Combination of Hypertension Along with a High Fat and Cholesterol Diet Induces Severe Hepatic Inflammation in Rats via a Signaling Network Comprising NF-κB, MAPK, and Nrf2 Pathways

Abstract
:1. Introduction
2. Materials and Methods
2.1. Animals and Experimental Protocol
2.2. Biochemical Analyses
2.3. Histopathology
2.4. Western Blotting
2.5. Enzyme-Linked Immunosorbent Assay (ELISA)
2.6. SOD Activity Assay
2.7. Statistical Analysis
3. Results
3.1. Body and Liver Weights
3.2. Serum and Hepatic Levels of Lipids and Liver Function Indices
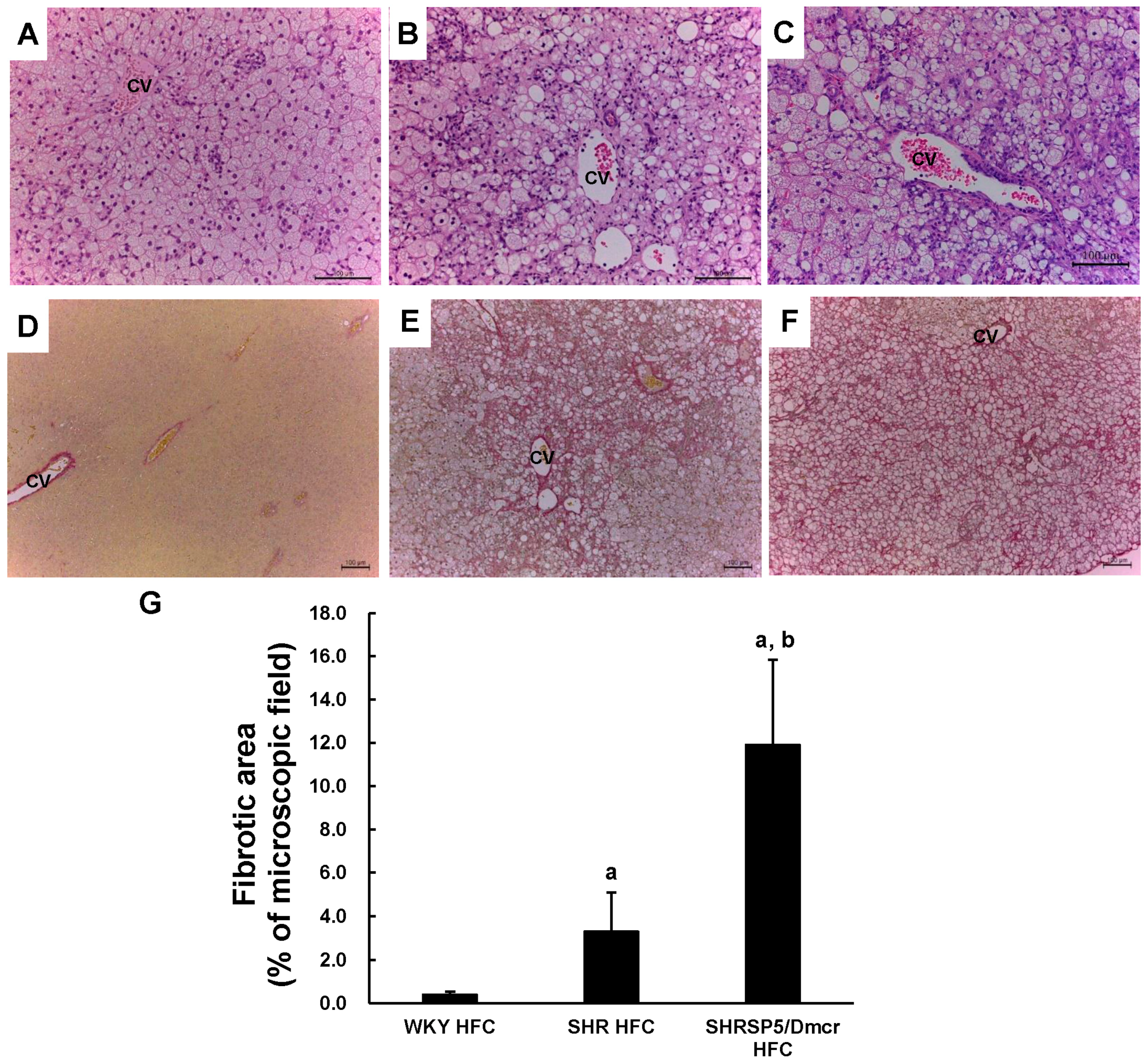
3.3. Histopathological Changes in the Liver
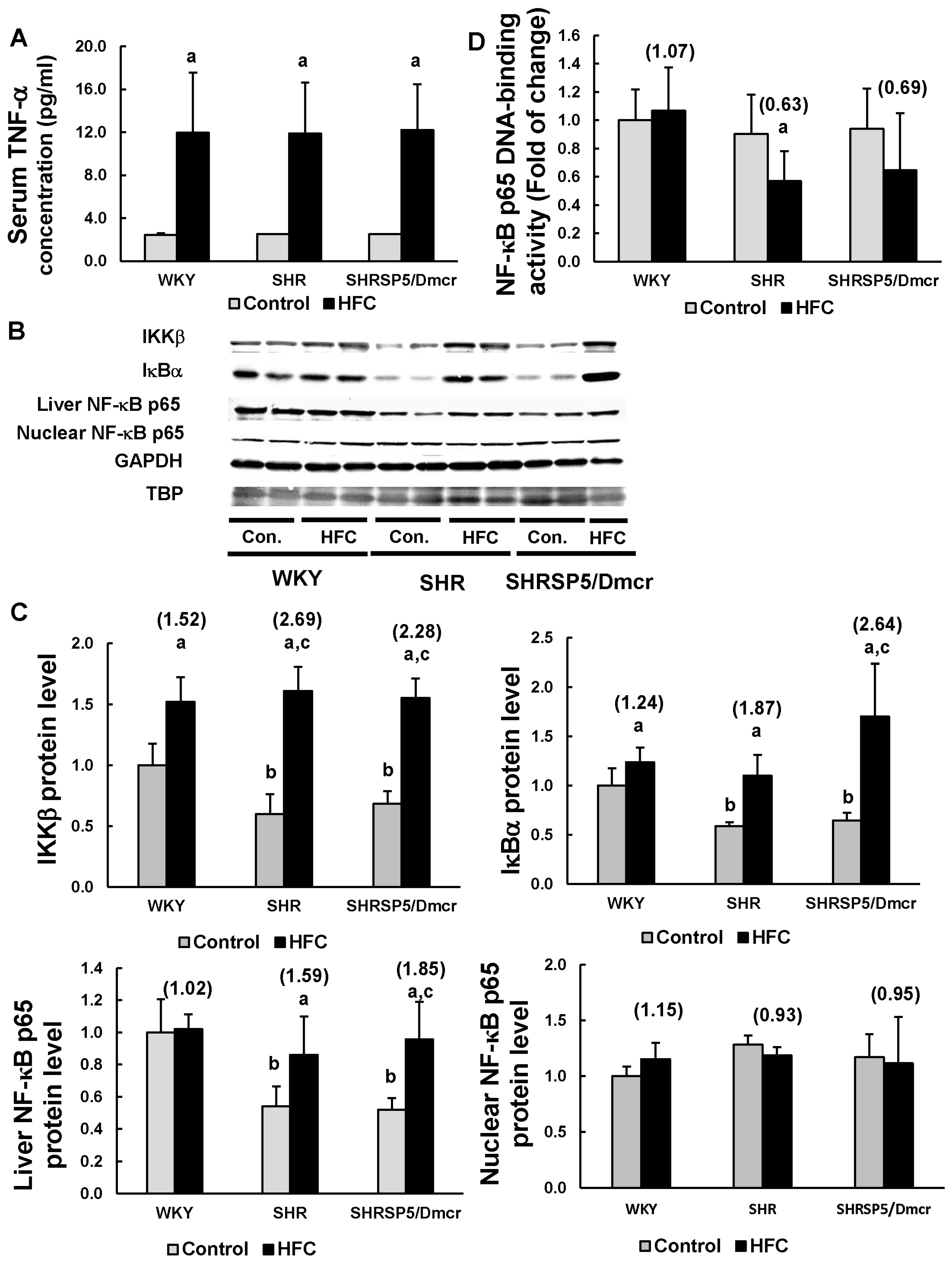
3.4. TNF-α/ NF-κB Pathway
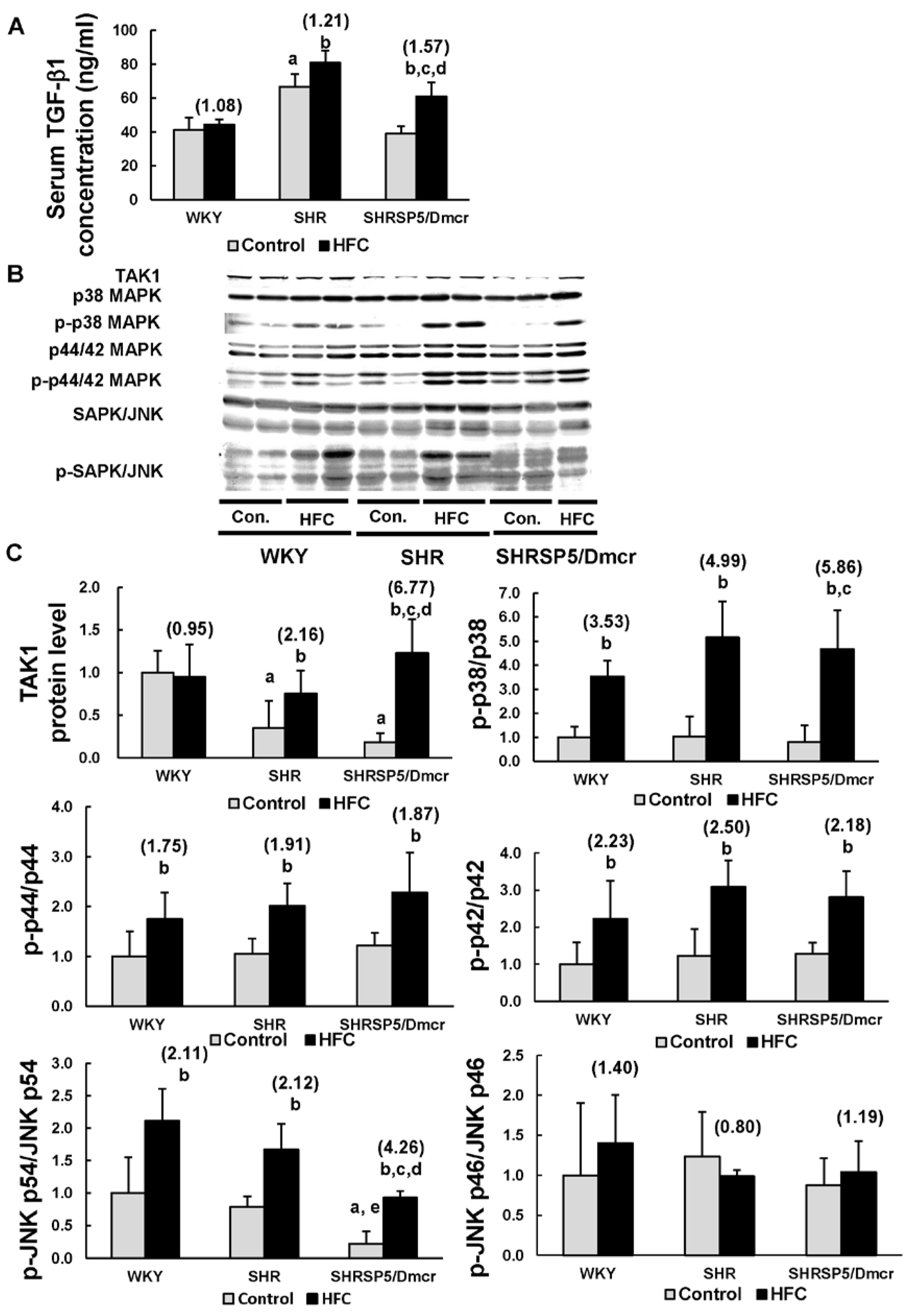
3.5. TGF-β1/MAPK Pathway
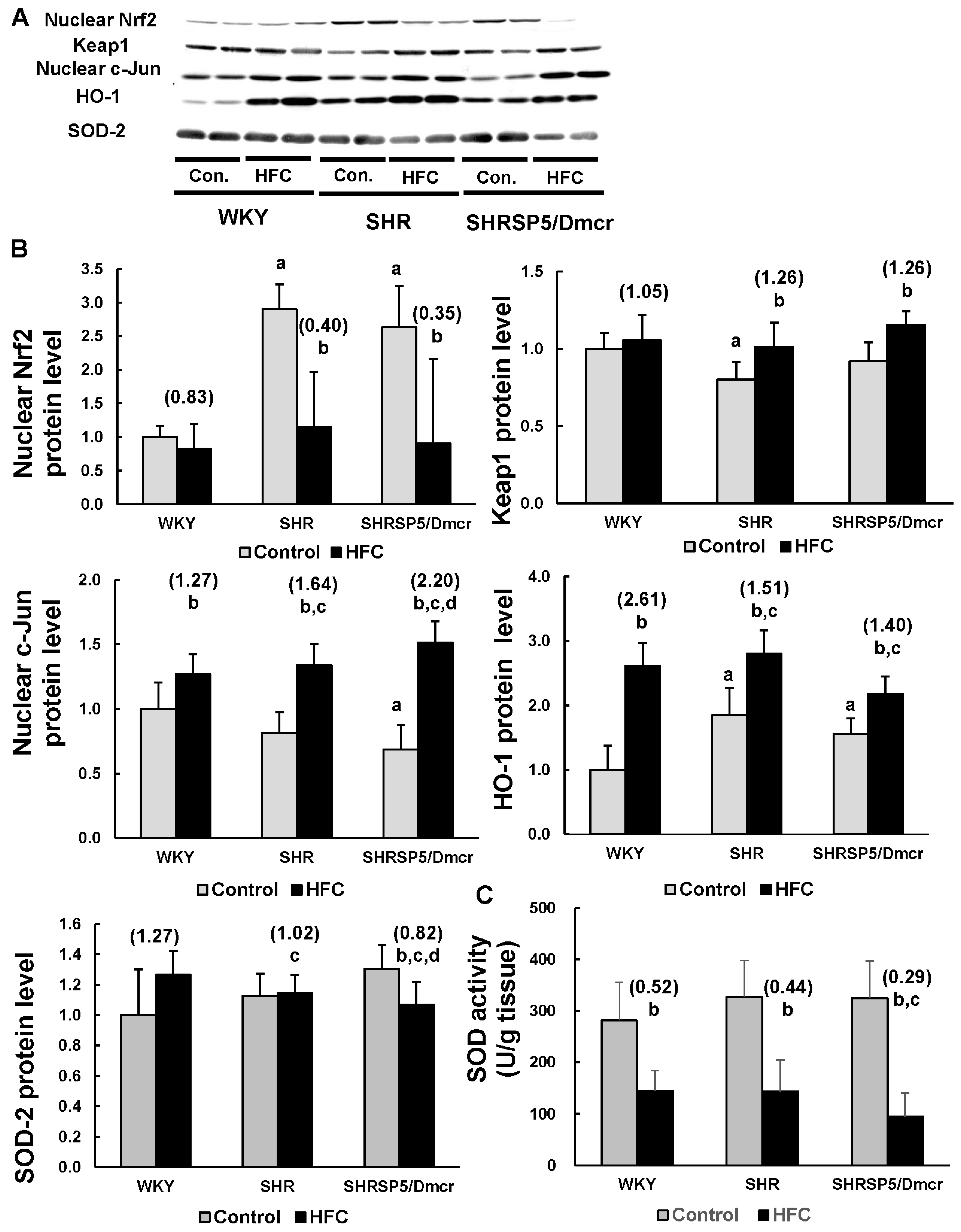
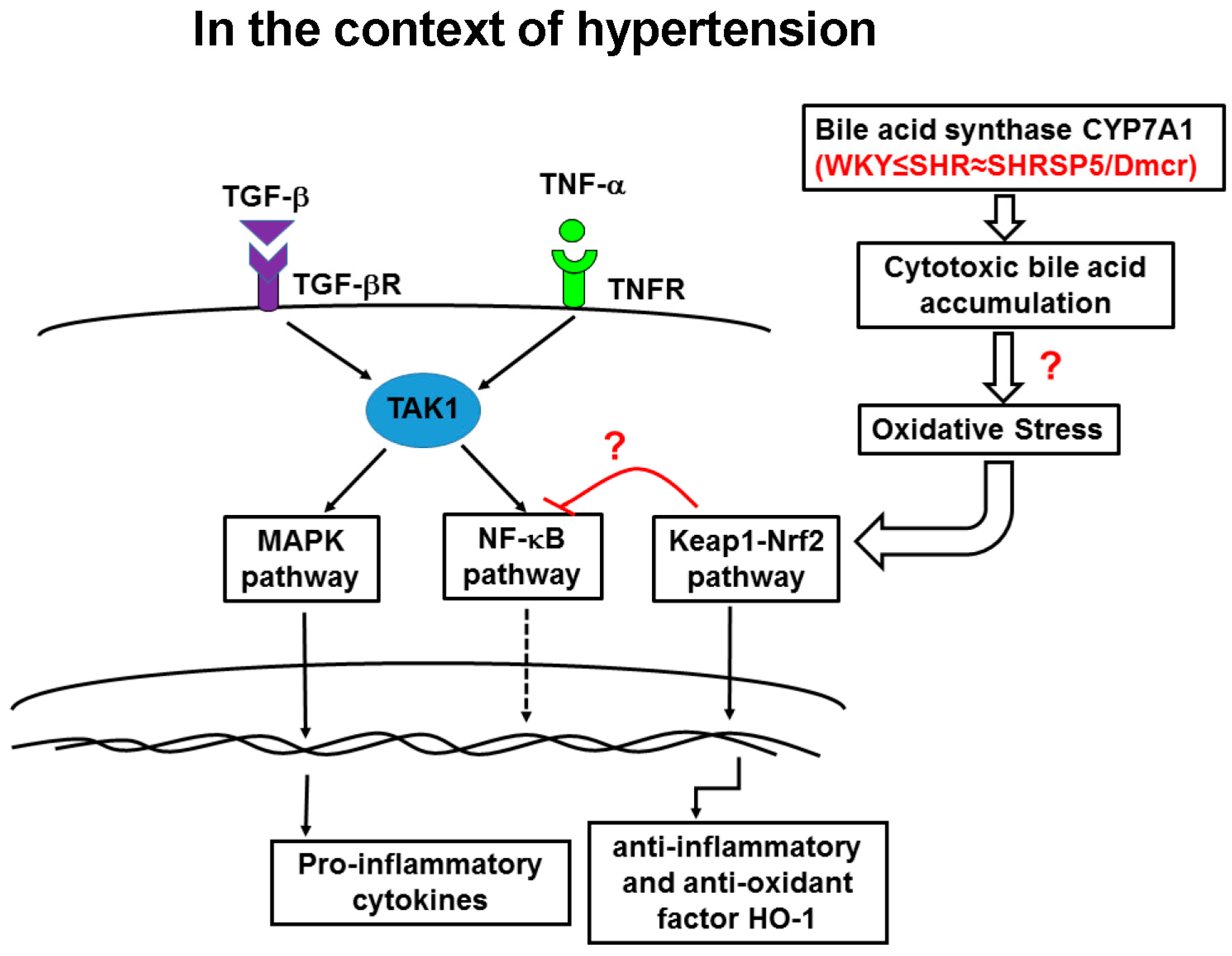
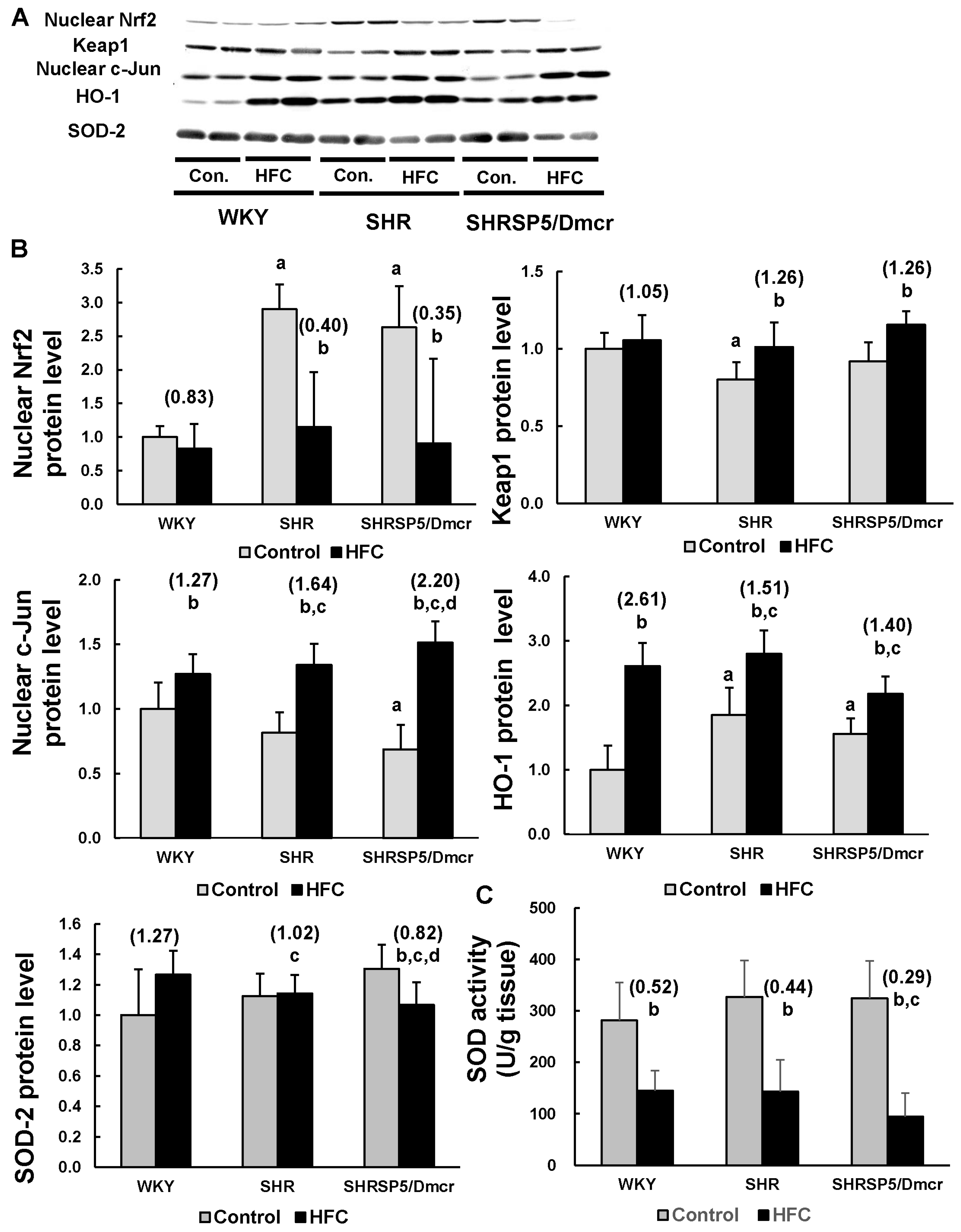
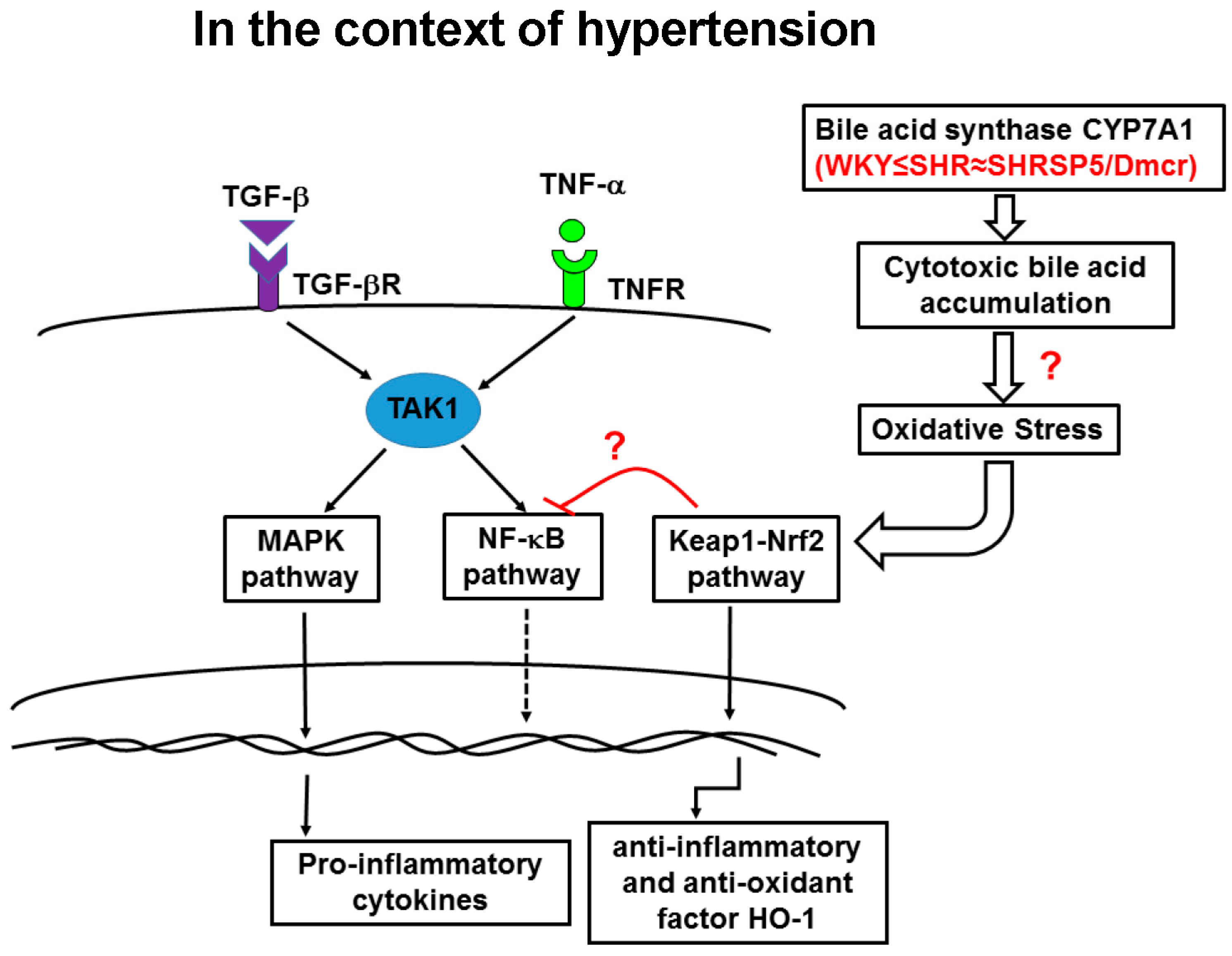
3.6. Nrf2/Keap1 Pathway
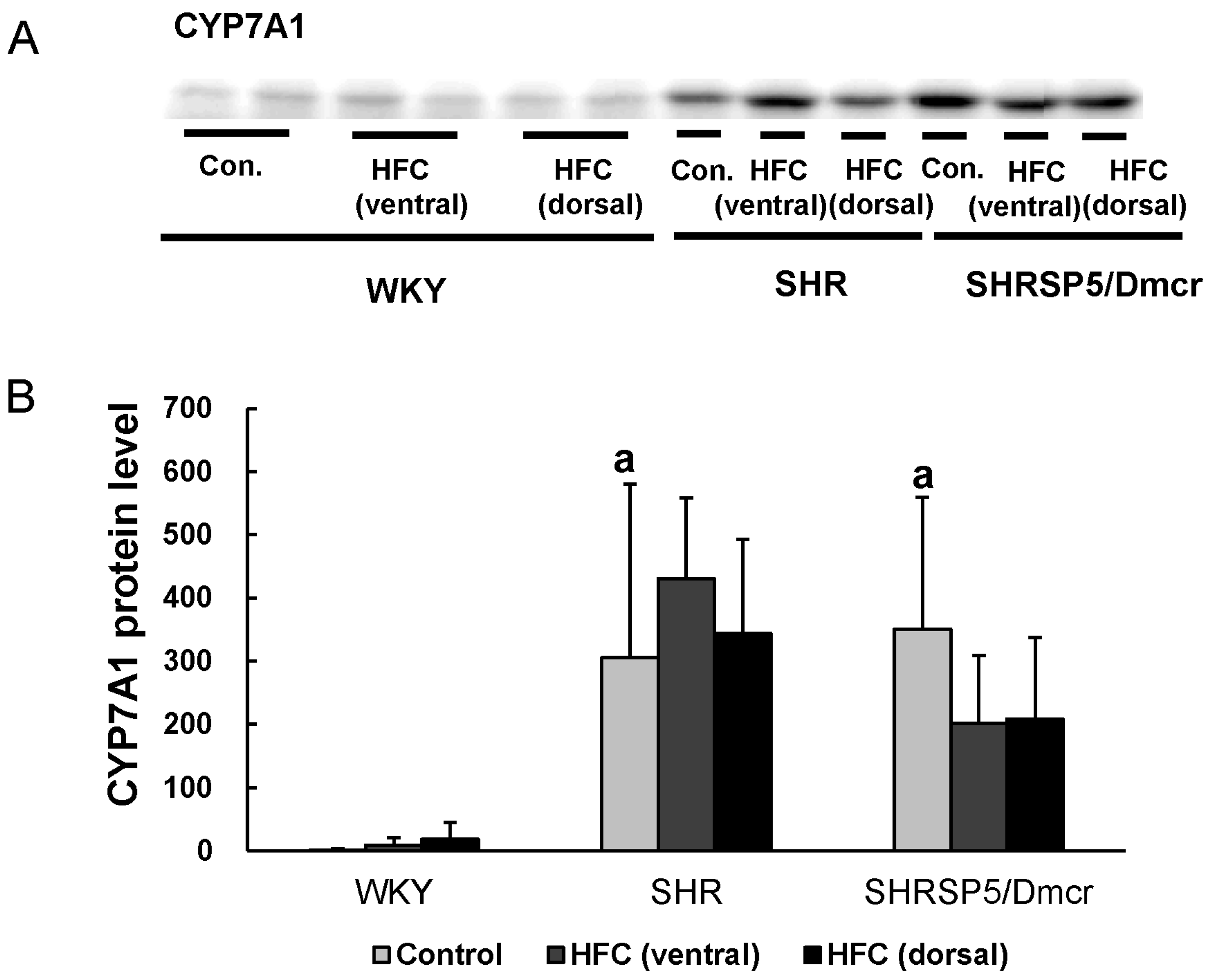
3.7. CYP7A1
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Bugianesi, E.; Lenzi, M.; McCullough, A.J.; Natale, S.; Forlani, G.; Melchionda, N. Nonalcoholic fatty liver disease: A feature of the metabolic syndrome. Diabetes 2001, 50, 1844–1850. [Google Scholar] [CrossRef] [PubMed]
- Matteoni, C.A.; Younossi, Z.M.; Gramlich, T.; Boparai, N.; Liu, Y.C.; McCullough, A.J. Nonalcoholic fatty liver disease: A spectrum of clinical and pathological severity. Gastroenterology 1999, 116, 1413–1419. [Google Scholar] [CrossRef]
- Angulo, P. Gi epidemiology: Nonalcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2007, 25, 883–889. [Google Scholar] [CrossRef] [PubMed]
- Donati, G.; Stagni, B.; Piscaglia, F.; Venturoli, N.; Morselli-Labate, A.M.; Rasciti, L.; Bolondi, L. Increased prevalence of fatty liver in arterial hypertensive patients with normal liver enzymes: Role of insulin resistance. Gut 2004, 53, 1020–1023. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zeng, Y.; Lin, C.; Chen, Z. Hypertension and non-alcoholic fatty liver disease proven by transient elastography. Hepatol. Res. 2016, 46, 1304–1310. [Google Scholar] [CrossRef] [PubMed]
- Ikuta, T.; Kanno, K.; Arihiro, K.; Matsuda, S.; Kishikawa, N.; Fujita, K.; Tazuma, S. Spontaneously hypertensive rats develop pronounced hepatic steatosis induced by choline-deficient diet: Evidence for hypertension as a potential enhancer in non-alcoholic steatohepatitis. Hepatol. Res. 2012, 42, 310–320. [Google Scholar] [CrossRef] [PubMed]
- Brookes, M.J.; Cooper, B.T. Hypertension and fatty liver: Guilty by association? J. Hum. Hypertens. 2007, 21, 264–270. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zeng, X.Y.; Zhou, X.; Wang, H.; Jo, E.; Robinson, S.R.; Xu, A.; Ye, J.M. Dietary cholesterol induces hepatic inflammation and blunts mitochondrial function in the liver of high-fat-fed mice. J. Nutr. Biochem. 2016, 27, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Kitamori, K.; Naito, H.; Tamada, H.; Kobayashi, M.; Miyazawa, D.; Yasui, Y.; Sonoda, K.; Tsuchikura, S.; Yasui, N.; Ikeda, K.; et al. Development of novel rat model for high-fat and high-cholesterol diet-induced steatohepatitis and severe fibrosis progression in shrsp5/Dmcr. Environ. Health Prev. Med. 2012, 17, 173–182. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Naito, H.; Yetti, H.; Tamada, H.; Kitamori, K.; Hayashi, Y.; Yamagishi, N.; Wang, D.; Yanagiba, Y.; Ito, Y.; et al. The modulation of hepatic adenosine triphosphate and inflammation by eicosapentaenoic acid during severe fibrotic progression in the shrsp5/Dmcr rat model. Life Sci. 2012, 90, 934–943. [Google Scholar] [CrossRef] [PubMed]
- Yamori, Y. Selection of arteriolipidosis-prone rats (ALR). Jpn. Heart J. 1977, 18, 602–603. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, K.; Aoki, K. Development of a strain of spontaneously hypertensive rats. Jpn. Circ. J. 1963, 27, 282–293. [Google Scholar] [CrossRef] [PubMed]
- Oeckinghaus, A.; Hayden, M.S.; Ghosh, S. Crosstalk in nf-kappab signaling pathways. Nat. Immunol. 2011, 12, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. Tgfbeta signalling in context. Nat. Rev. Mol. Cell. Biol. 2012, 13, 616–630. [Google Scholar] [CrossRef] [PubMed]
- Farrell, G.C.; van Rooyen, D.; Gan, L.; Chitturi, S. Nash is an inflammatory disorder: Pathogenic, prognostic and therapeutic implications. Gut Liver 2012, 6, 149–171. [Google Scholar] [CrossRef] [PubMed]
- Freudlsperger, C.; Bian, Y.; Contag Wise, S.; Burnett, J.; Coupar, J.; Yang, X.; Chen, Z.; Van Waes, C. Tgf-beta and nf-kappab signal pathway cross-talk is mediated through tak1 and smad7 in a subset of head and neck cancers. Oncogene 2013, 32, 1549–1559. [Google Scholar] [CrossRef] [PubMed]
- Sumida, Y.; Niki, E.; Naito, Y.; Yoshikawa, T. Involvement of free radicals and oxidative stress in NAFLD/NASH. Free Radic. Res. 2013, 47, 869–880. [Google Scholar] [CrossRef] [PubMed]
- Grossman, E. Does increased oxidative stress cause hypertension? Diabetes Care 2008, 31, S185–S189. [Google Scholar] [CrossRef] [PubMed]
- Baradaran, A.; Nasri, H.; Rafieian-Kopaei, M. Oxidative stress and hypertension: Possibility of hypertension therapy with antioxidants. J. Res. Med. Sci. 2014, 19, 358–367. [Google Scholar] [PubMed]
- Jaramillo, M.C.; Zhang, D.D. The emerging role of the NRF2-keap1 signaling pathway in cancer. Genes Dev. 2013, 27, 2179–2191. [Google Scholar] [CrossRef] [PubMed]
- Paine, A.; Eiz-Vesper, B.; Blasczyk, R.; Immenschuh, S. Signaling to heme oxygenase-1 and its anti-inflammatory therapeutic potential. Biochem. Pharmacol. 2010, 80, 1895–1903. [Google Scholar] [CrossRef] [PubMed]
- Gozzelino, R.; Jeney, V.; Soares, M.P. Mechanisms of cell protection by heme oxygenase-1. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 323–354. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Naito, H.; Jia, X.; Yetti, H.; Yanagiba, Y.; Tamada, H.; Kitamori, K.; Hayashi, Y.; Wang, D.; Kato, M.; Ishii, A.; et al. Importance of detoxifying enzymes in differentiating fibrotic development between shrsp5/Dmcr and shrsp rats. Environ. Health Prev. Med. 2016, 21, 368–381. [Google Scholar] [CrossRef] [PubMed]
- Dooley, S.; Ten Dijke, P. Tgf-beta in progression of liver disease. Cell Tissue Res. 2012, 347, 245–256. [Google Scholar] [CrossRef] [PubMed]
- Arthur, J.S.; Ley, S.C. Mitogen-activated protein kinases in innate immunity. Nat. Rev. Immunol. 2013, 13, 679–692. [Google Scholar] [CrossRef] [PubMed]
- Shaulian, E.; Karin, M. Ap-1 as a regulator of cell life and death. Nat. Cell Biol. 2002, 4, E131–E136. [Google Scholar] [CrossRef] [PubMed]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Jia, X.; Naito, H.; Yetti, H.; Tamada, H.; Kitamori, K.; Hayashi, Y.; Wang, D.; Yanagiba, Y.; Wang, J.; Ikeda, K.; et al. Dysregulated bile acid synthesis, metabolism and excretion in a high fat-cholesterol diet-induced fibrotic steatohepatitis in rats. Dig. Dis. Sci. 2013, 58, 2212–2222. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Bile acids: Regulation of synthesis. J. Lipid Res. 2009, 50, 1955–1966. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.; Fukusato, T. Histopathology of nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. World J. Gastroenterol. 2014, 20, 15539–15548. [Google Scholar] [CrossRef] [PubMed]
- Seki, E.; Schwabe, R.F. Hepatic inflammation and fibrosis: Functional links and key pathways. Hepatology 2015, 61, 1066–1079. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Esparza, M.; Tristan-Manzano, M.; Ruiz-Alcaraz, A.J.; Garcia-Penarrubia, P. Inflammatory status in human hepatic cirrhosis. World J. Gastroenterol. 2015, 21, 11522–11541. [Google Scholar] [CrossRef] [PubMed]
- Bishayee, A. The role of inflammation and liver cancer. Adv. Exp. Med. Biol. 2014, 816, 401–435. [Google Scholar] [PubMed]
- Arrese, M.; Cabrera, D.; Kalergis, A.M.; Feldstein, A.E. Innate immunity and inflammation in nafld/nash. Dig. Dis. Sci. 2016, 61, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Crespo, J.; Cayon, A.; Fernandez-Gil, P.; Hernandez-Guerra, M.; Mayorga, M.; Dominguez-Diez, A.; Fernandez-Escalante, J.C.; Pons-Romero, F. Gene expression of tumor necrosis factor alpha and TNF-receptors, p55 and p75, in nonalcoholic steatohepatitis patients. Hepatology 2001, 34, 1158–1163. [Google Scholar] [CrossRef] [PubMed]
- Hui, J.M.; Hodge, A.; Farrell, G.C.; Kench, J.G.; Kriketos, A.; George, J. Beyond insulin resistance in NASH: TNF-alpha or adiponectin? Hepatology 2004, 40, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.; Liu, Z.; Chen, Y. Regulation of tgf-beta signaling by SMAD7. Acta Biochim. Biophys. Sin. 2009, 41, 263–272. [Google Scholar] [CrossRef] [PubMed]
- Wrana, J.L. Signaling by the tgfbeta superfamily. Cold Spring Harb. Perspect Biol. 2013, 5, a011197. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Deeg, M.A.; Crabb, D.W. Systemic levels of lipid peroxidation and its metabolic and dietary correlates in patients with nonalcoholic steatohepatitis. Am. J. Gastroenterol. 2004, 99, 1497–1502. [Google Scholar] [CrossRef] [PubMed]
- Kojima, H.; Sakurai, S.; Uemura, M.; Fukui, H.; Morimoto, H.; Tamagawa, Y. Mitochondrial abnormality and oxidative stress in nonalcoholic steatohepatitis. Alcohol. Clin. Exp. Res. 2007, 31, S61–S66. [Google Scholar] [CrossRef] [PubMed]
- Malaguarnera, L.; Madeddu, R.; Palio, E.; Arena, N.; Malaguarnera, M. Heme oxygenase-1 levels and oxidative stress-related parameters in non-alcoholic fatty liver disease patients. J. Hepatol. 2005, 42, 585–591. [Google Scholar] [CrossRef] [PubMed]
- Ganesh Yerra, V.; Negi, G.; Sharma, S.S.; Kumar, A. Potential therapeutic effects of the simultaneous targeting of the nrf2 and nf-kappab pathways in diabetic neuropathy. Redox Biol. 2013, 1, 394–397. [Google Scholar] [CrossRef] [PubMed]
- Wardyn, J.D.; Ponsford, A.H.; Sanderson, C.M. Dissecting molecular cross-talk between Nrf2 and NF-KAPPAB response pathways. Biochem. Soc. Trans. 2015, 43, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Moriya, T.; Kitamori, K.; Naito, H.; Yanagiba, Y.; Ito, Y.; Yamagishi, N.; Tamada, H.; Jia, X.; Tsuchikura, S.; Ikeda, K.; et al. Simultaneous changes in high-fat and high-cholesterol diet-induced steatohepatitis and severe fibrosis and those underlying molecular mechanisms in novel SHRSP5/Dmcr rat. Environ. Health Prev. Med. 2012, 17, 444–456. [Google Scholar] [CrossRef] [PubMed]
- Saez, G.T.; Tormos, C.; Giner, V.; Chaves, J.; Lozano, J.V.; Iradi, A.; Redon, J. Factors related to the impact of antihypertensive treatment in antioxidant activities and oxidative stress by-products in human hypertension. Am. J. Hypertens. 2004, 17, 809–816. [Google Scholar] [CrossRef] [PubMed]
- Ghiadoni, L.; Magagna, A.; Versari, D.; Kardasz, I.; Huang, Y.; Taddei, S.; Salvetti, A. Different effect of antihypertensive drugs on conduit artery endothelial function. Hypertension 2003, 41, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Howden, R. Nrf2 and cardiovascular defense. Oxid. Med. Cell. Longev. 2013, 2013, 104308. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, M.; Bonkovsky, H.L. Increased heme oxygenase-1 gene expression in liver cells and splanchnic organs from portal hypertensive rats. Hepatology 1999, 29, 1672–1679. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.M.; Li, J.; Liu, L.; Fan, L.; Li, X.Y.; Wang, Y.T.; Abraham, N.G.; Cao, J. Effects of heme oxygenase-1 upregulation on blood pressure and cardiac function in an animal model of hypertensive myocardial infarction. Int. J. Mol. Sci. 2013, 14, 2684–2706. [Google Scholar] [CrossRef] [PubMed]
- Kamisako, T.; Tanaka, Y.; Kishino, Y.; Ikeda, T.; Yamamoto, K.; Masuda, S.; Ogawa, H. Role of nrf2 in the alteration of cholesterol and bile acid metabolism-related gene expression by dietary cholesterol in high fat-fed mice. J. Clin. Biochem. Nutr. 2014, 54, 90–94. [Google Scholar] [CrossRef] [PubMed]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [PubMed]
- Li, T.; Matozel, M.; Boehme, S.; Kong, B.; Nilsson, L.M.; Guo, G.; Ellis, E.; Chiang, J.Y. Overexpression of cholesterol 7alpha-hydroxylase promotes hepatic bile acid synthesis and secretion and maintains cholesterol homeostasis. Hepatology 2011, 53, 996–1006. [Google Scholar] [CrossRef] [PubMed]
- Erickson, S.K.; Lear, S.R.; Deane, S.; Dubrac, S.; Huling, S.L.; Nguyen, L.; Bollineni, J.S.; Shefer, S.; Hyogo, H.; Cohen, D.E.; et al. Hypercholesterolemia and changes in lipid and bile acid metabolism in male and female cyp7a1-deficient mice. J. Lipid Res. 2003, 44, 1001–1009. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rat strains | WKY | SHR | SHRSP5/Dmcr | |||
|---|---|---|---|---|---|---|
| Control | HFC | Control | HFC | Control | HFC | |
| Body weight (g) | 365 ± 22.6 | 331 ± 7.8 a | 339 ± 14.5 | 301 ± 10.8 a | 283 ± 16.4 | 260 ± 9.1 a |
| Liver weight (g) | 9.1 ± 0.6 | 23.2 ± 1.4 a (2.5) | 9.8 ± 0.4 | 28.5 ± 2.5 a (2.9) b | 8.0 ± 0.4 | 27.4 ± 1.8 a (3.4) b,d |
| Liver/body weight(%) | 2.5 ± 0.1 | 7.0 ± 0.3 a (2.8) | 2.9 ± 0.0 c | 9.5 ± 0.8 a (3.3) b | 2.8 ± 0.1 c | 10.6 ± 0.8 a (3.7) b,d |
| Serum | ||||||
| TG (mg/dL) | 12.3 ± 1.9 | 12.2 ± 2.6(1.0) | 17.7 ± 3.3 c | 17.0 ± 2.8(1.0) | 25.5 ± 4.1 c | 17.7 ± 4.6 a (0.7) b,d |
| TC (mg/dL) | 103 ± 7.8 | 140.3 ± 17.4 a (1.4) | 61.3 ± 5.4 c | 134 ± 19.6 a (2.2) | 53.5 ± 2.6 c | 149.2 ± 50.1 a (2.8) b |
| AST (IU/L) | 168.7 ± 47.6 | 424 ± 94.5 a (2.5) | 245.7 ± 49.1 c | 479.2 ± 93.9 a (2.0) | 177.8 ± 30.6 e | 633.8 ± 81.8 a (3.7) b,d |
| ALT (IU/L) | 33.8 ± 1.7 | 134.2 ± 50.0 a (4.0) | 51.7 ± 6.3 c | 205.7 ± 47.5 a (4.0) | 61 ± 11.0 c | 318.5 ± 55.0 a (5.2) |
| GGT (IU/L) | 1.5 | 1.8 ± 0.6(1.2) | 1.5 | 3.6 ± 1.6 a (2.4) | 1.5 | 4.8 ± 1.0 a (3.2) |
| Liver | ||||||
| TG (mg/dL) | 23.9 ± 3.1 | 64.6 ± 9.4 a (2.7) | 29.6 ± 7.1 | 63.6 ± 8.0 a (2.1) | 26.8 ± 7.3 | 68.6 ± 15.8 a (2.6) |
| TC (mg/dL) | 3.4 ± 0.4 | 176.2 ± 9.1 a (51.8) | 3.9 ± 0.6 | 167 ± 10.7 a (42.8) b | 3.5 ± 1.0 | 150.9 ± 15.4 a (43.1) b |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yuan, Y.; Naito, H.; Jia, X.; Kitamori, K.; Nakajima, T. Combination of Hypertension Along with a High Fat and Cholesterol Diet Induces Severe Hepatic Inflammation in Rats via a Signaling Network Comprising NF-κB, MAPK, and Nrf2 Pathways. Nutrients 2017, 9, 1018. https://doi.org/10.3390/nu9091018
Yuan Y, Naito H, Jia X, Kitamori K, Nakajima T. Combination of Hypertension Along with a High Fat and Cholesterol Diet Induces Severe Hepatic Inflammation in Rats via a Signaling Network Comprising NF-κB, MAPK, and Nrf2 Pathways. Nutrients. 2017; 9(9):1018. https://doi.org/10.3390/nu9091018
Chicago/Turabian StyleYuan, Yuan, Hisao Naito, Xiaofang Jia, Kazuya Kitamori, and Tamie Nakajima. 2017. "Combination of Hypertension Along with a High Fat and Cholesterol Diet Induces Severe Hepatic Inflammation in Rats via a Signaling Network Comprising NF-κB, MAPK, and Nrf2 Pathways" Nutrients 9, no. 9: 1018. https://doi.org/10.3390/nu9091018