Inhibitory Effect of Lycopene on Amyloid-β-Induced Apoptosis in Neuronal Cells



Abstract
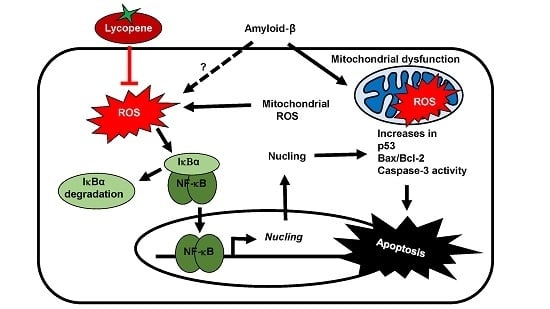
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Cell Line and Culture Condition
2.3. Small Interfering RNA (siRNA) Targeting Nucling
2.4. Experimental Protocol
2.5. Measurement of Cell Viability
2.6. Preparation of Cell Extracts
2.7. Measurement of Intracellular ROS and Mitochondrial ROS Levels
2.8. Western Blot Analysis for Bax, Bcl-2, caspase-3, p53, IκBα, p-IκBα, NF-κB, and Nucling
2.9. Electrophoretic Mobility Shift Assay (EMSA)
2.10. Measurement of Mitochondrial Membrane Potential (MMP)
2.11. Measurement of Oxygen Consumption Rate (OCR)
2.12. Statistical Analysis
3. Results
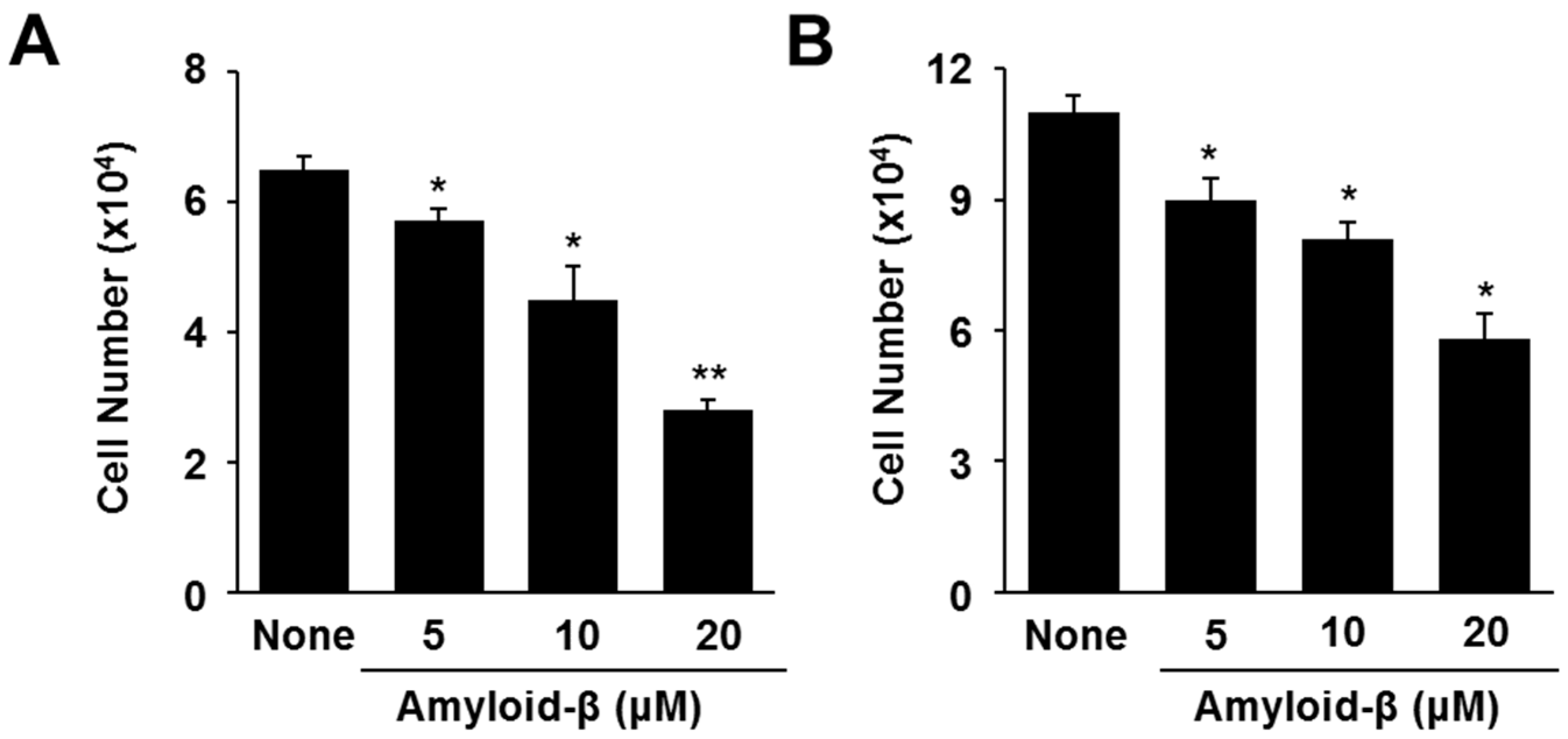
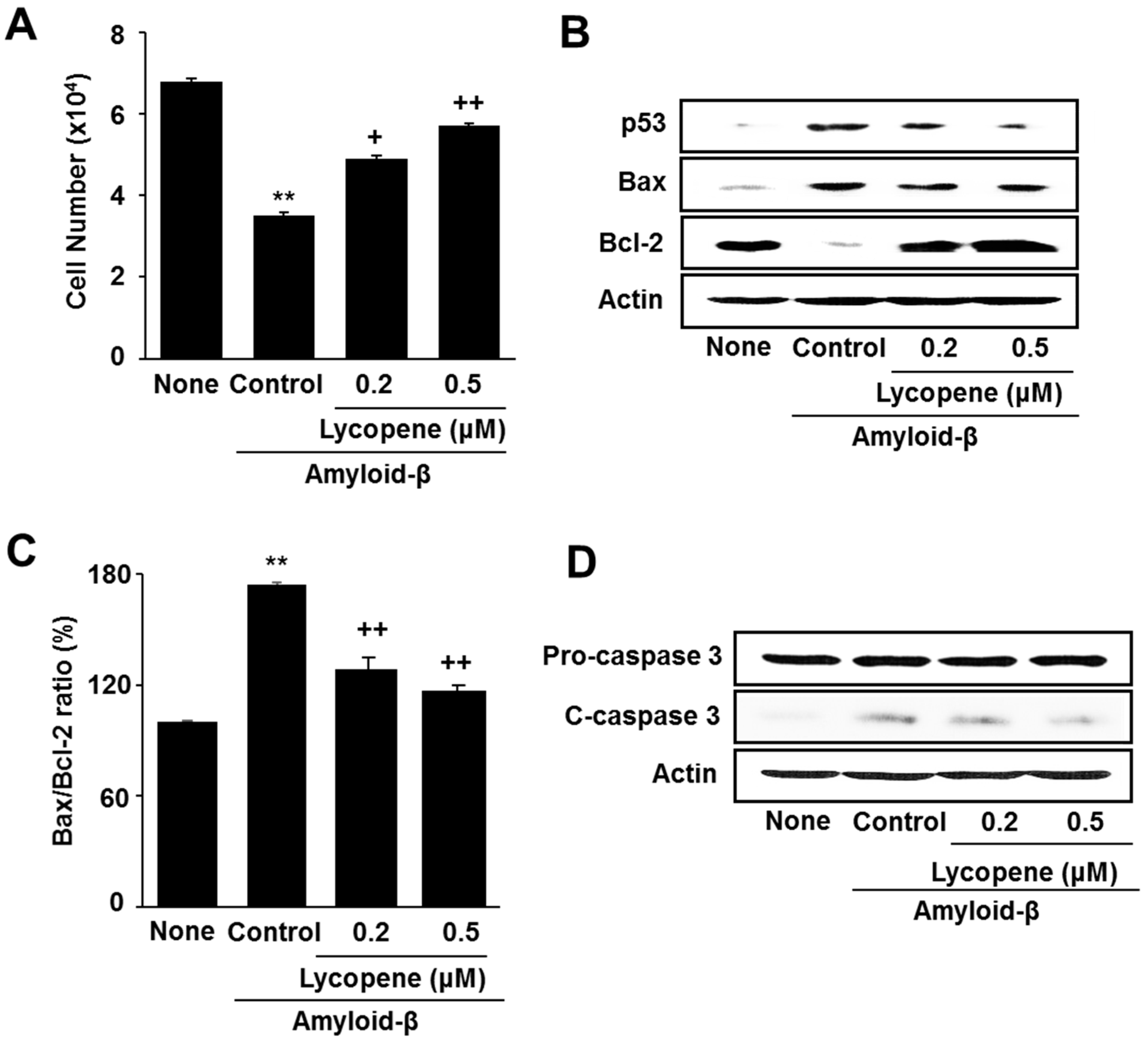
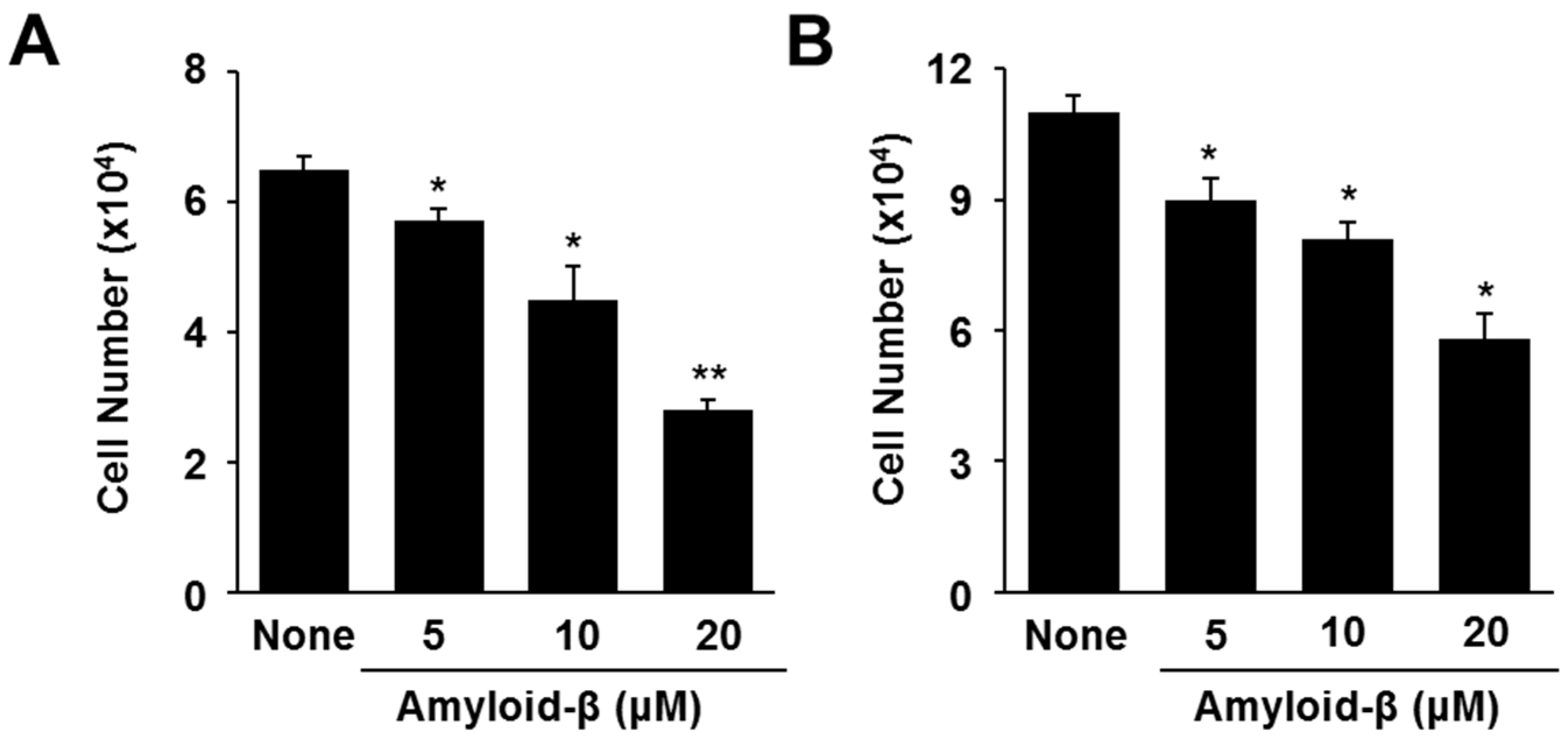
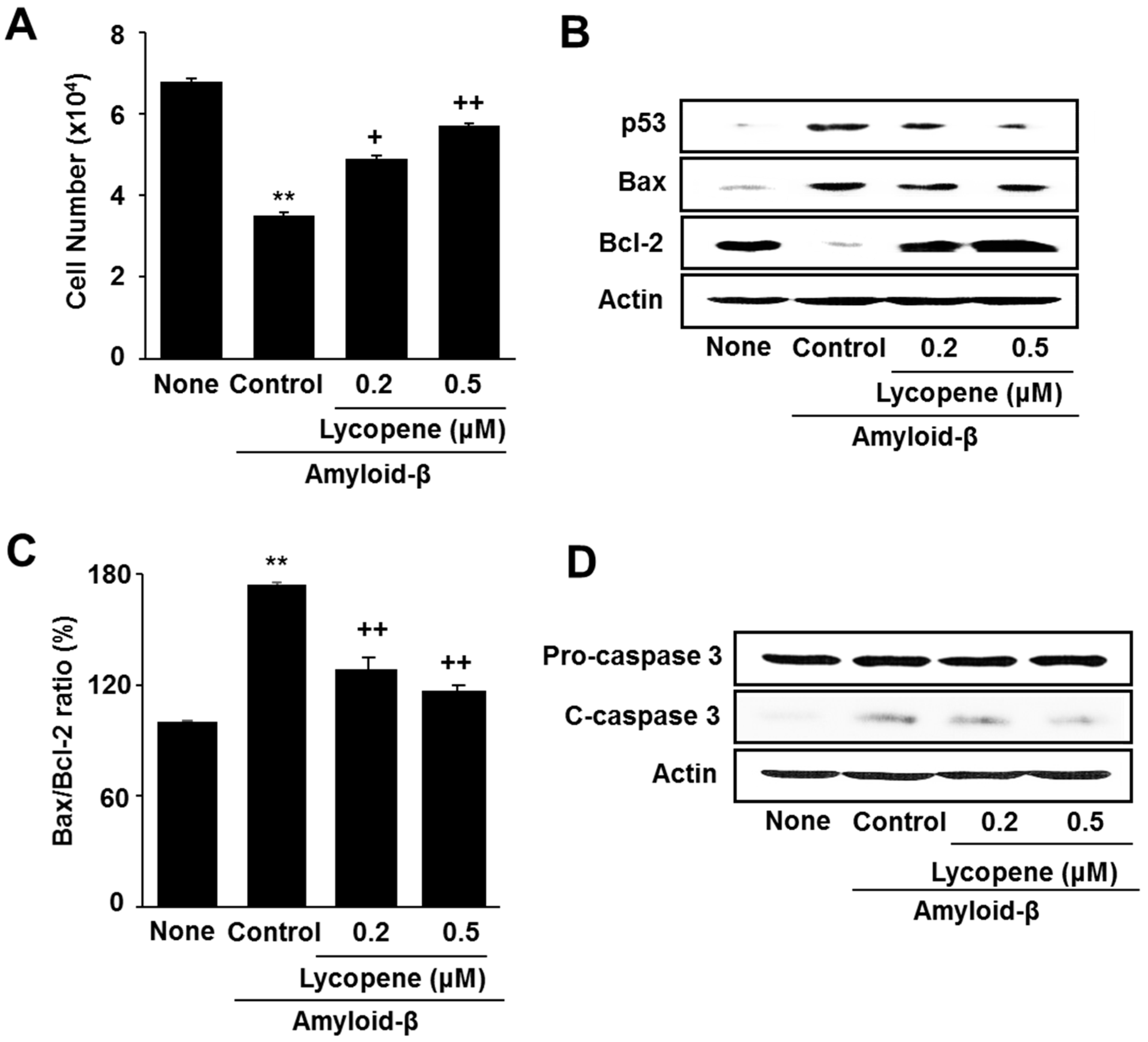
3.1. Lycopene Inhibits Amyloid-β-Induced Cell Death and Increase in Apoptotic Indices (p53, Bax/Bcl-2 Ratio, Caspase-3 Cleavage) in SH-SY5Y Cells
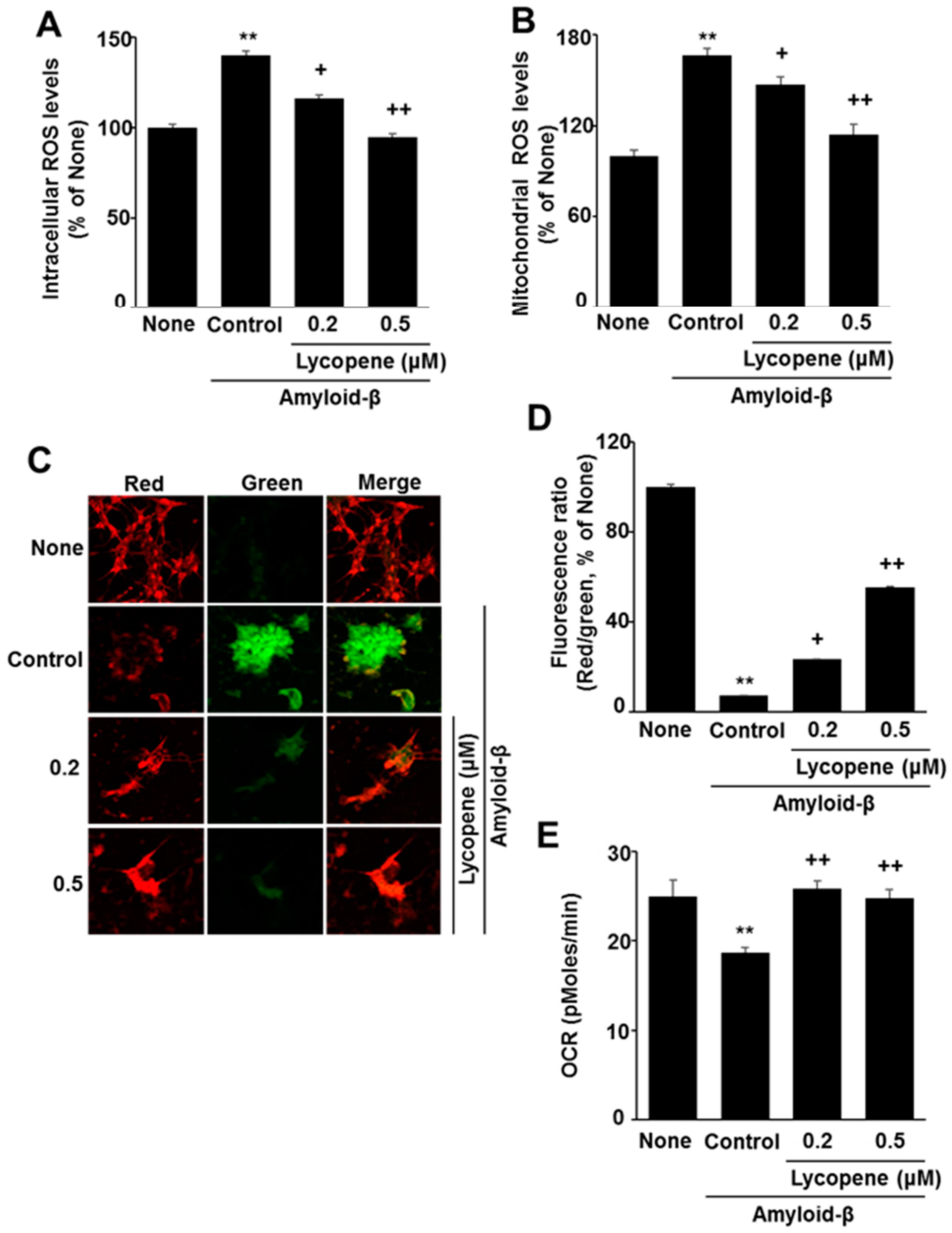
3.2. Lycopene Reduces ROS Levels and Inhibits Mitochondrial Dysfunction in Amyloid-β-Stimulated SH-SY5Y Cells
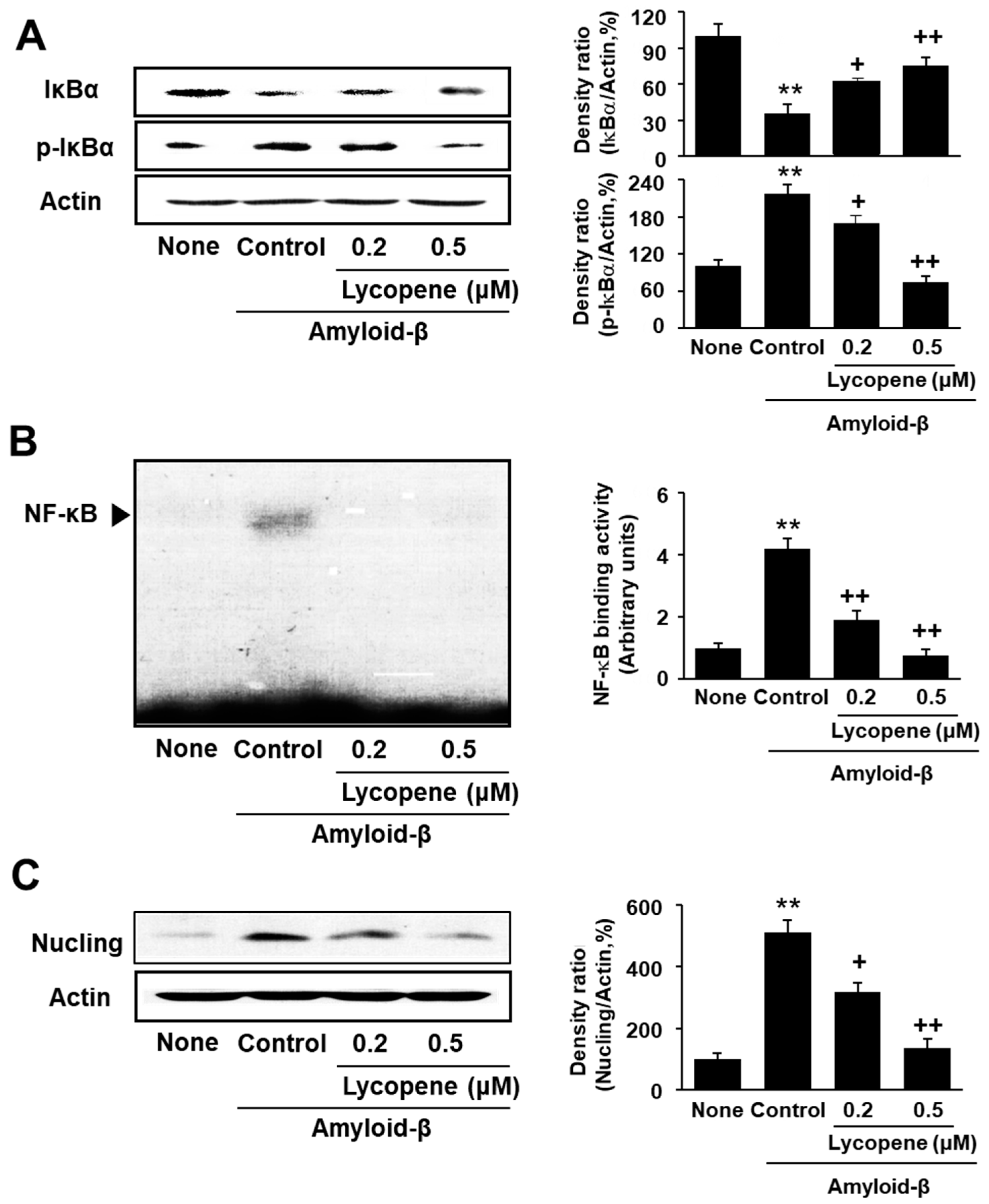
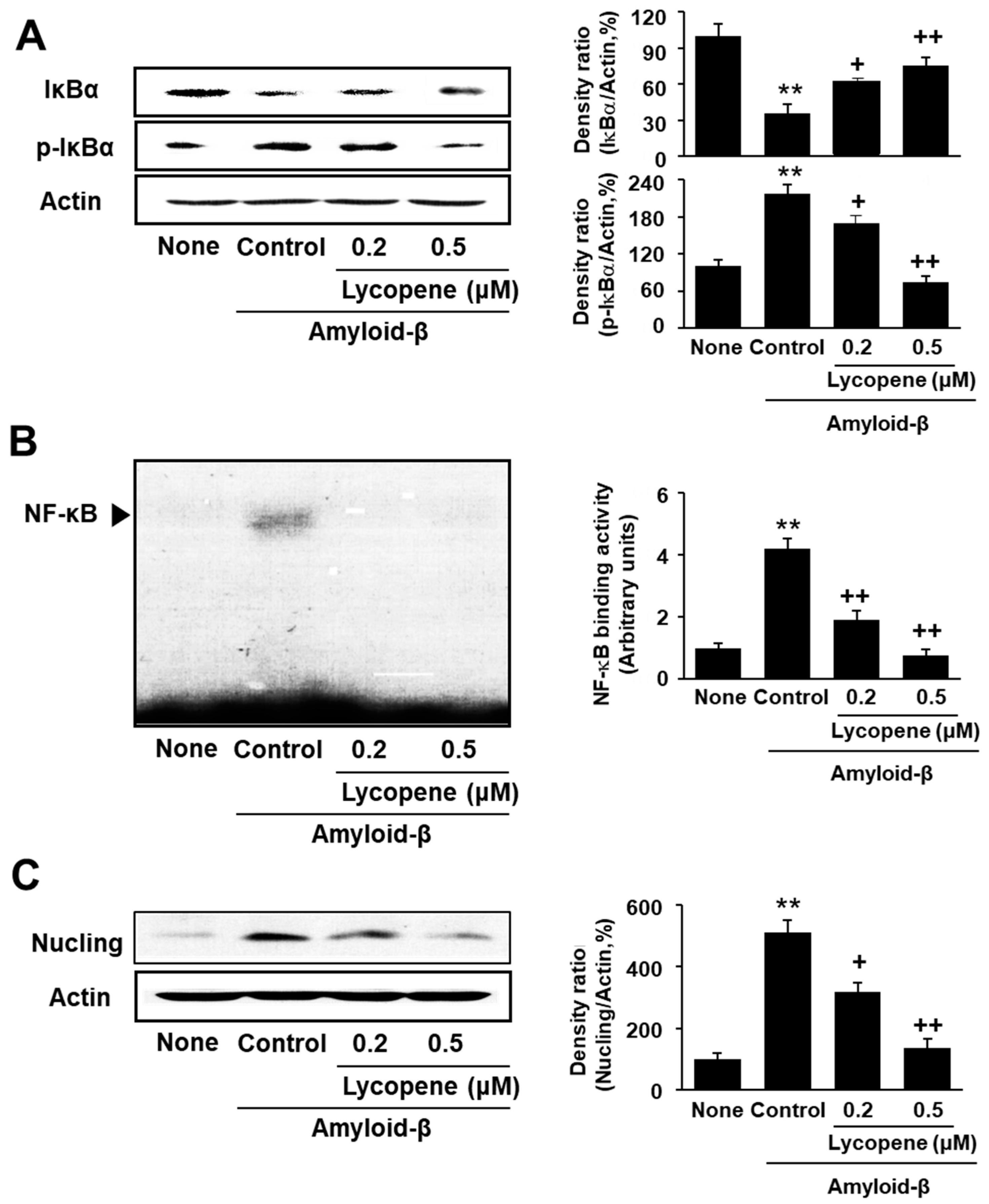
3.3. Lycopene Inhibits Phosphorylation of IκBα, NF-kB Activation, and Nucling Induction in Amyloid-β-Stimulated SH-SY5Y Cells
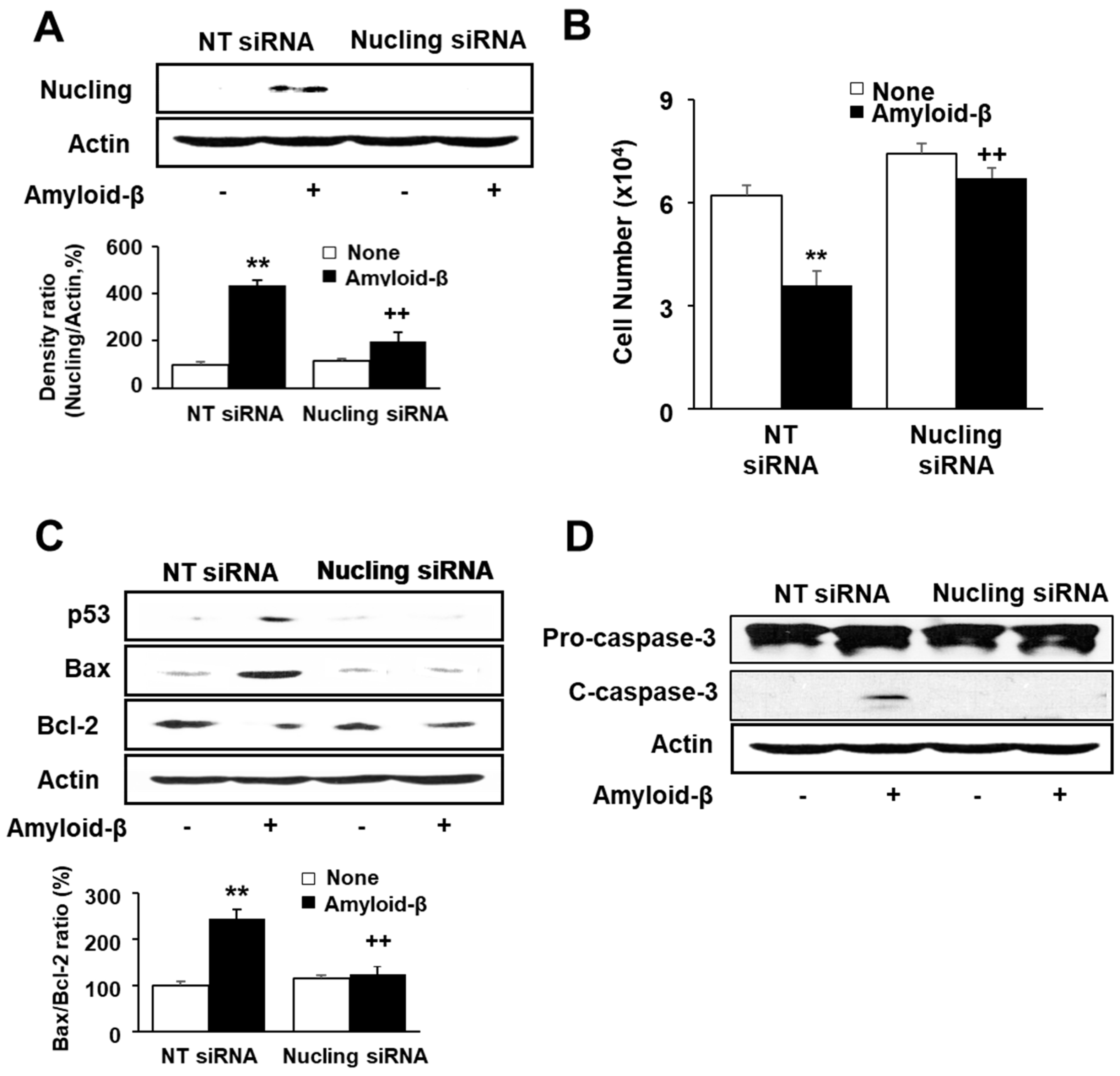
3.4. Transfection of Nucling siRNA Inhibits Cell Death and Increase in Apoptotic Indices in Amyloid-β-Stimulated SH-SY5Y Cells
4. Discussion
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Agostinho, P.; Cunha, R.A.; Oliveira, C. Neuroinflammation, oxidative stress and the pathogenesis of Alzheimer’s disease. Curr. Pharm. Des. 2010, 16, 2766–2778. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association. 2013 Alzheimer’s disease facts and figures. Alzheimers Dement. 2013, 9, 208–245. [Google Scholar]
- Bagyinszky, E.; Youn, Y.C.; An, S.S.; Kim, S. Mutations, associated with early-onset Alzheimer’s disease, discovered in Asian countries. Clin. Interv. Aging 2016, 11, 1467–1488. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Ohki, Y.; Tomita, T.; Osawa, S.; Reed, B.R.; Jagust, W.; Van Berlo, V.; Jin, L.W.; Chui, H.C.; Coppola, G.; et al. Two novel mutations in the first tansmembrane domain of Presenilin1 cause young-onset Alzheimer’s disease. J. Alzheimers Dis. 2017, 58, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- Bao, F.; Wicklund, L.; Klein, W.L.; Nordberg, A.; Marutle, A. Different amyloid-β oligomer assemblies in Alzheimer brains correlate with age of disease onset and impaired cholinergic activity. Neurobiol. Aging 2012, 33, 825. [Google Scholar] [CrossRef] [PubMed]
- Esparza, T.J.; Zhao, H.; Cirrito, J.R.; Cirrito, J.R.; Cairns, N.J.; Bateman, R.J.; Holtzman, D.M.; Brody, D.L. Amyloidbeta oligomerization in Alzheimer dementia versus high-pathology controls. Ann. Neurol. 2013, 73, 104–119. [Google Scholar] [CrossRef] [PubMed]
- Sadigh-Eteghad, S.; Sabermarouf, B.; Majdi, A.; Talebi, M.; Farhoudi, M.; Mahmoudi, J. Amyloid-beta: A crucial factor in Alzheimer’s disease. Med. Princ. Pract. 2015, 24, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Piemontese, L. New approaches for prevention and treatment of Alzheimer’s disease: A fascinating challenge. Neural Regen. Res. 2017, 12, 405–406. [Google Scholar] [CrossRef] [PubMed]
- Libro, R.; Giacoppo, S.; Soundara Rajan, T.; Bramanti, P.; Mazzon, E. Natural phytochemicals in the treatment and prevention of dementia: An overview. Molecules 2016, 21, 518. [Google Scholar] [CrossRef] [PubMed]
- Otaegui-Arrazola, A.; Amiano, P.; Elbusto, A.; Urdaneta, E.; Martínez-Lage, P. Diet, cognition, and Alzheimer’s disease: Food for thought. Eur. J. Nutr. 2014, 53, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Piemontese, L. Plant food supplements with antioxidant properties for the treatment of chronic and neurodegenerative diseases: Benefits or risks? J. Diet. Suppl. 2017, 14, 478–484. [Google Scholar] [CrossRef] [PubMed]
- Fiorini, A.; Sultana, R.; Barone, E.; Cenini, G.; Perluigi, M.; Mancuso, C.; Cai, J.; Klein, J.B.; St Clair, D.; Butterfield, D.A. Lack of p53 affects the expression of several brain mitochondrial proteins: Insights from proteomics into important pathways regulated by p53. PLoS ONE 2012, 7, e49846. [Google Scholar] [CrossRef] [PubMed]
- Gilgun-Sherki, Y.; Melamed, E.; Offen, D. Oxidative stress induced-neurodegenerative diseases: The need for antioxidants that penetrate the blood brain barrier. Neuropharmacology 2001, 40, 959–975. [Google Scholar] [CrossRef]
- Reeve, A.K.; Krishnan, K.J.; Turnbull, D. Mitochondrial DNA mutations in disease, aging, and neurodegeneration. Ann. N. Y. Acad. Sci. 2008, 1147, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Gleichmann, M.; Cheng, A. Mitochondria in neuroplasticity and neurological disorders. Neuron 2008, 60, 748–766. [Google Scholar] [CrossRef] [PubMed]
- Aronis, A.; Melendez, J.A.; Golan, O.; Shilo, S.; Dicter, N.; Tirosh, O. Potentiation of Fas-mediated apoptosis by attenuated production of mitochondria-derived reactive oxygen species. Cell Death Differ. 2003, 10, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Rockenstein, E.; Crews, L.; Masliah, E. Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer’s and Parkinson’s diseases. Neuromol. Med. 2003, 4, 21–35. [Google Scholar] [CrossRef]
- Karin, M.; Cao, Y.; Greten, F.R.; Li, Z.W. NF-κB in cancer: From innocent bystander to major culprit. Nat. Rev. Cancer 2002, 2, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Radhakrishnan, S.K.; Kamalakaran, S. Pro-apoptotic role of NF-κB: Implications for cancer therapy. Biochim. Biophys. Acta 2006, 176, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Sakai, T.; Liu, L.; Teng, X.; Mukai-Sakai, R.; Shimada, H.; Kaji, R.; Mitani, T.; Matsumoto, M.; Toida, K.; Ishimura, K.; et al. Nucling recruits Apaf-1/pro-caspase-9 complex for the induction of stress-induced apoptosis. J. Biol. Chem. 2004, 279, 41131–41140. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.H.; Sakai, T.; Kim, S.M.; Fukui, K. NF-κB regulates the expression of Nucling, a novel apoptosis regulator, with involvement of proteasome and caspase for its degradation. J. Biochem. 2010, 148, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Willcox, J.K.; Catignani, G.L.; Lazarus, S. Tomatoes and cardiovascular health. Crit. Rev. Food Sci. 2003, 43, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Di Mascio, P.; Kaiser, S.; Sies, H. Lycopene as the most efficient biological carotenoid singlet oxygen quencher. Arch. Biochem. Biophys. 1989, 274, 532–538. [Google Scholar] [CrossRef]
- Stahl, W.; Sies, H. Perspectives in biochemistry and biophysics, lycopene: A biologically important carotenoid in humans? Arch. Biochem. Biophys. 1996, 336, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mayne, S.T. Beta-carotene, carotenoids, and disease prevention in humans. FASEB J. 1996, 10, 690–701. [Google Scholar] [PubMed]
- Gerster, H. The potential role of lycopene for human health. J. Am. Coll. Nutr. 1997, 16, 109–126. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.V.; Agarwal, S. Role of antioxidant lycopene in cancer and heart disease. J. Am. Coll. Nutr. 2000, 19, 563–569. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Song, Y.; Zhang, L. Effect of lycopene supplementation on oxidative stress: An exploratory systematic review and meta-analysis of randomized controlled trials. J. Med. Food 2013, 16, 361–374. [Google Scholar] [CrossRef] [PubMed]
- Gartner, C.; Stahl, W.; Sies, H. Lycopene is more bioavailable from tomato paste than from fresh tomatoes. Am. J. Clin. Nutr. 1997, 66, 116–122. [Google Scholar] [PubMed]
- Srivastava, S.; Srivastava, A.K. Lycopene; chemistry, biosynthesis, metabolism and degradation under various abiotic parameters. J. Food Sci. Technol. 2015, 52, 41–53. [Google Scholar] [CrossRef]
- Wu, A.; Liu, R.; Dai, W.; Jie, Y.; Yu, G.; Fan, X.; Huang, Q. Lycopene attenuates early brain injury and inflammation following subarachnoid hemorrhage in rats. Int. J. Clin. Exp. Med. 2015, 8, 14316–14322. [Google Scholar] [PubMed]
- Khachik, F.; Carvalho, L.; Bernstein, P.S.; Muir, G.J.; Zhao, D.Y.; Katz, N.B. Chemistry, distribution, and metabolism of tomato carotenoids and their impact on human health. Exp. Biol. Med. 2002, 227, 845–851. [Google Scholar] [CrossRef]
- Hsiao, G.; Fong, T.H.; Tzu, N.H.; Lin, K.H.; Chou, D.S.; Sheu, J.R. A potent antioxidant, lycopene, affords neuroprotection against microglia activation and focal cerebral ischemia in rats. In Vivo 2004, 18, 351–356. [Google Scholar] [PubMed]
- Qu, M.; Li, L.; Chen, C.; Li, M.; Pei, L.; Chu, F.; Yang, J.; Yu, Z.; Wang, D.; Zhou, Z. Protective effects of lycopene against amyloid β-induced neurotoxicity in cultured rat cortical neurons. Neurosci. Lett. 2011, 505, 286–290. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Kalonia, H.; Kumar, A. Lycopene modulates nitric oxide pathways against 3-nitropropionic acid-induced neurotoxicity. Life Sci. 2009, 85, 711–718. [Google Scholar] [CrossRef] [PubMed]
- Pema, A.; Janakiraman, U.; Manivasagam, T.; Thenmozhi, A.J. Neuroprotective effect of lycopene against MPTP induced experimental Parkinson’s disease in mice. Neurosci. Lett. 2015, 599, 12–19. [Google Scholar]
- Nunomura, A.; Castellani, R.J.; Zhu, X.; Moreira, P.I.; Perry, G.; Smith, M.A. Involvement of oxidative stress in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2006, 65, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Palozza, P.; Parrone, N.; Simone, R.E.; Catalano, A. Lycopene in atherosclerosis prevention: An integrated scheme of the potential mechanisms of action from cell culture studies. Arch. Biochem. Biophys. 2010, 504, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Jiao, G.; Song, C.; Gu, S.; Brown, R.E.; Zhang, J.; Zhang, P.; Gagnon, J.; Locke, S.; Stefanova, R.; et al. An extract from shrimp processing by-products protects SH-SY5Y cells from neurotoxicity induced by Aβ25–35. Mar. Drugs 2017, 15, 83. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C. Mechanisms of apoptosis. Am. J. Pathol. 2000, 157, 1415–1430. [Google Scholar] [CrossRef]
- Chan, P.H. Mitochondria and neuronal death/survival signaling pathways in cerebral ischemia. Neurochem. Res. 2004, 29, 1943–1949. [Google Scholar] [CrossRef] [PubMed]
- Starkov, A.A. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N. Y. Acad. Sci. 2008, 1147, 37–52. [Google Scholar] [CrossRef] [PubMed]
- Maes, M.; Fišar, Z.; Medina, M.; Scapagnini, G.; Nowak, G.; Berk, M. New drug targets in depression: Inflammatory, cell-mediated immune, oxidative and nitrosative stress, mitochondrial, antioxidant, and neuroprogressive pathways. And new drug candidates—Nrf2 activators and GSK-3 inhibitors. Inflammopharmacology 2012, 20, 127–150. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Hwang, S.W.; Yu, J.H.; Lim, J.W.; Kim, H. Lycopene inhibits regulator of calcineurin 1-mediated apoptosis by reducing oxidative stress and down-regulating Nucling in neuronal cells. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
- Medin, A.C.; Carlsen, M.H.; Andersen, L.F. Associations between reported intakes of carotenoid-rich foods and concentrations of carotenoids in plasma: A validation study of a web-based food recall for children and adolescents. Public Health Nutr. 2016, 19, 3265–3275. [Google Scholar] [CrossRef] [PubMed]

© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hwang, S.; Lim, J.W.; Kim, H. Inhibitory Effect of Lycopene on Amyloid-β-Induced Apoptosis in Neuronal Cells. Nutrients 2017, 9, 883. https://doi.org/10.3390/nu9080883
Hwang S, Lim JW, Kim H. Inhibitory Effect of Lycopene on Amyloid-β-Induced Apoptosis in Neuronal Cells. Nutrients. 2017; 9(8):883. https://doi.org/10.3390/nu9080883
Chicago/Turabian StyleHwang, Sinwoo, Joo Weon Lim, and Hyeyoung Kim. 2017. "Inhibitory Effect of Lycopene on Amyloid-β-Induced Apoptosis in Neuronal Cells" Nutrients 9, no. 8: 883. https://doi.org/10.3390/nu9080883
APA StyleHwang, S., Lim, J. W., & Kim, H. (2017). Inhibitory Effect of Lycopene on Amyloid-β-Induced Apoptosis in Neuronal Cells. Nutrients, 9(8), 883. https://doi.org/10.3390/nu9080883