Crystal Structure of Botulinum Neurotoxin A2 in Complex with the Human Protein Receptor SV2C Reveals Plasticity in Receptor Binding


Abstract
1. Introduction
2. Results and Discussion
2.1. Overall Structure of HcA2 and SV2C
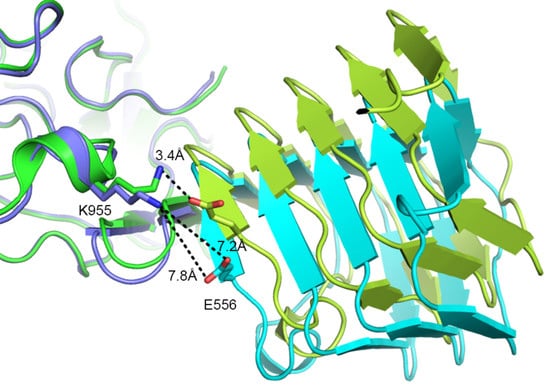
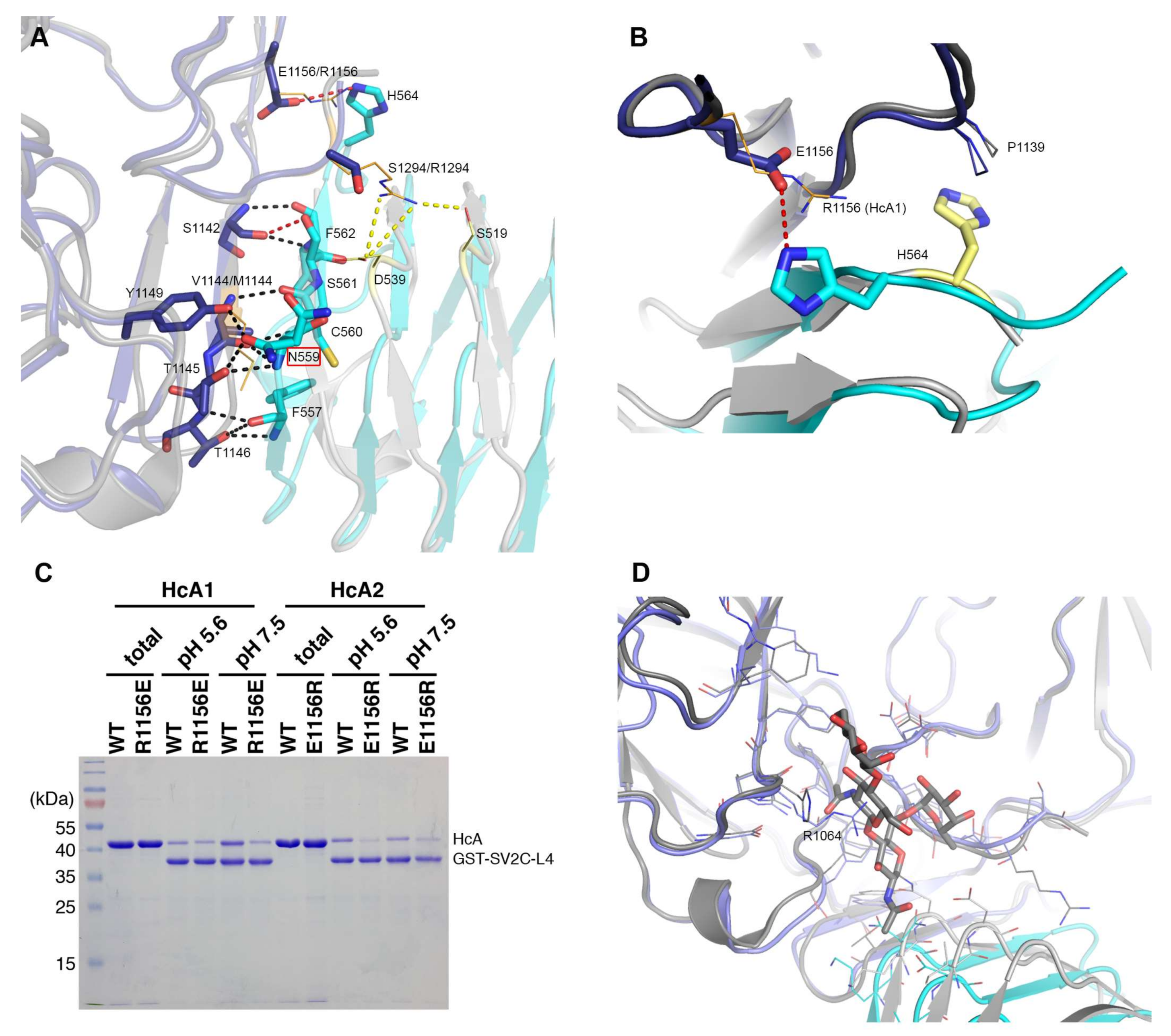
2.2. Comparison to Reported HcA2-SV2C-L4 Structure
2.3. Comparison to Reported HcA1-SV2C-L4 Structure
2.4. SV2 Sequence Comparison
2.5. In Vitro Binding Assay
3. Materials and Methods
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Hill, K.K.; Smith, T.J.; Helma, C.H.; Ticknor, L.O.; Foley, B.T.; Svensson, R.T.; Brown, J.L.; Johnson, E.A.; Smith, L.A.; Okinaka, R.T.; et al. Genetic diversity among botulinum neurotoxin-producing clostridial strains. J. Bacteriol. 2007, 189, 818–832. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Masuyer, G.; Zhang, J.; Shen, Y.; Lundin, D.; Henriksson, L.; Miyashita, S.I.; Martinez-Carranza, M.; Dong, M.; Stenmark, P. Identification and characterization of a novel botulinum neurotoxin. Nat. Commun. 2017, 8, 14130. [Google Scholar] [CrossRef] [PubMed]
- Montal, M. Botulinum neurotoxin: A marvel of protein design. Annu. Rev. Biochem. 2010, 79, 591–617. [Google Scholar] [CrossRef] [PubMed]
- Fischer, A.; Sambashivan, S.; Brunger, A.T.; Montal, M. Beltless translocation domain of botulinum neurotoxin A embodies a minimum ion-conductive channel. J. Biol. Chem. 2012, 287, 1657–1661. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, P.; Dupuy, J.; Imamura, A.; Kiso, M.; Stevens, R.C. Crystal structure of botulinum neurotoxin type A in complex with the cell surface co-receptor GT1b-insight into the toxin-neuron interaction. PLoS Pathog. 2008, 4, e1000129. [Google Scholar] [CrossRef] [PubMed]
- Bomba-Warczak, E.; Vevea, J.D.; Brittain, J.M.; Figueroa-Bernier, A.; Tepp, W.H.; Johnson, E.A.; Yeh, F.L.; Chapman, E.R. Interneuronal transfer and distal action of tetanus toxin and botulinum neurotoxins A and D in central neurons. Cell Rep. 2016, 16, 1974–1987. [Google Scholar] [CrossRef] [PubMed]
- Benoit, R.M.; Frey, D.; Hilbert, M.; Kevenaar, J.T.; Wieser, M.M.; Stirnimann, C.U.; McMillan, D.; Ceska, T.; Lebon, F.; Jaussi, R.; et al. Structural basis for recognition of synaptic vesicle protein 2C by botulinum neurotoxin A. Nature 2014, 505, 108–111. [Google Scholar] [CrossRef] [PubMed]
- Berntsson, R.P.; Peng, L.; Svensson, L.M.; Dong, M.; Stenmark, P. Crystal structures of botulinum neurotoxin DC in complex with its protein receptors synaptotagmin I and II. Structure 2013, 21, 1602–1611. [Google Scholar] [CrossRef] [PubMed]
- Chai, Q.; Arndt, J.W.; Dong, M.; Tepp, W.H.; Johnson, E.A.; Chapman, E.R.; Stevens, R.C. Structural basis of cell surface receptor recognition by botulinum neurotoxin B. Nature 2006, 444, 1096–1100. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Liu, H.; Tepp, W.H.; Johnson, E.A.; Janz, R.; Chapman, E.R. Glycosylated SV2A and SV2B mediate the entry of botulinum neurotoxin E into neurons. Mol. Biol. Cell 2008, 19, 5226–5237. [Google Scholar] [CrossRef] [PubMed]
- Dong, M.; Yeh, F.; Tepp, W.H.; Dean, C.; Johnson, E.A.; Janz, R.; Chapman, E.R. SV2 is the protein receptor for botulinum neurotoxin A. Science 2006, 312, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Fu, Z.; Chen, C.; Barbieri, J.T.; Kim, J.J.; Baldwin, M.R. Glycosylated SV2 and gangliosides as dual receptors for botulinum neurotoxin serotype F. Biochemistry 2009, 48, 5631–5641. [Google Scholar] [CrossRef] [PubMed]
- Jin, R.; Rummel, A.; Binz, T.; Brunger, A.T. Botulinum neurotoxin B recognizes its protein receptor with high affinity and specificity. Nature 2006, 444, 1092–1095. [Google Scholar] [CrossRef] [PubMed]
- Mahrhold, S.; Rummel, A.; Bigalke, H.; Davletov, B.; Binz, T. The synaptic vesicle protein 2C mediates the uptake of botulinum neurotoxin A into phrenic nerves. FEBS Lett. 2006, 580, 2011–2014. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Berntsson, R.P.; Tepp, W.H.; Pitkin, R.M.; Johnson, E.A.; Stenmark, P.; Dong, M. Botulinum neurotoxin D-C uses synaptotagmin I and II as receptors, and human synaptotagmin II is not an effective receptor for type B, D-C and G toxins. J. Cell Sci. 2012, 125, 3233–3242. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Tepp, W.H.; Johnson, E.A.; Dong, M. Botulinum neurotoxin D uses synaptic vesicle protein SV2 and gangliosides as receptors. PLoS Pathog. 2011, 7, e1002008. [Google Scholar] [CrossRef] [PubMed]
- Rummel, A.; Hafner, K.; Mahrhold, S.; Darashchonak, N.; Holt, M.; Jahn, R.; Beermann, S.; Karnath, T.; Bigalke, H.; Binz, T. Botulinum neurotoxins C, E and F bind gangliosides via a conserved binding site prior to stimulation-dependent uptake with botulinum neurotoxin F utilising the three isoforms of SV2 as second receptor. J. Neurochem. 2009, 110, 1942–1954. [Google Scholar] [CrossRef] [PubMed]
- Rummel, A.; Karnath, T.; Henke, T.; Bigalke, H.; Binz, T. Synaptotagmins I and II act as nerve cell receptors for botulinum neurotoxin G. J. Biol. Chem. 2004, 279, 30865–30870. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, J.; Karalewitz, A.; Benefield, D.A.; Mushrush, D.J.; Pruitt, R.N.; Spiller, B.W.; Barbieri, J.T.; Lacy, D.B. Structural analysis of botulinum neurotoxin type G receptor binding. Biochemistry 2010, 49, 5200–5205. [Google Scholar] [CrossRef] [PubMed]
- Yao, G.; Zhang, S.; Mahrhold, S.; Lam, K.H.; Stern, D.; Bagramyan, K.; Perry, K.; Kalkum, M.; Rummel, A.; Dong, M.; et al. N-linked glycosylation of SV2 is required for binding and uptake of botulinum neurotoxin A. Nat. Struct. Mol. Biol. 2016, 23, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.K.; Smith, T.J. Genetic diversity within Clostridium botulinum serotypes, botulinum neurotoxin gene clusters and toxin subtypes. Curr. Top. Microbiol. Immunol. 2013, 364, 1–20. [Google Scholar] [PubMed]
- Peck, M.W.; Smith, T.J.; Anniballi, F.; Austin, J.W.; Bano, L.; Bradshaw, M.; Cuervo, P.; Cheng, L.W.; Derman, Y.; Dorner, B.G.; et al. Historical perspectives and guidelines for botulinum neurotoxin subtype nomenclature. Toxins 2017, 9, 38. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Peng, L.; Berntsson, R.P.; Liu, S.M.; Park, S.; Yu, F.; Boone, C.; Palan, S.; Beard, M.; Chabrier, P.E.; et al. Engineered botulinum neurotoxin B with improved efficacy for targeting human receptors. Nat. Commun. 2017, 8, 53. [Google Scholar] [CrossRef] [PubMed]
- Mukai, Y.; Shimatani, Y.; Sako, W.; Asanuma, K.; Nodera, H.; Sakamoto, T.; Izumi, Y.; Kohda, T.; Kozaki, S.; Kaji, R. Comparison between botulinum neurotoxin type A2 and type A1 by electrophysiological study in healthy individuals. Toxicon 2014, 81, 32–36. [Google Scholar] [CrossRef] [PubMed]
- Kaji, R. Clinical differences between A1 and A2 botulinum toxin subtypes. Toxicon 2015, 107, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Benoit, R.M.; Scharer, M.A.; Wieser, M.M.; Li, X.; Frey, D.; Kammerer, R.A. Crystal structure of the BoNT/A2 receptor-binding domain in complex with the luminal domain of its neuronal receptor SV2C. Sci. Rep. 2017, 7, 43588. [Google Scholar] [CrossRef] [PubMed]
- Hegde, S.S.; Vetting, M.W.; Roderick, S.L.; Mitchenall, L.A.; Maxwell, A.; Takiff, H.E.; Blanchard, J.S. A fluoroquinolone resistance protein from Mycobacterium tuberculosis that mimics DNA. Science 2005, 308, 1480–1483. [Google Scholar] [CrossRef] [PubMed]
- Vetting, M.W.; Hegde, S.S.; Wang, M.; Jacoby, G.A.; Hooper, D.C.; Blanchard, J.S. Structure of QnrB1, a plasmid-mediated fluoroquinolone resistance factor. J. Biol. Chem. 2011, 286, 25265–25273. [Google Scholar] [CrossRef] [PubMed]
- Hamark, C.; Berntsson, R.P.; Masuyer, G.; Henriksson, L.M.; Gustafsson, R.; Stenmark, P.; Widmalm, G. Glycans confer specificity to the recognition of ganglioside receptors by botulinum neurotoxin A. J. Am. Chem. Soc. 2017, 139, 218–230. [Google Scholar] [CrossRef] [PubMed]
- Mahrhold, S.; Bergstrom, T.; Stern, D.; Dorner, B.G.; Astot, C.; Rummel, A. Only the complex N559-glycan in the synaptic vesicle glycoprotein 2C mediates high affinity binding to botulinum neurotoxin serotype A1. Biochem. J. 2016, 473, 2645–2654. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Lacy, D.B.; Tepp, W.; Cohen, A.C.; DasGupta, B.R.; Stevens, R.C. Crystal structure of botulinum neurotoxin type A and implications for toxicity. Nat. Struct. Biol. 1998, 5, 898–902. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Vagin, A.A.; Dodson, E.J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997, 53, 240–255. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B., 3rd; Snoeyink, J.; Richardson, J.S.; et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E. Crystal contacts as nature’s docking solutions. J. Comput. Chem. 2010, 31, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Detection of Protein Assemblies in Crystals; Springer: Berlin/Heidelberg, Germany, 2005; Volume 3695, pp. 163–174. [Google Scholar]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Krissinel, E.; Henrick, K. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2256–2268. [Google Scholar] [CrossRef] [PubMed]
- Robert, X.; Gouet, P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; McWilliam, H.; Remmert, M.; Soding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection | HcA2 + SV2C-L4 Complex |
|---|---|
| Space group | P21212 |
| Cell dimensions | |
| a, b, c (Å) | 100.7, 122.1, 47.7 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution (Å) | 47.66–2.00 (2.05–2.00) |
| Rmerge (%) | 24.2 (123.7) |
| I/σ (I) | 9.4 (2.2) |
| Completeness (%) | 99.8 (97.7) |
| CC (1/2) (%) | 99.6 (48.1) |
| Redundancy | 13.2 (13.4) |
| Refinement | |
| Resolution (Å) | 47.66–2.00 |
| No. unique reflections | 40,692 (2897) |
| Rwork/Rfree | 18.14/22.97 |
| Wilson B factor (Å2) | 24.7 |
| No. atoms | |
| HcA2 | 3471 |
| SV2C-L4 | 813 |
| Water | 359 |
| Acetate | 4 |
| B-factors (Å2) | |
| HcA2 | 27.9 |
| SV2C-L4 | 27.2 |
| Water | 36.2 |
| Acetate | 39.9 |
| RMS deviations | |
| Bond lengths (Å) | 0.0147 |
| Bond angles (°) | 1.6628 |
| Ramachandran plot, residues in (%) | |
| Most favourable region | 96.09 |
| Additional allowed region | 3.91 |
| Protein | Residue | Distance (Å) | Angle (°) * |
|---|---|---|---|
| HcA2 | G1141 | 0.5 | |
| S1142 | 0.5 | ||
| V1143 | 0.3 | ||
| V1144 | 0.7 | ||
| T1145 | 1.2 | ||
| T1146 | 1.8 | ||
| N1147 | 1.8 | ||
| I1148 | 0.9 | ||
| Y1149 | 0.5 | ||
| L1150 | 0.5 | ||
| SV2C-L4 | E556 | 3.9 | 15.6 |
| F557 | 2.4 | 12.4 | |
| K558 | 2.3 | 10.6 | |
| N559 | 1.8 | 9.6 | |
| C560 | 1.4 | 11.2 | |
| S561 | 0.9 | 12.1 | |
| F562 | 0.4 | Not applicable | |
| F563 | 1.1 | 16.1 | |
| H564 | 2.6 | 21.3 | |
| N565 | 2.8 | 22.8 | |
| K566 | 2.6 | 13.6 | |
| D546 | 3.0 | 16.8 | |
| F547 | 3.0 | 18.7 | |
| E548 | 4.0 | 22.4 | |
| P549 | 4.5 | 23.1 | |
| Y550 | 5.5 | 21.5 | |
| K551 | 4.9 | 20.1 | |
| F552 | 4.7 | 19.6 | |
| I553 | 5.9 | 19.8 | |
| D554 | 5.9 | 18.5 | |
| S555 | 4.7 | 17.4 | |
| N480 | 6.6 | 18.0 | |
| F481 | 6.1 | 17.0 | |
| T482 | 4.8 | 14.4 | |
| I483 | 4.5 | 13.6 | |
| N484 | 3.0 | 8.8 | |
| F485 | 4.3 | 11.2 | |
| T486 | 4.4 | 11.8 | |
| M487 | 5.7 | 14.7 | |
| E488 | 6.3 | 15.4 | |
| N489 | 7.4 | 16.8 | |
| Q490 | 7.7 | 17.9 | |
| I491 | 7.8 | 18.7 | |
| H492 | 7.3 | 18.4 | |
| T493 | 8.0 | 19.1 | |
| G494 | 8.1 | 19.3 | |
| M495 | 7.2 | 18.6 | |
| E496 | 6.5 | 18.3 | |
| Y497 | 5.9 | 18.4 |
| Structure | Reference | Space Group | Cell Parameters a, b, c (Å) | Cell Parameters α, β, γ (°) | HcA Buffer pH | SV2C Buffer pH | Crystal Condition pH |
|---|---|---|---|---|---|---|---|
| 6ES1 | This work | P21212 | 100.7, 122.1, 47.7 | 90.0, 90.0, 90.0 | 7.4 | 7.5 | 6.0 |
| 5MOY | [26] | P212121 | 47.7, 91.6, 147.7 | 90.0, 90.0, 90.0 | 7.5 | 7.5 | 7.5 |
| 5JMC | [20] | P1211 | 88.7, 144.0, 110.9 | 90.0, 93.6, 90.0 | 7.5 | 7.5 | 6.5 |
| 5JLV | [20] | C121 | 109.0, 111.9, 126.3 | 90.0, 101.3, 90.0 | 7.5 | 7.5 | 4.6 |
| 4JRA | [7] | C121 * | 115.4, 105.3, 128.0 | 90.0, 90.02, 90.0 | ** | ** | 7.5 |
| Structure | HcA Construct (Start → End) | HcA His-Tag Cleaved? | SV2C-L4 Construct (Start → End) | SV2C-L4 His-Tag Cleaved? |
|---|---|---|---|---|
| 6ES1 | 874–1296 | No | 474–567 | No |
| 5MOY | 871–1296 | Yes | 456–574 | No |
| 5JMC | 872–1296 | Yes | 455–577 (rat) | Yes |
| 5JLV | 872–1296 | Yes | 473–567 | Yes |
| 4JRA | 871–1296 | No | 456–574 | No |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gustafsson, R.; Zhang, S.; Masuyer, G.; Dong, M.; Stenmark, P. Crystal Structure of Botulinum Neurotoxin A2 in Complex with the Human Protein Receptor SV2C Reveals Plasticity in Receptor Binding. Toxins 2018, 10, 153. https://doi.org/10.3390/toxins10040153
Gustafsson R, Zhang S, Masuyer G, Dong M, Stenmark P. Crystal Structure of Botulinum Neurotoxin A2 in Complex with the Human Protein Receptor SV2C Reveals Plasticity in Receptor Binding. Toxins. 2018; 10(4):153. https://doi.org/10.3390/toxins10040153
Chicago/Turabian StyleGustafsson, Robert, Sicai Zhang, Geoffrey Masuyer, Min Dong, and Pål Stenmark. 2018. "Crystal Structure of Botulinum Neurotoxin A2 in Complex with the Human Protein Receptor SV2C Reveals Plasticity in Receptor Binding" Toxins 10, no. 4: 153. https://doi.org/10.3390/toxins10040153
APA StyleGustafsson, R., Zhang, S., Masuyer, G., Dong, M., & Stenmark, P. (2018). Crystal Structure of Botulinum Neurotoxin A2 in Complex with the Human Protein Receptor SV2C Reveals Plasticity in Receptor Binding. Toxins, 10(4), 153. https://doi.org/10.3390/toxins10040153