Proteomic Methods of Detection and Quantification of Protein Toxins


Abstract
1. Introduction
2. Methods of Detection of Protein Toxins
2.1. Biological Assays
2.2. Proteomic Methods
2.2.1. Immunological Assays
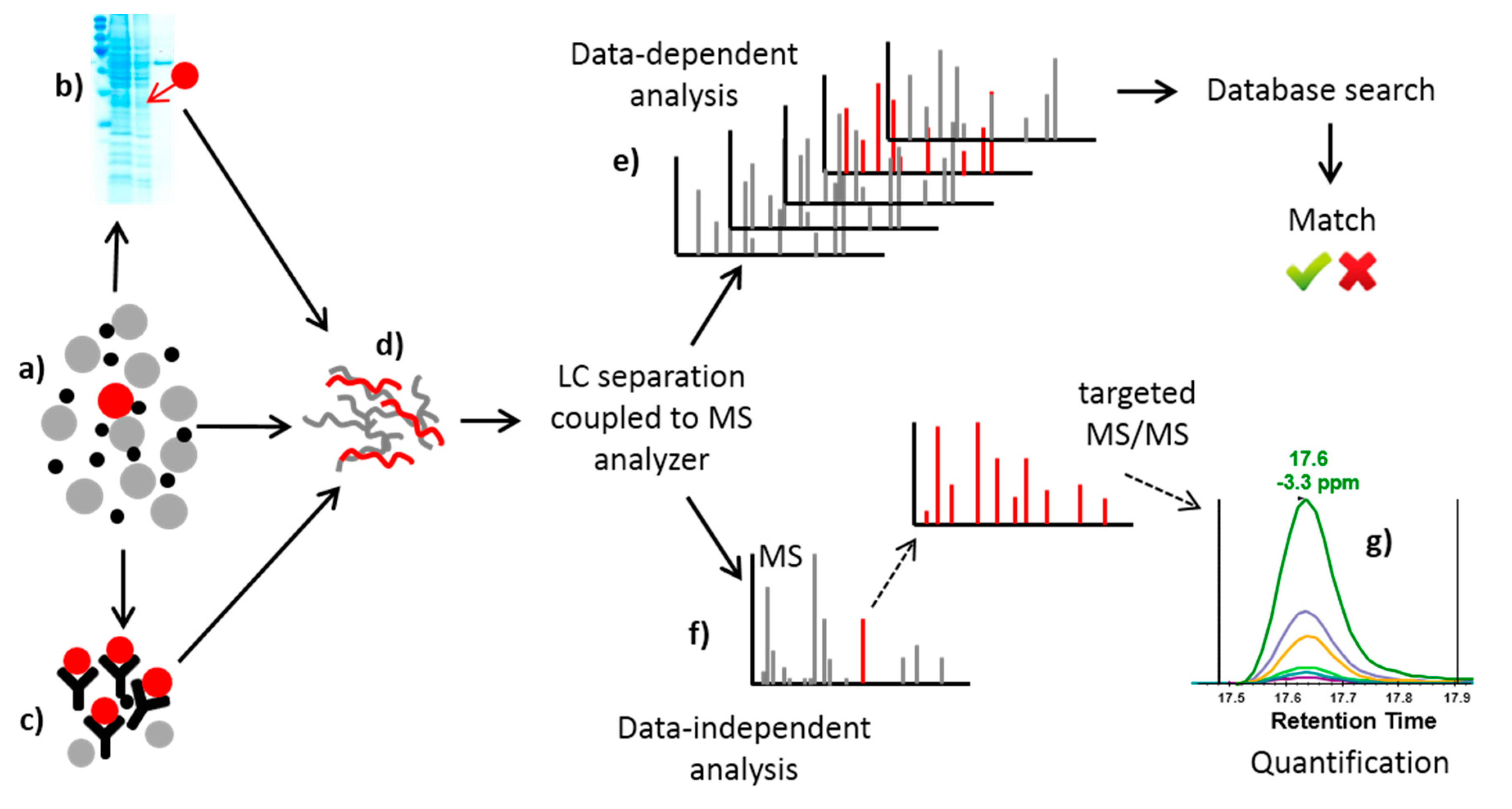
2.2.2. Mass Spectrometry-Based Methods
3. Detection of Selected Protein Toxins
3.1. Staphylococcal Toxins
3.2. Bacillus anthracis Toxins
3.3. Clostridium Toxins
3.4. Selected Plant Protein Toxins
3.5. Additional Underestimated Protein Toxins
4. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Pal, M.; Tsegaye, M.; Girzaw, F.; Bedada, H.; Godishala, V.; Kandi, V. An Overview on Biological Weapons and Bioterrorism. Am. J. Biomed. Res. 2017, 5, 24–34. [Google Scholar] [CrossRef]
- Jansen, H.J.; Breeveld, F.J.; Stijnis, C.; Grobusch, M.P. Biological warfare, bioterrorism, and biocrime. Clin. Microbiol. Infect. 2014, 20, 488–496. [Google Scholar] [CrossRef] [PubMed]
- NIAID Emerging Infectious Diseases/Pathogens | NIH: National Institute of Allergy and Infectious Diseases. Available online: https://www.niaid.nih.gov/research/emerging-infectious-diseases-pathogens (accessed on 12 June 2017).
- COMMISSION DELEGATED REGULATION (EU) 2016/1969 of 12 September 2016 Amending Council Regulation (EC) No 428/2009 Setting up a Community Regime for the Control of Exports, Transfer, Brokering and Transit of Dual-Use Items. Available online: https://danishbusinessauthority.dk/sites/default/files/media/reg._2016-1969_new_control_list.pdf (accessed on 1 November 2017).
- Lubran, M.M. Bacterial toxins. Ann. Clin. Lab. Sci. 1988, 18, 58–71. [Google Scholar] [PubMed]
- Sandvig, K.; Torgersen, M.L.; Engedal, N.; Skotland, T.; Iversen, T.-G. Protein toxins from plants and bacteria: Probes for intracellular transport and tools in medicine. FEBS Lett. 2010, 584, 2626–2634. [Google Scholar] [CrossRef] [PubMed]
- Dal Peraro, M.; van der Goot, F.G. Pore-forming toxins: Ancient, but never really out of fashion. Nat. Rev. Microbiol. 2016, 14, 77–92. [Google Scholar] [CrossRef] [PubMed]
- Duriez, E.; Armengaud, J.; Fenaille, F.; Ezan, E. Mass spectrometry for the detection of bioterrorism agents: From environmental to clinical applications. J. Mass Spectrom. JMS 2016, 51, 183–199. [Google Scholar] [CrossRef] [PubMed]
- Dorner, B.G.; Zeleny, R.; Harju, K.; Hennekinne, J.-A.; Vanninen, P.; Schimmel, H.; Rummel, A. Biological toxins of potential bioterrorism risk: Current status of detection and identification technology. TrAC Trends Anal. Chem. 2016, 85, 89–102. [Google Scholar] [CrossRef]
- Demirev, P.A.; Fenselau, C. Mass spectrometry in biodefense. J. Mass Spectrom. JMS 2008, 43, 1441–1457. [Google Scholar] [CrossRef] [PubMed]
- OPCW Hosts Series of Science and Technology Meetings. Available online: https://www.opcw.org/news/article/opcw-hosts-series-of-science-and-technology-meetings/ (accessed on 6 November 2017).
- Wilson, I.G.; Cooper, J.E.; Gilmour, A. Detection of enterotoxigenic Staphylococcus aureus in dried skimmed milk: Use of the polymerase chain reaction for amplification and detection of staphylococcal enterotoxin genes entB and entC1 and the thermonuclease gene nuc. Appl. Environ. Microbiol. 1991, 57, 1793–1798. [Google Scholar] [PubMed]
- Jay, M.J.; Loessner, J.M.; Golden, A.D. Bioassay and Related Methods. In Modern Food Microbiology; Food Science Text Series; Springer: New York, NY, USA, 2005; pp. 285–298. ISBN 978-0-387-23180-8. [Google Scholar]
- Lindström, M.; Korkeala, H. Laboratory diagnostics of botulism. Clin. Microbiol. Rev. 2006, 19, 298–314. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.; Singh, B.R.; Sharma, S. Botulism diagnostics: From clinical symptoms to in vitro assays. Crit. Rev. Microbiol. 2007, 33, 109–125. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Whiting, R.C. Methods for detection of Clostridium botulinum toxin in foods. J. Food Prot. 2005, 68, 1256–1263. [Google Scholar] [CrossRef] [PubMed]
- Solberg, M.; Post, L.S.; Furgang, D.; Graham, C. Bovine serum eliminates rapid nonspecific toxic reactions during bioassay of stored fish for Clostridium botulinum toxin. Appl. Environ. Microbiol. 1985, 49, 644–649. [Google Scholar] [PubMed]
- Dezfulian, M.; Bartlett, J.G. Detection of Clostridium botulinum type B toxin in the presence of a lethal substance interfering with toxin neutralization. Diagn. Microbiol. Infect. Dis. 1985, 3, 105–112. [Google Scholar] [CrossRef]
- Aydin, S. A short history, principles, and types of ELISA, and our laboratory experience with peptide/protein analyses using ELISA. Peptides 2015, 72, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Lewis, G.E.; Kulinski, S.S.; Reichard, D.W.; Metzger, J.F. Detection of Clostridium botulinum type G toxin by enzyme-linked immunosorbent assay. Appl. Environ. Microbiol. 1981, 42, 1018–1022. [Google Scholar] [PubMed]
- Park, C.E.; Akhtar, M.; Rayman, M.K. Nonspecific reactions of a commercial enzyme-linked immunosorbent assay kit (TECRA) for detection of staphylococcal enterotoxins in foods. Appl. Environ. Microbiol. 1992, 58, 2509–2512. [Google Scholar] [PubMed]
- Wieneke, A.A. Comparison of four kits for the detection of staphylococcal enterotoxin in foods from outbreaks of food poisoning. Int. J. Food Microbiol. 1991, 14, 305–312. [Google Scholar] [CrossRef]
- Yalow, R.S.; Berson, S.A. Immunoassay of endogenous plasma insulin in man. J. Clin. Investig. 1960, 39, 1157–1175. [Google Scholar] [CrossRef] [PubMed]
- Goldsmith, S.J. Radioimmunoassay: Review of basic principles. Semin. Nucl. Med. 1975, 5, 125–152. [Google Scholar] [CrossRef]
- Gan, S.D.; Patel, K.R. Enzyme Immunoassay and Enzyme-Linked Immunosorbent Assay. J. Investig. Dermatol. 2013, 133, e12. [Google Scholar] [CrossRef] [PubMed]
- Zhu, K.; Dietrich, R.; Didier, A.; Doyscher, D.; Märtlbauer, E. Recent Developments in Antibody-Based Assays for the Detection of Bacterial Toxins. Toxins 2014, 6, 1325–1348. [Google Scholar] [CrossRef] [PubMed]
- Hejtmancik, K.E.; Peterson, J.W.; Markel, D.E.; Kurosky, A. Radioimmunoassay for the antigenic determinants of cholera toxin and its components. Infect. Immun. 1977, 17, 621–628. [Google Scholar] [PubMed]
- Harris, C.C.; Yolken, R.H.; Krokan, H.; Hsu, I.C. Ultrasensitive enzymatic radioimmunoassay: Application to detection of cholera toxin and rotavirus. Proc. Natl. Acad. Sci. USA 1979, 76, 5336–5339. [Google Scholar] [CrossRef] [PubMed]
- Godal, A.; Olsnes, S.; Pihl, A. Radioimmunoassays of abrin and ricin in blood. J. Toxicol. Environ. Health 1981, 8, 409–417. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, S.; Eagle, M.R.; Houston, L.L. Radioimmunoassay of ricin A- and B-chains applied to samples of ricin A-chain prepared by chromatofocusing and by DEAE Bio-Gel A chromatography. Biochim. Biophys. Acta 1982, 719, 341–348. [Google Scholar] [CrossRef]
- Kurien, B.T.; Scofield, R.H. Western blotting. Methods 2006, 38, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Ladhani, S.; Robbie, S.; Garratt, R.C.; Chapple, D.S.; Joannou, C.L.; Evans, R.W. Development and Evaluation of Detection Systems for Staphylococcal Exfoliative Toxin A Responsible for Scalded-Skin Syndrome. J. Clin. Microbiol. 2001, 39, 2050–2054. [Google Scholar] [CrossRef] [PubMed]
- Lian, W.; Wu, D.; Lim, D.V.; Jin, S. Sensitive detection of multiplex toxins using antibody microarray. Anal. Biochem. 2010, 401, 271–279. [Google Scholar] [CrossRef] [PubMed]
- Skinner, C.; Patfield, S.; Stanker, L.H.; Fratamico, P.; He, X. New High-Affinity Monoclonal Antibodies against Shiga Toxin 1 Facilitate the Detection of Hybrid Stx1/Stx2 In Vivo. PLoS ONE 2014, 9, e99854. [Google Scholar] [CrossRef] [PubMed]
- Boyer, A.E.; Gallegos-Candela, M.; Lins, R.C.; Kuklenyik, Z.; Woolfitt, A.; Moura, H.; Kalb, S.; Quinn, C.P.; Barr, J.R. Quantitative Mass Spectrometry for Bacterial Protein Toxins—A Sensitive, Specific, High-Throughput Tool for Detection and Diagnosis. Molecules 2011, 16, 2391–2413. [Google Scholar] [CrossRef] [PubMed]
- Lange, V.; Picotti, P.; Domon, B.; Aebersold, R. Selected reaction monitoring for quantitative proteomics: A tutorial. Mol. Syst. Biol. 2008, 4, 222. [Google Scholar] [CrossRef] [PubMed]
- Bourmaud, A.; Gallien, S.; Domon, B. Parallel reaction monitoring using quadrupole-Orbitrap mass spectrometer: Principle and applications. Proteomics 2016, 16, 2146–2159. [Google Scholar] [CrossRef] [PubMed]
- MacLean, B.; Tomazela, D.M.; Shulman, N.; Chambers, M.; Finney, G.L.; Frewen, B.; Kern, R.; Tabb, D.L.; Liebler, D.C.; MacCoss, M.J. Skyline: An open source document editor for creating and analyzing targeted proteomics experiments. Bioinform. Oxf. Engl. 2010, 26, 966–968. [Google Scholar] [CrossRef] [PubMed]
- Desiderio, D.M.; Kai, M. Preparation of stable isotope-incorporated peptide internal standards for field desorption mass spectrometry quantification of peptides in biologic tissue. Biomed. Mass Spectrom. 1983, 10, 471–479. [Google Scholar] [CrossRef] [PubMed]
- Gallien, S.; Duriez, E.; Crone, C.; Kellmann, M.; Moehring, T.; Domon, B. Targeted Proteomic Quantification on Quadrupole-Orbitrap Mass Spectrometer. Mol. Cell. Proteom. MCP 2012, 11, 1709–1723. [Google Scholar] [CrossRef] [PubMed]
- Bereman, M.S.; MacLean, B.; Tomazela, D.M.; Liebler, D.C.; MacCoss, M.J. The development of selected reaction monitoring methods for targeted proteomics via empirical refinement. Proteomics 2012, 12, 1134–1141. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, A.; Hennekinne, J.-A.; Garin, J.; Brun, V. Protein Standard Absolute Quantification (PSAQ) for improved investigation of staphylococcal food poisoning outbreaks. Proteomics 2008, 8, 4633–4636. [Google Scholar] [CrossRef] [PubMed]
- Adrait, A.; Lebert, D.; Trauchessec, M.; Dupuis, A.; Louwagie, M.; Masselon, C.; Jaquinod, M.; Chevalier, B.; Vandenesch, F.; Garin, J.; et al. Development of a Protein Standard Absolute Quantification (PSAQ™) assay for the quantification of Staphylococcus aureus enterotoxin A in serum. J. Proteom. 2012, 75, 3041–3049. [Google Scholar] [CrossRef] [PubMed]
- Dupré, M.; Gilquin, B.; Fenaille, F.; Feraudet-Tarisse, C.; Dano, J.; Ferro, M.; Simon, S.; Junot, C.; Brun, V.; Becher, F. Multiplex quantification of protein toxins in human biofluids and food matrices using immunoextraction and high-resolution targeted mass spectrometry. Anal. Chem. 2015, 87, 8473–8480. [Google Scholar] [CrossRef] [PubMed]
- Kalb, S.R.; Boyer, A.E.; Barr, J.R. Mass Spectrometric Detection of Bacterial Protein Toxins and Their Enzymatic Activity. Toxins 2015, 7, 3497–3511. [Google Scholar] [CrossRef] [PubMed]
- Boyer, A.E.; Moura, H.; Woolfitt, A.R.; Kalb, S.R.; McWilliams, L.G.; Pavlopoulos, A.; Schmidt, J.G.; Ashley, D.L.; Barr, J.R. From the mouse to the mass spectrometer: Detection and differentiation of the endoproteinase activities of botulinum neurotoxins A-G by mass spectrometry. Anal. Chem. 2005, 77, 3916–3924. [Google Scholar] [CrossRef] [PubMed]
- Otto, M. Staphylococcus aureus toxins. Curr. Opin. Microbiol. 2014, 17, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Masalha, M.; Borovok, I.; Schreiber, R.; Aharonowitz, Y.; Cohen, G. Analysis of Transcription of the Staphylococcus aureus Aerobic Class Ib and Anaerobic Class III Ribonucleotide Reductase Genes in Response to Oxygen. J. Bacteriol. 2001, 183, 7260–7272. [Google Scholar] [CrossRef] [PubMed]
- Schlievert, P.M.; Case, L.C. Molecular analysis of staphylococcal superantigens. Methods Mol. Biol. 2007, 391, 113–126. [Google Scholar] [CrossRef] [PubMed]
- Pinchuk, I.V.; Beswick, E.J.; Reyes, V.E. Staphylococcal Enterotoxins. Toxins 2010, 2, 2177–2197. [Google Scholar] [CrossRef] [PubMed]
- Fries, B.C.; Varshney, A.K. Bacterial Toxins-Staphylococcal Enterotoxin B. Microbiol. Spectr. 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Zapor, M.; Fishbain, J.T. Aerosolized biologic toxins as agents of warfare and terrorism. Respir. Care Clin. N. Am. 2004, 10, 111–122. [Google Scholar] [CrossRef]
- Fulton, F. Staphylococcal Enterotoxin—With Special Reference to the Kitten Test. Br. J. Exp. Pathol. 1943, 24, 65–72. [Google Scholar]
- Scheuber, P.H.; Mossmann, H.; Beck, G.; Hammer, D.K. Direct skin test in highly sensitized guinea pigs for rapid and sensitive determination of staphylococcal enterotoxin B. Appl. Environ. Microbiol. 1983, 46, 1351–1356. [Google Scholar] [PubMed]
- Wu, S.; Duan, N.; Gu, H.; Hao, L.; Ye, H.; Gong, W.; Wang, Z. A Review of the Methods for Detection of Staphylococcus aureus Enterotoxins. Toxins 2016, 8, 176. [Google Scholar] [CrossRef] [PubMed]
- Saunders, G.C.; Bartlett, M.L. Double-antibody solid-phase enzyme immunoassay for the detection of staphylococcal enterotoxin A. Appl. Environ. Microbiol. 1977, 34, 518–522. [Google Scholar] [PubMed]
- Notermans, S.; Verjans, H.L.; Bol, J.; van Schothorst, M. Enzyme linked immunosorbent assay (ELISA) for determination of Staphylococcus aureus enterotoxin type B. Health Lab. Sci. 1978, 15, 28–31. [Google Scholar] [PubMed]
- Stiffler-Rosenberg, G.; Fey, H. Simple assay for staphylococcal enterotoxins A, B, and C: Modification of enzyme-linked immunosorbent assay. J. Clin. Microbiol. 1978, 8, 473–479. [Google Scholar] [PubMed]
- Fey, H.; Pfister, H.; Rüegg, O. Comparative evaluation of different enzyme-linked immunosorbent assay systems for the detection of staphylococcal enterotoxins A, B, C, and D. J. Clin. Microbiol. 1984, 19, 34–38. [Google Scholar] [PubMed]
- Wieneke, A.A.; Gilbert, R.J. The use of a sandwich ELISA for the detection of staphylococcal enterotoxin A in foods from outbreaks of food poisoning. Epidemiol. Infect. 1985, 95, 131–138. [Google Scholar] [CrossRef]
- Hahn, I.F.; Pickenhahn, P.; Lenz, W.; Brandis, H. An avidin-biotin ELISA for the detection of staphylococcal enterotoxins A and B. J. Immunol. Methods 1986, 92, 25–29. [Google Scholar] [CrossRef]
- Nia, Y.; Rodriguez, M.; Zeleny, R.; Herbin, S.; Auvray, F.; Fiebig, U.; Avondet, M.-A.; Munoz, A.; Hennekinne, J.-A. Organization and ELISA-Based Results of the First Proficiency Testing to Evaluate the Ability of European Union Laboratories to Detect Staphylococcal Enterotoxin Type B (SEB) in Buffer and Milk. Toxins 2016, 8, 268. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.S.; Cao, C.J.; Thompson, R.G.; Valdes, J.J. A simple and rapid fluorescence-based immunoassay for the detection of staphylococcal enterotoxin B. Mol. Cell. Probes 2003, 17, 125–126. [Google Scholar] [CrossRef]
- Hun, X.; Zhang, Z. A novel sensitive staphylococcal enterotoxin C1 fluoroimmunoassay based on functionalized fluorescent core-shell nanoparticle labels. Food Chem. 2007, 105, 1623–1629. [Google Scholar] [CrossRef]
- Vinayaka, A.C.; Thakur, M.S. An immunoreactor-based competitive fluoroimmunoassay for monitoring staphylococcal enterotoxin B using bioconjugated quantum dots. Analyst 2012, 137, 4343–4348. [Google Scholar] [CrossRef] [PubMed]
- Szkola, A.; Linares, E.M.; Worbs, S.; Dorner, B.G.; Dietrich, R.; Märtlbauer, E.; Niessner, R.; Seidel, M. Rapid and simultaneous detection of ricin, staphylococcal enterotoxin B and saxitoxin by chemiluminescence-based microarray immunoassay. Analyst 2014, 139, 5885–5892. [Google Scholar] [CrossRef] [PubMed]
- Sun, S.; Yang, M.; Kostov, Y.; Rasooly, A. ELISA-LOC: Lab-on-a-chip for enzyme-linked immunodetection. Lab Chip 2010, 10, 2093–2100. [Google Scholar] [CrossRef] [PubMed]
- Sd, S.; Rc, S.; Ap, L.; Ce, F. Surface plasmon resonance detection using antibody-linked magnetic nanoparticles for analyte capture, purification, concentration, and signal amplification., Surface Plasmon Resonance (SPR) Detection Using Antibody-Linked Magnetic Nanoparticles for Analyte Capture, Purification, Concentration and Signal Amplification. Anal. Chem. 2009, 81, 2357–2363. [Google Scholar] [CrossRef]
- Tang, D.; Tang, J.; Su, B.; Chen, G. Ultrasensitive electrochemical immunoassay of staphylococcal enterotoxin B in food using enzyme-nanosilica-doped carbon nanotubes for signal amplification. J. Agric. Food Chem. 2010, 58, 10824–10830. [Google Scholar] [CrossRef] [PubMed]
- Park, C.E.; Szabo, R. Evaluation of the reversed passive latex agglutination (RPLA) test kits for detection of staphylococcal enterotoxins A, B, C, and D in foods. Can. J. Microbiol. 1986, 32, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Fujikawa, H.; Igarashi, H. Rapid latex agglutination test for detection of staphylococcal enterotoxins A to E that uses high-density latex particles. Appl. Environ. Microbiol. 1988, 54, 2345–2348. [Google Scholar] [PubMed]
- Rose, S.A.; Bankes, P.; Stringer, M.F. Detection of staphylococcal enterotoxins in dairy products by the reversed passive latex agglutination (SET-RPLA) kit. Int. J. Food Microbiol. 1989, 8, 65–72. [Google Scholar] [CrossRef]
- Pereira, M.L.; Heneine, L.G.; Santos, E.J.; Carmo, L.S.; Pereira, J.L.; Bergdoll, M.S. Prevention of nonspecific reactions on reversed passive latex agglutination assay (RPLA) for detecting low amounts of staphylococcal enterotoxins. Rev. Latinoam. Microbiol. 1997, 39, 57–63. [Google Scholar] [PubMed]
- Hall, H.E.; Angelotti, R.; Lewis, K.H. Quantitative detection of staphylococcal Enterotoxin B in food by gel-diffusion methods. Public Health Rep. 1963, 78, 1089–1098. [Google Scholar] [CrossRef] [PubMed]
- Salomon, L.L.; Tew, R.W. Assay of staphylococcal enterotoxin B by latex agglutination. Proc. Soc. Exp. Biol. Med. Soc. Exp. Biol. Med. 1968, 129, 539–542. [Google Scholar] [CrossRef]
- Lee, C.L.; Lin, C.C. Detection of staphylococcal enterotoxin by latex agglutination inhibition test. Zhonghua Min Guo Wei Sheng Wu Ji Mian Yi Xue Za Zhi 1984, 17, 77–80. [Google Scholar] [PubMed]
- Read, R.B.; Bradshaw, J.; Pritchard, W.L.; Black, L.A. Assay of Staphylococcal Enterotoxin from Cheese. J. Dairy Sci. 1965, 48, 420–424. [Google Scholar] [CrossRef]
- Kientz, C.E.; Hulst, A.G.; Wils, E.R. Determination of staphylococcal enterotoxin B by on-line (micro) liquid chromatography-electrospray mass spectrometry. J. Chromatogr. A 1997, 757, 51–64. [Google Scholar] [CrossRef]
- Kawano, Y.; Ito, Y.; Yamakawa, Y.; Yamashino, T.; Horii, T.; Hasegawa, T.; Ohta, M. Rapid isolation and identification of staphylococcal exoproteins by reverse phase capillary high performance liquid chromatography-electrospray ionization mass spectrometry. FEMS Microbiol. Lett. 2000, 189, 103–108. [Google Scholar] [CrossRef]
- Nedelkov, D.; Rasooly, A.; Nelson, R.W. Multitoxin biosensor-mass spectrometry analysis: A new approach for rapid, real-time, sensitive analysis of staphylococcal toxins in food. Int. J. Food Microbiol. 2000, 60, 1–13. [Google Scholar] [CrossRef]
- Callahan, J.H.; Shefcheck, K.J.; Williams, T.L.; Musser, S.M. Detection, confirmation, and quantification of staphylococcal enterotoxin B in food matrixes using liquid chromatography–mass spectrometry. Anal. Chem. 2006, 78, 1789–1800. [Google Scholar] [CrossRef] [PubMed]
- Bao, K.D.; Letellier, A.; Beaudry, F. Analysis of Staphylococcus enterotoxin B using differential isotopic tags and liquid chromatography quadrupole ion trap mass spectrometry. Biomed. Chromatogr. BMC 2012, 26, 1049–1057. [Google Scholar] [CrossRef] [PubMed]
- Zuberovic Muratovic, A.; Hagström, T.; Rosén, J.; Granelli, K.; Hellenäs, K.-E. Quantitative Analysis of Staphylococcal Enterotoxins A and B in Food Matrices Using Ultra High-Performance Liquid Chromatography Tandem Mass Spectrometry (UPLC-MS/MS). Toxins 2015, 7, 3637–3656. [Google Scholar] [CrossRef] [PubMed]
- Andjelkovic, M.; Tsilia, V.; Rajkovic, A.; De Cremer, K.; Van Loco, J. Application of LC-MS/MS MRM to Determine Staphylococcal Enterotoxins (SEB and SEA) in Milk. Toxins 2016, 8, 118. [Google Scholar] [CrossRef] [PubMed]
- Gilquin, B.; Jaquinod, M.; Louwagie, M.; Kieffer-Jaquinod, S.; Kraut, A.; Ferro, M.; Becher, F.; Brun, V. A proteomics assay to detect eight CBRN-relevant toxins in food. Proteomics 2017, 17, 1600357. [Google Scholar] [CrossRef] [PubMed]
- Edwards, K.A.; Clancy, H.A.; Baeumner, A.J. Bacillus anthracis: Toxicology, epidemiology and current rapid-detection methods. Anal. Bioanal. Chem. 2006, 384, 73–84. [Google Scholar] [CrossRef] [PubMed]
- CDC—Biosafety Home. Available online: https://www.cdc.gov/biosafety/ (accessed on 4 July 2017).
- Collier, R.J. Membrane translocation by anthrax toxin. Mol. Asp. Med. 2009, 30, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Panchal, R.G.; Halverson, K.M.; Ribot, W.; Lane, D.; Kenny, T.; Abshire, T.G.; Ezzell, J.W.; Hoover, T.A.; Powell, B.; Little, S.; et al. Purified Bacillus anthracis lethal toxin complex formed in vitro and during infection exhibits functional and biological activity. J. Biol. Chem. 2005, 280, 10834–10839. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Peng, D.; Song, X.; Ruan, L.; Mahillon, J.; Sun, M. Differentiation of Bacillus anthracis, B. cereus, and B. thuringiensis on the Basis of the csaB Gene Reflects Host Source. Appl. Environ. Microbiol. 2013, 79, 3860–3863. [Google Scholar] [CrossRef] [PubMed]
- Hurtle, W.; Bode, E.; Kulesh, D.A.; Kaplan, R.S.; Garrison, J.; Bridge, D.; House, M.; Frye, M.S.; Loveless, B.; Norwood, D. Detection of the Bacillus anthracis gyrA gene by using a minor groove binder probe. J. Clin. Microbiol. 2004, 42, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Acharya, G.; Doorneweerd, D.D.; Chang, C.-L.; Henne, W.A.; Low, P.S.; Savran, C.A. Label-free optical detection of anthrax-causing spores. J. Am. Chem. Soc. 2007, 129, 732–733. [Google Scholar] [CrossRef] [PubMed]
- Zahavy, E.; Heleg-Shabtai, V.; Zafrani, Y.; Marciano, D.; Yitzhaki, S. Application of fluorescent nanocrystals (q-dots) for the detection of pathogenic bacteria by flow-cytometry. J. Fluoresc. 2010, 20, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Krebs, M.D.; Mansfield, B.; Yip, P.; Cohen, S.J.; Sonenshein, A.L.; Hitt, B.A.; Davis, C.E. Novel technology for rapid species-specific detection of Bacillus spores. Biomol. Eng. 2006, 23, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Young, M.A.; Lyandres, O.; Van Duyne, R.P. Rapid detection of an anthrax biomarker by surface-enhanced Raman spectroscopy. J. Am. Chem. Soc. 2005, 127, 4484–4489. [Google Scholar] [CrossRef] [PubMed]
- Farrell, S.; Halsall, H.B.; Heineman, W.R. Immunoassay for B. globigii spores as a model for detecting B. anthracis spores in finished water. Analyst 2005, 130, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Zahavy, E.; Fisher, M.; Bromberg, A.; Olshevsky, U. Detection of frequency resonance energy transfer pair on double-labeled microsphere and Bacillus anthracis spores by flow cytometry. Appl. Environ. Microbiol. 2003, 69, 2330–2339. [Google Scholar] [CrossRef] [PubMed]
- Makino, S.I.; Cheun, H.I.; Watarai, M.; Uchida, I.; Takeshi, K. Detection of anthrax spores from the air by real-time PCR. Lett. Appl. Microbiol. 2001, 33, 237–240. [Google Scholar] [CrossRef] [PubMed]
- Dang, J.L.; Heroux, K.; Kearney, J.; Arasteh, A.; Gostomski, M.; Emanuel, P.A. Bacillus Spore Inactivation Methods Affect Detection Assays. Appl. Environ. Microbiol. 2001, 67, 3665–3670. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Stopa, P.J. The flow cytometry of Bacillus anthracis spores revisited. Cytometry 2000, 41, 237–244. [Google Scholar] [CrossRef]
- Gatto-Menking, D.L.; Yu, H.; Bruno, J.G.; Goode, M.T.; Miller, M.; Zulich, A.W. Sensitive detection of biotoxoids and bacterial spores using an immunomagnetic electrochemiluminescence sensor. Biosens. Bioelectron. 1995, 10, 501–507. [Google Scholar] [CrossRef]
- Phillips, A.P.; Martin, K.L. Comparison of direct and indirect immunoradiometric assays (IRMA) for Bacillus anthracis spores immobilised on multispot microscope slides. J. Appl. Bacteriol. 1983, 55, 315–324. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Moayeri, M.; Chen, Z.; Harma, H.; Zhao, J.; Hu, H.; Purcell, R.H.; Leppla, S.H.; Hewlett, I.K. Detection of Anthrax Toxin by an Ultrasensitive Immunoassay Using Europium Nanoparticles. Clin. Vaccine Immunol. CVI 2009, 16, 408–413. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, Z.P.; Sirisena, M. Development of automated amperometric detection of antibodies against Bacillus anthracis protective antigen. Anal. Bioanal. Chem. 2007, 389, 507–515. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-H.; Zhang, J.-B.; Zhang, Z.-P.; Zhou, Y.-F.; Yang, R.-F.; Chen, J.; Guo, Y.-C.; You, F.; Zhang, X.-E. Construction of single chain variable fragment (ScFv) and BiscFv-alkaline phosphatase fusion protein for detection of Bacillus anthracis. Anal. Chem. 2006, 78, 997–1004. [Google Scholar] [CrossRef] [PubMed]
- Rucker, V.C.; Havenstrite, K.L.; Herr, A.E. Antibody microarrays for native toxin detection. Anal. Biochem. 2005, 339, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Sastry, K.S.R.; Tuteja, U.; Batra, H.V. Generation and characterization of monoclonal antibodies to protective antigen of Bacillus anthracis. Indian J. Exp. Biol. 2003, 41, 123–128. [Google Scholar] [PubMed]
- Bell, C.A.; Uhl, J.R.; Hadfield, T.L.; David, J.C.; Meyer, R.F.; Smith, T.F.; Cockerill, F.R. Detection of Bacillus anthracis DNA by LightCycler PCR. J. Clin. Microbiol. 2002, 40, 2897–2902. [Google Scholar] [CrossRef] [PubMed]
- Wilson, W.J.; Erler, A.M.; Nasarabadi, S.L.; Skowronski, E.W.; Imbro, P.M. A multiplexed PCR-coupled liquid bead array for the simultaneous detection of four biothreat agents. Mol. Cell. Probes 2005, 19, 137–144. [Google Scholar] [CrossRef] [PubMed]
- Castro, A.; Okinaka, R.T. Ultrasensitive, direct detection of a specific DNAsequence of Bacillus anthracis in solution. Analyst 2000, 125, 9–11. [Google Scholar] [CrossRef] [PubMed]
- Boyer, A.E.; Quinn, C.P.; Woolfitt, A.R.; Pirkle, J.L.; McWilliams, L.G.; Stamey, K.L.; Bagarozzi, D.A.; Hart, J.C.; Barr, J.R. Detection and quantification of anthrax lethal factor in serum by mass spectrometry. Anal. Chem. 2007, 79, 8463–8470. [Google Scholar] [CrossRef] [PubMed]
- Harrison, L.H.; Ezzell, J.W.; Abshire, T.G.; Kidd, S.; Kaufmann, A.F. Evaluation of serologic tests for diagnosis of anthrax after an outbreak of cutaneous anthrax in Paraguay. J. Infect. Dis. 1989, 160, 706–710. [Google Scholar] [CrossRef] [PubMed]
- Quinn, C.P.; Semenova, V.A.; Elie, C.M.; Romero-Steiner, S.; Greene, C.; Li, H.; Stamey, K.; Steward-Clark, E.; Schmidt, D.S.; Mothershed, E.; et al. Specific, sensitive, and quantitative enzyme-linked immunosorbent assay for human immunoglobulin G antibodies to anthrax toxin protective antigen. Emerg. Infect. Dis. 2002, 8, 1103–1110. [Google Scholar] [CrossRef] [PubMed]
- Kuklenyik, Z.; Boyer, A.E.; Lins, R.; Quinn, C.P.; Gallegos-Candela, M.; Woolfitt, A.; Pirkle, J.L.; Barr, J.R. Comparison of MALDI-TOF-MS and HPLC-ESI-MS/MS for endopeptidase activity-based quantification of Anthrax lethal factor in serum. Anal. Chem. 2011, 83, 1760–1765. [Google Scholar] [CrossRef] [PubMed]
- Boyer, A.E.; Gallegos-Candela, M.; Quinn, C.P.; Woolfitt, A.R.; Brumlow, J.O.; Isbell, K.; Hoffmaster, A.R.; Lins, R.C.; Barr, J.R. High-sensitivity MALDI-TOF MS quantification of anthrax lethal toxin for diagnostics and evaluation of medical countermeasures. Anal. Bioanal. Chem. 2015, 407, 2847–2858. [Google Scholar] [CrossRef] [PubMed]
- Klaubert, B.; Vujtovic-Ockenga, N.; Wermter, R.; Schad, K.; von Meyer, L. Determination of botulinum toxins after peptic sample pre-treatment by multidimensional nanoscale liquid chromatography and nano-electrospray ion-trap mass spectrometry. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2009, 877, 1084–1092. [Google Scholar] [CrossRef] [PubMed]
- Pellizzari, R.; Rossetto, O.; Schiavo, G.; Montecucco, C. Tetanus and botulinum neurotoxins: Mechanism of action and therapeutic uses. Philos. Trans. R. Soc. B Biol. Sci. 1999, 354, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.K.; Smith, T.J.; Helma, C.H.; Ticknor, L.O.; Foley, B.T.; Svensson, R.T.; Brown, J.L.; Johnson, E.A.; Smith, L.A.; Okinaka, R.T.; et al. Genetic Diversity among Botulinum Neurotoxin-Producing Clostridial Strains. J. Bacteriol. 2007, 189, 818–832. [Google Scholar] [CrossRef] [PubMed]
- Pearce, L.B.; Borodic, G.E.; First, E.R.; Maccallum, R.D. Measurement of Botulinum Toxin Activity: Evaluation of the Lethality Assay. Toxicol. Appl. Pharmacol. 1994, 128, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Kameyama, S.; Sakaguchi, G. Assay in mice for low levels of Clostridium botulinum toxin. Int. J. Food Microbiol. 1990, 11, 271–277. [Google Scholar] [CrossRef]
- Adler, M.; Scovill, J.; Parker, G.; Lebeda, F.J.; Piotrowski, J.; Deshpande, S.S. Antagonism of botulinum toxin-induced muscle weakness by 3,4-diaminopyridine in rat phrenic nerve-hemidiaphragm preparations. Toxicon 1995, 33, 527–537. [Google Scholar] [CrossRef]
- Deshpande, S.S.; Sheridan, R.E.; Adler, M. A study of zinc-dependent metalloendopeptidase inhibitors as pharmacological antagonists in botulinum neurotoxin poisoning. Toxicon 1995, 33, 551–557. [Google Scholar] [CrossRef]
- Torii, Y.; Goto, Y.; Takahashi, M.; Ishida, S.; Harakawa, T.; Sakamoto, T.; Kaji, R.; Kozaki, S.; Ginnaga, A. Quantitative determination of biological activity of botulinum toxins utilizing compound muscle action potentials (CMAP), and comparison of neuromuscular transmission blockage and muscle flaccidity among toxins. Toxicon 2010, 55, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Lindström, M.; Keto, R.; Markkula, A.; Nevas, M.; Hielm, S.; Korkeala, H. Multiplex PCR assay for detection and identification of Clostridium botulinum types A, B, E, and F in food and fecal material. Appl. Environ. Microbiol. 2001, 67, 5694–5699. [Google Scholar] [CrossRef] [PubMed]
- Fach, P.; Gibert, M.; Griffais, R.; Guillou, J.P.; Popoff, M.R. PCR and gene probe identification of botulinum neurotoxin A-, B-, E-, F-, and G-producing Clostridium spp. and evaluation in food samples. Appl. Environ. Microbiol. 1995, 61, 389–392. [Google Scholar] [PubMed]
- Takeshi, K.; Fujinaga, Y.; Inoue, K.; Nakajima, H.; Oguma, K.; Ueno, T.; Sunagawa, H.; Ohyama, T. Simple method for detection of Clostridium botulinum type A to F neurotoxin genes by ploymerase chain reaction. Microbiol. Immunol. 1996, 40, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Fach, P.; Micheau, P.; Mazuet, C.; Perelle, S.; Popoff, M. Development of real-time PCR tests for detecting botulinum neurotoxins A, B, E, F producing Clostridium botulinum, Clostridium baratii and Clostridium butyricum. J. Appl. Microbiol. 2009, 107, 465–473. [Google Scholar] [CrossRef] [PubMed]
- Kirchner, S.; Krämer, K.M.; Schulze, M.; Pauly, D.; Jacob, D.; Gessler, F.; Nitsche, A.; Dorner, B.G.; Dorner, M.B. Pentaplexed quantitative real-time PCR assay for the simultaneous detection and quantification of botulinum neurotoxin-producing clostridia in food and clinical samples. Appl. Environ. Microbiol. 2010, 76, 4387–4395. [Google Scholar] [CrossRef] [PubMed]
- Boroff, D.A.; Shu-Chen, G. Radioimmunoassay for Type A Toxin of Clostridium botulinum. Appl. Microbiol. 1973, 25, 545–549. [Google Scholar] [PubMed]
- Ashton, A.C.; Crowther, J.S.; Dolly, J.O. A sensitive and useful radioimmunoassay for neurotoxin and its haemagglutinin complex from Clostridium botulinum. Toxicon 1985, 23, 235–246. [Google Scholar] [CrossRef]
- Sonnenschein, B. Use of the reversed passive hemagglutination in detection of Clostridium botulinum type A, B, and E toxin (author’s transl). Zentralbl. Bakteriol. Orig. A 1978, 240, 221–234. [Google Scholar]
- Johnson, H.M.; Brenner, K.; Angelotti, R.; Hall, H.E. Serological Studies of Types A, B, and E Botulinal Toxins by Passive Hemagglutination and Bentonite Flocculation. J. Bacteriol. 1966, 91, 967–974. [Google Scholar] [PubMed]
- Mestrandrea, L.W. Rapid Detection of Clostridium botulinum Toxin by Capillary Tube Diffusion. Appl. Microbiol. 1974, 27, 1017–1022. [Google Scholar] [PubMed]
- Kozaki, S.; Dufrenne, J.; Hagenaars, A.M.; Notermans, S. Enzyme linked immunosorbent assay (ELISA) for detection of Clostridium botulinum type B toxin. Jpn. J. Med. Sci. Biol. 1979, 32, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Notermans, S.; Dufrenne, J.; Kozaki, S. Enzyme-linked immunosorbent assay for detection of Clostridium botulinum type E toxin. Appl. Environ. Microbiol. 1979, 37, 1173–1175. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, J.L. Comparison of amplified ELISA and mouse bioassay procedures for determination of botulinal toxins A, B, E, and F. J. AOAC Int. 2001, 84, 85–88. [Google Scholar] [PubMed]
- Ferreira, J.L.; Eliasberg, S.J.; Harrison, M.A.; Edmonds, P. Detection of preformed type A botulinal toxin in hash brown potatoes by using the mouse bioasssay and a modified ELISA test. J. AOAC Int. 2001, 84, 1460–1464. [Google Scholar] [PubMed]
- Ferreira, J.L.; Maslanka, S.; Johnson, E.; Goodnough, M. Detection of botulinal neurotoxins A, B, E, and F by amplified enzyme-linked immunosorbent assay: Collaborative study. J. AOAC Int. 2003, 86, 314–331. [Google Scholar] [PubMed]
- Ferreira, J.L.; Eliasberg, S.J.; Edmonds, P.; Harrison, M.A. Comparison of the mouse bioassay and enzyme-linked immunosorbent assay procedures for the detection of type A botulinal toxin in food. J. Food Prot. 2004, 67, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Dezfulian, M.; Bartlett, J.G. Selective isolation and rapid identification of Clostridium botulinum types A and B by toxin detection. J. Clin. Microbiol. 1985, 21, 231–233. [Google Scholar] [PubMed]
- Sharma, S.K.; Ferreira, J.L.; Eblen, B.S.; Whiting, R.C. Detection of type A, B, E, and F Clostridium botulinum neurotoxins in foods by using an amplified enzyme-linked immunosorbent assay with digoxigenin-labeled antibodies. Appl. Environ. Microbiol. 2006, 72, 1231–1238. [Google Scholar] [CrossRef] [PubMed]
- Poli, M.A.; Rivera, V.R.; Neal, D. Development of sensitive colorimetric capture ELISAs for Clostridium botulinum neurotoxin serotypes E and F. Toxicon 2002, 40, 797–802. [Google Scholar] [CrossRef]
- Dezfulian, M.; Hatheway, C.L.; Yolken, R.H.; Bartlett, J.G. Enzyme-linked immunosorbent assay for detection of Clostridium botulinum type A and type B toxins in stool samples of infants with botulism. J. Clin. Microbiol. 1984, 20, 379–383. [Google Scholar] [PubMed]
- Smith, T.J.; Lou, J.; Geren, I.N.; Forsyth, C.M.; Tsai, R.; LaPorte, S.L.; Tepp, W.H.; Bradshaw, M.; Johnson, E.A.; Smith, L.A.; et al. Sequence Variation within Botulinum Neurotoxin Serotypes Impacts Antibody Binding and Neutralization. Infect. Immun. 2005, 73, 5450–5457. [Google Scholar] [CrossRef] [PubMed]
- Stanker, L.H.; Merrill, P.; Scotcher, M.C.; Cheng, L.W. Development and partial characterization of high-affinity monoclonal antibodies for botulinum toxin type A and their use in analysis of milk by sandwich ELISA. J. Immunol. Methods 2008, 336, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Guo, J.; Xu, C.; Li, X.; Chen, S. A Simple, Rapid and Sensitive FRET Assay for Botulinum Neurotoxin Serotype B Detection. PLoS ONE 2014, 9, e114124. [Google Scholar] [CrossRef] [PubMed]
- Poras, H.; Ouimet, T.; Orng, S.-V.; Fournié-Zaluski, M.-C.; Popoff, M.R.; Roques, B.P. Detection and Quantification of Botulinum Neurotoxin Type A by a Novel Rapid In Vitro Fluorimetric Assay. Appl. Environ. Microbiol. 2009, 75, 4382–4390. [Google Scholar] [CrossRef] [PubMed]
- Rasooly, R.; Stanker, L.H.; Carter, J.M.; Do, P.M.; Cheng, L.W.; He, X.; Brandon, D.L. Detection of botulinum neurotoxin-A activity in food by peptide cleavage assay. Int. J. Food Microbiol. 2008, 126, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Hallis, B.; James, B.A.; Shone, C.C. Development of novel assays for botulinum type A and B neurotoxins based on their endopeptidase activities. J. Clin. Microbiol. 1996, 34, 1934–1938. [Google Scholar] [PubMed]
- Barr, J.R.; Moura, H.; Boyer, A.E.; Woolfitt, A.R.; Kalb, S.R.; Pavlopoulos, A.; McWilliams, L.G.; Schmidt, J.G.; Martinez, R.A.; Ashley, D.L. Botulinum Neurotoxin Detection and Differentiation by Mass Spectrometry. Emerg. Infect. Dis. 2005, 11, 1578–1583. [Google Scholar] [CrossRef] [PubMed]
- Kalb, S.R.; Krilich, J.C.; Dykes, J.K.; Lúquez, C.; Maslanka, S.E.; Barr, J.R. Detection of Botulinum Toxins A, B, E, and F in Foods by Endopep-MS. J. Agric. Food Chem. 2015, 63, 1133–1141. [Google Scholar] [CrossRef] [PubMed]
- Rosen, O.; Feldberg, L.; Gura, S.; Brosh-Nissimov, T.; Guri, A.; Zimhony, O.; Shapiro, E.; Beth-Din, A.; Stein, D.; Ozeri, E.; et al. Early, Real-Time Medical Diagnosis of Botulism by Endopeptidase-Mass Spectrometry. Clin. Infect. Dis. 2015, 61, e58–e61. [Google Scholar] [CrossRef] [PubMed]
- Van Baar, B.L.M.; Hulst, A.G.; de Jong, A.L.; Wils, E.R.J. Characterisation of botulinum toxins type A and B, by matrix-assisted laser desorption ionisation and electrospray mass spectrometry. J. Chromatogr. A 2002, 970, 95–115. [Google Scholar] [CrossRef]
- Van Baar, B.L.M.; Hulst, A.G.; de Jong, A.L.; Wils, E.R.J. Characterisation of botulinum toxins type C, D, E, and F by matrix-assisted laser desorption ionisation and electrospray mass spectrometry. J. Chromatogr. A 2004, 1035, 97–114. [Google Scholar] [CrossRef] [PubMed]
- Hines, H.B.; Lebeda, F.; Hale, M.; Brueggemann, E.E. Characterization of Botulinum Progenitor Toxins by Mass Spectrometry. Appl. Environ. Microbiol. 2005, 71, 4478–4486. [Google Scholar] [CrossRef] [PubMed]
- Rasetti-Escargueil, C.; Liu, Y.; Rigsby, P.; Jones, R.G.A.; Sesardic, D. Phrenic nerve-hemidiaphragm as a highly sensitive replacement assay for determination of functional botulinum toxin antibodies. Toxicon 2011, 57, 1008–1016. [Google Scholar] [CrossRef] [PubMed]
- Bigalke, H.; Rummel, A. Botulinum Neurotoxins: Qualitative and Quantitative Analysis Using the Mouse Phrenic Nerve Hemidiaphragm Assay (MPN). Toxins 2015, 7, 4895–4905. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.R.A.; Moreira, G.M.S.G.; da Cunha, C.E.P.; Mendonça, M.; Salvarani, F.M.; Moreira, Â.N.; Conceição, F.R. Recombinant Alpha, Beta, and Epsilon Toxins of Clostridium perfringens: Production Strategies and Applications as Veterinary Vaccines. Toxins 2016, 8, 340. [Google Scholar] [CrossRef] [PubMed]
- Petit, L.; Gibert, M.; Popoff, M.R. Clostridium perfringens: Toxinotype and genotype. Trends Microbiol. 1999, 7, 104–110. [Google Scholar] [CrossRef]
- Sakurai, J.; Nagahama, M.; Oda, M. Clostridium perfringens alpha-toxin: Characterization and mode of action. J. Biochem. 2004, 136, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Steinthorsdottir, V.; Halldórsson, H.; Andrésson, O.S. Clostridium perfringens beta-toxin forms multimeric transmembrane pores in human endothelial cells. Microb. Pathog. 2000, 28, 45–50. [Google Scholar] [CrossRef] [PubMed]
- Stiles, B.G.; Barth, G.; Barth, H.; Popoff, M.R. Clostridium perfringens Epsilon Toxin: A Malevolent Molecule for Animals and Man? Toxins 2013, 5, 2138–2160. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, J.; Nagahama, M.; Oda, M.; Tsuge, H.; Kobayashi, K. Clostridium perfringens Iota-Toxin: Structure and Function. Toxins 2009, 1, 208–228. [Google Scholar] [CrossRef] [PubMed]
- Farzan, A.; Kircanski, J.; DeLay, J.; Soltes, G.; Songer, J.G.; Friendship, R.; Prescott, J.F. An investigation into the association between cpb2-encoding Clostridium perfringens type A and diarrhea in neonatal piglets. Can. J. Vet. Res. 2013, 77, 45–53. [Google Scholar] [PubMed]
- Hamad, M.A.; Habra, N.; Allouz, A.K. Biotyping of Clostridium perfringens strains isolated from enterotoxemia cases in sheep using ELISA technique. Iraqi J. Vet. Sci. 2010, 24, 17–22. [Google Scholar]
- Tansuphasiri, U. Development of duplex PCR assay for rapid detection of enterotoxigenic isolates of Clostridium perfringens. Southeast Asian J. Trop. Med. Public Health 2001, 32, 105–113. [Google Scholar] [PubMed]
- Kull, S.; Pauly, D.; Störmann, B.; Kirchner, S.; Stämmler, M.; Dorner, M.B.; Lasch, P.; Naumann, D.; Dorner, B.G. Multiplex detection of microbial and plant toxins by immunoaffinity enrichment and matrix-assisted laser desorption/ionization mass spectrometry. Anal. Chem. 2010, 82, 2916–2924. [Google Scholar] [CrossRef] [PubMed]
- Alam, S.I.; Kumar, B.; Kamboj, D.V. Multiplex detection of protein toxins using MALDI-TOF-TOF tandem mass spectrometry: Application in unambiguous toxin detection from bioaerosol. Anal. Chem. 2012, 84, 10500–10507. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.; Kumar, M.; Kanaujia, P.K.; Virdi, J.S. MALDI-TOF mass spectrometry: An emerging technology for microbial identification and diagnosis. Front. Microbiol. 2015, 6, 791. [Google Scholar] [CrossRef] [PubMed]
- Brodzik, C.; Augustynowicz, E.; Korzeniowska-Kowal, A.; Lutyhska, A. Application of the MALDI-TOF for identification of Clostridium perfringens strains. Medycyna Doswiadczalna I Mikrobiologia 2016, 68, 13–21. [Google Scholar] [PubMed]
- Krt, B. Development and evaluation of various enzyme-linked immunosorbent assays for the detection of Clostridium perfringens beta anti-toxins. FEMS Immunol. Med. Microbiol. 1999, 24, 293–297. [Google Scholar] [PubMed]
- Baums, C.G.; Schotte, U.; Amtsberg, G.; Goethe, R. Diagnostic multiplex PCR for toxin genotyping of Clostridium perfringens isolates. Vet. Microbiol. 2004, 100, 11–16. [Google Scholar] [CrossRef]
- Albini, S.; Brodard, I.; Jaussi, A.; Wollschlaeger, N.; Frey, J.; Miserez, R.; Abril, C. Real-time multiplex PCR assays for reliable detection of Clostridium perfringens toxin genes in animal isolates. Vet. Microbiol. 2008, 127, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Hernández, M.; López-Enríquez, L.; Rodríguez-Lázaro, D. Quantitative Detection of Clostridium perfringens by Real-Time PCR in Raw Milk. Food Anal. Methods 2017, 10, 1139–1147. [Google Scholar] [CrossRef]
- McClane, B.A.; Strouse, R.J. Rapid detection of Clostridium perfringens type A enterotoxin by enzyme-linked immunosorbent assay. J. Clin. Microbiol. 1984, 19, 112–115. [Google Scholar] [PubMed]
- Nagahama, M.; Kobayashi, K.; Ochi, S.; Sakurai, J. Enzyme-linked immunosorbent assay for rapid detection of toxins from Clostridium perfringens. FEMS Microbiol. Lett. 1991, 84, 41–44. [Google Scholar] [CrossRef]
- Everley, R.A.; Mott, T.M.; Toney, D.M.; Croley, T.R. Characterization of Clostridium species utilizing liquid chromatography/mass spectrometry of intact proteins. J. Microbiol. Methods 2009, 77, 152–158. [Google Scholar] [CrossRef] [PubMed]
- Naylor, R.D.; Martin, P.K.; Sharpe, R.T. Detection of Clostridium perfringens epsilon toxin by ELISA. Res. Vet. Sci. 1987, 42, 255–256. [Google Scholar] [PubMed]
- Layana, J.E.; Fernandez Miyakawa, M.E.; Uzal, F.A. Evaluation of different fluids for detection of Clostridium perfringens type D epsilon toxin in sheep with experimental enterotoxemia. Anaerobe 2006, 12, 204–206. [Google Scholar] [CrossRef] [PubMed]
- Uzal, F.A.; Plumb, J.J.; Blackall, L.L.; Kelly, W.R. PCR detection of Clostridium perfringens producing different toxins in faeces of goats. Lett. Appl. Microbiol. 1997, 25, 339–344. [Google Scholar] [CrossRef] [PubMed]
- Seyer, A.; Fenaille, F.; Féraudet-Tarisse, C.; Volland, H.; Popoff, M.R.; Tabet, J.-C.; Junot, C.; Becher, F. Rapid quantification of clostridial epsilon toxin in complex food and biological matrixes by immunopurification and ultraperformance liquid chromatography-tandem mass spectrometry. Anal. Chem. 2012, 84, 5103–5109. [Google Scholar] [CrossRef] [PubMed]
- Payne, D.W.; Williamson, E.D.; Havard, H.; Modi, N.; Brown, J. Evaluation of a new cytotoxicity assay for Clostridium perfringens type D epsilon toxin. FEMS Microbiol. Lett. 1994, 116, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Fredriksson, S.-Å.; Artursson, E.; Bergström, T.; Östin, A.; Nilsson, C.; Åstot, C. Identification of RIP-II toxins by affinity enrichment, enzymatic digestion and LC-MS. Anal. Chem. 2015, 87, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Hale, M.L. Microtiter-based assay for evaluating the biological activity of ribosome-inactivating proteins. Pharmacol. Toxicol. 2001, 88, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Becher, F.; Duriez, E.; Volland, H.; Tabet, J.C.; Ezan, E. Detection of Functional Ricin by Immunoaffinity and Liquid Chromatography–Tandem Mass Spectrometry. Anal. Chem. 2007, 79, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Pauly, D.; Worbs, S.; Kirchner, S.; Shatohina, O.; Dorner, M.B.; Dorner, B.G. Real-time cytotoxicity assay for rapid and sensitive detection of ricin from complex matrices. PLoS ONE 2012, 7, e35360. [Google Scholar] [CrossRef] [PubMed]
- Poli, M.A.; Rivera, V.R.; Hewetson, J.F.; Merrill, G.A. Detection of ricin by colorimetric and chemiluminescence ELISA. Toxicon 1994, 32, 1371–1377. [Google Scholar] [CrossRef]
- Cook, D.L.; David, J.; Griffiths, G.D. Retrospective identification of ricin in animal tissues following administration by pulmonary and oral routes. Toxicology 2006, 223, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Worbs, S.; Köhler, K.; Pauly, D.; Avondet, M.-A.; Schaer, M.; Dorner, M.B.; Dorner, B.G. Ricinus communis Intoxications in Human and Veterinary Medicine-A Summary of Real Cases. Toxins 2011, 3, 1332–1372. [Google Scholar] [CrossRef] [PubMed]
- Bozza, W.P.; Tolleson, W.H.; Rivera Rosado, L.A.; Zhang, B. Ricin detection: Tracking active toxin. Biotechnol. Adv. 2015, 33, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Assiri, A.S. Ricin poisoning causing death after ingestion of herbal medicine. Ann. Saudi Med. 2012, 32, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Beyer, N.H.; Kogutowska, E.; Hansen, J.J.; Engelhart Illigen, K.E.; Heegaard, N.H.H. A mouse model for ricin poisoning and for evaluating protective effects of antiricin antibodies. Clin. Toxicol. 2009, 47, 219–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Baudys, J.; Barr, J.R.; Kalb, S.R. Improved Sensitivity for the Qualitative and Quantitative Analysis of Active Ricin by MALDI-TOF Mass Spectrometry. Anal. Chem. 2016, 88, 6867–6872. [Google Scholar] [CrossRef] [PubMed]
- Sturm, M.B.; Schramm, V.L. Detecting Ricin: Sensitive Luminescent Assay for Ricin A-Chain Ribosome Depurination Kinetics. Anal. Chem. 2009, 81, 2847–2853. [Google Scholar] [CrossRef] [PubMed]
- Kalb, S.R.; Barr, J.R. Mass spectrometric detection of ricin and its activity in food and clinical samples. Anal. Chem. 2009, 81, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, G.D. Understanding Ricin from a Defensive Viewpoint. Toxins 2011, 3, 1373–1392. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.M.; Lamb, F.I.; Pappin, D.J.; Lord, J.M. The primary sequence of Ricinus communis agglutinin. Comparison with ricin. J. Biol. Chem. 1985, 260, 15682–15686. [Google Scholar] [PubMed]
- Kumar, O.; Pradhan, S.; Sehgal, P.; Singh, Y.; Vijayaraghavan, R. Denatured ricin can be detected as native ricin by immunological methods, but nontoxic in vivo. J. Forensic Sci. 2010, 55, 801–807. [Google Scholar] [CrossRef] [PubMed]
- Mouser, P.; Filigenzi, M.S.; Puschner, B.; Johnson, V.; Miller, M.A.; Hooser, S.B. Fatal ricin toxicosis in a puppy confirmed by liquid chromatography/mass spectrometry when using ricinine as a marker. J. Vet. Diagn. Investig. 2007, 19, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Hines, H.B.; Brueggemann, E.E.; Hale, M.L. High-performance liquid chromatography-mass selective detection assay for adenine released from a synthetic RNA substrate by ricin A chain. Anal. Biochem. 2004, 330, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Bevilacqua, V.L.H.; Nilles, J.M.; Rice, J.S.; Connell, T.R.; Schenning, A.M.; Reilly, L.M.; Durst, H.D. Ricin activity assay by direct analysis in real time mass spectrometry detection of adenine release. Anal. Chem. 2010, 82, 798–800. [Google Scholar] [CrossRef] [PubMed]
- McGrath, S.C.; Schieltz, D.M.; McWilliams, L.G.; Pirkle, J.L.; Barr, J.R. Detection and Quantification of Ricin in Beverages Using Isotope Dilution Tandem Mass Spectrometry. Anal. Chem. 2011, 83, 2897–2905. [Google Scholar] [CrossRef] [PubMed]
- Schieltz, D.M.; McGrath, S.C.; McWilliams, L.G.; Rees, J.; Bowen, M.D.; Kools, J.J.; Dauphin, L.A.; Gomez-Saladin, E.; Newton, B.N.; Stang, H.L.; et al. Analysis of active ricin and castor bean proteins in a ricin preparation, castor bean extract, and surface swabs from a public health investigation. Forensic Sci. Int. 2011, 209, 70–79. [Google Scholar] [CrossRef] [PubMed]
- Felder, E.; Mossbrugger, I.; Lange, M.; Wölfel, R. Simultaneous detection of ricin and abrin DNA by real-time PCR (qPCR). Toxins 2012, 4, 633–642. [Google Scholar] [CrossRef] [PubMed]
- Pauly, D.; Kirchner, S.; Stoermann, B.; Schreiber, T.; Kaulfuss, S.; Schade, R.; Zbinden, R.; Avondet, M.-A.; Dorner, M.B.; Dorner, B.G. Simultaneous quantification of five bacterial and plant toxins from complex matrices using a multiplexed fluorescent magnetic suspension assay. Analyst 2009, 134, 2028–2039. [Google Scholar] [CrossRef] [PubMed]
- Garber, E.A.E.; Venkateswaran, K.V.; O’Brien, T.W. Simultaneous multiplex detection and confirmation of the proteinaceous toxins abrin, ricin, botulinum toxins, and Staphylococcus enterotoxins A, B, and C in food. J. Agric. Food Chem. 2010, 58, 6600–6607. [Google Scholar] [CrossRef] [PubMed]
- Hegde, R.; Maiti, T.K.; Podder, S.K. Purification and characterization of three toxins and two agglutinins from Abrus precatorius seed by using lactamyl-Sepharose affinity chromatography. Anal. Biochem. 1991, 194, 101–109. [Google Scholar] [CrossRef]
- Hung, C.H.; Lee, M.C.; Lee, T.C.; Lin, J.Y. Primary structure of three distinct isoabrins determined by cDNA sequencing. Conservation and significance. J. Mol. Biol. 1993, 229, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Hegde, R.; Podder, S.K. A- and B-subunit variant distribution in the holoprotein variants of protein toxin abrin: Variants of abrins I and III have constant toxic A subunits and variant lectin B subunits. Arch. Biochem. Biophys. 1997, 344, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.L.; Tsai, C.C.; Lin, S.C.; Wang, L.I.; Hsu, C.I.; Hwang, M.J.; Lin, J.Y. Primary structure and function analysis of the Abrus precatorius agglutinin A chain by site-directed mutagenesis. Pro(199) of amphiphilic alpha-helix H impairs protein synthesis inhibitory activity. J. Biol. Chem. 2000, 275, 1897–1901. [Google Scholar] [CrossRef] [PubMed]
- Bagaria, A.; Surendranath, K.; Ramagopal, U.A.; Ramakumar, S.; Karande, A.A. Structure-function analysis and insights into the reduced toxicity of Abrus precatorius agglutinin I in relation to abrin. J. Biol. Chem. 2006, 281, 34465–34474. [Google Scholar] [CrossRef] [PubMed]
- Garber, E.A.E. Toxicity and detection of ricin and abrin in beverages. J. Food Prot. 2008, 71, 1875–1883. [Google Scholar] [CrossRef] [PubMed]
- Garber, E.A.E.; Walker, J.L.; O’Brien, T.W. Detection of abrin in food using enzyme-linked immunosorbent assay and electrochemiluminescence technologies. J. Food Prot. 2008, 71, 1868–1874. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Tian, X.-L.; Li, Y.-S.; Pan, F.-G.; Zhang, Y.-Y.; Zhang, J.-H.; Wang, X.-R.; Ren, H.-L.; Lu, S.-Y.; Li, Z.-H.; et al. Development of a monoclonal antibody-based sandwich-type enzyme-linked immunosorbent assay (ELISA) for detection of abrin in food samples. Food Chem. 2012, 135, 2661–2665. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Nie, C.; Wang, J.; Wang, J.; Kang, L.; Zhou, Y.; Wang, J.-L. Colloidal gold-based immunochromatographic test strip for rapid detection of abrin in food samples. J. Food Prot. 2012, 75, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Li, X.; Liu, G.; Zhang, B.; Zhang, Y.; Kong, T.; Tang, J.; Li, D.; Wang, Z. A colloidal gold probe-based silver enhancement immunochromatographic assay for the rapid detection of abrin-a. Biosens. Bioelectron. 2011, 26, 3710–3713. [Google Scholar] [CrossRef] [PubMed]
- Ramage, J.G.; Prentice, K.W.; Morse, S.A.; Carter, A.J.; Datta, S.; Drumgoole, R.; Gargis, S.R.; Griffin-Thomas, L.; Hastings, R.; Masri, H.P.; et al. Comprehensive laboratory evaluation of a specific lateral flow assay for the presumptive identification of abrin in suspicious white powders and environmental samples. Biosecur. Bioterror. Biodef. Strategy Pract. Sci. 2014, 12, 49–62. [Google Scholar] [CrossRef] [PubMed]
- Owens, J.; Koester, C. Quantitation of Abrine, an Indole Alkaloid Marker of the Toxic Glycoproteins Abrin, by Liquid Chromatography/Tandem Mass Spectrometry When Spiked into Various Beverages. J. Agric. Food Chem. 2008, 56, 11139–11143. [Google Scholar] [CrossRef] [PubMed]
- Olsnes, S.; Stirpe, F.; Sandvig, K.; Pihl, A. Isolation and characterization of viscumin, a toxic lectin from Viscum album L. (mistletoe). J. Biol. Chem. 1982, 257, 13263–13270. [Google Scholar] [PubMed]
- Rubina, A.Y.; Dyukova, V.I.; Dementieva, E.I.; Stomakhin, A.A.; Nesmeyanov, V.A.; Grishin, E.V.; Zasedatelev, A.S. Quantitative immunoassay of biotoxins on hydrogel-based protein microchips. Anal. Biochem. 2005, 340, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Jäggy, C.; Musielski, H.; Urech, K.; Schaller, G. Quantitative determination of lectins in mistletoe preparations. Arzneimittel-Forschung 1995, 45, 905–909. [Google Scholar] [PubMed]
- Layer, R.T.; McIntosh, J.M. Conotoxins: Therapeutic Potential and Application. Mar. Drugs 2006, 4, 119–142. [Google Scholar] [CrossRef]
- Cestèle, S.; Catterall, W.A. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie 2000, 82, 883–892. [Google Scholar] [CrossRef]
- Clark, R.J.; Jensen, J.; Nevin, S.T.; Callaghan, B.P.; Adams, D.J.; Craik, D.J. The engineering of an orally active conotoxin for the treatment of neuropathic pain. Angew. Chem. Int. Ed. Engl. 2010, 49, 6545–6548. [Google Scholar] [CrossRef] [PubMed]
- Olivera, B.M.; Imperial, J.S.; Concepcion, G.P. Snail Peptides. In Handbook of Biologically Active Peptides, 2nd ed.; Kastin, A.J., Ed.; Academic Press: Boston, MA, USA, 2013; Chapter 61; pp. 437–450. ISBN 978-0-12-385095-9. [Google Scholar]
- Ashcom, J.D.; Stiles, B.G. Characterization of alpha-conotoxin interactions with the nicotinic acetylcholine receptor and monoclonal antibodies. Biochem. J. 1997, 328 Pt 1, 245–250. [Google Scholar] [CrossRef] [PubMed]
- Loughnan, M.L.; Nicke, A.; Jones, A.; Adams, D.J.; Alewood, P.F.; Lewis, R.J. Chemical and functional identification and characterization of novel sulfated alpha-conotoxins from the cone snail Conus anemone. J. Med. Chem. 2004, 47, 1234–1241. [Google Scholar] [CrossRef] [PubMed]
- Prasarnpun, S.; Walsh, J.; Awad, S.S.; Harris, J.B. Envenoming bites by kraits: The biological basis of treatment-resistant neuromuscular paralysis. Brain 2005, 128, 2987–2996. [Google Scholar] [CrossRef] [PubMed]
- Shan, L.-L.; Gao, J.-F.; Zhang, Y.-X.; Shen, S.-S.; He, Y.; Wang, J.; Ma, X.-M.; Ji, X. Proteomic characterization and comparison of venoms from two elapid snakes (Bungarus multicinctus and Naja atra) from China. J. Proteom. 2016, 138, 83–94. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.S.; Yang, C.C. Separation and Characterization of the A Chain and B Chain in β1-Bungarotoxin from Bungarus Multicinctus (Taiwan Banded Krait) Venom. J. Protein Chem. 1993, 12, 469–475. [Google Scholar] [CrossRef] [PubMed]
- Selvanayagam, Z.E.; Gopalakrishnakone, P. Tests for detection of snake venoms, toxins and venom antibodies: Review on recent trends (1987–1997). Toxicon 1999, 37, 565–586. [Google Scholar] [CrossRef]
- Dong, L.V.; Selvanayagam, Z.E.; Gopalakrishnakone, P.; Eng, K.H. A new avidin–biotin optical immunoassay for the detection of beta-bungarotoxin and application in diagnosis of experimental snake envenomation. J. Immunol. Methods 2002, 260, 125–136. [Google Scholar] [CrossRef]
- Selvanayagam, Z.E.; Neuzil, P.; Gopalakrishnakone, P.; Sridhar, U.; Singh, M.; Ho, L.C. An ISFET-based immunosensor for the detection of β-Bungarotoxin. Biosens. Bioelectron. 2002, 17, 821–826. [Google Scholar] [CrossRef]


{kind=link}
| Target Protein | Method of Detection | References |
|---|---|---|
| SEs | Kitten emesis test | [53] |
| Reversed passive latex agglutination kit | [70,71,72,73] | |
| Enzyme-linked immunosorbent assay | [58,59] | |
| SEA | Double-antibody solid-phase enzyme immunoassay | [56] |
| Enzyme-linked immunosorbent assay | [60] | |
| SEB | Direct skin test | [54] |
| Single-gel diffusion test | [74] | |
| Latex agglutination test | [75] | |
| Latex agglutination inhibition test | [76] | |
| Enzyme-linked immunosorbent assay | [57,62] | |
| Fluorescence-based immunoassay | [63] | |
| Immunoreactor-based competitive fluoroimmunoassay | [65] | |
| ELISA-Lab-on-a-chip | [67] | |
| Surface plasmon resonance | [68] | |
| Electrochemical immunoassay using enzyme-nanosilica-doped carbon nanotubes for signal amplification | [69] | |
| SEA, SEB | Double-gel diffusion assay | [77] |
| Avidin-biotin ELISA | [61] | |
| SEC | Fluoroimmunoassay based on functionalized fluorescent core-shell nanoparticle labels | [64] |
| Staphylococcal enterotoxin genes | Polymerase chain reaction | [12] |
| Toxin | Matrix | Detection (QqQ) | Standards (PSAQ) | LOD | LOQ * | Reference |
|---|---|---|---|---|---|---|
| SEB | Apple juice | (QTOF) | - | 60 ng/mL | N/A | [81] |
| SEA | Chinese dessert (Coco-pearls) | + | + | N/A | N/A | [42] |
| SEB | Chicken meat | (QIT) | - | N/A | N/A | [82] |
| SEA | Milk | + | + | SEA: 2.5 ng/g | Milk: 2.5 ppb | [83] |
| SEB | Shrimp | SEB: 10 ng/g | Shrimp: 5 ppm | |||
| SEA | Milk | + | + | SEA: 4 ng/g | N/A | [84] |
| SEB | SEB: 8 ng/g | |||||
| SEA | Soup | + | + | SEA: 78 ng/mL | N/A | [85] |
| SEB | SEB: 141 ng/mL | |||||
| SED | SED: 48 ng/mL |
| Target of Method | Method of Detection | Reference |
|---|---|---|
| Gene | RT-PCR-F | [91] |
| Spores | Light transmission | [92] |
| Fluorescence | [93] | |
| MS | [94] | |
| SERS | [95] | |
| Bead-based sandwich immunoassay | [96] | |
| FRET-flow cytometry | [97] | |
| RT-PCR-F | [98] | |
| ELISA flow cytometry | [99] | |
| Flow cytometry | [100] | |
| ECL | [101] | |
| Immunoradiometric assay | [102] | |
| Protective antigen | Fluorescence | [103] |
| ENIA | [103] | |
| AM | [104] | |
| AFM | [105] | |
| Antibody microarray | [106] | |
| ELISA | [107] | |
| DNA | RT-PCR-F | [108] |
| PCR | [109] | |
| Fluorescence | [110] | |
| Lethal factor | MS | [111] |
| Antibody microarray | [106] |
| Target of Method | Technique | Reference |
|---|---|---|
| Botulinum toxin activity | Mouse lethality assay | [119,120] |
| Rat compound muscle action potentials test | [123] | |
| Spores | Polymerase chain reaction | [124] |
| Genes | Polymerase chain reaction | [125,126,127,128] |
| BoNT | Radioimmunoassay | [129,130] |
| Passive hemagglutination | [131,132] | |
| Gel diffusion assay | [133] | |
| Enzyme-linked immune sorbent assay | [136,137,138,139,140,141,142,143,144,145] | |
| Fluorescence resonance energy transfer technology | [146] | |
| In vitro fluorimetric assay | [147] | |
| Endopeptidase assay | [149] | |
| Endopeptidase-MS assay | [150,151,152] | |
| Mass spectrometry | [116,153,154,155] |
| Toxinotype | Major Toxins | Minor Toxins | Associated Disease | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| α | β | ε | ι | CPE | λ | θ | δ | Humans | Animals | |
| A | ++ | − | − | − | + | − | + | − | Gangrene, GI diseases | Diarrhea (foals, pig) NE in fowl |
| B | + | + | + | − | − | + | − | + | NE | Dysentery in newborn lambs Hemorrhagic enteritis in neonatal calves and foals Enterotoxemia in sheep |
| C | + | + | − | − | + | − | − | + | NE in piglets Enterotoxemia in sheep | |
| D | + | − | + | − | + | + | − | − | Enterotoxemia in lambs, sheep, calves and goats | |
| E | + | − | − | + | + | + | − | − | Enterotoxemia in calves | |
| Target of Method | Technique | Reference |
|---|---|---|
| C. perfringens strains | ELISA | [165] |
| MS | [170,177] | |
| Gene | PCR | [166,170,172,173,174] |
| Toxins | MS | [167,168,181] |
| EIA | [171,175,176] | |
| ELISA | [178,179] | |
| PCR | [180] | |
| Clostridium toxin activity | Cytotoxicity assay | [182] |
| Target of Method | Technique | Reference |
|---|---|---|
| RIPs | MS | [183] |
| Multiplex fluorescent magnetic suspension assay | [186] | |
| ELISA | [212] | |
| Hydrogen-based protein microchips | [220] | |
| Biological activity of RIP toxins | Microtiter-based assay (luciferase luminescence test) | [184] |
| Ricin | MS | [85,185,193,195,200,201,202,203] |
| ELISA | [187,188,198] | |
| Mouse model | [192] | |
| Luminescent assay | [194] | |
| Chemiluminescence-based microarray immunoassay | [66] | |
| Ricinine | MS | [199] |
| Genes (abrin) | PCR | [204] |
| Abrin | ELISA | [213,214] |
| Immunochromatographic assay | [215,216] | |
| MS | [218] | |
| Viscumin | Enzyme-linked lectin assay | [221] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Duracova, M.; Klimentova, J.; Fucikova, A.; Dresler, J. Proteomic Methods of Detection and Quantification of Protein Toxins. Toxins 2018, 10, 99. https://doi.org/10.3390/toxins10030099
Duracova M, Klimentova J, Fucikova A, Dresler J. Proteomic Methods of Detection and Quantification of Protein Toxins. Toxins. 2018; 10(3):99. https://doi.org/10.3390/toxins10030099
Chicago/Turabian StyleDuracova, Miloslava, Jana Klimentova, Alena Fucikova, and Jiri Dresler. 2018. "Proteomic Methods of Detection and Quantification of Protein Toxins" Toxins 10, no. 3: 99. https://doi.org/10.3390/toxins10030099
APA StyleDuracova, M., Klimentova, J., Fucikova, A., & Dresler, J. (2018). Proteomic Methods of Detection and Quantification of Protein Toxins. Toxins, 10(3), 99. https://doi.org/10.3390/toxins10030099