Relationships between the Toxicities of Radix Aconiti Lateralis Preparata (Fuzi) and the Toxicokinetics of Its Main Diester-Diterpenoid Alkaloids

Abstract
:1. Introduction
2. Classification of Aconitum Alkaloids
3. Toxicities of the Main Diester-Diterpenoid Alkaloids in Fuzi
3.1. Clinical Toxicities of Fuzi
3.2. Toxicities of Fuzi and Its Main Diester-Diterpenoid Alkaloids in Pre-Clinical Models
4. Toxicokinetic Characteristics of the Main Diester-Diterpenoid Alkaloids in Fuzi
4.1. Toxicokinetic Profiles of the Main Diester-Diterpenoid Alkaloids in Humans after Ingestion of Fuzi
4.2. Toxicokinetic Characteristics of the Main Diester-Diterpenoid Alkaloids of Fuzi in Pre-Clinical Models
4.2.1. Absorption
4.2.2. Distribution
4.2.3. Metabolism
4.2.4. Excretion
4.2.5. Modulation of the Transporters and Enzymes
5. Relationships between Toxicities of Fuzi Extract and the Toxicokinetic Profiles of Its Main Diester-Diterpenoid Alkaloids
5.1. Relationship between the Cardiac Toxicity and the Toxicokinetic Profiles of the Main Diester-Diterpenoid Alkaloids after Oral Intake of Their Pure Compounds
5.1.1. Relationship between the Acute Cardiac Toxicity and Toxicokinetic Profiles of the Main Diester-Diterpenoid Alkaloids after a Single Oral Dose
5.1.2. Relationship between the Sub-Chronic Toxicity and Toxicokinetic Profiles of the Aconitine after Multiple Oral Doses
5.2. Relationship between the Heart, Liver, and Kidney Toxicities of Fuzi and the Toxicokinetics of Its Main Diester-Diterpenoid Alkaloids after Oral Intake of Its Extract
5.2.1. Dose-Dependent Toxicities of Fuzi and the Toxicokinetic Profiles of Its Main Diester-Diterpenoid Alkaloids after Single Oral Administration
AC-Related Toxicokinetic Profiles at the Toxic Level of Fuzi
HA-Related Toxicokinetic Profiles at the Toxic Level of Fuzi
MA-Related Toxicokinetic Profiles at the Toxic Level of Fuzi
5.2.2. Dose-Dependent Toxicity of Fuzi and the Toxicokinetic Profile of Its Main Diester-Diterpenoid Alkaloids after Long-Term Oral Administration
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Zhou, G.; Tang, L.; Zhou, X.; Wang, T.; Kou, Z.; Wang, Z. A review on phytochemistry and pharmacological activities of the processed lateral root of Aconitum carmichaelii Debeaux. J. Ethnopharmacol. 2015, 160, 173–193. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.Y.K. Aconite poisoning. Clin. Toxicol. 2009, 47, 279–285. [Google Scholar] [CrossRef] [PubMed]
- Singhuber, J.; Zhu, M.; Prinz, S.; Kopp, B. Aconitum in Traditional Chinese Medicine—A valuable drug or an unpredictable risk? J. Ethnopharmacol. 2009, 126, 18–30. [Google Scholar] [CrossRef] [PubMed]
- Tai, C.J.; El-Shazly, M.; Wu, T.Y.; Lee, K.T.; Csupor, D.; Hohmann, J.; Chang, F.R.; Wu, Y.C. Clinical Aspects of Aconitum Preparations. Planta Med. 2015, 81, 1017–1028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chinese Pharmacopoeia Commission. Chinese Pharmacopoeia; China Medical Science Press: Beijing, China, 2015; Volume 1, pp. 191–193. [Google Scholar]
- Li, H.; Liu, L.; Zhu, S.; Liu, Q. Case reports of aconite poisoning in mainland China from 2004–2015: A retrospective analysis. J. Forensic Leg. Med. 2016, 42, 68–73. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.Y.K. Aconitum Alkaloid Poisoning because of Contamination of Herbs by Aconite Roots. Phyther. Res. 2016, 30, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Chan, T.Y.K. Contributory factors in herb-induced fatal aconite poisoning. Forensic Sci. Int. 2012, 223, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Chan, T. Aconitum Alkaloid Poisoning Related to the Culinary Uses of Aconite Roots. Toxins 2014, 6, 2605–2611. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moritz, F.; Compagnon, P.; Kaliszczak, I.G.; Kaliszczak, Y.; Caliskan, V.; Girault, C. Severe acute poisoning with homemade Aconitum napellus capsules: Toxicokinetic and clinical data. Clin. Toxicol. 2005, 43, 873–876. [Google Scholar] [CrossRef]
- Lin, C.C.; Phua, D.H.; Deng, J.F.; Yang, C.C. Aconitine intoxication mimicking acute myocardial infarction. Hum. Exp. Toxicol. 2011, 30, 782–785. [Google Scholar] [CrossRef] [PubMed]
- Fujita, Y.; Terui, K.; Fujita, M.; Kakizaki, A.; Sato, N.; Oikawa, K.; Aoki, H.; Takahashi, K.; Endo, S. Five cases of aconite poisoning: Toxicokinetics of aconitines. J. Anal. Toxicol. 2007, 31, 132–137. [Google Scholar] [CrossRef] [PubMed]
- Niitsu, H.; Fujita, Y.; Fujita, S.; Kumagai, R.; Takamiya, M.; Aoki, Y.; Dewa, K. Distribution of Aconitum alkaloids in autopsy cases of aconite poisoning. Forensic Sci. Int. 2013, 227, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Nyirimigabo, E.; Xu, Y.; Li, Y.; Wang, Y.; Agyemang, K.; Zhang, Y. A review on phytochemistry, pharmacology and toxicology studies of Aconitum. J. Pharm. Pharmacol. 2015, 67, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Jiang, B.; Lin, S.; Zhu, C.; Wang, S.; Wang, Y.; Chen, M.; Zhang, J.; Hu, J.; Chen, N.; Yang, Y.; et al. Diterpenoid alkaloids from the lateral root of Aconitum carmichaelii. J. Nat. Prod. 2012, 75, 1145–1159. [Google Scholar] [CrossRef] [PubMed]
- Bello-Ramírez, A.M.; Nava-Ocampo, A.A. A QSAR analysis of toxicity of Aconitum alkaloids. Fundam. Clin. Pharmacol. 2004, 18, 699–704. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Kong, D.; Du, Q.; Zhao, J.; Li, Q.; Zhang, J.; Li, T.; Ren, L. A conscious rat model involving bradycardia and hypotension after oral administration: A toxicokinetical study of aconitine. Xenobiotica 2017, 47, 515–525. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Guo, Z.Z.; Zhu, Y.F.; Huang, Z.J.; Gong, X.; Li, Y.H.; Son, W.J.; Li, X.Y.; Lou, Y.M.; Zhu, L.J.; et al. A systematic review of pharmacokinetic studies on herbal drug Fuzi: Implications for Fuzi as personalized medicine. Phytomedicine 2018, 15, 187–203. [Google Scholar] [CrossRef] [PubMed]
- Pereira, F. Polypharmacology of Aconitum and Delphinium sp. Diterpene Alkaloids: Antiarrhythmic, Analgesic and Anti-Inflammatory Effects. Mini Rev. Org. Chem. 2017, 14. [Google Scholar] [CrossRef]
- Liu, S.; Li, F.; Li, Y.; Li, W.; Xu, J.; Du, H. A review of traditional and current methods used to potentially reduce toxicity of Aconitum roots in Traditional Chinese Medicine. J. Ethnopharmacol. 2017, 207, 237–250. [Google Scholar] [CrossRef] [PubMed]
- Wada, K.; Nihira, M.; Hayakawa, H.; Tomita, Y.; Hayashida, M.; Ohno, Y. Effects of long-term administrations of aconitine on electrocardiogram and tissue concentrations of aconitine and its metabolites in mice. Forensic Sci. Int. 2005, 148, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Zhang, M.; Zhang, Q.; Ma, K.; Li, H.; Li, F.; Dong, F.; Yan, X. Metabonomics study of the effects of pretreatment with glycyrrhetinic acid on mesaconitine-induced toxicity in rats. J. Ethnopharmacol. 2014, 154, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Ko, J.; Liu, X.; Lu, C.; Li, J.; Xiao, C.; Li, L.; Niu, X.; Jiang, M.; He, X.; et al. Serum metabolomics reveals betaine and phosphatidylcholine as potential biomarkers for the toxic responses of processed Aconitum carmichaelii Debx. Mol. Biosyst. 2014, 10, 2305–2316. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Gao, Y.; Tan, G.; Wu, S.; Dong, X.; Lou, Z.; Zhu, Z.; Chai, Y. Myocardial lipidomics profiling delineate the toxicity of traditional Chinese medicine Aconiti Lateralis radix praeparata. J. Ethnopharmacol. 2013, 147, 349–356. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.B.; Sun, H.; Meng, X.B.; Hu, J.; Zhang, Q.; Liu, B.; Wang, M.; Xu, H.B.; Sun, X.B. Aconitine-induced Ca2+overload causes arrhythmia and triggers apoptosis through p38 MAPK signaling pathway in rats. Toxicol. Appl. Pharmacol. 2014, 279, 8–22. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Zhang, P.; Hou, Z.; Xie, J.; Wang, Y.; Yang, B.; Xu, Y.; Li, Y. Research on the relationships between endogenous biomarkers and exogenous toxic substances of acute toxicity in Radix Aconiti. Molecules 2016, 21, 1623. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.; Hoshi, T.; Kameyama, S. Effects of aconitine on the cardiac membrane potential of the dog. Jpn. J. Physiol. 1959, 9, 419–429. [Google Scholar] [CrossRef]
- Winslow, E. Hemodynamic and arrhythmogenic effects of aconitine applied to the left atria of anesthetized cats. Effects of amiodarone and atropine. J. Cardiovasc. Pharmacol. 1981, 3, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Huang, C.X.; Jiang, H.; Tang, Q.Z.; Yang, B.; Li, G.S. Effects of BmKIM on sodium current of isolated cardiomyocytes, transmembrane action potential and aconitine induced arrhythmia in vivo in rabbits. Zhonghua Xin Xue Guan Bing Za Zhi 2009, 37, 102–107. [Google Scholar] [PubMed]
- Ameri, A. The effects of Aconitum alkaloids on the central nervous system. Prog. Neurobiol. 1998, 56, 211–235. [Google Scholar] [CrossRef]
- Fu, M.; Wu, M.; Wang, J.F.; Qiao, Y.J.; Wang, Z. Disruption of the intracellular Ca2+ homeostasis in the cardiac excitation-contraction coupling is a crucial mechanism of arrhythmic toxicity in aconitine-induced cardiomyocytes. Biochem. Biophys. Res. Commun. 2007, 354, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Li, R.X.; Fan, L.; He, G.W.; Thornburg, K.L.; Wang, Z. Sarcoplasmic reticulum Ca2+ release channel ryanodine receptor (RyR2) plays a crucial role in aconitine-induced arrhythmias. Biochem. Pharmacol. 2008, 75, 2147–2156. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Zheng, T.; Yang, F.; Li, Y.X.; Zhang, D.K. Study of neurotoxic effects and underlying mechanisms of aconitine on cerebral cortex neuron cells. Arch. Pharm. Res. 2009, 32, 1533–1543. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Wang, L.; Wang, Y.H.; Li, Y.X.; Pan, Y. The toxicity of aconitine, emodin on ICC cell and the antagonist effect of the compatibility. Eur. J. Drug Metab. Pharmacokinet. 2009, 34, 213–220. [Google Scholar] [PubMed]
- Zhang, S.W.; Liu, Y.; Huang, G.Z.; Liu, L. Aconitine alters connexin43 phosphorylation status and [Ca2+] oscillation patterns in cultured ventricular myocytes of neonatal rats. Toxicol. In Vitro 2007, 21, 1476–1485. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Wang, X.; Chung, Y.Y.; Koh, C.H.; Liu, Z.; Guo, H.; Yuan, Q.; Wang, C.; Su, S.; Wei, H. L-type calcium channel inhibition contributes to the proarrhythmic effects of aconitine in human cardiomyocytes. PLoS ONE 2017, 12, e0168435. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.H.; Piao, X.M.; Liu, X.; Liang, H.H.; Wang, L.M.; Xiong, X.H.; Wang, L.; Lu, Y.J.; Shan, H.L. Arrhythmogenesis toxicity of aconitine is related to intracellular Ca2+ signals. Int. J. Med. Sci. 2013, 10, 1242–1249. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Du, S.; Li, F.; Huang, F.; Deng, R.; Zhou, J.; Chen, D. Histone deacetylase-high mobility group box-1 pathway targeted by hypaconitine suppresses the apoptosis of endothelial cells. Exp. Biol. Med. 2017, 242, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Voss, L.J.; Voss, J.M.; McLeay, L.; Sleigh, J.W. Aconitine induces prolonged seizure-like events in rat neocortical brain slices. Eur. J. Pharmacol. 2008, 584, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Jia, Y.; Liu, A.; Dai, R.; Huang, L. Hypaconitine-induced QT prolongation mediated through inhibition of KCNH2 (hERG) potassium channels in conscious dogs. J. Ethnopharmacol. 2015, 166, 375–379. [Google Scholar] [CrossRef] [PubMed]
- Shu, H.; Yi-Ming, W.; Xu, L.P.; Miao, C.Y.; Su, D.F. Increased susceptibility of ventricular arrhythmias to aconitine in anaesthetized rats is attributed to the inhibition of baroreflex. Clin. Exp. Pharmacol. Physiol. 2004, 31, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.G.; Sun, Y.; Duan, M.Y.; Chen, Y.J.; Zhong, D.F.; Zhang, H.Q. Separation and identification of Aconitum alkaloids and their metabolites in human urine. Toxicon 2005, 46, 500–506. [Google Scholar] [CrossRef] [PubMed]
- Ito, K.; Tanaka, S.; Funayama, M.; Mizugaki, M. Distribution of Aconitum alkaloids in body fluids and tissues in a suicidal case of aconite ingestion. J. Anal. Toxicol. 2000, 24, 348–353. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Gong, Y.; Lv, C.; Ye, L.; Liu, L.; Liu, Z. Pharmacokinetics of aconitine as the targeted marker of Fuzi (Aconitum carmichaelii) following single and multiple oral administrations of Fuzi extracts in rat by UPLC/MS/MS. J. Ethnopharmacol. 2012, 141, 736–741. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Li, Z.; Zhang, T.; Liu, F.; Ruan, J.; Zhang, Z. Transcellular transport of aconitine across human intestinal Caco-2 cells. Food Chem. Toxicol. 2013, 57, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Zhang, T.; Li, Z.; Xu, L.; Liu, F.; Ruan, J.; Liu, K.; Zhang, Z. P-glycoprotein is responsible for the poor intestinal absorption and low toxicity of oral aconitine: In vitro, in situ, in vivo and in silico studies. Toxicol. Appl. Pharmacol. 2013, 273, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lin, N.; Li, F.; Zhang, G.; He, S.; Zhu, Y.; Ou, R.; Li, N.; Liu, S.; Feng, L.; et al. Induction of P-glycoprotein expression and activity by Aconitum alkaloids: Implication for clinical drug-drug interactions. Sci. Rep. 2016, 6, 25343. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Yang, X.; Yang, Z.; Gao, S.; Yin, T.; Liu, W.; Wang, F.; Hu, M.; Liu, Z. The role of efflux transporters on the transport of highly toxic aconitine, mesaconitine, hypaconitine, and their hydrolysates, as determined in cultured Caco-2 and transfected MDCKII cells. Toxicol. Lett. 2013, 216, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wu, J.; Zhao, M.; Song, W.; Qi, X.; Wang, Y.; Lu, L.; Liu, Z. Mdr1a plays a crucial role in regulating the analgesic effect and toxicity of aconitine by altering its pharmacokinetic characteristics. Toxicol. Appl. Pharmacol. 2017, 320, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Sun, S.; Zhang, W.; Xie, X.; Zhu, Z.; Chai, Y.; Zhang, G. Biological activities and pharmacokinetics of aconitine, benzoylaconine, and aconine after oral administration in rats. Drug Test. Anal. 2016, 8, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wang, S.; Liu, Y.; Yan, L.; Dou, G.; Gao, Y. Characterization of metabolites and cytochrome P450 isoforms involved in the microsomal metabolism of aconitine. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2006, 844, 292–300. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Ye, L.; Lv, C.; Zheng, Z.; Gong, Y.; Liu, Z. Involvement of CYP3A4/5 and CYP2D6 in the metabolism of aconitine using human liver microsomes and recombinant CYP450 enzymes. Toxicol. Lett. 2011, 202, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Peng, C.S.; Li, X.B. Human intestine and liver microsomal metabolic differences between C19-diester and monoester diterpenoid alkaloids from the roots of Aconitum carmichaelii Debx. Toxicol. In Vitro 2017, 45, 318–333. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Tang, L.; Gong, Y.; Lv, C.; Zheng, Z.; Jiang, Z.; Liu, Z. Characterization of metabolites and human P450 isoforms involved in the microsomal metabolism of mesaconitine. Xenobiotica 2011, 41, 46–58. [Google Scholar] [CrossRef] [PubMed]
- Sui, Z.; Li, N.; Liu, Z.; Yan, J.; Liu, Z. Metabolite profile analysis of aconitine in rabbit stomach after oral administration by liquid chromatography/electrospray ionization/multiple-stage tandem mass spectrometry. Xenobiotica 2013, 43, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Peng, C.S.; Li, X.B. In vivo and in vitro metabolites from the main diester and monoester diterpenoid alkaloids in a traditional Chinese herb, the aconitum species. Evid.-Based Complement. Altern. Med. 2015, 2015, 252434. [Google Scholar] [CrossRef]
- He, H.; Yan, F. Relative quantification of the metabolite of aconitine in rat urine by LC-ESI-MS/MS and its application to pharmacokinetics. Anal. Sci. 2012, 28, 1203–1205. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.J.; Zhu, Y.F.; Guo, Z.Z.; Lou, Y.M.; He, S.G.; Guan, Y.; Zhu, L.J.; Liu, Z.Q.; Lu, L.L.; Liu, L. Aconitum alkaloids, the major components of Aconitum species, affect expression of multidrug resistance-associated protein 2 and breast cancer resistance protein by activating the Nrf2-mediated signalling pathway. Phytomedicine 2017, 44, 87–97. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Yang, X.; Zhou, J.; Tang, L.; Xia, B.; Hu, M.; Zhou, F.; Liu, Z. The exposure of highly toxic aconitine does not significantly impact the activity and expression of cytochrome P450 3A in rats determined by a novel ultra performance liquid chromatography-tandem mass spectrometric method of a specific probe buspirone. Food Chem. Toxicol. 2013, 51, 396–403. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Lu, L.; Guo, E.; Wu, J.; Wang, Y.; Hu, M.; Liu, Z. The Influences of Aconitine, an Active/Toxic Alkaloid from Aconitum, on the Oral Pharmacokinetics of CYP3A Probe Drug Buspirone in Rats. Drug Metab. Lett. 2014, 8, 135–144. [Google Scholar] [CrossRef] [PubMed]
- Miao, X.; Wang, J.; Meng, M.; Chen, Y. Influence of hypaconitine and liquirtin used alone and together on mRNA expression and activity of CYP450 enzymes in rat liver. China J. Tradit. Chin. Med. Pharm. 2016, 31, 313–316. [Google Scholar]
- Wu, J.; Cheng, Z.; Zhu, L.; Lu, L.; Zhang, G.; Wang, Y.; Xu, Y.; Lin, N.; Liu, Z. Coadministration of Pinellia ternata can significantly reduce Aconitum carmichaelii to inhibit CYP3A activity in rats. Evid. Based Complement. Altern. Med. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J. Pharmacokinetics and Cardiac Distribution of Hypaconitine in Rats. Master’s Thesis, Beijing University of Chinese Medicine, Beijing, China, 2010. [Google Scholar]
- U.S. Food and Drug Administration. Guidance for Industry Estimating the Maximum Safe Starting Dose in Initial Clinical Trials for Therapeutics in Adult Healthy Volunteers; U.S. Food and Drug Administration, Center for Drug Evaluation and Research (CDER): Rockville, MD, USA, 2005; p. 7.
- Wada, K.; Nihira, M.; Ohno, Y. Effects of chronic administrations of aconitine on body weight and rectal temperature in mice. J. Ethnopharmacol. 2006, 105, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.F.; Xie, Y.; Liu, L.; Ho, H.M.; Wong, Y.F.; Liu, Z.Q.; Zhou, H. Paeoniflorin reduced acute toxicity of aconitine in rats is associated with the pharmacokinetic alteration of aconitine. J. Ethnopharmacol. 2012, 141, 701–708. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Wen, J.; He, Y. Research on pharmacokinetics of aconitine by LC-MS. J. Instrum. Anal. 2004, 23, 51–53. [Google Scholar]
- Wang, R. Quality Evaluation of Fuzi and Pharmacokinetic Study of Aconitine. Ph.D. Thesis, Beijing University of Chinese Medicine, Beijing, China, 2007. [Google Scholar]
- Liu, J.; Li, Q.; Yin, Y.; Liu, R.; Xu, H.; Bi, K. Ultra-fast LC-ESI-MS/MS method for the simultaneous determination of six highly toxic Aconitum alkaloids from Aconiti kusnezoffii radix in rat plasma and its application to a pharmacokinetic study. J. Sep. Sci. 2014, 37, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Peng, W.W.; Li, W.; Li, J.S.; Cui, X.B.; Zhang, Y.X.; Yang, G.M.; Wen, H.M.; Cai, B.C. The effects of Rhizoma Zingiberis on pharmacokinetics of six Aconitum alkaloids in herb couple of Radix Aconiti Lateralis-Rhizoma Zingiberis. J. Ethnopharmacol. 2013, 148, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Y.X.; Zhao, M.J.; Yuan, A.; Gong, X.H.; Zhao, M.J.; Peng, C. The Effects of Rheum palmatum L. on the Pharmacokinetic of Major Diterpene Alkaloids of Aconitum carmichaelii Debx. in Rats. Eur. J. Drug Metab. Pharmacokinet. 2017, 42, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Sun, B.; Zhang, Q.; Fang, J.; Ma, K.; Li, Y.; Chen, H.; Dong, F.; Gao, Y.; Li, F.; et al. Metabonomic study on the toxicity of Hei-Shun-Pian, the processed lateral root of Aconitum carmichaelii Debx. (Ranunculaceae). J. Ethnopharmacol. 2008, 116, 561–568. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.P.; Liu, W.H. Review and reappraise on researches of Fuzi’s effect on cardiovarscular system, part one. Pharmacol. Clin. Chin. Mater. Med. 2013, 2, 198–205. [Google Scholar]
- Xiao, R.P.; Lai, X.P.; Zhao, Y.; Yu, L.W.; Zhu, Y.L.; Li, G. Pharmacokinetic study of six aconitine alkaloids in aconiti lateralis radix praeparata in beagle dogs. J. Chin. Med. Mater. 2014, 37, 284–287. [Google Scholar]
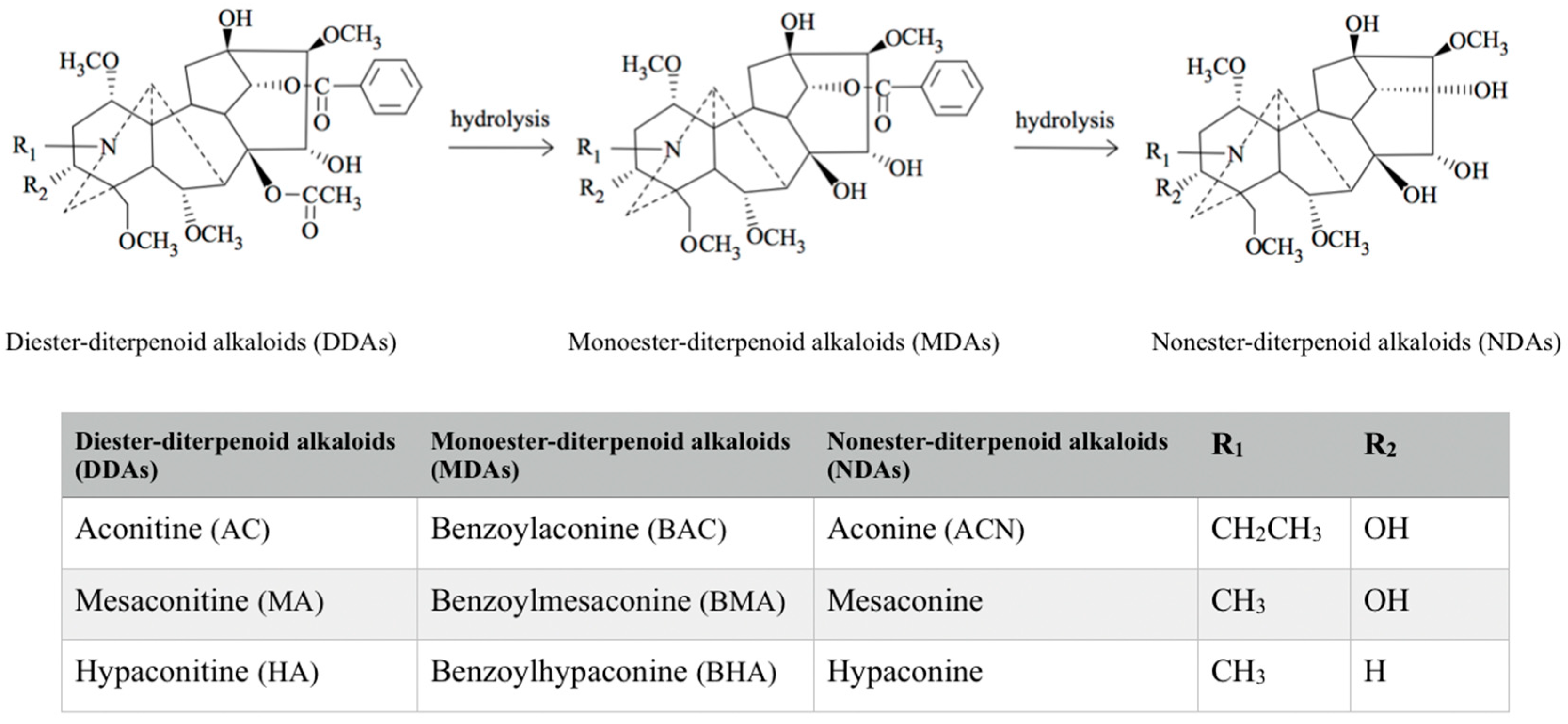

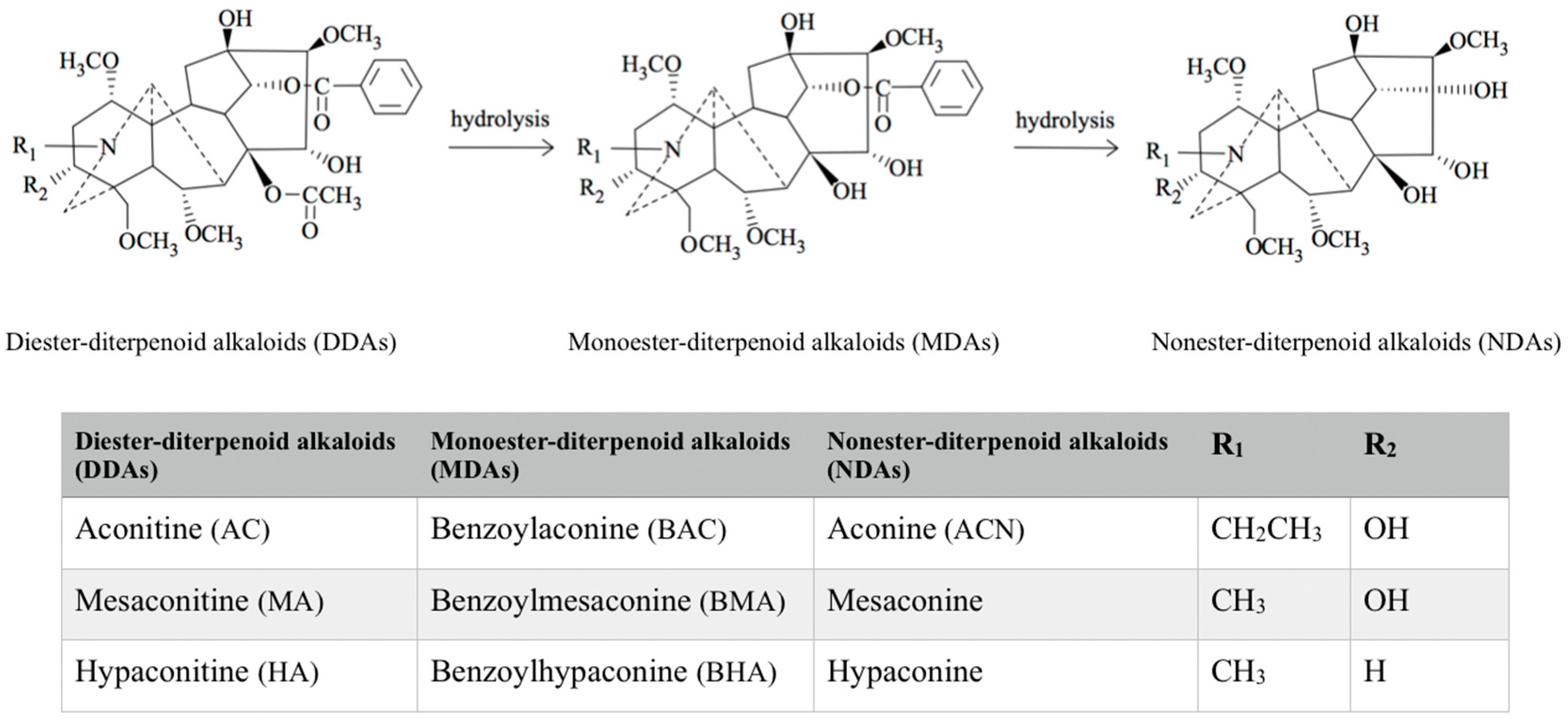{kind=link}
| Compound | Species | Duration/Dose | Toxicokinetic Parameters | Pharmacokinetics in Heart | Toxicity Measurement | Reference | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Tmax (min) | Cmax (ng/mL) | T1/2 (min) | AUC0-last (min·ng/mL) | Tmax (min) | Cmax (ng/g) | Serum Biomarker | Heart Histology | ECG | Behavior | ||||
| AC | Mice | 7 days/0.146 mg/kg | NR | NR | NR | NR | NR | NR | ↑ CK, AST, LDH | Hyper-chromatic nuclei, condensed cytoplasm | Ventricular tachycardia | NR | [25] |
| 22 days/1 mg/kg | NR | ↓compared to single dose | NR | NR | NR | ↓compared to single dose | NR | NR | NR | ↓ Body weight ↓ Rectal temperature | [65] | ||
| Single dose/0.1 mg/kg | 23.3 ± 2.1 | 6.5 ± 0.5 | 58.8 ± 5.5 | 671.4 ± 63.0 | 10 | 10.6 ± 2.7 | NS vs. control on CK, s-100 β | NS vs. control | NR | NS vs. control | [49] | ||
| Single dose/0.2 mg/kg | 35.0 ± 6.0 | 13.0 ± 0.9 | 69.1 ± 8.2 | 1578.4 ± 118.8 | 30 | 14.9 ± 2.6 | NR | NR | NR | Inactive, abnormal pulsation and breathing | [49] | ||
| Single dose/1 mg/kg | 15 | 8.5 ± 0.4 | NR | NR | 14.85 | 71.3 ± 5.5 | NR | NR | Arrhythmias | 14% Dead | [21] | ||
| Rats | 7 days/0.504 mg/kg | 25.0 ± 4.4 | 10.1 ± 2.1 | 211.8 ± 20.0 | 3329.4 ± 1199.0 | NR | NR | NR | NR | NR | NR | [44] | |
| Single dose/0.1 mg/kg | 37.5 ± 3.0 | 6.9 ± 0.8 | NR | 1485.7 ± 144.3 | NR | NR | NR | NR | NS vs. control on heart rate | NR | [17] | ||
| Single dose/0.2 mg/kg | 52.2 ± 13.2 | 39.4 ± 3.2 | 100.2 ± 12.6 | 9043.8 ± 725.4 | NR | NR | NR | NR | NR | NR | [46] | ||
| Single dose/0.2 mg/kg | 46.0 ± 15.0 | 9.7 ± 1.9 | 77.2 ± 8.4 | 1650.3 ± 359.2 | NR | NR | NR | NR | NR | NR | [66] | ||
| Single dose/0.2 mg/kg | 25.5 ± 5.8 | 7.5 ± 0.8 | NR | 1582.9 ± 125.1 | NR | NR | NR | NR | ↓ Heart rate | NR | [17] | ||
| Single dose/0.4 mg/kg | 130.7 ± 10.0 | 7.7 ± 0.9 | NR | 2884.7 ± 135.9 | NR | NR | NR | NR | ↓ Heart rate | ↓ Blood pressure | [17] | ||
| Single dose/0.5 mg/kg | 30.1 ± 9.7 | 8.7 ± 5.3 | 223.2 ± 53.0 | 2913.5 ± 981.1 | NR | NR | NR | NR | NR | NR | [44] | ||
| Single dose/0.5 mg/kg | 47.0 ± 8.4 | 44.3 ± 13.0 | 56.1 ± 15.6 | 10,092.0 ± 964.8 | NR | NR | NR | NR | NR | NR | [67] | ||
| HA | Rats | Single dose/2 mg/kg | 240 | 2.73 | 145.38 | 834.03 | 15 | 4.6 ± 5.9 | NR | NR | NR | 5% dead | [63] |
| Dog | Single dose/0.05 mg/kg | 60 | 1.53 | NR | NR | NR | NR | NR | NR | Prolonged QT interval | NR | [40] | |
| Single dose/0.15 mg/kg | 60 | 5.74 | NR | NR | NR | NR | NR | NR | Prolonged QT interval | NR | [40] | ||
| Single dose/0.45 mg/kg | 60 | 10.11 | NR | NR | NR | NR | NR | NR | Prolonged QT interval | NR | [40] | ||
| Species | Extract Dose | Dose of Components (mg/kg) | Targeted Compound | Toxicokinetic Parameters | Toxicity Measurements | Reference | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DDAs | AC | HA | MA | Tmax (min) | Cmax (ng/mL) | T1/2 (min) | AUC0-last (min·ng/mL) | Serum | Heart | Liver | Kidney | ||||
| Beagle dog | Fuzi extract 1.5 g/kg | 0.940 | 0.121 | 0.406 | 0.413 | AC | 70 ± 9 | 14.1 ± 0.8 | 273 ± 5 | 5514 ± 76 | NR | NR | NR | NR | [74] |
| HA | 70 ± 9 | 43.2 ± 1.5 | 292 ± 14 | 18,891 ± 455 | NR | NR | NR | NR | |||||||
| MA | 70 ± 9 | 45.4 ± 1.8 | 375 ± 6 | 21,638 ± 144 | NR | NR | NR | NR | |||||||
| Rats | Fuzi extract 0.2 g/kg | 0.356 | 0.037 | 0.181 | 0.138 | AC | 120 ± 60 | 1.0 ± 0.1 | 84 ± 16 | 257 ± 45 | NR | NR | NR | NR | [68] |
| HA | 160 ± 92 | 5.3 ± 0.2 | 104 ± 2 | 1485 ± 243 | NR | NR | NR | NR | |||||||
| MA | 132 ± 96 | 1.9 ± 0.3 | 89 ± 5 | 477 ± 175 | NR | NR | NR | NR | |||||||
| Chuanwu extract NR | 0.46 | 0.06 | 0.10 | 0.30 | AC | 105 ± 16 | 1.3 ± 0.5 | 294 ± 312 | 353 ± 113 | NR | NR | NR | NR | [69] | |
| HA | 105 ± 18 | 3.0 ± 0.6 | 180 ± 81 | 786 ± 180 | NR | NR | NR | NR | |||||||
| MA | 105 ± 18 | 3.3 ± 1.0 | 251 ± 198 | 840 ± 204 | NR | NR | NR | NR | |||||||
| 0.66 | 0.06 | 0.30 | 0.30 | AC | 20 ± 8 | 2.1 ± 1.3 | 462 ± 292 | 498 ± 84 | NR | NR | NR | NR | |||
| HA | 20 ± 11 | 7.5 ± 3.2 | 310 ± 102 | 2184 ± 708 | NR | NR | NR | NR | |||||||
| MA | 25 ± 12 | 6.1 ± 3.8 | 636 ± 210 | 1200 ± 252 | NR | NR | NR | NR | |||||||
| Fuzi extract 5.4 g/kg | 2.951 | 0.078 | 2.856 | 0.017 | AC | 41 ± 14 | 0.9 ± 0.1 | 220 ± 27 | 340 ± 40 | NR | NR | NR | NR | [70] | |
| HA | 71 ± 14 | 31.7 ± 1.6 | 253 ± 67 | 13,910 ± 2504 | NR | NR | NR | NR | |||||||
| MA | 56 ± 8 | 1.0 ± 0.1 | 192 ± 49 | 380 ± 31 | NR | NR | NR | NR | |||||||
| Fuzi extract 0.0384 g/kg | 4.900 | 0.177 | 2.918 | 1.805 | AC | 60 ± 0 | 10.2 ± 1.5 | 644 ± 29 | 4297 ± 1570 | NR | NR | NR | NR | [71] | |
| HA | 60 ± 0 | 60.2 ± 4.3 | 559 ± 62 | 24,635 ± 100 | NR | NR | NR | NR | |||||||
| MA | 60 ± 0 | 24.8 ± 4.2 | 617 ± 23 | 10,988 ± 2192 | NR | NR | NR | NR | |||||||
| Fuzi extract 4.5 g/kg | NR | 0.118 | NR | NR | AC | 58 ± 22 | 3.2 ± 0.4 | 218 ± 86 | 640 ± 107 | NR | NR | NR | NR | [44] | |
| Fuzi extract 2 g/kg | 1.169 | 0.116 | 0.586 | 0.467 | NA | NR | NR | NR | NR | NS vs. control on CK, LDH, ALT, AST, and Urea | Nuclear varies in size | Mild edema | NS vs. control | [26] | |
| Fuzi extract 5 g/kg | 2.924 | 0.290 | 1.466 | 1.168 | NA | NR | NR | NR | NR | ↑ LDH | Dilated blood vessels | Mild edema | Scattered lymphocytes | ||
| Fuzi extract 10 g/kg | 5.848 | 0.580 | 2.932 | 2.336 | NA | NR | NR | NR | NR | ↑ CK, LDH, AST, Urea | Nuclear varies in size | Edema | Scattered atrophy | ||
| Duration | Extract Dosage | Dose of Components (mg/kg) | Targeted Compound | Toxicokinetic Parameters | Toxicity Measurements | Reference | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DDAs | AC | HA | MA | Tmax (min) | Cmax (ng/mL) | T1/2 (min) | AUC0-last (min·ng/mL) | Serum | Heart | Liver | Kidney | ||||
| 7 days | Fuzi 4.5 g/kg | 0.118 | 0.118 | NR | NR | AC | 20 ± 9 | 2.6 ± 1.0 | 384 ± 97 | 989 ± 67 | NR | NR | NR | NR | [44] |
| Fuzi 17.6 g/kg | 2.066 | 0.000 | 0.804 | 1.262 | NA | NR | NR | NR | NR | NS vs. control on BUN | NR | NR | NR | [72] | |
| Fuzi 35.6 g/kg | 4.180 | 0.000 | 1.627 | 2.553 | NA | NR | NR | NR | NR | NS vs. control on BUN | NR | NR | NR | ||
| Fuzi 88.1 g/kg | 10.343 | 0.000 | 4.026 | 6.317 | NA | NR | NR | NR | NR | ↓ BUN | NR | NR | NR | ||
| 15 days | Baifupian 0.32 g/kg | 0.175 | 0.005 | 0.162 | 0.008 | NA | NR | NR | NR | NR | NS vs. control on Creatine, BUN, ALT, CK, and LDH | NR | NR | NR | [23] |
| Baifupian 0.64 g/kg | 0.055 | 0.010 | 0.324 | 0.016 | NA | NR | NR | NR | NR | NS vs. control on Creatine, BUN, ALT, CK and LDH | NR | NR | NR | ||
| Baifupian 1.28 g/kg | 0.11 | 0.02 | 0.647 | 0.032 | NA | NR | NR | NR | NR | ↑ Creatine, BUN, ALT, CK, and LDH | NR | NR | NR | ||
| Baifupian 2.56 g/kg | 0.22 | 0.04 | 1.294 | 0.064 | NA | NR | NR | NR | NR | ↑ Creatine, BUN, ALT, CK, and LDH | Inflammatory infiltration edema | Necrosis inflammation | Vascular dilatation | ||
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, M.; Ji, X.; Zuo, Z. Relationships between the Toxicities of Radix Aconiti Lateralis Preparata (Fuzi) and the Toxicokinetics of Its Main Diester-Diterpenoid Alkaloids. Toxins 2018, 10, 391. https://doi.org/10.3390/toxins10100391
Yang M, Ji X, Zuo Z. Relationships between the Toxicities of Radix Aconiti Lateralis Preparata (Fuzi) and the Toxicokinetics of Its Main Diester-Diterpenoid Alkaloids. Toxins. 2018; 10(10):391. https://doi.org/10.3390/toxins10100391
Chicago/Turabian StyleYang, Mengbi, Xiaoyu Ji, and Zhong Zuo. 2018. "Relationships between the Toxicities of Radix Aconiti Lateralis Preparata (Fuzi) and the Toxicokinetics of Its Main Diester-Diterpenoid Alkaloids" Toxins 10, no. 10: 391. https://doi.org/10.3390/toxins10100391
APA StyleYang, M., Ji, X., & Zuo, Z. (2018). Relationships between the Toxicities of Radix Aconiti Lateralis Preparata (Fuzi) and the Toxicokinetics of Its Main Diester-Diterpenoid Alkaloids. Toxins, 10(10), 391. https://doi.org/10.3390/toxins10100391