Garritsen, H.; Hock, B.; Toleikis, L.; et al. Generation and analysis of the improved human HAL9/10 antibody phage display libraries. BMC Biotechnol. 2015, 15, 10. [Google Scholar] [CrossRef] [PubMed]
- Russo, G.; Meier, D.; Helmsing, S.; Wenzel, E.; Oberle, F.; Frenzel, A.; Hust, M. Parallelized antibody selection in microtiter plates. Methods Mol. Biol. 2018, 1701, 273–284. [Google Scholar] [PubMed]
- Jager, V.; Bussow, K.; Wagner, A.; Weber, S.; Hust, M.; Frenzel, A.; Schirrmann, T. High level transient production of recombinant antibodies and antibody fusion proteins in HEK293 cells. BMC Biotechnol. 2013, 13, 52. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Base Sequence (3’-5’) | |
|---|---|---|
| pGEX-2T constructs | CDTa (aa 1–463) s | TAGGATCCAAAAAATTTAGGAAACATAAAAGGATTAG |
| CDTa (aa 44–463) s | TTAGGATCCGTTTGCAATACTACTTACAAGGC | |
| CDTa (aa 1–463) a | ATGAATTCTTAAGGTATCAATGTTGCATCAAC | |
| CDTb (aa 1–876) s | TAAGATCTAAAATACAAATGAGGAATAAAAAGGTATTAAG | |
| CDTb (aa 43–876) s | TAAGATCTGAAATTGTAAATGAAGATATACTCCC | |
| CDTb (aa 1–876) a | ATGAATTCCTAATCAACACTAAGAACTAATAACTCTC | |
| pQE30 constructs | CDTaN Bam s | AATGGATCCGTTTGCAATACTACTTACAAGG |
| CDTaN Kpn a | AATGGTACCATCATCTTTAAAATCAAGACTATTTAC | |
| GTD TcdB Kpn s | AATGGTACCATGAGTTTAGTTAATAGAAAACAGTTAG | |
| GTD TcdB Hind a | AATAAGCTTTTAAAGAGAACCTTCAAAATAATTCCTTTTATATTC | |
| GTD TcdA Kpn s | AATGGTACCATGTCTTTAATATCTAAAGAAGAG | |
| GTD TcdA Hind a | AATAABCTTTTAAAGAGATCCACCAGTATAATCTC | |
| GTD TcnA Kpn s | AATGGTACCATGCTTATAACAAGAGAACAATTAATG | |
| GTD TcnA Hind a | AATAAGCTTTTAGAGAGTTCTTCCTATATAAGTTTTTATC | |
| GTD TcsH Kpn s | AATGGTACCATGTCTTTAATATCTAAAGATGAATTAATAAAAC | |
| GTD TcsH Hind a | AATAAGCTTTTAAAGAGATTCCTGAGTATAATCTCTTAC | |
| GTD TcsL Kpn s | AATGGTACCATGAACTTAGTTAACAAAGCCC | |
| GTD TcsL Hind a | AATAAGCTTTTAAAGTGCACCTTCAAAATAACC |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Beer, L.-A.; Tatge, H.; Schneider, C.; Ruschig, M.; Hust, M.; Barton, J.; Thiemann, S.; Fühner, V.; Russo, G.; Gerhard, R. The Binary Toxin CDT of Clostridium difficile as a Tool for Intracellular Delivery of Bacterial Glucosyltransferase Domains. Toxins 2018, 10, 225. https://doi.org/10.3390/toxins10060225
Beer L-A, Tatge H, Schneider C, Ruschig M, Hust M, Barton J, Thiemann S, Fühner V, Russo G, Gerhard R. The Binary Toxin CDT of Clostridium difficile as a Tool for Intracellular Delivery of Bacterial Glucosyltransferase Domains. Toxins. 2018; 10(6):225. https://doi.org/10.3390/toxins10060225
Chicago/Turabian StyleBeer, Lara-Antonia, Helma Tatge, Carmen Schneider, Maximilian Ruschig, Michael Hust, Jessica Barton, Stefan Thiemann, Viola Fühner, Giulio Russo, and Ralf Gerhard. 2018. "The Binary Toxin CDT of Clostridium difficile as a Tool for Intracellular Delivery of Bacterial Glucosyltransferase Domains" Toxins 10, no. 6: 225. https://doi.org/10.3390/toxins10060225
APA StyleBeer, L.-A., Tatge, H., Schneider, C., Ruschig, M., Hust, M., Barton, J., Thiemann, S., Fühner, V., Russo, G., & Gerhard, R. (2018). The Binary Toxin CDT of Clostridium difficile as a Tool for Intracellular Delivery of Bacterial Glucosyltransferase Domains. Toxins, 10(6), 225. https://doi.org/10.3390/toxins10060225