Botulinum Toxin for Central Neuropathic Pain

Abstract
1. Introduction
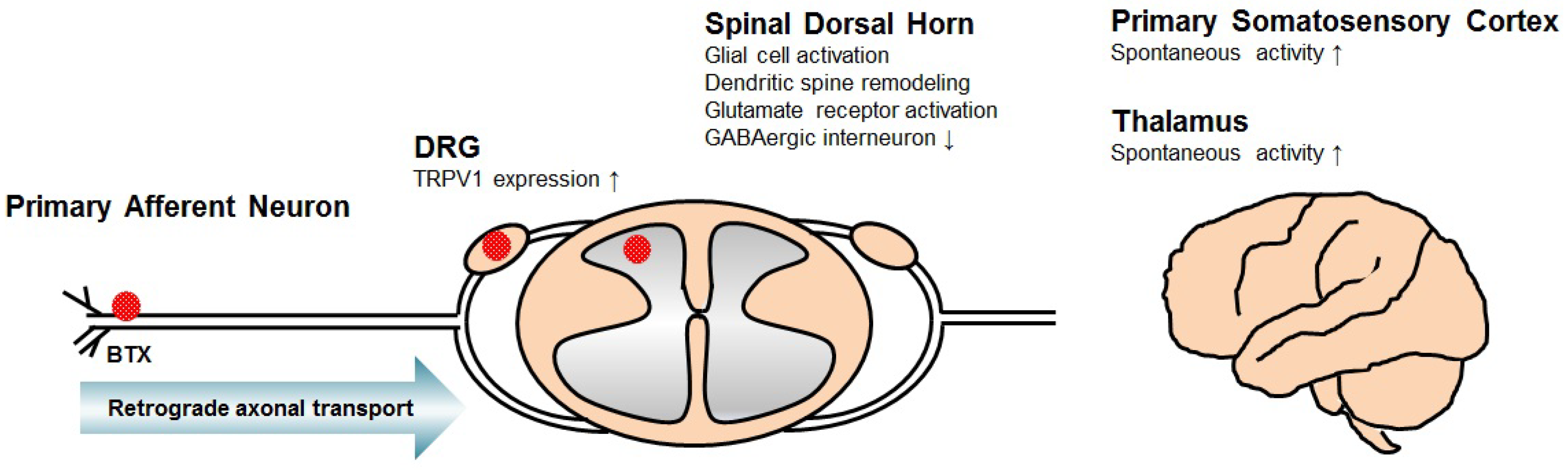
2. Mechanism of Central Neuropathic Pain
3. Mechanism of BTX for Central Neuropathic Pain
4. Clinical Studies of BTX for Central Neuropathic Pain
4.1. Neuropathic Pain after Spinal Cord Injury
4.2. Post-Stroke Shoulder Pain
4.3. Multiple Sclerosis
4.4. Complex Regional Pain Syndrome
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Simpson, L.L. The origin, structure, and pharmacological activity of botulinum toxin. Pharmacol. Rev. 1981, 33, 155–188. [Google Scholar] [PubMed]
- Tian, J.H.; Wu, Z.X.; Unzicker, M.; Lu, L.; Cai, Q.; Li, C.; Schirra, C.; Matti, U.; Stevens, D.; Deng, C.; et al. The role of Snapin in neurosecretion: Snapin knock-out mice exhibit impaired calcium-dependent exocytosis of large dense-core vesicles in chromaffin cells. J. Neurosci. 2005, 25, 10546–10555. [Google Scholar] [CrossRef] [PubMed]
- Ilardi, J.M.; Mochida, S.; Sheng, Z.H. Snapin: A SNARE-associated protein implicated in synaptic transmission. Nat. Neurosci. 1999, 2, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Matak, I.; Lacković, Z. Botulinum toxin A, brain and pain. Prog. Neurobiol. 2014, 119–120, 39–59. [Google Scholar] [CrossRef] [PubMed]
- Evans, D.M.; Williams, R.S.; Shone, C.C.; Hambleton, P.; Melling, J.; Dolly, J.O. Botulinum neurotoxin type B. Its purification, radioiodination and interaction with rat-brain synaptosomal membranes. Eur. J. Biochem. 1986, 154, 409–416. [Google Scholar] [CrossRef] [PubMed]
- Black, J.D. Interaction of 125I-labeled botulinum neurotoxins with nerve terminals. II. Autoradiographic evidence for its uptake into motor nerves by acceptor-mediated endocytosis. J. Cell Biol. 1986, 103, 535–544. [Google Scholar] [CrossRef] [PubMed]
- Simpson, L.L. The binding fragment from tetanus toxin antagonizes the neuromuscular blocking actions of botulinum toxin. J. Pharmacol. Exp. Ther. 1984, 229, 182–187. [Google Scholar] [PubMed]
- Castiglione, A.; Bagnato, S.; Boccagni, C.; Romano, M.C.; Galardi, G. Efficacy of intra-articular injection of botulinum toxin type A in refractory hemiplegic shoulder pain. Arch. Phys. Med. Rehabil. 2011, 92, 1034–1037. [Google Scholar] [CrossRef] [PubMed]
- Safarpour, Y.; Jabbari, B. Botulinum toxin treatment of pain syndromes -an evidence based review. Toxicon 2018. [Google Scholar] [CrossRef] [PubMed]
- Merskey, H.B.N. (Ed.) Classification of Chronic Pain: Descriptions of Chronic Pain Syndromes and Definitions of Pain Terms, 2nd ed.; IASP Press: Seattle, WA, USA, 1994. [Google Scholar]
- Reeh, P.W.; Kress, M. Molecular physiology of proton transduction in nociceptors. Curr. Opin. Pharmacol. 2001, 1, 45–51. [Google Scholar] [CrossRef]
- Bedi, S.S.; Yang, Q.; Crook, R.J.; Du, J.; Wu, Z.; Fishman, H.M.; Grill, R.J.; Carlton, S.M.; Walters, E.T. Chronic spontaneous activity generated in the somata of primary nociceptors is associated with pain-related behavior after spinal cord injury. J. Neurosci. 2010, 30, 14870–14882. [Google Scholar] [CrossRef] [PubMed]
- Ramer, L.M.; van Stolk, A.P.; Inskip, J.A.; Ramer, M.S.; Krassioukov, A.V. Plasticity of TRPV1-Expressing Sensory Neurons Mediating Autonomic Dysreflexia Following Spinal Cord Injury. Front. Physiol. 2012, 3, 257. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xie, W.; Xie, Y. Spinal cord injury triggers sensitization of wide dynamic range dorsal horn neurons in segments rostral to the injury. Brain Res. 2005, 1055, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Gwak, Y.S.; Kang, J.; Leem, J.W.; Hulsebosch, C.E. Spinal AMPA receptor inhibition attenuates mechanical allodynia and neuronal hyperexcitability following spinal cord injury in rats. J. Neurosci. Res. 2007, 85, 2352–2359. [Google Scholar] [CrossRef] [PubMed]
- Putatunda, R.; Hala, T.J.; Chin, J.; Lepore, A.C. Chronic at-level thermal hyperalgesia following rat cervical contusion spinal cord injury is accompanied by neuronal and astrocyte activation and loss of the astrocyte glutamate transporter, GLT1, in superficial dorsal horn. Brain Res. 2014, 1581, 64–79. [Google Scholar] [CrossRef] [PubMed]
- Gwak, Y.S.; Kang, J.; Unabia, G.C.; Hulsebosch, C.E. Spatial and temporal activation of spinal glial cells: Role of gliopathy in central neuropathic pain following spinal cord injury in rats. Exp. Neurol. 2012, 234, 362–372. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.M.; Stamboulian, S.; Chang, Y.W.; Zhao, P.; Hains, A.B.; Waxman, S.G.; Hains, B.C. Neuropathic pain memory is maintained by Rac1-regulated dendritic spine remodeling after spinal cord injury. J. Neurosci. 2008, 28, 13173–13183. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.; Hill, M.; Liu, S.; Chen, L.; Bangalore, L.; Waxman, S.G.; Tan, A.M. Dendritic spine remodeling following early and late Rac1 inhibition after spinal cord injury: Evidence for a pain biomarker. J. Neurophysiol. 2016, 115, 2893–2910. [Google Scholar] [CrossRef] [PubMed]
- Leem, J.W.; Kim, H.K.; Hulsebosch, C.E.; Gwak, Y.S. Ionotropic glutamate receptors contribute to maintained neuronal hyperexcitability following spinal cord injury in rats. Exp. Neurol. 2010, 224, 321–324. [Google Scholar] [CrossRef] [PubMed]
- Meisner, J.G.; Marsh, A.D.; Marsh, D.R. Loss of GABAergic interneurons in laminae I-III of the spinal cord dorsal horn contributes to reduced GABAergic tone and neuropathic pain after spinal cord injury. J. Neurotrauma 2010, 27, 729–737. [Google Scholar] [CrossRef] [PubMed]
- Berrocal, Y.A.; Almeida, V.W.; Puentes, R.; Knott, E.P.; Hechtman, J.F.; Garland, M.; Pearse, D.D. Loss of central inhibition: Implications for behavioral hypersensitivity after contusive spinal cord injury in rats. Pain Res. Treat. 2014, 2014, 178278. [Google Scholar] [CrossRef] [PubMed]
- Hains, B.C.; Johnson, K.M.; Eaton, M.J.; Willis, W.D.; Hulsebosch, C.E. Serotonergic neural precursor cell grafts attenuate bilateral hyperexcitability of dorsal horn neurons after spinal hemisection in rat. Neuroscience 2003, 116, 1097–1110. [Google Scholar] [CrossRef]
- Lin, C.Y.; Lee, Y.S.; Lin, V.W.; Silver, J. Fibronectin inhibits chronic pain development after spinal cord injury. J. Neurotrauma. 2012, 29, 589–599. [Google Scholar] [CrossRef] [PubMed]
- Boroujerdi, A.; Zeng, J.; Sharp, K.; Kim, D.; Steward, O.; Luo, Z.D. Calcium channel alpha-2-delta-1 protein upregulation in dorsal spinal cord mediates spinal cord injury-induced neuropathic pain states. Pain 2011, 152, 649–655. [Google Scholar] [CrossRef] [PubMed]
- Lenz, F.A.; Kwan, H.C.; Dostrovsky, J.O.; Tasker, R.R. Characteristics of the bursting pattern of action potentials that occurs in the thalamus of patients with central pain. Brain Res. 1989, 496, 357–360. [Google Scholar] [CrossRef]
- Gerke, M.B.; Duggan, A.W.; Xu, L.; Siddall, P.J. Thalamic neuronal activity in rats with mechanical allodynia following contusive spinal cord injury. Neuroscience 2003, 117, 715–722. [Google Scholar] [CrossRef]
- Quiton, R.L.; Masri, R.; Thompson, S.M.; Keller, A. Abnormal activity of primary somatosensory cortex in central pain syndrome. J. Neurophysiol. 2010, 104, 1717–1725. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Voulalas, P.; Ji, Y.; Masri, R. Post-translational modification of cortical GluA receptors in rodents following spinal cord lesion. Neuroscience 2016, 316, 122–129. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, R.A.; de Andrade, D.C.; Machado, A.G.; Teixeira, M.J. Central poststroke pain: Somatosensory abnormalities and the presence of associated myofascial pain syndrome. BMC Neurol. 2012, 12, 89. [Google Scholar] [CrossRef] [PubMed]
- Kalita, J.; Kumar, B.; Misra, U.K.; Pradhan, P.K. Central post stroke pain: Clinical, MRI, and SPECT correlation. Pain Med. 2011, 12, 282–288. [Google Scholar] [CrossRef] [PubMed]
- Landerholm, A.H.; Hansson, P.T. Mechanisms of dynamic mechanical allodynia and dysesthesia in patients with peripheral and central neuropathic pain. Eur. J. Pain (Lond. Engl.) 2011, 15, 498–503. [Google Scholar] [CrossRef] [PubMed]
- Sprenger, T.; Seifert, C.L.; Valet, M.; Andreou, A.P.; Foerschler, A.; Zimmer, C.; Collins, D.L.; Goadsby, P.J.; Tolle, T.R.; Chakravarty, M.M. Assessing the risk of central post-stroke pain of thalamic origin by lesion mapping. Brain J. Neurol. 2012, 135, 2536–2545. [Google Scholar] [CrossRef] [PubMed]
- Hong, J.H.; Bai, D.S.; Jeong, J.Y.; Choi, B.Y.; Chang, C.H.; Kim, S.H.; Ahn, S.H.; Jang, S.H. Injury of the spino-thalamo-cortical pathway is necessary for central post-stroke pain. Eur. Neurol. 2010, 64, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Krause, T.; Brunecker, P.; Pittl, S.; Taskin, B.; Laubisch, D.; Winter, B.; Lentza, M.E.; Malzahn, U.; Villringer, K.; Villringer, A.; et al. Thalamic sensory strokes with and without pain: Differences in lesion patterns in the ventral posterior thalamus. J. Neurol. Neurosurg. Psychiatry 2012, 83, 776–784. [Google Scholar] [CrossRef] [PubMed]
- Blasi, F.; Herisson, F.; Wang, S.; Mao, J.; Ayata, C. Late-onset thermal hypersensitivity after focal ischemic thalamic infarcts as a model for central post-stroke pain in rats. J. Cereb. Blood Flow Metab. 2015, 35, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.C.; Chang, W.J.; Kuan, Y.H.; Huang, A.C.; Shyu, B.C. A [14C]iodoantipyrine study of inter-regional correlations of neural substrates following central post-stroke pain in rats. Mol. Pain 2015, 11, 9. [Google Scholar] [CrossRef] [PubMed]
- Takami, K.; Fujita-Hamabe, W.; Harada, S.; Tokuyama, S. Abeta and Adelta but not C-fibres are involved in stroke related pain and allodynia: An experimental study in mice. J. Pharmacy Pharmacol. 2011, 63, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Kuan, Y.H.; Shih, H.C.; Tang, S.C.; Jeng, J.S.; Shyu, B.C. Targeting P(2)X(7) receptor for the treatment of central post-stroke pain in a rodent model. Neurobiol. Dis. 2015, 78, 134–145. [Google Scholar] [CrossRef] [PubMed]
- McMahon, H.T.; Foran, P.; Dolly, J.O.; Verhage, M.; Wiegant, V.M.; Nicholls, D.G. Tetanus toxin and botulinum toxins type A and B inhibit glutamate, gamma-aminobutyric acid, aspartate, and met-enkephalin release from synaptosomes. Clues to the locus of action. J. Biol. Chem. 1992, 267, 21338–21343. [Google Scholar] [PubMed]
- Welch, M.J.; Purkiss, J.R.; Foster, K.A. Sensitivity of embryonic rat dorsal root ganglia neurons to Clostridium botulinum neurotoxins. Toxicon 2000, 38, 245–258. [Google Scholar] [CrossRef]
- Durham, P.L.; Cady, R.; Cady, R. Regulation of calcitonin gene-related peptide secretion from trigeminal nerve cells by botulinum toxin type A: Implications for migraine therapy. Headache 2004, 44, 35–42; discussion 33–42. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Cheng, J.; Zhuang, Y.; Qu, W.; Muir, J.; Liang, H.; Zhang, D. Botulinum toxin type A reduces hyperalgesia and TRPV1 expression in rats with neuropathic pain. Pain Med. 2013, 14, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Xiao, L.; Cheng, J.; Dai, J.; Zhang, D. Botulinum toxin decreases hyperalgesia and inhibits P2X3 receptor over-expression in sensory neurons induced by ventral root transection in rats. Pain Med. 2011, 12, 1385–1394. [Google Scholar] [CrossRef] [PubMed]
- Antonucci, F.; Rossi, C.; Gianfranceschi, L.; Rossetto, O.; Caleo, M. Long-distance retrograde effects of botulinum neurotoxin A. J. Neurosci. 2008, 28, 3689–3696. [Google Scholar] [CrossRef] [PubMed]
- Restani, L.; Antonucci, F.; Gianfranceschi, L.; Rossi, C.; Rossetto, O.; Caleo, M. Evidence for anterograde transport and transcytosis of botulinum neurotoxin A (BoNT/A). J. Neurosci. 2011, 31, 15650–15659. [Google Scholar] [CrossRef] [PubMed]
- Matak, I.; Riederer, P.; Lackovic, Z. Botulinum toxin’s axonal transport from periphery to the spinal cord. Neurochem. Int. 2012, 61, 236–239. [Google Scholar] [CrossRef] [PubMed]
- Koizumi, H.; Goto, S.; Okita, S.; Morigaki, R.; Akaike, N.; Torii, Y.; Harakawa, T.; Ginnaga, A.; Kaji, R. Spinal Central Effects of Peripherally Applied Botulinum Neurotoxin A in Comparison between Its Subtypes A1 and A2. Front. Neurol. 2014, 5, 98. [Google Scholar] [CrossRef] [PubMed]
- Matak, I.; Bach-Rojecky, L.; Filipovic, B.; Lackovic, Z. Behavioral and immunohistochemical evidence for central antinociceptive activity of botulinum toxin A. Neuroscience 2011, 186, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Martin, S.; Papadopulos, A.; Harper, C.B.; Mavlyutov, T.A.; Niranjan, D.; Glass, N.R.; Cooper-White, J.J.; Sibarita, J.B.; Choquet, D.; et al. Control of autophagosome axonal retrograde flux by presynaptic activity unveiled using botulinum neurotoxin type A. J. Neurosci. 2015, 35, 6179–6194. [Google Scholar] [CrossRef] [PubMed]
- Restani, L.; Giribaldi, F.; Manich, M.; Bercsenyi, K.; Menendez, G.; Rossetto, O.; Caleo, M.; Schiavo, G. Botulinum neurotoxins A and E undergo retrograde axonal transport in primary motor neurons. PLoS Pathog. 2012, 8, e1003087. [Google Scholar] [CrossRef] [PubMed]
- Bach-Rojecky, L.; Salkovic-Petrisic, M.; Lackovic, Z. Botulinum toxin type A reduces pain supersensitivity in experimental diabetic neuropathy: Bilateral effect after unilateral injection. Eur. J. Pharmacol. 2010, 633, 10–14. [Google Scholar] [CrossRef] [PubMed]
- Favre-Guilmard, C.; Auguet, M.; Chabrier, P.E. Different antinociceptive effects of botulinum toxin type A in inflammatory and peripheral polyneuropathic rat models. Eur. J. Pharmacol. 2009, 617, 48–53. [Google Scholar] [CrossRef] [PubMed]
- Bach-Rojecky, L.; Lackovic, Z. Central origin of the antinociceptive action of botulinum toxin type A. Pharmacol. Biochem. Behav. 2009, 94, 234–238. [Google Scholar] [CrossRef] [PubMed]
- Jabbari, B.; Maher, N.; Difazio, M.P. Botulinum toxin a improved burning pain and allodynia in two patients with spinal cord pathology. Pain Med. 2003, 4, 206–210. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.A.; Song, D.H.; Chung, M.E. Effect of subcutaneous injection of botulinum toxin A on spinal cord injury-associated neuropathic pain. Spinal Cord. 2014, 52 (Suppl. 1), S5–S6. [Google Scholar] [CrossRef] [PubMed]
- Han, Z.A.; Song, D.H.; Oh, H.M.; Chung, M.E. Botulinum toxin type A for neuropathic pain in patients with spinal cord injury. Ann Neurol. 2016, 79, 569–578. [Google Scholar] [CrossRef] [PubMed]
- Yelnik, A.P.; Colle, F.M.; Bonan, I.V.; Vicaut, E. Treatment of shoulder pain in spastic hemiplegia by reducing spasticity of the subscapular muscle: A randomised, double blind, placebo controlled study of botulinum toxin A. J. Neurol. Neurosurg. Psychiatry 2007, 78, 845–848. [Google Scholar] [CrossRef] [PubMed]
- Marco, E.; Duarte, E.; Vila, J.; Tejero, M.; Guillen, A.; Boza, R.; Escalada, F.; Espadaler, J.M. Is botulinum toxin type A effective in the treatment of spastic shoulder pain in patients after stroke? A double-blind randomized clinical trial. J. Rehabil. Med. 2007, 39, 440–447. [Google Scholar] [CrossRef] [PubMed]
- Kong, K.H.; Neo, J.J.; Chua, K.S. A randomized controlled study of botulinum toxin A in the treatment of hemiplegic shoulder pain associated with spasticity. Clin. Rehabil. 2007, 21, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.; Koh, J.H.; Paik, N.J. Intramuscular botulinum toxin-A reduces hemiplegic shoulder pain: A randomized, double-blind, comparative study versus intraarticular triamcinolone acetonide. Stroke 2008, 39, 126–131. [Google Scholar] [CrossRef] [PubMed]
- De Boer, K.S.; Arwert, H.J.; de Groot, J.H.; Meskers, C.G.; Mishre, A.D.; Arendzen, J.H. Shoulder pain and external rotation in spastic hemiplegia do not improve by injection of botulinum toxin A into the subscapular muscle. J. Neurol. Neurosurg. Psychiatry 2008, 79, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Shaw, L.C.; Price, C.I.; van Wijck, F.M.; Shackley, P.; Steen, N.; Barnes, M.P.; Ford, G.A.; Graham, L.A.; Rodgers, H.; Bo, T.I. Botulinum Toxin for the Upper Limb after Stroke (BoTULS) Trial: Effect on impairment, activity limitation, and pain. Stroke 2011, 42, 1371–1379. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, C.M.; Harvey, R.L.; Gagnon, C.M.; Duraski, S.A.; Denby, F.A.; McCarty, S.; Bravi, L.A.; Polo, K.M.; Fierstein, K.M. Does botulinum toxin type A decrease pain and lessen disability in hemiplegic survivors of stroke with shoulder pain and spasticity?: A randomized, double-blind, placebo-controlled trial. Am. J. Phys. Med. Rehabil. 2012, 91, 1007–1019. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.G.; Shin, J.H.; Kim, B.R. Botulinum Toxin A Injection into the Subscapularis Muscle to Treat Intractable Hemiplegic Shoulder Pain. Ann. Rehabil. Med. 2016, 40, 592–599. [Google Scholar] [CrossRef] [PubMed]
- Carroll, I.; Clark, J.D.; Mackey, S. Sympathetic block with botulinum toxin to treat complex regional pain syndrome. Ann. Neurol. 2009, 65, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Safarpour, D.; Salardini, A.; Richardson, D.; Jabbari, B. Botulinum toxin A for treatment of allodynia of complex regional pain syndrome: A pilot study. Pain Med. 2010, 11, 1411–1414. [Google Scholar] [CrossRef] [PubMed]
- Kharkar, S.; Ambady, P.; Venkatesh, Y.; Schwartzman, R.J. Intramuscular botulinum toxin in complex regional pain syndrome: Case series and literature review. Pain Phys. 2011, 14, 419–424. [Google Scholar]
- Safarpour, D.; Jabbari, B. Botulinum toxin A (Botox) for treatment of proximal myofascial pain in complex regional pain syndrome: Two cases. Pain Med. 2010, 11, 1415–1418. [Google Scholar] [CrossRef] [PubMed]
- Birthi, P.; Sloan, P.; Salles, S. Subcutaneous botulinum toxin A for the treatment of refractory complex regional pain syndrome. PM R J. Inj. Funct. Rehabil. 2012, 4, 446–449. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.; Cho, C.W.; Kim, H.Y.; Lee, P.B.; Nahm, F.S. Lumbar Sympathetic Block with Botulinum Toxin Type B for Complex Regional Pain Syndrome: A Case Study. Pain Phys. 2015, 18, E911–E916. [Google Scholar]
- Buonocore, M.; Demartini, L.; Mandrini, S.; Dall’Angelo, A.; Dalla Toffola, E. Effect of Botulinum Toxin on Disabling Neuropathic Pain: A Case Presentation Suggesting a New Therapeutic Strategy. PM R J. Inj. Funct. Rehabil. 2017, 9, 200–203. [Google Scholar] [CrossRef] [PubMed]
- Klit, H.; Finnerup, N.B.; Andersen, G.; Jensen, T.S. Central poststroke pain: A population-based study. Pain 2011, 152, 818–824. [Google Scholar] [CrossRef] [PubMed]
- Andersen, G.; Vestergaard, K.; Ingeman-Nielsen, M.; Jensen, T.S. Incidence of central post-stroke pain. Pain 1995, 61, 187–193. [Google Scholar] [CrossRef]
- Leijon, G.; Boivie, J.; Johansson, I. Central post-stroke pain--neurological symptoms and pain characteristics. Pain 1989, 36, 13–25. [Google Scholar] [CrossRef]
- Dromerick, A.W.; Edwards, D.F.; Kumar, A. Hemiplegic shoulder pain syndrome: Frequency and characteristics during inpatient stroke rehabilitation. Arch. Phys. Med. Rehabil. 2008, 89, 1589–1593. [Google Scholar] [CrossRef] [PubMed]
- Kalichman, L.; Ratmansky, M. Underlying pathology and associated factors of hemiplegic shoulder pain. Am. J. Phys. Med. Rehabil. 2011, 90, 768–780. [Google Scholar] [CrossRef] [PubMed]
- Sheean, D.G. Is spasticity painful? Eur. J. Neurol. 2009, 16, 157–158. [Google Scholar] [CrossRef] [PubMed]
- Zeilig, G.; Rivel, M.; Weingarden, H.; Gaidoukov, E.; Defrin, R. Hemiplegic shoulder pain: Evidence of a neuropathic origin. Pain 2013, 154, 263–271. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.A.; Fitzgerald, P.M. Botulinum toxin for shoulder pain. Cochrane Database Syst. Rev. 2010. [Google Scholar] [CrossRef]
- Wu, T.; Fu, Y.; Song, H.X.; Ye, Y.; Dong, Y.; Li, J.H. Effectiveness of Botulinum Toxin for Shoulder Pain Treatment: A Systematic Review and Meta-Analysis. Arch. Phys. Med. Rehabil. 2015, 96, 2214–2220. [Google Scholar] [CrossRef] [PubMed]
- Truini, A.; Barbanti, P.; Pozzilli, C.; Cruccu, G. A mechanism-based classification of pain in multiple sclerosis. J. Neurol. 2013, 260, 351–367. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, A.; Phadke, C.P.; Ismail, F.; Boulias, C. Relationship Between Botulinum Toxin, Spasticity, and Pain: A Survey of Patient Perception. Can. J. Neurol. Sci. 2016, 43, 311–315. [Google Scholar] [CrossRef] [PubMed]
- Jänig, W.; Baron, R. Complex regional pain syndrome: Mystery explained? Lancet Neurol. 2003, 2, 687–697. [Google Scholar] [CrossRef]
- Woolf, C.J.; Thompson, S.W. The induction and maintenance of central sensitization is dependent on N-methyl-d-aspartic acid receptor activation; implications for the treatment of post-injury pain hypersensitivity states. Pain 1991, 44, 293–299. [Google Scholar] [CrossRef]
- Del Valle, L.; Schwartzman, R.J.; Alexander, G. Spinal cord histopathological alterations in a patient with longstanding complex regional pain syndrome. Brain Behav. Immun. 2009, 23, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Forss, N.; Kirveskari, E.; Gockel, M. Mirror-like spread of chronic pain. Neurology 2005, 65, 748–750. [Google Scholar] [CrossRef] [PubMed]
- Van Rijn, M.A.; Marinus, J.; Putter, H.; Bosselaar, S.R.; Moseley, G.L.; van Hilten, J.J. Spreading of complex regional pain syndrome: Not a random process. J. Neural Transm. 2011, 118, 1301–1309. [Google Scholar] [CrossRef] [PubMed]
- Aira, Z.; Buesa, I.; Gallego, M.; Garcia del Cano, G.; Mendiable, N.; Mingo, J.; Rada, D.; Bilbao, J.; Zimmermann, M.; Azkue, J.J. Time-dependent cross talk between spinal serotonin 5-HT2A receptor and mGluR1 subserves spinal hyperexcitability and neuropathic pain after nerve injury. J. Neurosci. 2012, 32, 13568–13581. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Author, Year | Study Design | Sample Size (N) | Diagnosis | Injection Site/Dose | Follow up | Pain Measure | Results |
|---|---|---|---|---|---|---|---|
| Jabbari, 2003 [55] | Case series | 2 | SCI | Subcutaneous injection at the site of allodynia/BTX-A 16–20 U/site | VAS | Pain was decreased; frequency of severe spontaneous pain was reduced | |
| Han, 2014 [56] | Case report | 1 | SCI | Subcutaneous injection in the painful foot/BTX-A | Week 4 | VAS | Pain severity and the frequency of burst was reduced |
| Han, 2016 [57] | Double-blind, randomized controlled study | 40 | SCI | Subcutaneous injection/BTX-A 200 U | Week 4, 8 | VAS (100 mm), McGill Pain Questionnaire | Pain was reduced significantly in BTX-A treated group |
| Yelnik, 2007 [58] | Double-blind, randomized controlled study | 20 | stroke | Subscapularis muscle/BTX-A 500 U/injection + physical therapy | Week 1, 2, 4 | verbal scale (10 point) | Pain improvement with BTX-A from first week |
| Marco, 2007 [59] | Double-blind, randomized controlled study | 31 | stroke | Pectoralis major muscle/BTX-A 500 U/injection + TENS for 6 weeks | Week 1, 4, 12, 24 | VAS (100 mm) | Significantly greater pain improvement from the first week in BTX group |
| Kong, 2007 [60] | Double-blind, randomized controlled study | 17 | stroke | Pectoralis major, biceps brachii muscles/BTX-A 500 U | Week 4, 8, 12 | VAS (0–10) | No difference in shoulder pain |
| Lim, 2008 [61] | Double-blind, randomized controlled study | 29 | stroke | Infraspinatus, pectoralis and subscapularis muscles + IA saline injection; IA triamcinolone (40 mg) injection + saline to the same muscles/BTX-A 100 U | Week 2, 6, 12 | NRS | Significantly greater pain improvement in the BTX-A–treated at 12 weeks |
| Boer, 2008 [62] | Double-blind, randomized controlled study | 22 | stroke | Subscapular muscle/BTX-A 50 U, twice | Week 6, 12 | VAS (vertical 100 mm) | No significant changes in pain |
| Shaw, 2011 [63] | Double-blind, randomized controlled study | 333 | stroke | Elbow, wrist and finger flexor muscles/ BTX-A, 4 times/injection + physical therapy 4 weeks | Week 4, 12, 48 | verbal scale, NRS | Significant decrease at 12 months in the BTX group |
| Castiglione, 2011 [8] | Pilot study | 5 | stroke | IA shoulder joint/BTX-A 500 or 100 units | Week 2, 8 | VAS | Decreased pain at 2 and 8 weeks after BTX-A injection |
| Marciniak, 2012 [64] | Double-blind, randomized controlled study | 21 | stroke | Pectoralis major ± teres major muscles/BTX-A 140–200 units | Week 2, 4, 12 | VAS | Decreased pain scores at 4 weeks |
| Choi, 2016 [65] | Retrospective, unblinded, uncontrolled study | 6 | stroke | Subscapularis muscle/BTX-A | Week 1, 2, 4, 8 | PI-NRS | Pain improvement with BTX-A injection |
| Carroll, 2009 [66] | Double-blind, randomized controlled study | 18 | CRPS | LSB/Bupivacaine 0.5% + 75 units of BTX-A | Week 4 | VAS (10 cm) | The rate of pain return was significantly lower after LSB with BTA |
| Safarpour, 2010 [67] | Double-blind, randomized controlled study Uncontrolled, unblinded, open-label study | 14 (6 control) | CRPS | Intradermally and subcutaneously into the allodynic area/ 5 units per site (total 40–200 units) | Week 3, 8 | Brief pain inventory, PIQ, McGill pain questionnaire, QST, patients satisfaction scale | No significant response after injection; study terminated prematurely because of intolerance |
| Kharkar, 2011 [68] | Retrospective, unblinded, uncontolled study | 37 | CRPS | Upper limb girdle muscles/BTX-A 10–20 units per muscle (total 100 units) | Week 4 | Likert scale (11 point) | 43% decrease in local pain scores |
| Safarpour, 2010 [69] | Case series | 2 | CRPS | Trigger point in the proximal muscle/BTX-A 20 units per site | □ | VAS (1–10) | Alleviate both myofascial pain syndrome and the distal allodynia, discoloration and, tissue swelling |
| Birthi, 2012 [70] | Case report | 1 | CRPS | Subcutaneous injection on the dorsum of the hand/BTX-A 5 units per site (total 100 units) | weekly, 12 weeks | McGill Pain Questionnaire | Able to decrease daily opioid medication by 20% at 8th week; pain returned to baseline at 12th week |
| Choi, 2015 [71] | Case series | 2 | CRPS | Lumbar sympathetic block/levovupivacaine 0.25% + 5000 units of BTX-B | Week 8 | VAS, LANSS | Pain intensity and LANSS score were significantly reduced |
| Buonocore, 2017 [72] | Case report | 1 | CRPS | TP, FDL, FHL muscles, tibial nerve around the tarsal tunnel/BTX-A 120 units per muscle, twice | Week 36 | □ | Significant decrease in the frequency of acute dysesthesias |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, J.; Chung, M.E. Botulinum Toxin for Central Neuropathic Pain. Toxins 2018, 10, 224. https://doi.org/10.3390/toxins10060224
Park J, Chung ME. Botulinum Toxin for Central Neuropathic Pain. Toxins. 2018; 10(6):224. https://doi.org/10.3390/toxins10060224
Chicago/Turabian StylePark, Jihye, and Myung Eun Chung. 2018. "Botulinum Toxin for Central Neuropathic Pain" Toxins 10, no. 6: 224. https://doi.org/10.3390/toxins10060224
APA StylePark, J., & Chung, M. E. (2018). Botulinum Toxin for Central Neuropathic Pain. Toxins, 10(6), 224. https://doi.org/10.3390/toxins10060224