Computational Studies of Snake Venom Toxins


 , , ,
, , ,
Abstract
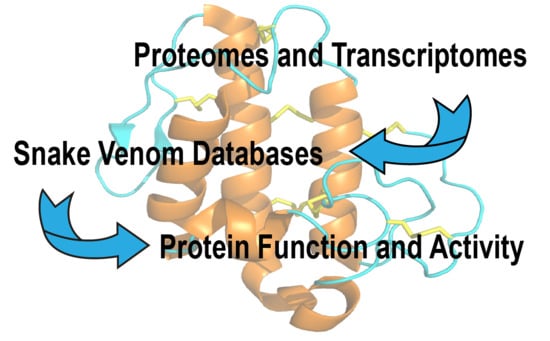
1. Introduction
2. Extent of Our Knowledge on Snake Toxins
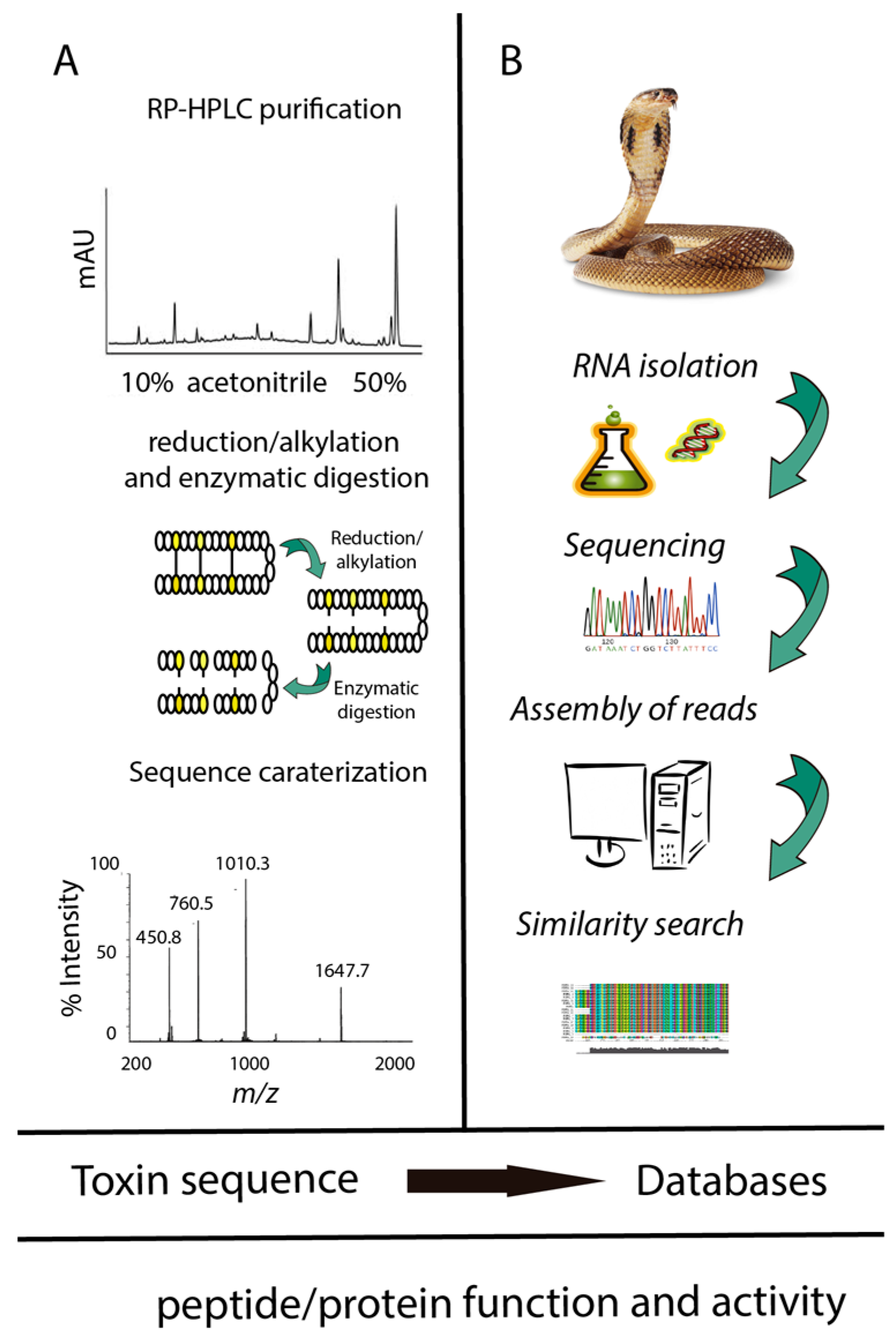
2.1. Transcriptomic Analyses of Peptides and Proteins from Snake Venom Glands
2.2. Proteomic Analyses of Peptides and Proteins from Snake Venom Glands
3. Snake Toxin Structures and Activities
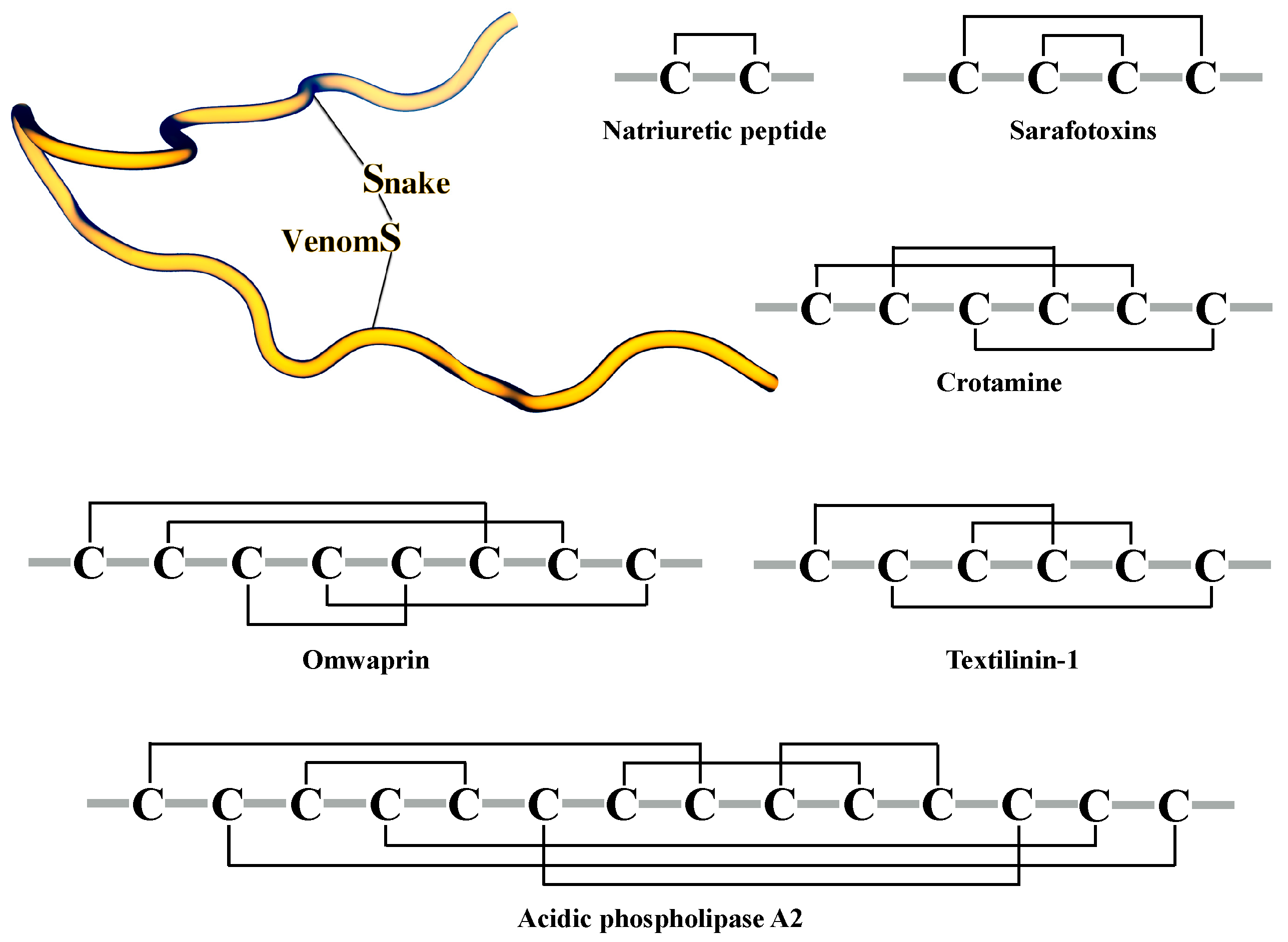
3.1. Classification of Snake Venom Toxins
3.2. Structures of Snake Venom Toxins
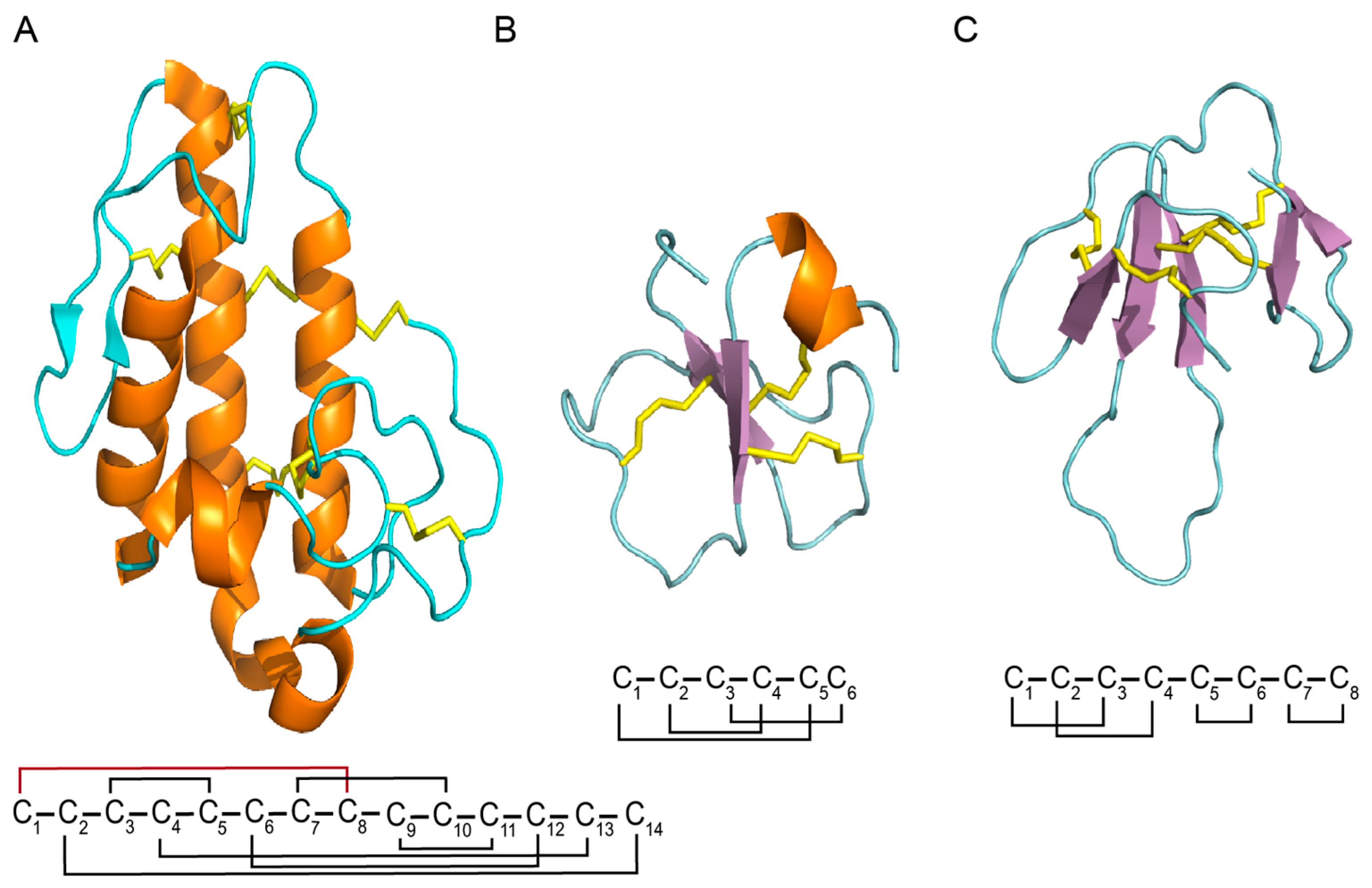
3.2.1. ICK Fold
3.2.2. α/β Fold
3.3. Molecular Modeling of Snake Toxin Structures
3.4. Molecular Modeling of Snake Toxin—Target Complexes
3.5. Molecular Modeling Applied to the Study of PLA2, Crotamine and Mambalgin
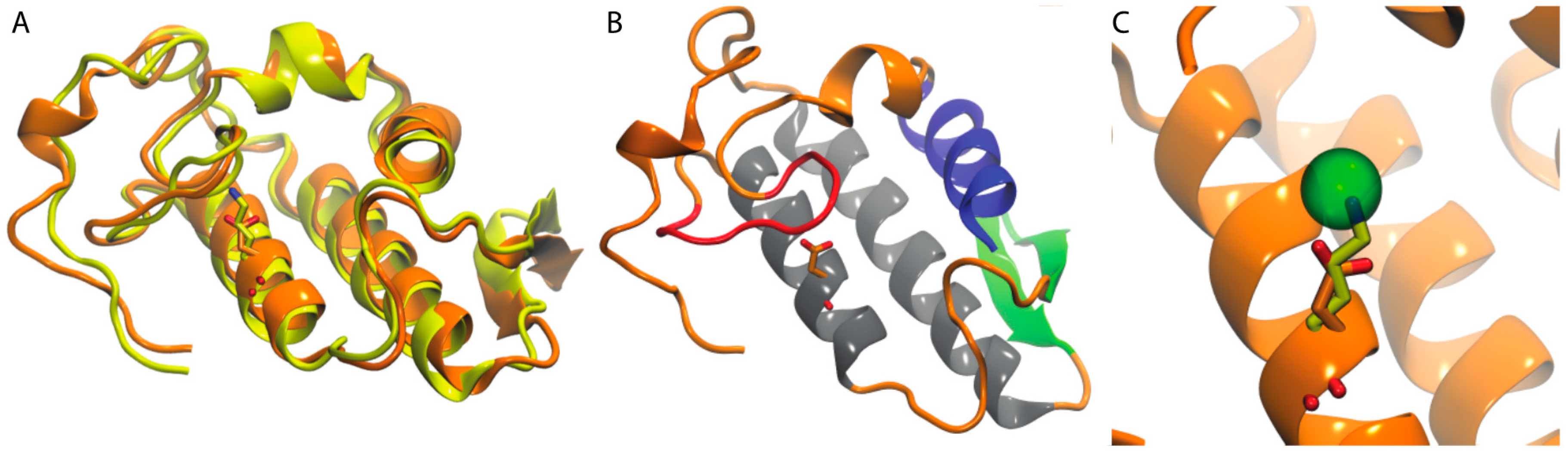
3.5.1. PLA2
3.5.2. Crotamine
3.5.3. Mambalgin-1 and -2
3.6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Reeks, T.A.; Fry, B.G.; Alewood, P.F. Privileged frameworks from snake venom. Cell. Mol. Life Sci. 2015, 72, 1939–1958. [Google Scholar] [CrossRef] [PubMed]
- Georgieva, D.; Arni, R.K.; Betzel, C. Proteome analysis of snake venom toxins: Pharmacological insights. Expert Rev. Proteom. 2008, 5, 787–797. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.S.; Cheung, R.C.; Xia, L.; Wong, J.H.; Ng, T.B.; Chan, W.Y. Snake venom toxins: Toxicity and medicinal applications. Appl. Microbiol. Biotechnol. 2016, 100, 6165–6181. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L. Toxins and drug discovery. Toxicon 2014, 92, 193–200. [Google Scholar] [CrossRef] [PubMed]
- Camargo, A.C.M.; Ianzer, D.; Guerreiro, J.R.; Serrano, S.M.T. Bradykinin-potentiating peptides: Beyond captopril. Toxicon 2012, 59, 516–523. [Google Scholar] [CrossRef] [PubMed]
- Cushman, D.W.; Cheung, H.S.; Sabo, E.F.; Ondetti, M.A. Design of potent competitive inhibitors of angiotensin-converting enzyme—Carboxyalkanoyl and mercaptoalkanoyl amino-acids. Biochemistry 1977, 16, 5484–5491. [Google Scholar] [CrossRef] [PubMed]
- Wojta, J. Cenderitide: A multivalent designer-peptide-agonist of particulate guanylyl cyclase receptors with considerable therapeutic potential in cardiorenal disease states. Eur. Heart J. Cardiovasc. 2016, 2, 106–107. [Google Scholar] [CrossRef] [PubMed]
- Zheng, L.; Mao, Y.; Li, M.; Dai, X.; Li, B.; Zheng, X.L. Therapeutic efficacy of anfibatide in a murine model of thrombotic thrombocytopenic purpura. Blood 2015, 126, 659. [Google Scholar] [CrossRef]
- Ferreira, R.S.; de Barros, L.C.; Abbade, L.P.F.; Barraviera, S.R.C.S.; Silvares, M.R.C.; de Pontes, L.G.; dos Santos, L.D.; Barraviera, B. Heterologous fibrin sealant derived from snake venom: From bench to bedside—An overview. J. Venom. Anim. Toxins 2017, 23, 21. [Google Scholar] [CrossRef] [PubMed]
- Bressan, E.; Touska, F.; Vetter, I.; Kistner, K.; Kichko, T.I.; Teixeira, N.B.; Picolo, G.; Cury, Y.; Lewis, R.J.; Fischer, M.J.M.; et al. Crotalphine desensitizes trpa1 ion channels to alleviate inflammatory hyperalgesia. Pain 2016, 157, 2504–2516. [Google Scholar] [CrossRef] [PubMed]
- Diochot, S.; Alloui, A.; Rodrigues, P.; Dauvois, M.; Friend, V.; Aissouni, Y.; Eschalier, A.; Lingueglia, E.; Baron, A. Analgesic effects of Mambalgin peptide inhibitors of acid-sensing ion channels in inflammatory and neuropathic pain. Pain 2016, 157, 552–559. [Google Scholar] [CrossRef] [PubMed]
- Calvete, J.J.; Sanz, L.; Angulo, Y.; Lomonte, B.; Gutierrez, J.M. Venoms, venomics, antivenomics. FEBS Lett. 2009, 583, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Calvete, J.J. Snake venomics: From the inventory of toxins to biology. Toxicon 2013, 75, 44–62. [Google Scholar] [CrossRef] [PubMed]
- Ducancel, F.; Durban, J.; Verdenaud, M. Transcriptomics and venomics: Implications for medicinal chemistry. Future Med. Chem. 2014, 6, 1629–1643. [Google Scholar] [CrossRef] [PubMed]
- Brahma, R.K.; McCleary, R.J.; Kini, R.M.; Doley, R. Venom gland transcriptomics for identifying, cataloging, and characterizing venom proteins in snakes. Toxicon 2015, 93, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Chatrath, S.T.; Chapeaurouge, A.; Lin, Q.; Lim, T.K.; Dunstan, N.; Mirtschin, P.; Kumar, P.P.; Kini, R.M. Identification of novel proteins from the venom of a cryptic snake drysdalia coronoides by a combined transcriptomics and proteomics approach. J. Proteome Res. 2011, 10, 739–750. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. Uniprot: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar]
- Jain, E.; Bairoch, A.; Duvaud, S.; Phan, I.; Redaschi, N.; Suzek, B.E.; Martin, M.J.; McGarvey, P.; Gasteiger, E. Infrastructure for the life sciences: Design and implementation of the uniprot website. BMC Bioinform. 2009, 10, 136. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. Genbank. Nucleic Acids Res. 2014, 42, D32–D37. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Gutmanas, A.; Alhroub, Y.; Battle, G.M.; Berrisford, J.M.; Bochet, E.; Conroy, M.J.; Dana, J.M.; Fernandez Montecelo, M.A.; van Ginkel, G.; Gore, S.P.; et al. Pdbe: Protein data bank in Europe. Nucleic Acids Res. 2014, 42, D285–D291. [Google Scholar] [CrossRef] [PubMed]
- Bultet, L.A.; Aguilar Rodriguez, J.; Ahrens, C.H.; Ahrne, E.L.; Ai, N.; Aimo, L.; Akalin, A.; Aleksiev, T.; Alocci, D.; Altenhoff, A.; et al. The SIB Swiss institute of bioinformatics’ resources: Focus on curated databases. Nucleic Acids Res. 2016, 44, D27–D37. [Google Scholar]
- Kaas, Q.; Yu, R.; Jin, A.H.; Dutertre, S.; Craik, D.J. Conoserver: Updated content, knowledge, and discovery tools in the conopeptide database. Nucleic Acids Res. 2012, 40, D325–D330. [Google Scholar] [CrossRef] [PubMed]
- Herzig, V.; Wood, D.L.; Newell, F.; Chaumeil, P.A.; Kaas, Q.; Binford, G.J.; Nicholson, G.M.; Gorse, D.; King, G.F. Arachnoserver 2.0, an updated online resource for spider toxin sequences and structures. Nucleic Acids Res. 2011, 39, D653–D657. [Google Scholar] [CrossRef] [PubMed]
- Roly, Z.Y.; Hakim, M.A.; Zahan, A.S.; Hossain, M.M.; Reza, M.A. ISOB: A database of indigenous snake species of Bangladesh with respective known venom composition. Bioinformation 2015, 11, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Ranko Gacesa, P.L. Toxclassifier, Version 1.0. Available online: http://bioserv7.bioinfo.pbf.hr/ToxClassifier/ (accessed on 20 December 2017).
- Gacesa, R.; Barlow, D.J.; Long, P.F. Machine learning can differentiate venom toxins from other proteins having non-toxic physiological functions. PeerJ Comput. Sci. 2016, 2, e90. [Google Scholar] [CrossRef]
- Castoe, T.A.; de Koning, A.P.; Hall, K.T.; Card, D.C.; Schield, D.R.; Fujita, M.K.; Ruggiero, R.P.; Degner, J.F.; Daza, J.M.; Gu, W.; et al. The Burmese python genome reveals the molecular basis for extreme adaptation in snakes. Proc. Natl. Acad. Sci. USA 2013, 110, 20645–20650. [Google Scholar] [CrossRef] [PubMed]
- Vonk, F.J.; Casewell, N.R.; Henkel, C.V.; Heimberg, A.M.; Jansen, H.J.; McCleary, R.J.; Kerkkamp, H.M.; Vos, R.A.; Guerreiro, I.; Calvete, J.J.; et al. The king cobra genome reveals dynamic gene evolution and adaptation in the snake venom system. Proc. Natl. Acad. Sci. USA 2013, 110, 20651–20656. [Google Scholar] [CrossRef] [PubMed]
- Ching, A.T.; Rocha, M.M.; Paes Leme, A.F.; Pimenta, D.C.; de Fatima, D.F.M.; Serrano, S.M.; Ho, P.L.; Junqueira-de-Azevedo, I.L. Some aspects of the venom proteome of the Colubridae snake Philodryas olfersii revealed from a duvernoy’s (venom) gland transcriptome. FEBS Lett. 2006, 580, 4417–4422. [Google Scholar] [CrossRef] [PubMed]
- Correa-Netto, C.; Junqueira-de-Azevedo Ide, L.; Silva, D.A.; Ho, P.L.; Leitao-de-Araujo, M.; Alves, M.L.; Sanz, L.; Foguel, D.; Zingali, R.B.; Calvete, J.J. Snake venomics and venom gland transcriptomic analysis of Brazilian coral snakes, Micrurus altirostris and M. corallinus. J. Proteom. 2011, 74, 1795–1809. [Google Scholar] [CrossRef] [PubMed]
- Suntravat, M.; Uzcategui, N.L.; Atphaisit, C.; Helmke, T.J.; Lucena, S.E.; Sanchez, E.E.; Acosta, A.R. Gene expression profiling of the venom gland from the Venezuelan mapanare (bothrops colombiensis) using expressed sequence tags (ESTS). BMC Mol. Biol. 2016, 17, 7. [Google Scholar] [CrossRef]
- Tan, K.Y.; Tan, C.H.; Chanhome, L.; Tan, N.H. Comparative venom gland transcriptomics of Naja kaouthia (monocled cobra) from Malaysia and Thailand: Elucidating geographical venom variation and insights into sequence novelty. PeerJ 2017, 5, e3142. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 2012, 251364. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Chopra, R.; Burow, G.; Farmer, A.; Mudge, J.; Simpson, C.E.; Burow, M.D. Comparisons of de novo transcriptome assemblers in diploid and polyploid species using peanut (Arachis spp.) RNA-seq data. PLoS ONE 2014, 9, e115055. [Google Scholar] [CrossRef] [PubMed]
- Francischetti, I.M.; My-Pham, V.; Harrison, J.; Garfield, M.K.; Ribeiro, J.M. Bitis gabonica (gaboon viper) snake venom gland: Toward a catalog for the full-length transcripts (CDNA) and proteins. Gene 2004, 337, 55–69. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Madan, A. Cap3: A DNA sequence assembly program. Genome Res. 1999, 9, 868–877. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.; Green, P. Consed: A graphical editor for next-generation sequencing. Bioinformatics 2013, 29, 2936–2937. [Google Scholar] [CrossRef] [PubMed]
- Swindell, S.R.; Plasterer, T.N. Seqman. Contig assembly. Methods Mol. Biol. 1997, 70, 75–89. [Google Scholar] [PubMed]
- Chevreux, B.; Pfisterer, T.; Drescher, B.; Driesel, A.J.; Muller, W.E.; Wetter, T.; Suhai, S. Using the miraest assembler for reliable and automated mRNA transcript assembly and SNP detection in sequenced ESTs. Genome Res. 2004, 14, 1147–1159. [Google Scholar] [CrossRef] [PubMed]
- Xu, N.; Zhao, H.Y.; Yin, Y.; Shen, S.S.; Shan, L.L.; Chen, C.X.; Zhang, Y.X.; Gao, J.F.; Ji, X. Combined venomics, antivenomics and venom gland transcriptome analysis of the monocoled cobra (Naja kaouthia) from China. J. Proteom. 2017, 159, 19–31. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Zhang, X.; Hu, T.; Zhou, W.; Cui, Q.; Tian, J.; Zheng, Y.; Fan, Q. Discovery of toxin-encoding genes from the false viper macropisthodon rudis, a rear-fanged snake, by transcriptome analysis of venom gland. Toxicon 2015, 106, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Goncalves-Machado, L.; Pla, D.; Sanz, L.; Jorge, R.J.B.; Leitao-De-Araujo, M.; Alves, M.L.M.; Alvares, D.J.; De Miranda, J.; Nowatzki, J.; de Morais-Zani, K.; et al. Combined venomics, venom gland transcriptomics, bioactivities, and antivenomics of two Bothrops jararaca populations from geographic isolated regions within the brazilian atlantic rainforest. J. Proteom. 2016, 135, 73–89. [Google Scholar] [CrossRef] [PubMed]
- Junqueira-de-Azevedo, I.L.; Bastos, C.M.; Ho, P.L.; Luna, M.S.; Yamanouye, N.; Casewell, N.R. Venom-related transcripts from Bothrops jararaca tissues provide novel molecular insights into the production and evolution of snake venom. Mol. Biol. Evol. 2015, 32, 754–766. [Google Scholar] [CrossRef] [PubMed]
- Aird, S.D.; Watanabe, Y.; Villar-Briones, A.; Roy, M.C.; Terada, K.; Mikheyev, A.S. Quantitative high-throughput profiling of snake venom gland transcriptomes and proteomes (Ovophis okinavensis and Protobothrops flavoviridis). BMC Genom. 2013, 14, 790. [Google Scholar] [CrossRef] [PubMed]
- Ramsden, J. Bioinformatics: An Introduction; Springer: London, UK, 2009. [Google Scholar]
- Aird, S.D.; Aggarwal, S.; Villar-Briones, A.; Tin, M.M.; Terada, K.; Mikheyev, A.S. Snake venoms are integrated systems, but abundant venom proteins evolve more rapidly. BMC Genom. 2015, 16, 647. [Google Scholar] [CrossRef] [PubMed]
- Leao, L.I.; Ho, P.L.; Junqueira-de-Azevedo Ide, L. Transcriptomic basis for an antiserum against Micrurus corallinus (coral snake) venom. BMC Genom. 2009, 10, 112. [Google Scholar] [CrossRef] [PubMed]
- Sanz, L.; Escolano, J.; Ferretti, M.; Biscoglio, M.J.; Rivera, E.; Crescenti, E.J.; Angulo, Y.; Lomonte, B.; Gutierrez, J.M.; Calvete, J.J. Snake venomics of the south and central American bushmasters. Comparison of the toxin composition of lachesis muta gathered from proteomic versus transcriptomic analysis. J. Proteom. 2008, 71, 46–60. [Google Scholar] [CrossRef] [PubMed]
- Tasoulis, T.; Isbister, G.K. A review and database of snake venom proteomes. Toxins 2017, 9, 290. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.W.; Serrano, S.M. Exploring snake venom proteomes: Multifaceted analyses for complex toxin mixtures. Proteomics 2008, 8, 909–920. [Google Scholar] [CrossRef] [PubMed]
- Klint, J.K.; Berecki, G.; Durek, T.; Mobli, M.; Knapp, O.; King, G.F.; Adams, D.J.; Alewood, P.F.; Rash, L.D. Isolation, synthesis and characterization of omega-trtx-cc1a, a novel tarantula venom peptide that selectively targets l-type cav channels. Biochem. Pharmacol. 2014, 89, 276–286. [Google Scholar] [CrossRef] [PubMed]
- Mobli, M.; King, G.F. NMR methods for determining disulfide-bond connectivities. Toxicon 2010, 56, 849–854. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The future of peptide-based drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S.; Hamilton, A.D. Targeting protein-protein interactions by rational design: Mimicry of protein surfaces. J. R. Soc. Interface 2006, 3, 215–233. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.T.; Chau, J.K.; Perry, N.A.; de Boer, L.; Zaat, S.A.; Vogel, H.J. Serum stabilities of short tryptophan- and arginine-rich antimicrobial peptide analogs. PLoS ONE 2010, 5, e12684. [Google Scholar] [CrossRef] [PubMed]
- Brunel, F.M.; Dawson, P.E. Synthesis of constrained helical peptides by thioether ligation: Application to analogs of gp41. Chem. Commun. 2005, 2552–2554. [Google Scholar] [CrossRef] [PubMed]
- Doley, R.; Kini, R.M. Protein complexes in snake venom. Cell. Mol. Life Sci. 2009, 66, 2851–2871. [Google Scholar] [CrossRef] [PubMed]
- Dufton, M.J.; Hider, R.C. Snake toxin secondary structure predictions. Structure activity relationships. J. Mol. Biol. 1977, 115, 177–193. [Google Scholar] [CrossRef]
- Tsernoglou, D.; Petsko, G.A.; Hudson, R.A. Structure and function of snake venom curarimimetic neurotoxins. Mol. Pharmacol. 1978, 14, 710–716. [Google Scholar] [PubMed]
- Blundell, T.L.; Jhoti, H.; Abell, C. High-throughput crystallography for lead discovery in drug design. Nat. Rev. Drug Discov. 2002, 1, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Bhat, T.N.; Bourne, P.E.; Feng, Z.; Gilliland, G.; Weissig, H.; Westbrook, J. The protein data bank and the challenge of structural genomics. Nat. Struct. Biol. 2000, 7, 957–959. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, D. Computational methods applied to rational drug design. Open Med. Chem. J. 2016, 10, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Fry, B.G. From genome to “venome”: Molecular origin and evolution of the snake venom proteome inferred from phylogenetic analysis of toxin sequences and related body proteins. Genome Res. 2005, 15, 403–420. [Google Scholar] [CrossRef] [PubMed]
- Haas, J.; Roth, S.; Arnold, K.; Kiefer, F.; Schmidt, T.; Bordoli, L.; Schwede, T. The protein model portal—A comprehensive resource for protein structure and model information. Database 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- UniProt Consortium. The universal protein resource (UniProt) 2009. Nucleic Acids Res. 2009, 37, D169–D174. [Google Scholar]
- Boutet, E.; Lieberherr, D.; Tognolli, M.; Schneider, M.; Bairoch, A. Uniprotkb/Swiss-Prot. Methods Mol. Biol. 2007, 406, 89–112. [Google Scholar] [PubMed]
- Herzig, V.; King, G.F. The cystine knot is responsible for the exceptional stability of the insecticidal spider toxin omega-hexatoxin-hv1a. Toxins 2015, 7, 4366–4380. [Google Scholar] [CrossRef] [PubMed]
- Poth, A.G.; Chan, L.Y.; Craik, D.J. Cyclotides as grafting frameworks for protein engineering and drug design applications. Biopolymers 2013, 100, 480–491. [Google Scholar] [CrossRef] [PubMed]
- Mouhat, S.; Jouirou, B.; Mosbah, A.; De Waard, M.; Sabatier, J.M. Diversity of folds in animal toxins acting on ion channels. Biochem. J. 2004, 378, 717–726. [Google Scholar] [CrossRef] [PubMed]
- Gelly, J.C.; Gracy, J.; Kaas, Q.; Le-Nguyen, D.; Heitz, A.; Chiche, L. The knottin website and database: A new information system dedicated to the knottin scaffold. Nucleic Acids Res. 2004, 32, D156–D159. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Daly, N.L.; Waine, C. The cystine knot motif in toxins and implications for drug design. Toxicon 2001, 39, 43–60. [Google Scholar] [CrossRef]
- Saez, N.J.; Senff, S.; Jensen, J.E.; Er, S.Y.; Herzig, V.; Rash, L.D.; King, G.F. Spider-venom peptides as therapeutics. Toxins 2010, 2, 2851–2871. [Google Scholar] [CrossRef] [PubMed]
- King, G.F.; Hardy, M.C. Spider-venom peptides: Structure, pharmacology, and potential for control of insect pests. Annu. Rev. Entomol. 2013, 58, 475–496. [Google Scholar] [CrossRef] [PubMed]
- King, G.F. Venoms as a platform for human drugs: Translating toxins into therapeutics. Expert Opin. Biol. Ther. 2011, 11, 1469–1484. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez de la Vega, R.C.; Possani, L.D. Current views on scorpion toxins specific for k+-channels. Toxicon 2004, 43, 865–875. [Google Scholar] [CrossRef] [PubMed]
- Sunagar, K.; Undheim, E.A.; Chan, A.H.; Koludarov, I.; Munoz-Gomez, S.A.; Antunes, A.; Fry, B.G. Evolution stings: The origin and diversification of scorpion toxin peptide scaffolds. Toxins 2013, 5, 2456–2487. [Google Scholar] [CrossRef] [PubMed]
- Saxena, A.; Singh, R.; Mishra, S. Fundamentals of homology modeling steps and comparison among important bioinformatics tools: An overview. Sci. Int. 2013, 1, 237–252. [Google Scholar]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM—A new method for protein modeling and design: Applications to docking and structure prediction from the distorted native conformation. J. Comput. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- Zhu, K.; Day, T.; Warshaviak, D.; Murrett, C.; Friesner, R.; Pearlman, D. Antibody structure determination using a combination of homology modeling, energy-based refinement, and loop prediction. Proteins 2014, 82, 1646–1655. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. Swiss-model and the Swiss-Pdbviewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-tasser server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, M.; Mortier, J.; Rakers, C.; Sydow, D.; Wolber, G. More than a look into a crystal ball: Protein structure elucidation guided by molecular dynamics simulations. Drug Discov. Today 2016, 21, 1799–1805. [Google Scholar] [CrossRef] [PubMed]
- Durrant, J.D.; McCammon, J.A. Molecular dynamics simulations and drug discovery. BMC Biol. 2011, 9, 71. [Google Scholar] [CrossRef] [PubMed]
- Dror, R.O.; Dirks, R.M.; Grossman, J.P.; Xu, H.; Shaw, D.E. Biomolecular simulation: A computational microscope for molecular biology. Annu. Rev. Biophys. 2012, 41, 429–452. [Google Scholar] [CrossRef] [PubMed]
- Sledz, P.; Caflisch, A. Protein structure-based drug design: From docking to molecular dynamics. Curr. Opin. Struct. Biol. 2017, 48, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935–949. [Google Scholar] [CrossRef] [PubMed]
- Leach, A.R. Molecular Modelling: Principles and Applications; Prentice Hall: Upper Saddle River, NJ, USA, 2001. [Google Scholar]
- Abiram, A.; Kolandaivel, P. Effect of piratoxin II and acutohaemolysin phospholipase (PLA2) proteins on myristic fatty acid—An oniom and DFT study. J. Mol. Model. 2010, 16, 1853–1865. [Google Scholar] [CrossRef] [PubMed]
- Alcântara, A.F.D.C.; Piló-Veloso, D.; Fernandes, A.J.D.N.; Dos-Santos, M.C. Theoretical investigation of the structural properties of two crotamines isolated from the venom of Crotalus durissus. Open Nat. Prod. J. 2011, 4, 16–20. [Google Scholar] [CrossRef]
- Ramirez, D.; Caballero, J. Is it reliable to use common molecular docking methods for comparing the binding affinities of enantiomer pairs for their protein target? Int. J. Mol. Sci. 2016, 17, 525. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.R.; Sun, Y. Improving docking accuracy through molecular mechanics generalized born optimization and scoring. J. Chem. Theory Comput. 2007, 3, 1106–1119. [Google Scholar] [CrossRef] [PubMed]
- Adasme-Carreno, F.; Munoz-Gutierrez, C.; Caballero, J.; Alzate-Morales, J.H. Performance of the MM/GBSA scoring using a binding site hydrogen bond network-based frame selection: The protein kinase case. Phys. Chem. Chem. Phys. 2014, 16, 14047–14058. [Google Scholar] [CrossRef] [PubMed]
- Mena-Ulecia, K.; Vergara-Jaque, A.; Poblete, H.; Tiznado, W.; Caballero, J. Study of the affinity between the protein kinase pka and peptide substrates derived from kemptide using molecular dynamics simulations and MM/GBSA. PLoS ONE 2014, 9, e109639. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, Y.A.; Gutierrez, M.; Ramirez, D.; Alzate-Morales, J.; Bernal, C.C.; Guiza, F.M.; Romero Bohorquez, A.R. Novel N-allyl/propargyl tetrahydroquinolines: Synthesis via three-component cationic imino Diels-alder reaction, binding prediction, and evaluation as cholinesterase inhibitors. Chem. Biol. Drug Des. 2016, 88, 498–510. [Google Scholar] [CrossRef] [PubMed]
- Chinnasamy, S.; Chinnasamy, S.; Muthusamy, K. High-affinity selective inhibitor against phospholipase a2 (pla2): A computational study. J. Recept. Signal Transduct. Res. 2016, 36, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Pak, Y.; Wang, S. Application of a molecular dynamics simulation method with a generalized effective potential to the flexible molecular docking problems. J. Phys. Chem. B 2000, 104, 354–359. [Google Scholar] [CrossRef]
- Caballero, J.; Alzate-Morales, J.H. Molecular dynamics of protein kinase-inhibitor complexes: A valid structural information. Curr. Pharm. Des. 2012, 18, 2946–2963. [Google Scholar] [CrossRef] [PubMed]
- Salam, N.K.; Nuti, R.; Sherman, W. Novel method for generating structure-based pharmacophores using energetic analysis. J. Chem. Inf. Model. 2009, 49, 2356–2368. [Google Scholar] [CrossRef] [PubMed]
- Six, D.A.; Dennis, E.A. The expanding superfamily of phospholipase a(2) enzymes: Classification and characterization. Biochim. Biophys. Acta 2000, 1488, 1–19. [Google Scholar] [CrossRef]
- Ramirez, F.; Jain, M.K. Phospholipase a2 at the bilayer interface. Proteins 1991, 9, 229–239. [Google Scholar] [CrossRef] [PubMed]
- Kini, R.M. Excitement ahead: Structure, function and mechanism of snake venom phospholipase a2 enzymes. Toxicon 2003, 42, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.A.; Borges, R.J.; Lomonte, B.; Fontes, M.R. A structure-based proposal for a comprehensive myotoxic mechanism of phospholipase a2-like proteins from viperid snake venoms. Biochim. Biophys. Acta 2014, 1844, 2265–2276. [Google Scholar] [CrossRef] [PubMed]
- Arni, R.K.; Ward, R.J. Phospholipase a2—A structural review. Toxicon 1996, 34, 827–841. [Google Scholar] [CrossRef]
- Arni, R.K.; Ward, R.J.; Gutierrez, J.M.; Tulinsky, A. Structure of a calcium-independent phospholipase-like myotoxic protein from Bothrops asper venom. Acta Crystallogr. D Biol. Crystallogr. 1995, 51, 311–317. [Google Scholar] [CrossRef] [PubMed]
- Nargotra, A.; Sharma, S.; Alam, M.I.; Ahmed, Z.; Bhagat, A.; Taneja, S.C.; Qazi, G.N.; Koul, S. In silico identification of viper phospholipasea2 inhibitors: Validation by in vitro, in vivo studies. J. Mol. Model. 2011, 17, 3063–3073. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.I.; Alam, M.A.; Alam, O.; Nargotra, A.; Taneja, S.C.; Koul, S. Molecular modeling and snake venom phospholipase a2 inhibition by phenolic compounds: Structure-activity relationship. Eur. J. Med. Chem. 2016, 114, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Anilkumar, N.C.; Sundaram, M.S.; Mohan, C.D.; Rangappa, S.; Bulusu, K.C.; Fuchs, J.E.; Girish, K.S.; Bender, A.; Rangappa, K.S. A one pot synthesis of novel bioactive tri-substitute-condensed-imidazopyridines that targets snake venom phospholipase a2. PLoS ONE 2015, 10, e0131896. [Google Scholar] [CrossRef] [PubMed]
- Yadava, U.; Singh, M.; Roychoudhury, M. Pyrazolo[3,4-d]pyrimidines as inhibitor of anti-coagulation and inflammation activities of phospholipase a2: Insight from molecular docking studies. J. Biol. Phys. 2013, 39, 419–438. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, C.; Joshi, V.; Joseph, J.M.; Vishwanath, B.S.; Velmurugan, D. Identification of novel inhibitors of daboia Russelli phospholipase a2 using the combined pharmacophore modeling approach. Chem. Biol. Drug Des. 2014, 84, 379–392. [Google Scholar] [CrossRef] [PubMed]
- Sivaramakrishnan, V.; Ilamathi, M.; Ghosh, K.S.; Sathish, S.; Gowda, T.V.; Vishwanath, B.S.; Rangappa, K.S.; Dhananjaya, B.L. Virtual analysis of structurally diverse synthetic analogs as inhibitors of snake venom secretory phospholipase a2. J. Mol. Recognit. 2016, 29, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Ramakrishnan, C.; Subramanian, V.; Velmurugan, D. Molecular dynamics study of secretory phospholipase a2 of Russell’s viper and bovine pancreatic sources. J. Phys. Chem. B 2010, 114, 13463–13472. [Google Scholar] [CrossRef] [PubMed]
- Pereanez, J.A.; Patino, A.C.; Nunez, V.; Osorio, E. The biflavonoid morelloflavone inhibits the enzymatic and biological activities of a snake venom phospholipase a2. Chem. Biol. Interact. 2014, 220, 94–101. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhong, L.; Zhou, B.; Chen, J.Y.; Li, C.M. Interaction of characteristic structural elements of persimmon tannin with Chinese cobra pla2. Toxicon 2013, 74, 34–43. [Google Scholar] [CrossRef] [PubMed]
- Chavan, S.G.; Deobagkar, D.D. In silico molecular interaction analysis of LTNF peptide-LT10 with snake venom enzymes. Protein Pept. Lett. 2014, 21, 646–656. [Google Scholar] [CrossRef] [PubMed]
- Chavanayarn, C.; Thanongsaksrikul, J.; Thueng-In, K.; Bangphoomi, K.; Sookrung, N.; Chaicumpa, W. Humanized-single domain antibodies (VH/VHH) that bound specifically to Naja Kaouthia phospholipase a2 and neutralized the enzymatic activity. Toxins 2012, 4, 554–567. [Google Scholar] [CrossRef] [PubMed]
- Hage-Melim, L.I.; da Silva, C.H.; Semighini, E.P.; Taft, C.A.; Sampaio, S.V. Computer-aided drug design of novel pla2 inhibitor candidates for treatment of snakebite. J. Biomol. Struct. Dyn. 2009, 27, 27–36. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.T.; Vicoti, M.M.; Abrego, J.R.; Lourenzoni, M.R.; Cintra, A.C.; Arruda, E.Z.; Tomaz, M.A.; Melo, P.A.; Arni, R.K. Interfacial surface charge and free accessibility to the pla2-active site-like region are essential requirements for the activity of lys49 pla2 homologues. Toxicon 2007, 49, 378–387. [Google Scholar] [CrossRef] [PubMed]
- Svensson, M.; Humbel, S.; Froese, R.D.J.; Matsubara, T.; Sieber, S.; Morokuma, K. Oniom: A multilayered integrated mo + mm method for geometry optimizations and single point energy predictions. A test for diels−alder reactions and pt(p(t-bu)3)2 + h2 oxidative addition. J. Phys. Chem. 1996, 100, 19357–19363. [Google Scholar] [CrossRef]
- De Oliveira, T.C.; de Amorim, H.L.N.; Guimarães, J.A. Interfacial activation of snake venom phospholipases a2 (SVPLA2) probed by molecular dynamics simulations. J. Mol. Struct. THEOCHEM 2007, 818, 31–41. [Google Scholar] [CrossRef]
- Vieira, L.F.; Magro, A.J.; Fernandes, C.A.; de Souza, B.M.; Cavalcante, W.L.; Palma, M.S.; Rosa, J.C.; Fuly, A.L.; Fontes, M.R.; Gallacci, M.; et al. Biochemical, functional, structural and phylogenetic studies on intercro, a new isoform phospholipase a2 from Crotalus durissus terrificus snake venom. Biochimie 2013, 95, 2365–2375. [Google Scholar] [CrossRef] [PubMed]
- Faure, G.; Xu, H.; Saul, F.A. Crystal structure of crotoxin reveals key residues involved in the stability and toxicity of this potent heterodimeric beta-neurotoxin. J. Mol. Biol. 2011, 412, 176–191. [Google Scholar] [CrossRef] [PubMed]
- Santoro, M.L.; Sousa-e-Silva, M.C.; Goncalves, L.R.; Almeida-Santos, S.M.; Cardoso, D.F.; Laporta-Ferreira, I.L.; Saiki, M.; Peres, C.A.; Sano-Martins, I.S. Comparison of the biological activities in venoms from three subspecies of the south American rattlesnake (Crotalus durissus terrificus, c. Durissus cascavella and c. Durissus collilineatus). Comp. Biochem. Physiol. C Pharmacol. Toxicol. Endocrinol. 1999, 122, 61–73. [Google Scholar] [PubMed]
- Matavel, A.C.; Ferreira-Alves, D.L.; Beirao, P.S.; Cruz, J.S. Tension generation and increase in voltage-activated Na+ current by crotamine. Eur. J. Pharmacol. 1998, 348, 167–173. [Google Scholar] [CrossRef]
- Toyama, M.H.; Carneiro, E.M.; Marangoni, S.; Barbosa, R.L.; Corso, G.; Boschero, A.C. Biochemical characterization of two crotamine isoforms isolated by a single step RP-HPLC from Crotalus durissus terrificus (South American rattlesnake) venom and their action on insulin secretion by pancreatic islets. Biochim. Biophys. Acta 2000, 1474, 56–60. [Google Scholar] [CrossRef]
- Beltran, J.R.; Mascarenhas, Y.P.; Craievich, A.F.; Laure, C.J. Saxs study of the snake toxin alpha-crotamine. Eur. Biophys. J. 1990, 17, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Giglio, J.R. Analytical studies on crotamine hydrochloride. Anal. Biochem. 1975, 69, 207–221. [Google Scholar] [CrossRef]
- Radis-Baptista, G.; Oguiura, N.; Hayashi, M.A.; Camargo, M.E.; Grego, K.F.; Oliveira, E.B.; Yamane, T. Nucleotide sequence of crotamine isoform precursors from a single South American rattlesnake (Crotalus durissus terrificus). Toxicon 1999, 37, 973–984. [Google Scholar] [CrossRef]
- Ownby, C.L. Structure, function and biophysical aspects of the myotoxins from snake venoms. J. Toxicol. Toxin Rev. 1998, 17, 213–238. [Google Scholar] [CrossRef]
- Oguiura, N.; Boni-Mitake, M.; Radis-Baptista, G. New view on crotamine, a small basic polypeptide myotoxin from South American rattlesnake venom. Toxicon 2005, 46, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Kerkis, I.; Hayashi, M.A.; Prieto da Silva, A.R.; Pereira, A.; De Sa Junior, P.L.; Zaharenko, A.J.; Radis-Baptista, G.; Kerkis, A.; Yamane, T. State of the art in the studies on crotamine, a cell penetrating peptide from South American rattlesnake. Biomed. Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Kerkis, A.; Kerkis, I.; Radis-Baptista, G.; Oliveira, E.B.; Vianna-Morgante, A.M.; Pereira, L.V.; Yamane, T. Crotamine is a novel cell-penetrating protein from the venom of rattlesnake Crotalus durissus terrificus. FASEB J. 2004, 18, 1407–1409. [Google Scholar] [CrossRef] [PubMed]
- Ruczynski, J.; Wierzbicki, P.M.; Kogut-Wierzbicka, M.; Mucha, P.; Siedlecka-Kroplewska, K.; Rekowski, P. Cell-penetrating peptides as a promising tool for delivery of various molecules into the cells. Folia Histochem. Cytobiol. 2014, 52, 257–269. [Google Scholar] [CrossRef] [PubMed]
- Siqueira, A.M.; Martins, N.F.; De Lima, M.E.; Diniz, C.R.; Cartier, A.; Brown, D.; Maigret, B. A proposed 3D structure for crotamine based on homology building, molecular simulations and circular dichroism. J. Mol. Graph. Model. 2002, 20, 389–398. [Google Scholar] [CrossRef]
- Nicastro, G.; Franzoni, L.; de Chiara, C.; Mancin, A.C.; Giglio, J.R.; Spisni, A. Solution structure of crotamine, a Na+ channel affecting toxin from Crotalus durissus terrificus venom. Eur. J. Biochem. 2003, 270, 1969–1979. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, G.R.; Legault, P.; Selsted, M.E.; Pardi, A. Solution structure of bovine neutrophil beta-defensin-12: The peptide fold of the beta-defensins is identical to that of the classical defensins. Biochemistry 1995, 34, 13663–13671. [Google Scholar] [CrossRef] [PubMed]
- Fadel, V.; Bettendorff, P.; Herrmann, T.; de Azevedo, W.F., Jr.; Oliveira, E.B.; Yamane, T.; Wuthrich, K. Automated NMR structure determination and disulfide bond identification of the myotoxin crotamine from Crotalus durissus terrificus. Toxicon 2005, 46, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Yount, N.Y.; Kupferwasser, D.; Spisni, A.; Dutz, S.M.; Ramjan, Z.H.; Sharma, S.; Waring, A.J.; Yeaman, M.R. Selective reciprocity in antimicrobial activity versus cytotoxicity of HBD-2 and crotamine. Proc. Natl. Acad. Sci. USA 2009, 106, 14972–14977. [Google Scholar] [CrossRef] [PubMed]
- Diochot, S.; Baron, A.; Salinas, M.; Douguet, D.; Scarzello, S.; Dabert-Gay, A.S.; Debayle, D.; Friend, V.; Alloui, A.; Lazdunski, M.; et al. Black mamba venom peptides target acid-sensing ion channels to abolish pain. Nature 2012, 490, 552–555. [Google Scholar] [CrossRef] [PubMed]
- Fiser, A.; Sali, A. Modeller: Generation and refinement of homology-based protein structure models. Methods Enzymol. 2003, 374, 461–491. [Google Scholar] [PubMed]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed]
- Schroeder, C.I.; Rash, L.D.; Vila-Farres, X.; Rosengren, K.J.; Mobli, M.; King, G.F.; Alewood, P.F.; Craik, D.J.; Durek, T. Chemical synthesis, 3D structure, and asic binding site of the toxin mambalgin-2. Angew. Chem. Int. Ed. Engl. 2014, 53, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Li, L.; Weng, Z. Zdock: An initial-stage protein-docking algorithm. Proteins 2003, 52, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Salinas, M.; Besson, T.; Delettre, Q.; Diochot, S.; Boulakirba, S.; Douguet, D.; Lingueglia, E. Binding site and inhibitory mechanism of the mambalgin-2 pain-relieving peptide on acid-sensing ion channel 1a. J. Biol. Chem. 2014, 289, 13363–13373. [Google Scholar] [CrossRef] [PubMed]
- Mourier, G.; Salinas, M.; Kessler, P.; Stura, E.A.; Leblanc, M.; Tepshi, L.; Besson, T.; Diochot, S.; Baron, A.; Douguet, D.; et al. Mambalgin-1 pain-relieving peptide, stepwise solid-phase synthesis, crystal structure, and functional domain for acid-sensing ion channel 1a inhibition. J. Biol. Chem. 2016, 291, 2616–2629. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Gao, Z.; Yang, H. Computer-aided drug discovery and design targeting ion channels. Curr. Top. Med. Chem. 2016, 16, 1819–1829. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira Junior, N.G.; Franco, O.L. Snake venoms: Attractive antimicrobial proteinaceous compounds for therapeutic purposes. Cell. Mol. Life Sci. 2013, 70, 4645–4658. [Google Scholar] [CrossRef] [PubMed]
- Gunasekaran, D.; Sridhar, J.; Suryanarayanan, V.; Manimaran, C.; Singh, S.K. Molecular modeling and structural analysis of NACHR variants uncovers the mechanism of resistance to snake toxins. J. Biomol. Struct. Dyn. 2017, 35, 1654–1671. [Google Scholar] [CrossRef] [PubMed]
- Moretti, R.; Fleishman, S.J.; Agius, R.; Torchala, M.; Bates, P.A.; Kastritis, P.L.; Rodrigues, J.P.G.L.M.; Trellet, M.; Bonvin, A.M.J.J.; Cui, M.; et al. Community-wide evaluation of methods for predicting the effect of mutations on protein-protein interactions. Proteins 2013, 81, 1980–1987. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.L.; Craik, D.J.; Kaas, Q. Blockade of neuronal alpha 7-nachr by alpha-conotoxin IMI explained by computational scanning and energy calculations. PLoS Comput. Biol. 2011, 7, e1002011. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zheng, Y.F.; Tang, H. Identifying the types of ion channel-targeted conotoxins by incorporating new properties of residues into pseudo amino acid composition. Biomed. Res. Int. 2016. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Deng, E.Z.; Yuan, L.F.; Liu, L.; Lin, H.; Chen, W.; Chou, K.C. ICTX-type: A sequence-based predictor for identifying the types of conotoxins in targeting ion channels. Biomed. Res. Int. 2014. [Google Scholar] [CrossRef] [PubMed]
- Yuan, L.F.; Ding, C.; Guo, S.H.; Ding, H.; Chen, W.; Lin, H. Prediction of the types of ion channel-targeted conotoxins based on radial basis function network. Toxicol. Vitro 2013, 27, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.N.; Zhang, C.J.; Gao, R.; Yang, R.T.; Song, Q. Using the smote technique and hybrid features to predict the types of ion channel-targeted conotoxins. J. Theor. Biol. 2016, 403, 75–84. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Snake Venom Protein Family | Entries | 3D Structure | ||
|---|---|---|---|---|
| X-ray | NMR | Model | ||
| 5′-nucleotidase | 3 | |||
| AB hydrolase superfamily, lipase family | 2 | |||
| AVIT (prokineticin) | 1 | 1 | ||
| Bradykinin-potentiating peptide | 65 | 2 | ||
| Cathelicidin | 10 | 1 | ||
| Complement C3 homolog | 5 | 5 | ||
| CRISP | 66 | 9 | ||
| Crotamine-myotoxin * | 17 | 1 | 2 | |
| Cystatin | 17 | |||
| Desintegrin * | 33 | 5 | 5 | |
| Endothelin/sarafotoxin | 6 | 3 | ||
| Flavin monoamine oxidase (l-amino acid oxidase) | 51 | 9 | ||
| Glycosyl hydrolase 56 (hyaluronidase) | 9 | |||
| Multicopper oxidase | 3 | 1 | ||
| Natriuretic peptide | 55 | 2 | 1 | |
| NGF-beta | 38 | 1 | ||
| Nucleotide pyrophosphatase/phosphodiesterase | 2 | |||
| Ohanin/vespryn | 4 | |||
| PDGF/VEGF growth factor | 20 | 2 | ||
| Peptidase S1 (serine protease) | 202 | 11 | 4 | |
| Phospholipase A2 | 469 | 187 | 2 | |
| Phospholipase B-like | 2 | |||
| pHpG (metalloprotease inhibitor) * | 8 | |||
| Snaclec * | 172 | 60 | 2 | |
| Snake three-finger toxin * | 505 | 65 | 65 | 18 |
| TCTP (translationally controlled tumor protein) | 3 | |||
| True venom lectin (C-type lectin) | 44 | 2 | 8 | |
| Type-B carboxylesterase/lipase | 2 | 1 | ||
| Venom, Kunitz-type | 143 | 8 | 6 | |
| Venom metalloproteinase (M12B) | 255 | 39 | 16 | 3 |
| Not in a family | 12 | |||
| Total | 2224 | 410 | 100 | 37 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ojeda, P.G.; Ramírez, D.; Alzate-Morales, J.; Caballero, J.; Kaas, Q.; González, W. Computational Studies of Snake Venom Toxins. Toxins 2018, 10, 8. https://doi.org/10.3390/toxins10010008
Ojeda PG, Ramírez D, Alzate-Morales J, Caballero J, Kaas Q, González W. Computational Studies of Snake Venom Toxins. Toxins. 2018; 10(1):8. https://doi.org/10.3390/toxins10010008
Chicago/Turabian StyleOjeda, Paola G., David Ramírez, Jans Alzate-Morales, Julio Caballero, Quentin Kaas, and Wendy González. 2018. "Computational Studies of Snake Venom Toxins" Toxins 10, no. 1: 8. https://doi.org/10.3390/toxins10010008
APA StyleOjeda, P. G., Ramírez, D., Alzate-Morales, J., Caballero, J., Kaas, Q., & González, W. (2018). Computational Studies of Snake Venom Toxins. Toxins, 10(1), 8. https://doi.org/10.3390/toxins10010008