Antioxidants Mediate Both Iron Homeostasis and Oxidative Stress


Abstract
:1. Overview of Iron Metabolism
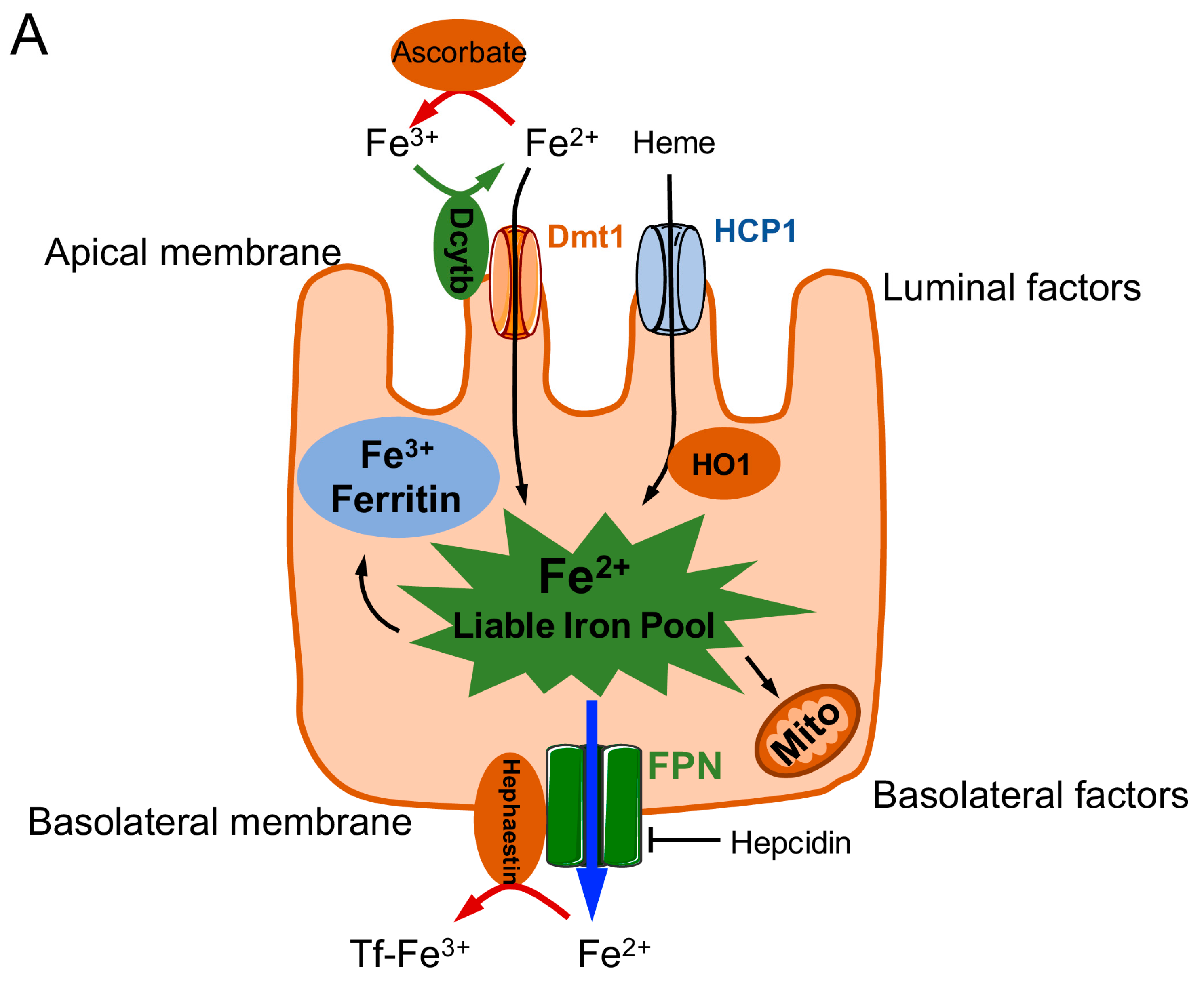
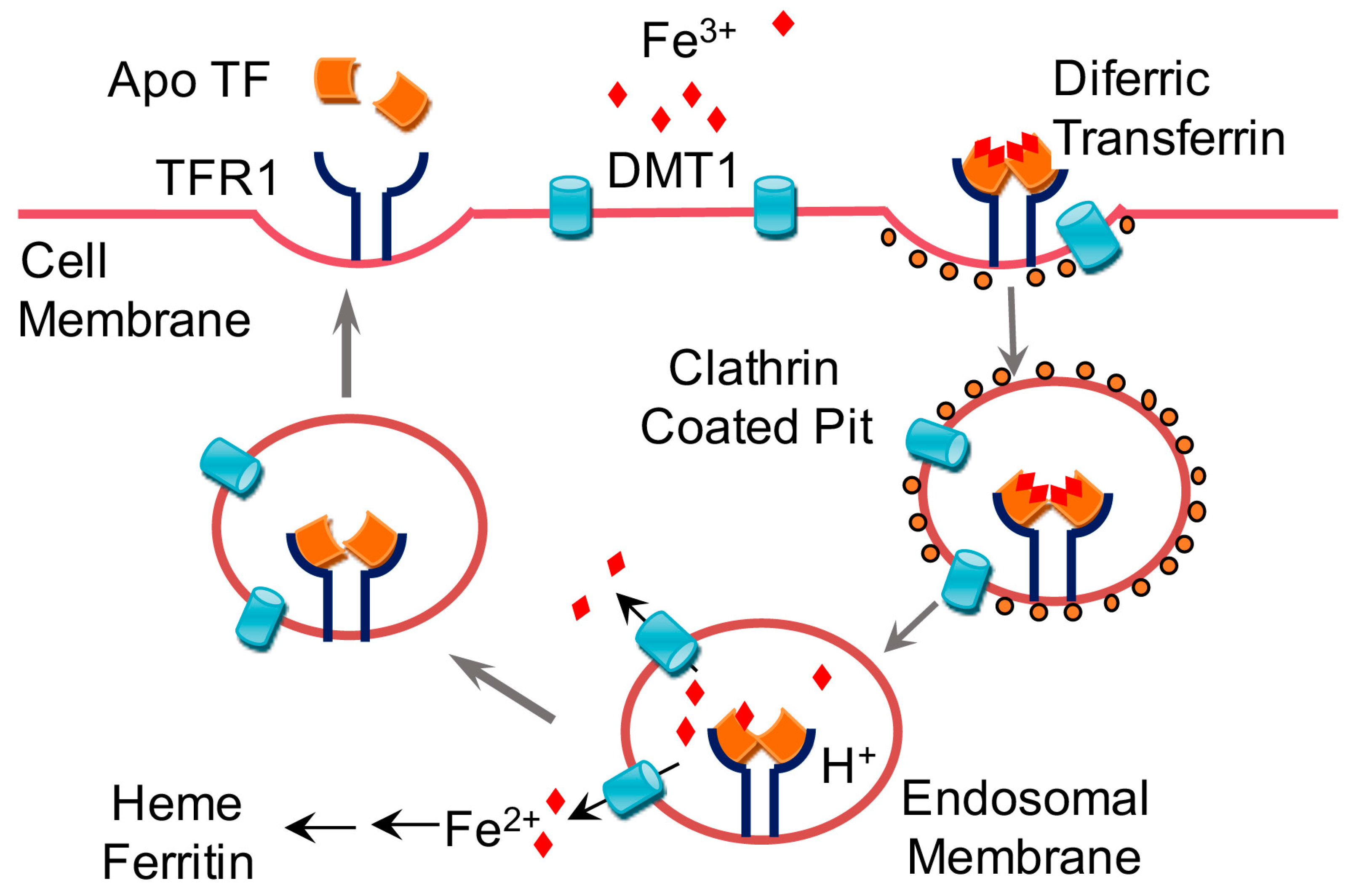
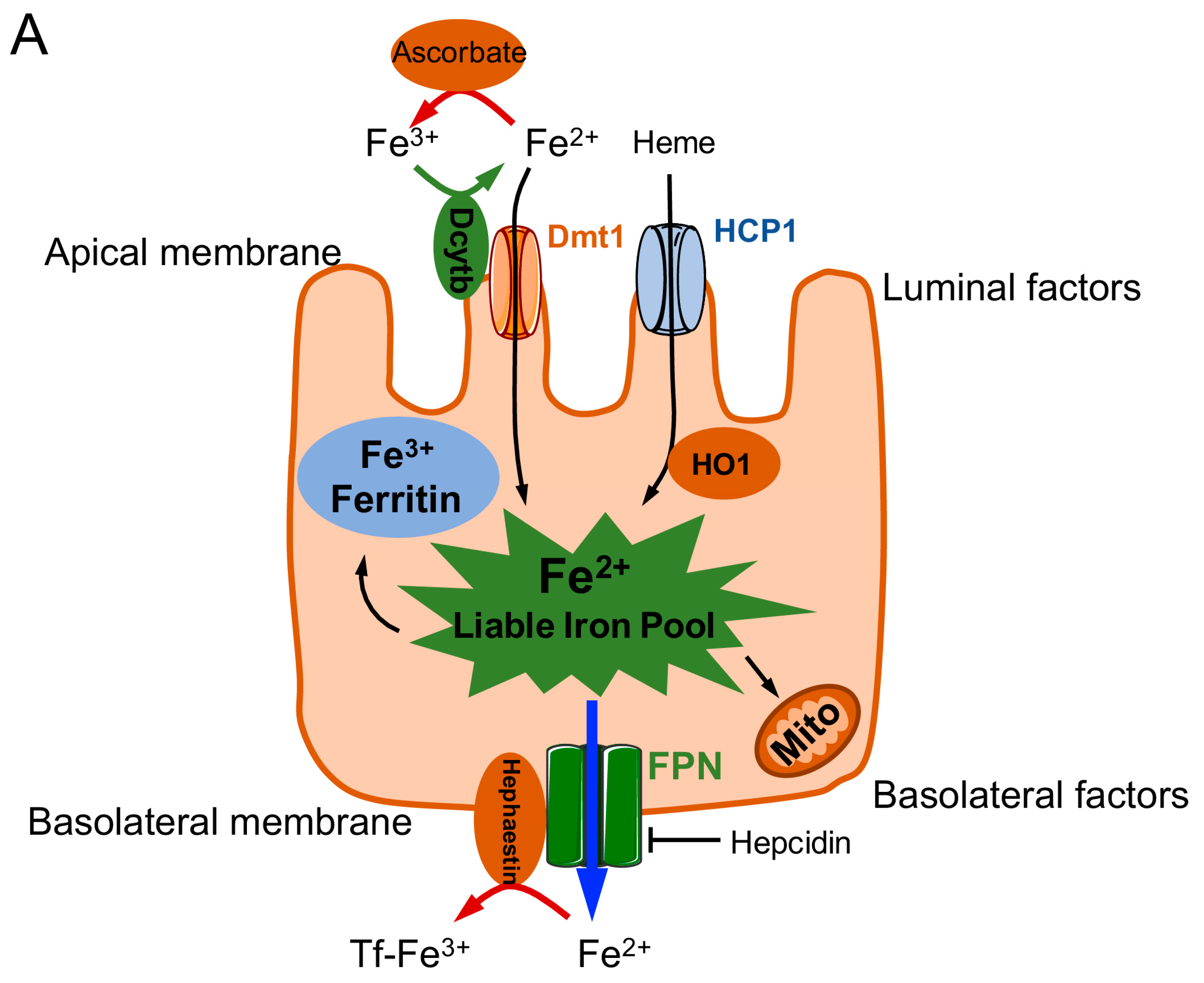
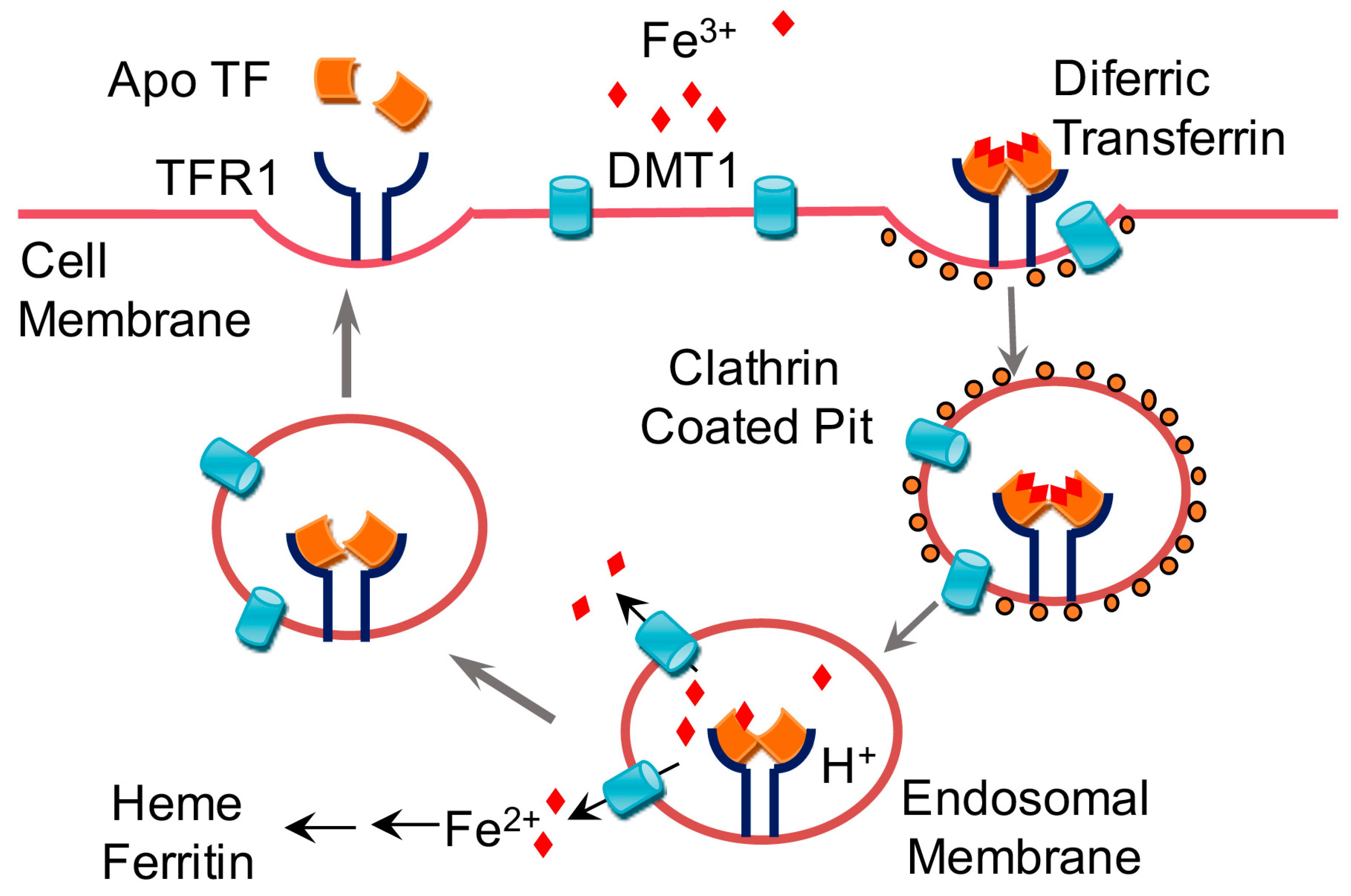
1.1. Dietary Requirements for Iron and Iron Absorption, Transport, Storage, and Utilization
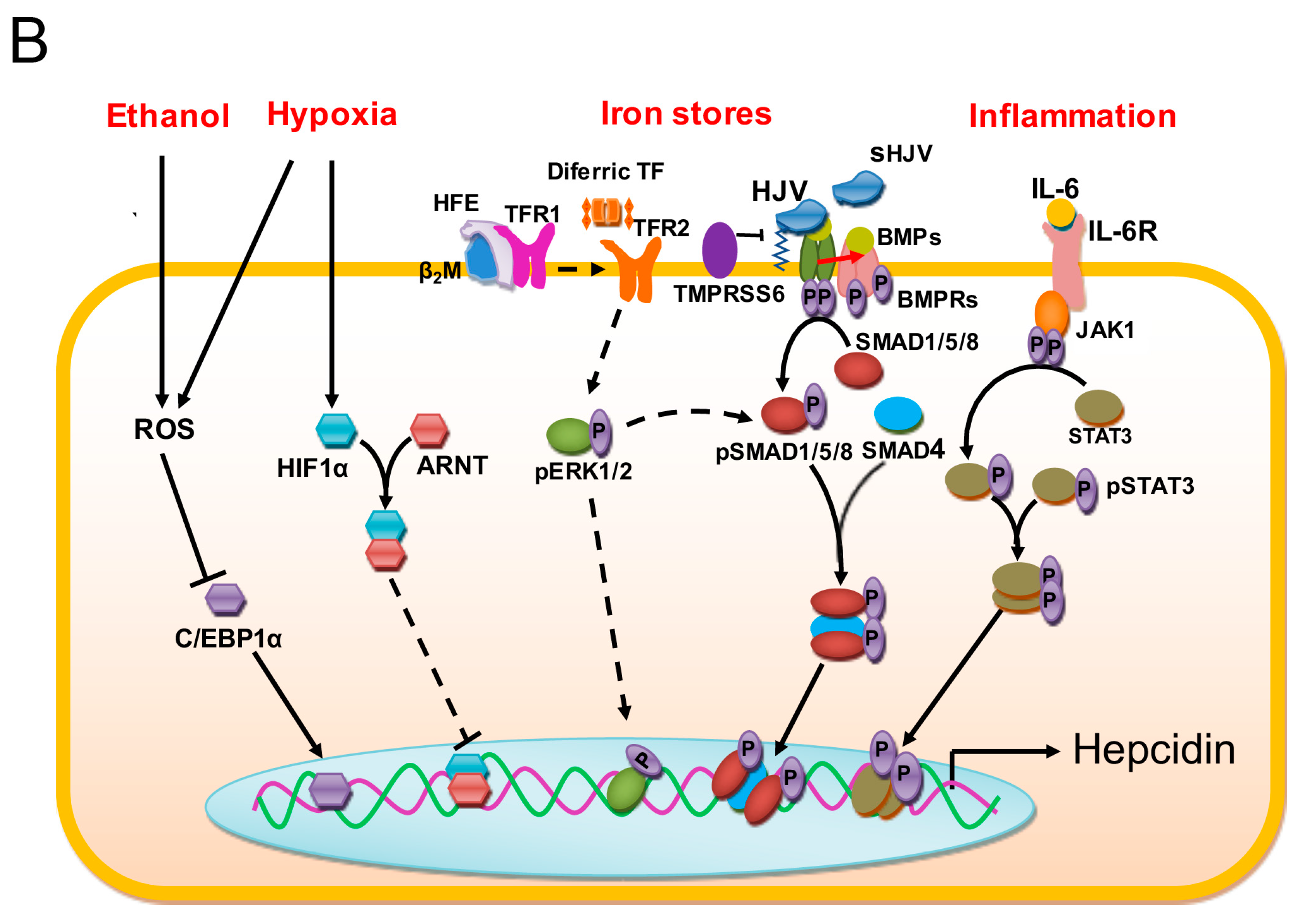
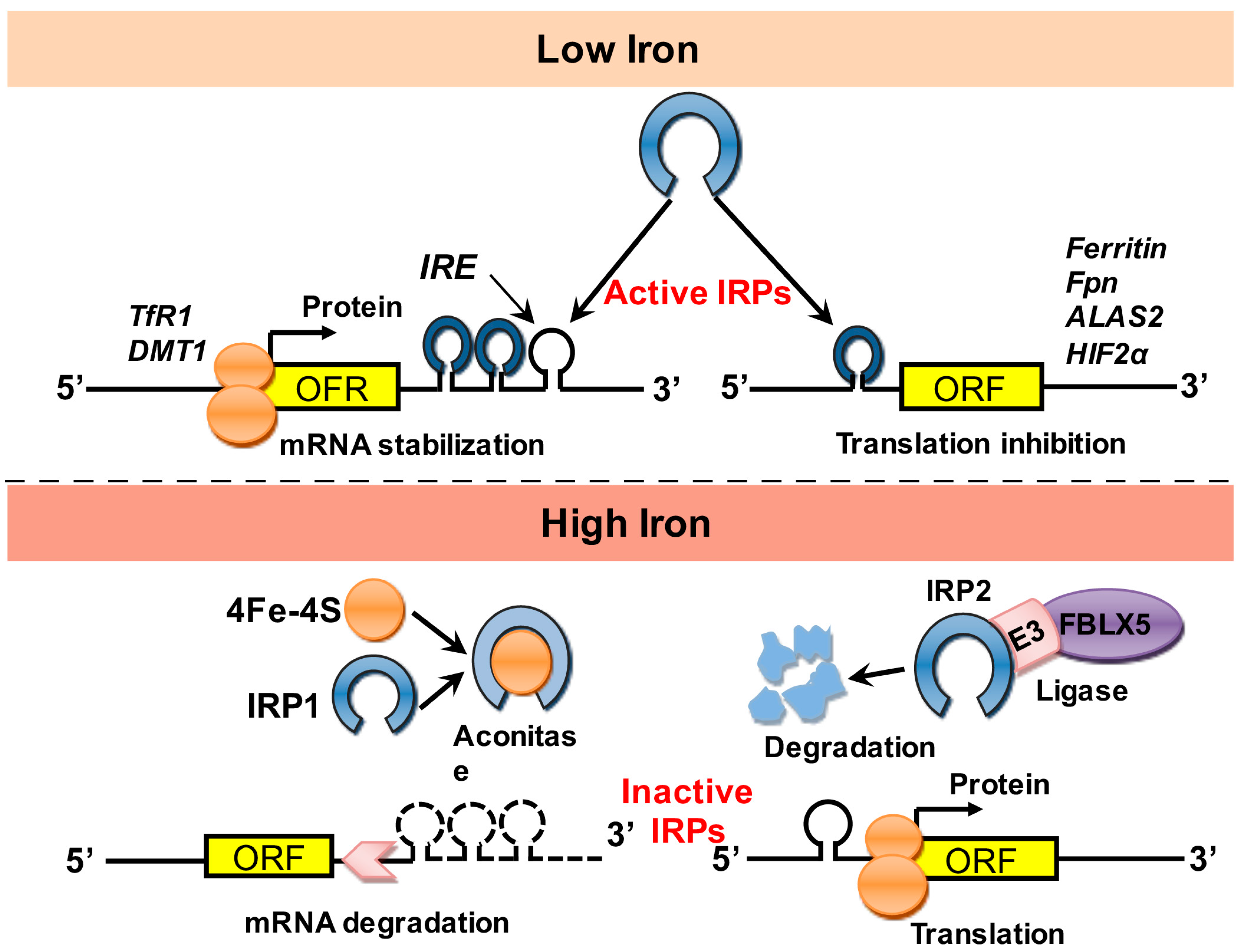
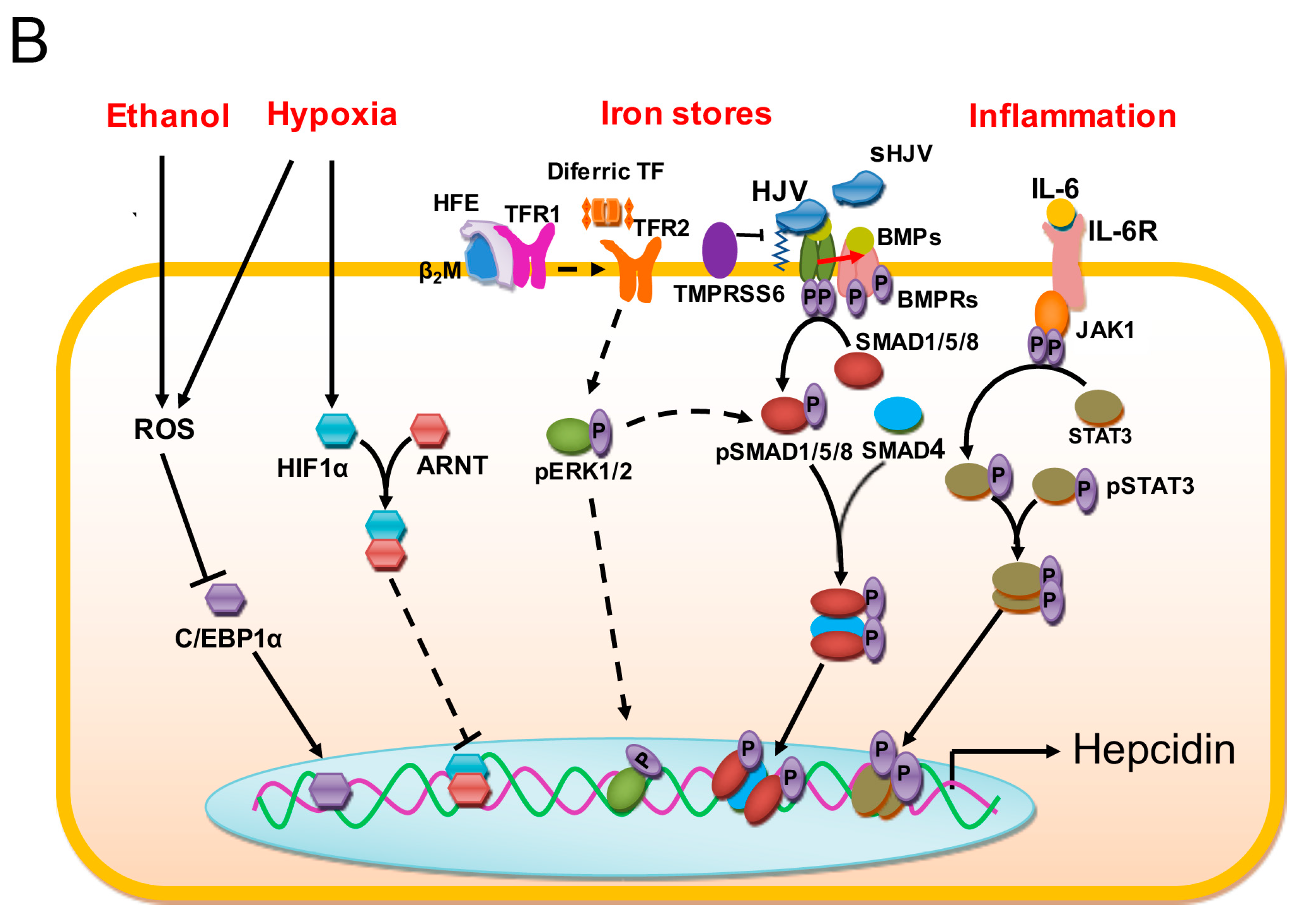
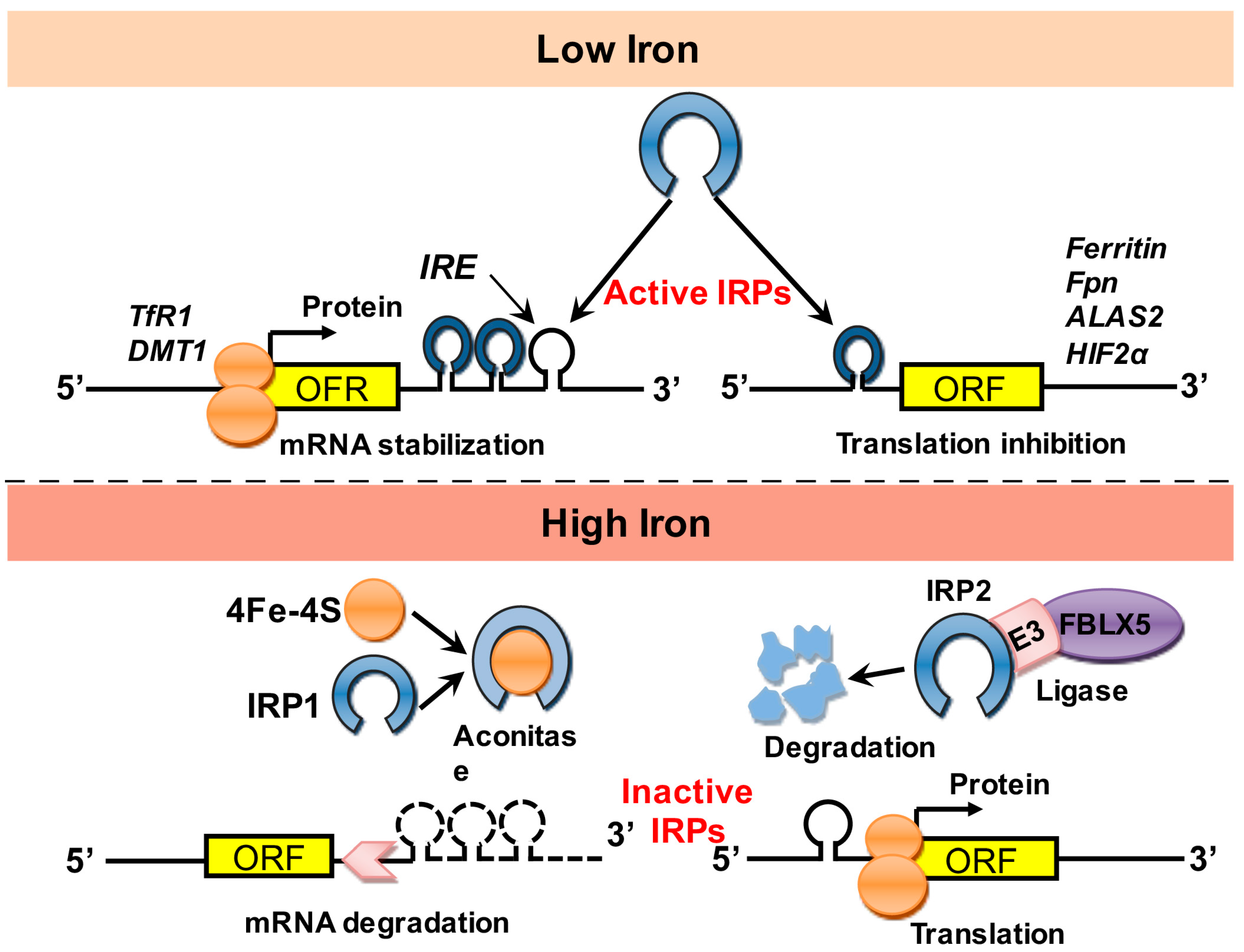
1.2. Regulation of Iron Stores
2. Iron Toxicity, Oxidative Stress, and Antioxidants
2.1. Iron Toxicity
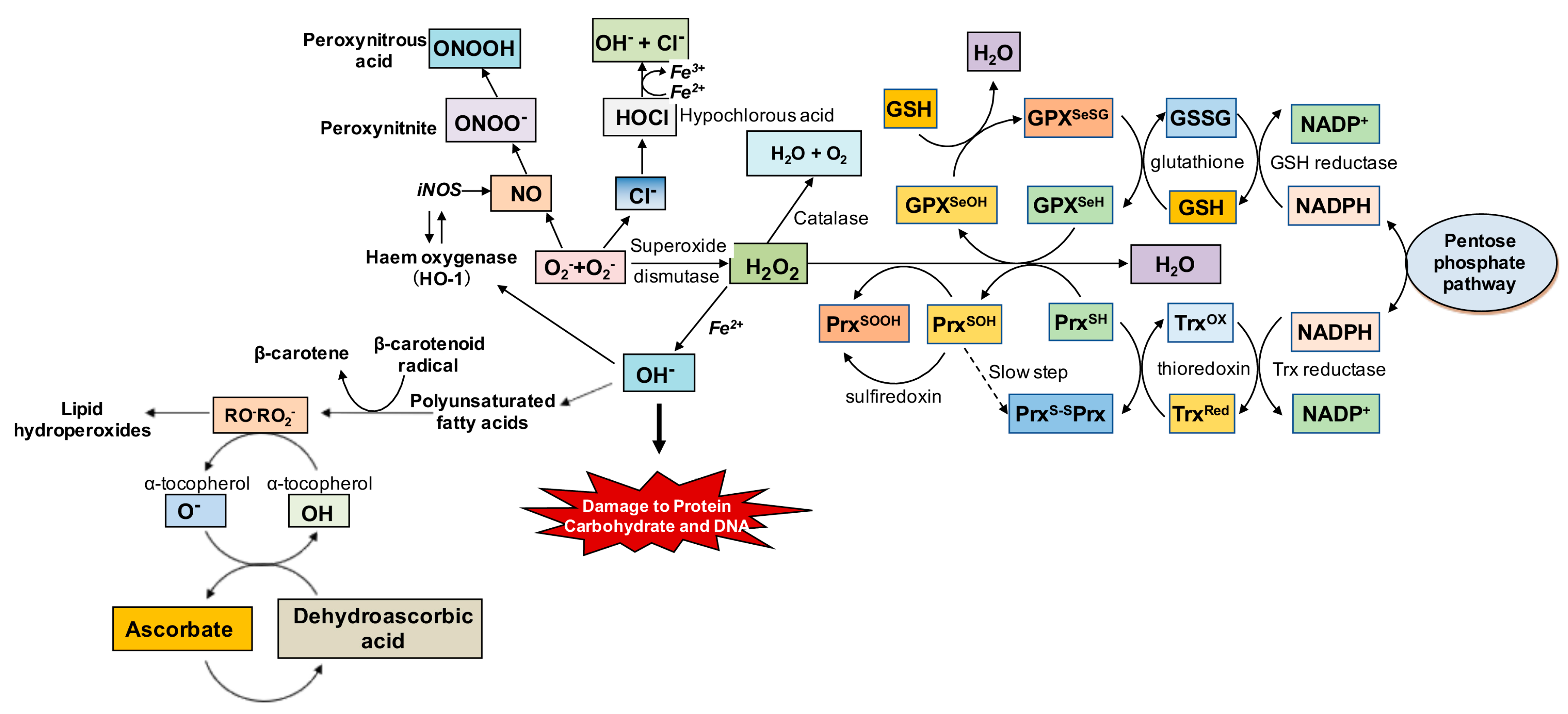
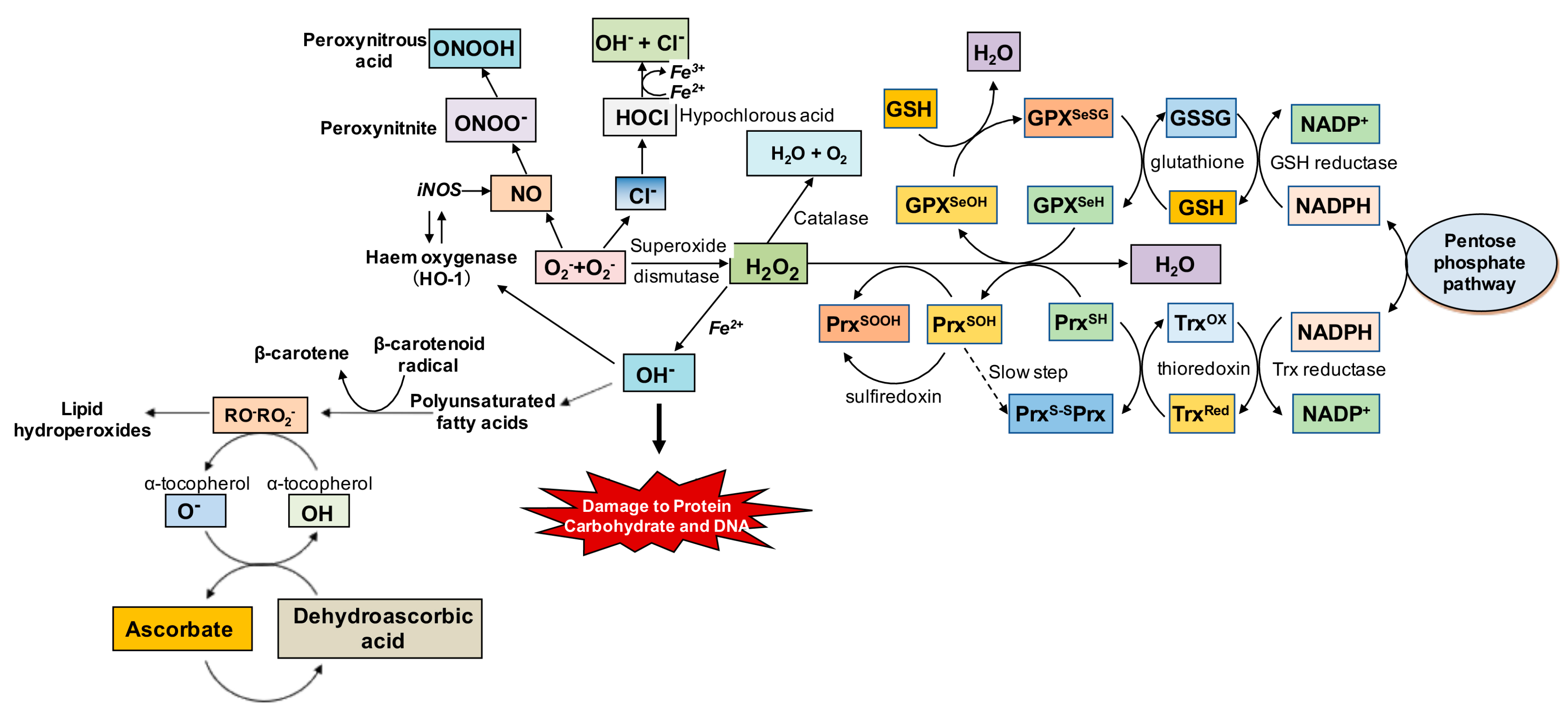
2.2. Oxidative Stress
2.2.1. Oxidative Stress and Iron Deficiency Anemia
2.2.2. Oxidative Stress and Iron Overload
2.2.3. Iron Overload and Cell Death
2.3. Antioxidants
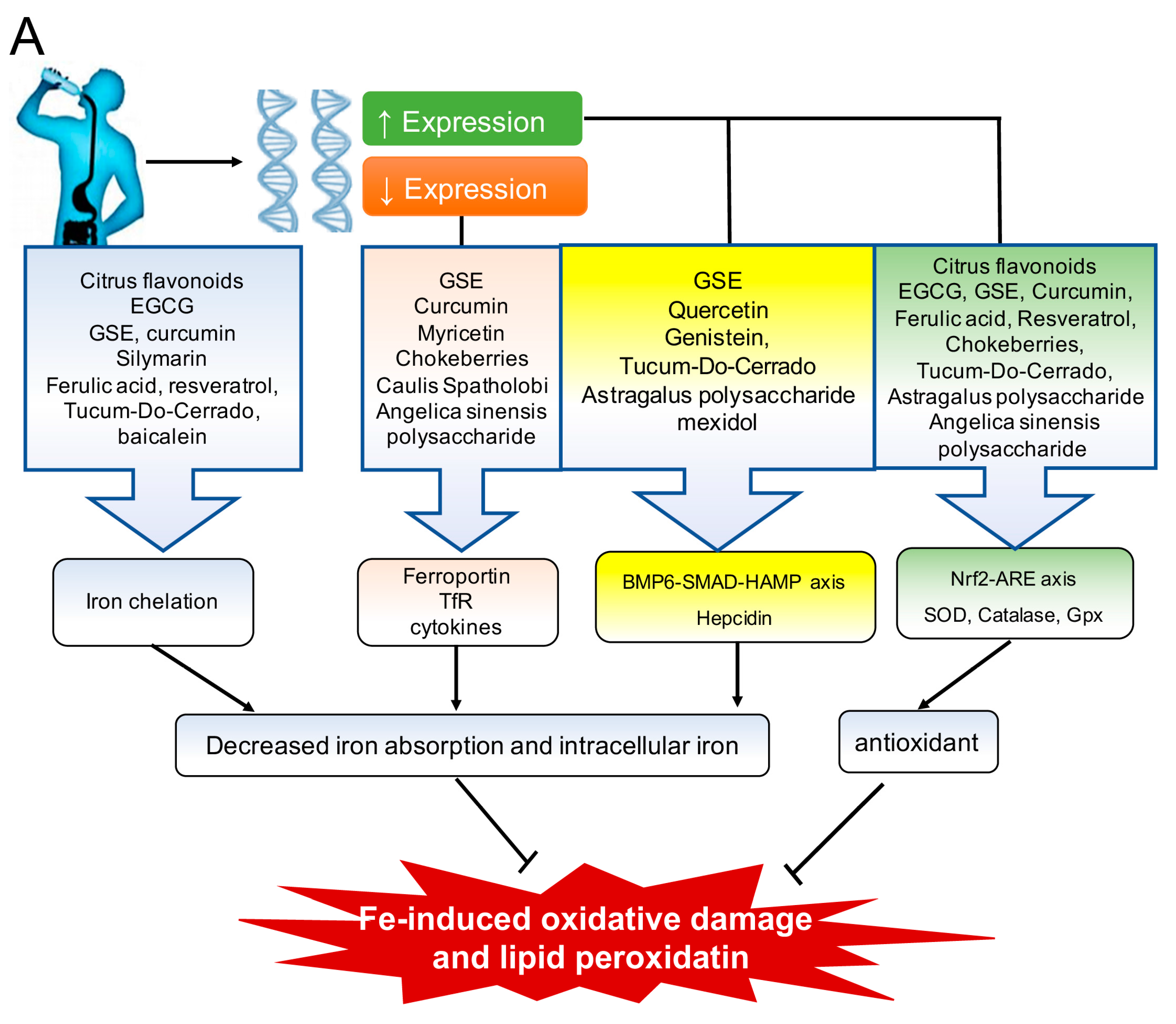
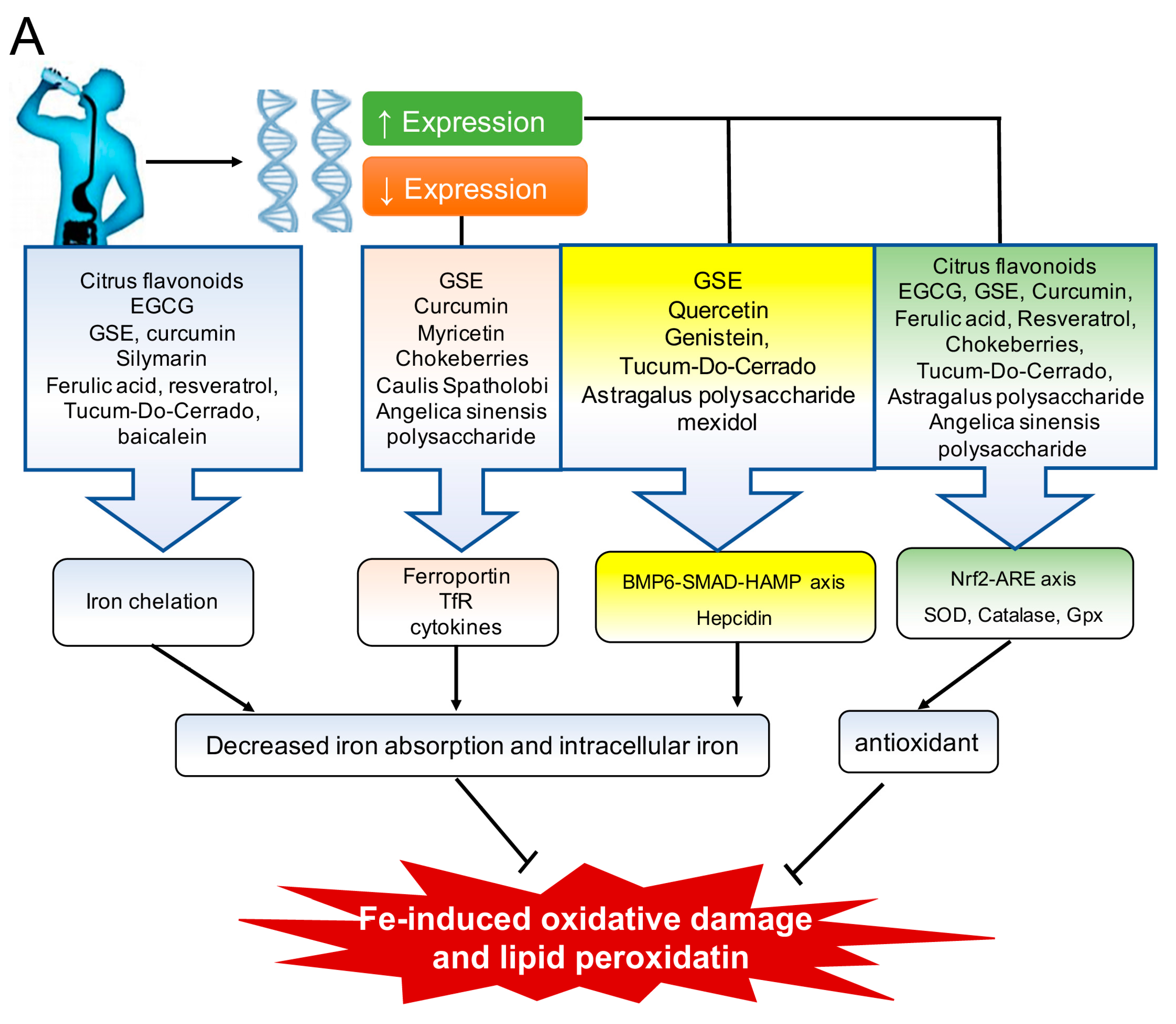
3. Bioactive Compounds That Regulate Oxidative Stress and Iron Metabolism
3.1. Polyphenols
3.1.1. The Flavonoids: Tea Catechins, Black Soy Bean Seed Anthocyanins and Myricetin, Citrus Flavonoids, Grape Seed Extract, Curcumin, Quercetin, Genistein, and Silymarin
3.1.2. Ferulic Acid
3.1.3. Resveratrol
3.1.4. Chokeberries
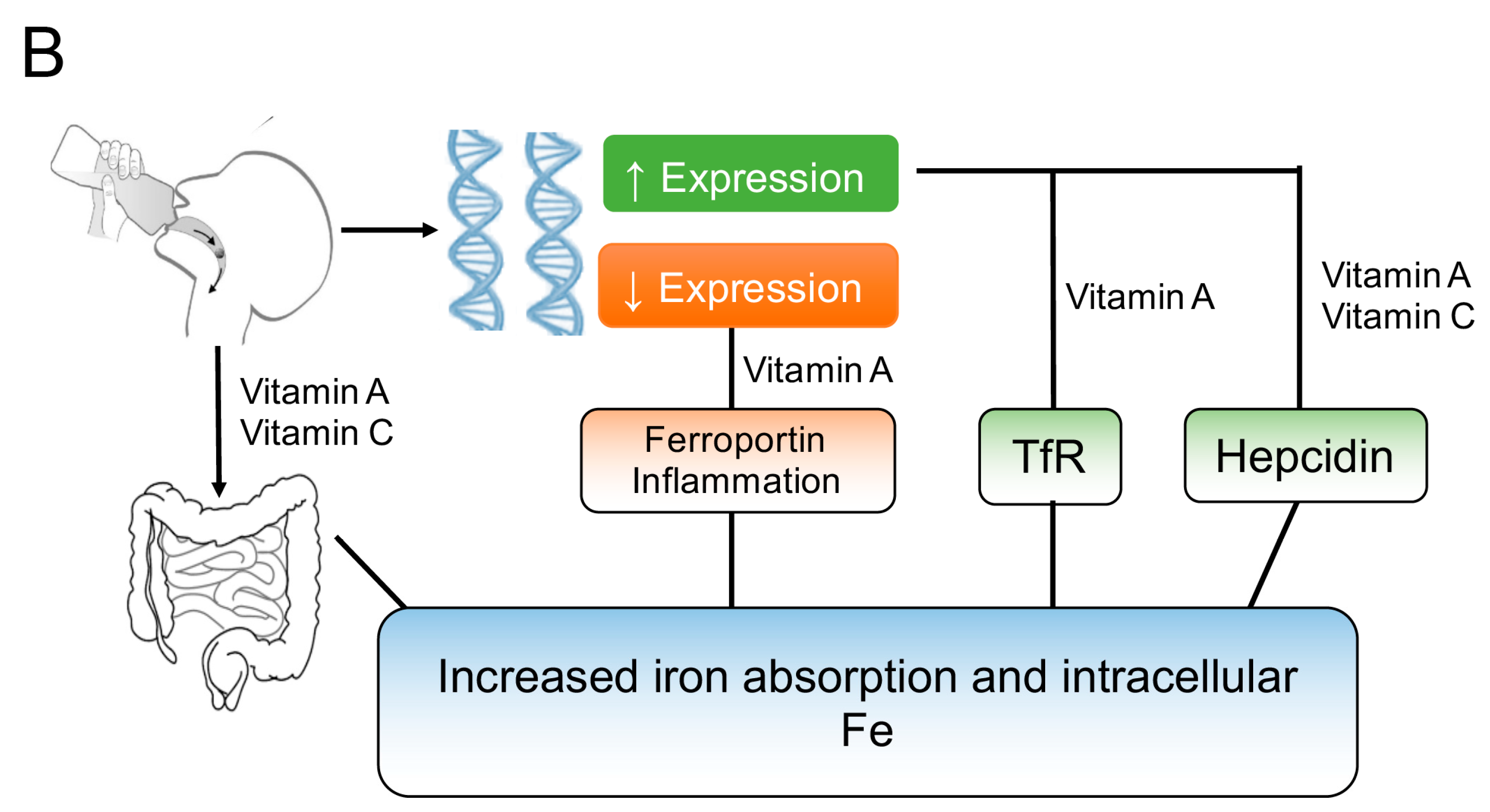
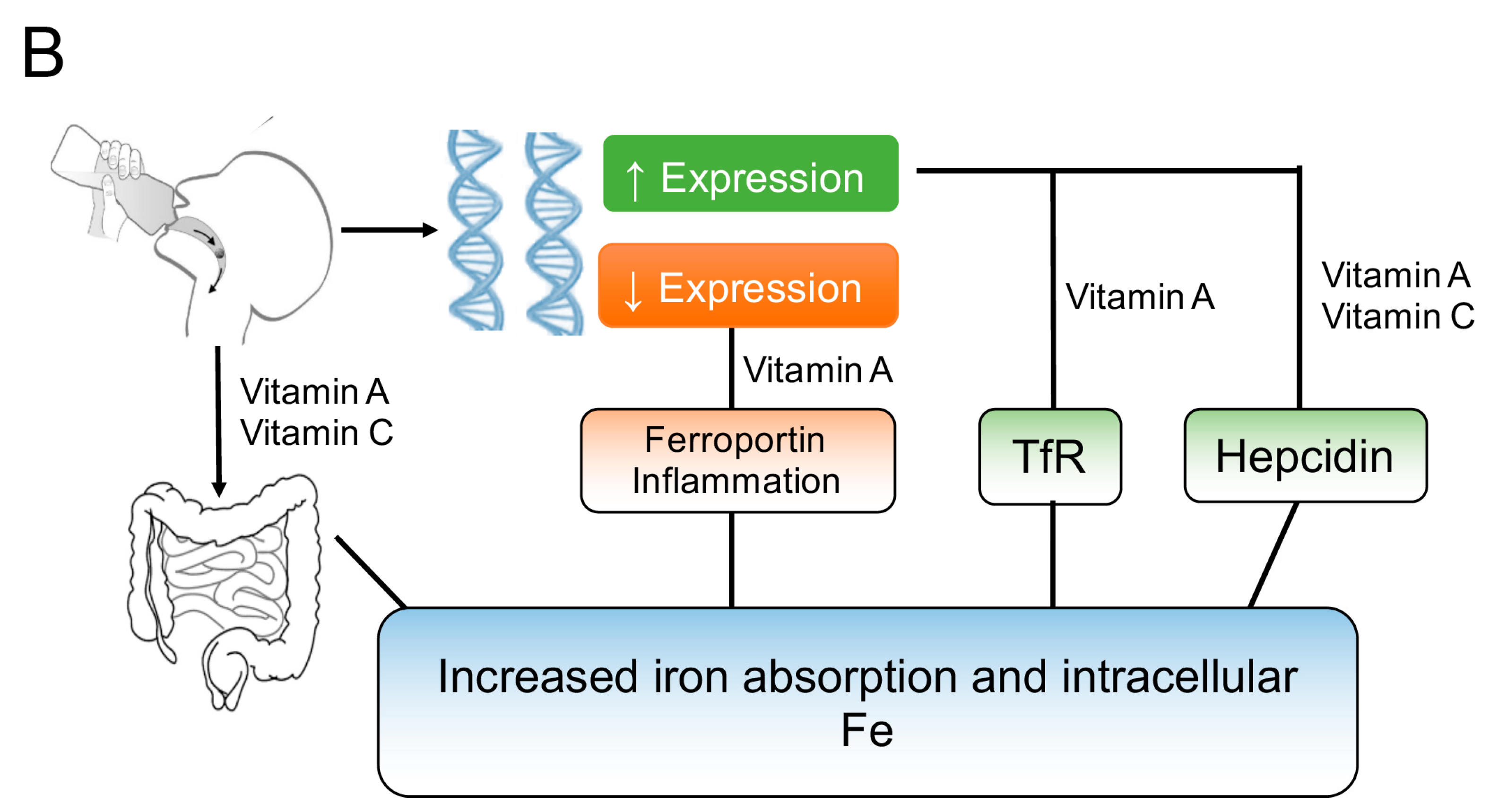
3.2. Vitamin A and Vitamin C
3.3. Other Plant Extracts
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kohgo, Y.; Ikuta, K.; Ohtake, T.; Torimoto, Y.; Kato, J. Body iron metabolism and pathophysiology of iron overload. Int. J. Hematol. 2008, 88, 7–15. [Google Scholar] [CrossRef] [PubMed]
- Beard, J.L.; Dawson, H.; Piñero, D.J. Iron metabolism: A comprehensive review. Nutr. Rev. 1998, 54, 295–317. [Google Scholar] [CrossRef]
- Papanikolaou, G.; Pantopoulos, K. Iron metabolism and toxicity. Toxicol. Appl. Pharm. 2005, 202, 199–211. [Google Scholar] [CrossRef] [PubMed]
- Bresgen, N.; Eckl, P.M. Oxidative stress and the homeodynamics of iron metabolism. Biomolecules 2015, 5, 808–847. [Google Scholar] [CrossRef] [PubMed]
- Emerit, J.; Beaumont, C.; Trivin, F. Iron metabolism, free radicals, and oxidative injury. Biomed. Pharmacother. 2001, 55, 333–339. [Google Scholar] [CrossRef]
- Rouault, T.A.; Tong, W.H. Iron–sulfur cluster biogenesis and human disease. Trends Genet. 2008, 24, 398–407. [Google Scholar] [CrossRef] [PubMed]
- Beinert, H.; Holm, R.H.; Münck, E. Iron-sulfur clusters: Nature’s modular, multipurpose structures. Science 1997, 277, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Oexle, H.; Gnaiger, E.; Weiss, G. Iron-dependent changes in cellular energy metabolism: Influence on citric acid cycle and oxidative phosphorylation. Biochim. Biophys. Acta 1999, 1413, 99–107. [Google Scholar] [CrossRef]
- Gropper, S.S.; Smith, J.L. Advanced Nutrition and Human Metabolism, 6th ed.; Wadsworth: Belmont, CA, USA, 2013; p. 481. [Google Scholar]
- Hambræus, L. Animal-and plant-food-based diets and iron status: Benefits and costs. Proc. Nutr. Soc. 1999, 58, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Martınez-Navarrete, N.; Camacho, M.M.; Martınez-Lahuerta, J.; Martınez-Monzó, J.; Fito, P. Iron deficiency and iron fortified foods—A review. Food Res. Int. 2002, 35, 225–231. [Google Scholar] [CrossRef]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef] [PubMed]
- West, A.P.; Bennett, M.J.; Sellers, V.M.; Andrews, N.C.; Enns, C.A.; Bjorkman, P.J. Comparison of the interactions of transferrin receptor and transferrin receptor 2 with transferrin and the hereditary hemochromatosis protein HFE. J. Biol. Chem. 2000, 275, 38135–38138. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Sheokand, N.; Mhadeshwar, M.A.; Raje, C.I.; Raje, M. Characterization of glyceraldehyde-3-phosphate dehydrogenase as a novel transferrin receptor. Int. J. Biochem. Cell Biol. 2012, 44, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Sheokand, N.; Kumar, S.; Malhotra, H.; Tillu, V.; Raje, C.I.; Raje, M. Secreted glyceraldehye-3-phosphate dehydrogenase is a multifunctional autocrine transferrin receptor for cellular iron acquisition. Biochim. Biophys. Acta 2013, 1830, 3816–3827. [Google Scholar] [CrossRef] [PubMed]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.R.; Merlot, A.M.; Huang, M.L.-H.; Bae, D.-H.; Jansson, P.J.; Sahni, S.; Kalinowski, D.S.; Richardson, D.R. Cellular iron uptake, trafficking and metabolism: Key molecules and mechanisms and their roles in disease. Biochim. Biophys. Acta 2015, 1853, 1130–1144. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Cellular iron: Ferroportin is the only way out. Cell Metab. 2005, 1, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Cook, J.D.; Lipschitz, D.A.; Miles, L.E.; Finch, C.A. Serum ferritin as a measure of iron stores in normal subjects. Am. J. Clin. Nutr. 1974, 27, 681–687. [Google Scholar] [PubMed]
- Theil, E.C.; Elizabeth, C. Ferritin protein nanocages—The story. Nanotechnol. Percept. 2012, 8, 7–16. [Google Scholar] [CrossRef] [PubMed]
- Theil, E.C. Ferritin: structure, gene regulation, and cellular function in animals, plants, and microorganisms. Annu. Rev. Biochem. 1987, 56, 289–315. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Mikhael, M.; Xu, D.; Li, Y.; Soe-Lin, S.; Ning, B.; Ponka, P. Lysosomal proteolysis is the primary degradation pathway for cytosolic ferritin and cytosolic ferritin degradation is necessary for iron exit. Antioxid. Redox Signal. 2010, 13, 999–1009. [Google Scholar] [CrossRef] [PubMed]
- Roeser, H.P.; Lee, G.R.; Nacht, S.; Cartwright, G.E. The role of ceruloplasmin in iron metabolism. J. Clin. Investig. 1970, 49, 2408. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Ganz, T. Regulation of iron metabolism by hepcidin. Annu. Rev. Nutr. 2006, 26, 323–342. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T.; Nemeth, E. Hepcidin and iron homeostasis. Biochim. Biophys. Acta 2012, 1823, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.J. Regulation of iron metabolism by hepcidin under conditions of inflammation. J. Biol. Chem. 2015, 290, 18975–18983. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Hepcidin, a key regulator of iron metabolism and mediator of anemia of inflammation. Blood 2003, 102, 783–788. [Google Scholar] [CrossRef] [PubMed]
- Pantopoulos, K. Iron metabolism and the IRE/IRP regulatory system: An update. Ann. N. Y. Acad. Sci. 2004, 1012, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Laurent, S.; Saei, A.A.; Behzadi, S.; Panahifar, A.; Mahmoudi, M. Superparamagnetic iron oxide nanoparticles for delivery of therapeutic agents: Opportunities and challenges. Expert Opin. Drug Deliv. 2014, 11, 1449–1470. [Google Scholar] [CrossRef] [PubMed]
- Cabantchik, Z.I.; Kakhlon, O.; Epsztejn, S.; Zanninelli, G.; Breuer, W. Intracellular and extracellular labile iron pools. Adv. Exp. Med. Biol. 2002, 509, 55–75. [Google Scholar] [PubMed]
- Finney, L.A.; O’Halloran, T.V. Transition metal speciation in the cell: Insights from the chemistry of metal ion receptors. Science 2003, 300, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Philpott, C.C.; Ryu, M.S. Special delivery: Distributing iron in the cytosol of mammalian cells. Front. Pharmacol. 2014, 5, 173. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Free radicals, antioxidants, and human disease curiosity, cause, or consequence? Lancet 1994, 344, 721–724. [Google Scholar] [CrossRef]
- Valko, M.; Leibfritz, D.; Moncol, J.; Cronin, M.T.D.; Mazur, M.; Telser, J. Free radicals and antioxidants in normal physiological functions and human disease. Int. J. Biochem. Cell Biol. 2007, 39, 44–84. [Google Scholar] [CrossRef] [PubMed]
- Mylonas, C.; Kouretas, D. Lipid peroxidation and tissue damage. In Vivo (Athens, Greece) 1999, 13, 295–309. [Google Scholar]
- Crichton, R.R.; Wilmet, S.; Legssyer, R.; Ward, R.J. Molecular and cellular mechanisms of iron homeostasis and toxicity in mammalian cells. J. Inorg. Biochem. 2002, 91, 9–18. [Google Scholar] [CrossRef]
- Camaschella, C. Iron deficiency anemia. N. Engl. J. Med. 2015, 372, 1832–1843. [Google Scholar] [CrossRef] [PubMed]
- Leung, A.K.; Chan, K.W. Iron deficiency anemia. Adv. Pediatr. 2000, 48, 385–408. [Google Scholar]
- Tsai, S.F.; Chen, S.J.; Yen, H.J.; Hung, G.Y.; Tsao, P.C.; Jeng, M.J.; Lee, Y.S.; Soong, W.J.; Tang, R.B. Iron deficiency anemia in predominantly breastfed young children. Pediatr. Neonatol. 2014, 55, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Allen, L.H. Anemia and iron deficiency: Effects on pregnancy outcome. Am. J. Clin. Nutr. 2000, 71, 1280s–1284s. [Google Scholar] [PubMed]
- Cassat, J.E.; Skaar, E.P. Iron in infection and immunity. Cell Host Microbe 2013, 13, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Grune, T.; Sommerburg, O.; Siems, W.G. Oxidative stress in anemia. Clin. Nephrol. 2000, 53, S18–S22. [Google Scholar] [PubMed]
- Fibach, E.; Rachmilewitz, E. The role of oxidative stress in hemolytic anemia. Curr. Mol. Med. 2008, 8, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Merryweather-Clarke, A.T.; Cadet, E.; Bomford, A.; Capron, D.; Viprakasit, V.; Miller, A.; Livesey, K.J. Digenic inheritance of mutations in HAMP and HFE results in different types of haemochromatosis. Hum. Mol. Genet. 2003, 12, 2241–2247. [Google Scholar] [CrossRef] [PubMed]
- Kruszewski, M. Labile iron pool: The main determinant of cellular response to oxidative stress. Mutat. Res. 2003, 531, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Lunova, M.; Goehring, C.; Kuscuoglu, D.; Mueller, K.; Chen, Y.; Walther, P.; Deschemin, J.C.; Vaulont, S.; Haybaeck, J.; Lackner, C.; et al. Hepcidin knockout mice fed with iron-rich diet develop chronic liver injury and liver fibrosis due to lysosomal iron overload. J. Hepatol. 2014, 61, 633–641. [Google Scholar] [CrossRef] [PubMed]
- Fortes, G.B.; Alves, L.S.; de Oliveira, R.; Dutra, F.F.; Rodrigues, D.; Fernandez, P.L.; Souto-Padron, T.; De Rosa, M.J.; Kelliher, M.; Golenbock, D.; et al. Heme induces programmed necrosis on macrophages through autocrine TNF and ROS production. Blood 2012, 119, 2368–2375. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Perez, C.; Roy, S.S.; Naghdi, S.; Lin, X.; Davies, E.; Hajnoczky, G. Bid-induced mitochondrial membrane permeabilization waves propagated by local reactive oxygen species (ROS) signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 4497–4502. [Google Scholar] [CrossRef] [PubMed]
- Dixon, S.J.; Lemberg, K.M.; Lamprecht, M.R.; Skouta, R.; Zaitsev, E.M.; Gleason, C.E.; Patel, D.N.; Bauer, A.J.; Cantley, A.M.; Yang, W.S.; et al. Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 2012, 149, 1060–1072. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; SriRamaratnam, R.; Welsch, M.E.; Shimada, K.; Skouta, R.; Viswanathan, V.S.; Cheah, J.H.; Clemons, P.A.; Shamji, A.F.; Clish, C.B.; et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014, 156, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Shimada, K.; Hayano, M.; Pagano, N.C.; Stockwell, B.R. Cell-line selectivity improves the predictive power of pharmacogenomicanalyses and helps identify NADPH as biomarker for ferroptosissensitivity. Cell Chem. Biol. 2016, 23, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Jiang, L.; Kon, N.; Li, T.; Wang, S.J.; Su, T.; Hibshoosh, H.; Baer, R.; Gu, W. Ferroptosis as a p53-mediated activity during tumour suppression. Nature 2015, 520, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; An, P.; Xie, E.; Wu, Q.; Fang, X.; Gao, H.; Zhang, Z.; Li, Y.; Wang, X.; Zhang, J.; et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology 2017. [Google Scholar] [CrossRef] [PubMed]
- Kwon, M.Y.; Park, E.; Lee, S.J.; Chung, S.W. Heme oxygenase-1 accelerates erastin-induced ferroptotic cell death. Oncotarget 2015, 6, 24393–24403. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Monian, P.; Quadri, N.; Ramasamy, R.; Jiang, X. Glutaminolysis and transferrin regulate ferroptosis. Mol. Cell 2015, 59, 298–308. [Google Scholar] [CrossRef] [PubMed]
- Hou, W.; Xie, Y.; Song, X.; Sun, X.; Lotze, M.T.; Zeh, H.J.; Kang, R.; Tang, D. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 2016, 12, 1425–1428. [Google Scholar] [CrossRef] [PubMed]
- Zelko, I.N.; Mariani, T.J.; Folz, R.J. Superoxide dismutase multigene family: a comparison of the CuZn-SOD (SOD1), Mn-SOD (SOD2), and EC-SOD (SOD3) gene structures, evolution, and expression. Free Radic. Biol. Med. 2002, 33, 337–349. [Google Scholar] [CrossRef]
- Wu, G.; Fang, Y.Z.; Yang, S.; Lupton, J.R.; Turner, N.D. Glutathione metabolism and its implications for health. J. Nutr. 2004, 134, 489–492. [Google Scholar] [PubMed]
- Chae, H.Z.; Kim, H.J.; Kang, S.W.; Rhee, S.G. Characterization of three isoforms of mammalian peroxiredoxin that reduce peroxides in the presence of thioredoxin. Diabetes Res. Clin. Pract. 1999, 45, 101–112. [Google Scholar] [CrossRef]
- Willcox, J.K.; Ash, S.L.; Catignani, G.L. Antioxidants and Prevention of Chronic Disease. Crit. Rev. Food Sci. Nutr. 2010, 4, 275–295. [Google Scholar] [CrossRef] [PubMed]
- Temple, N.J. Antioxidants and disease: More questions than answers. Nutr. Res. 2000, 20, 449–459. [Google Scholar] [CrossRef]
- Sripetchwandee, J.; Pipatpiboon, N.; Chattipakorn, N.; Chattipakorn, S. Combined therapy of iron chelator and antioxidant completely restores brain dysfunction induced by iron toxicity. PLoS ONE 2014, 9, e85115. [Google Scholar] [CrossRef] [PubMed]
- Wongjaikam, S.; Kumfu, S.; Khamseekaew, J.; Sripetchwandee, J.; Srichairatanakool, S.; Fucharoen, S.; Chattipakorn, N. Combined iron chelator and antioxidant exerted greater efficacy on cardioprotection than monotherapy in iron-overloaded rats. PLoS ONE 2016, 11, e0159414. [Google Scholar] [CrossRef] [PubMed]
- Galati, G.; O’brien, P.J. Potential toxicity of flavonoids and other dietary phenolics: Significance for their chemopreventive and anticancer properties. Free Radic. Biol. Med. 2004, 37, 287–303. [Google Scholar] [CrossRef] [PubMed]
- Ferlazzo, N.; Visalli, G.; Cirmi, S.; Lombardo, G.E.; Laganà, P.; Di Pietro, A.; Navarra, M. Natural iron chelators: Protective role in A549 cells of flavonoids-rich extracts of Citrus juices in Fe3+—Induced oxidative stress. Environ. Toxicol. Pharmacol. 2016, 43, 248–256. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Kim, E.Y.; Lindsay, E.A.; Han, O. Bioactive dietary polyphenols inhibit heme iron absorption in a dose-dependent manner in human intestinal Caco-2 Cells. J. Food Sci. 2011, 76, H143–H150. [Google Scholar] [CrossRef] [PubMed]
- Shin, J.H.; Jeon, H.J.; Park, J.; Chang, M.S. Epigallocatechin-3-gallate prevents oxidative stress-induced cellular senescence in human mesenchymal stem cells via Nrf2. Int. J. Mol. Med. 2016, 38, 1075–1082. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Wilkinson, J.; Di, X.; Wang, W.; Hatcher, H.; Kock, N.D.; Torti, S.V. Curcumin, a cancer chemopreventive and chemotherapeutic agent, is a biologically active iron chelator. Blood 2009, 113, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Qian, K.; Xiong, J.; Ma, K.; Wang, A.; Zou, Y. Curcumin alleviates lipopolysaccharide induced sepsis and liver failure by suppression of oxidative stress-related inflammation via PI3K/AKT and NF-κB related signaling. Biomed. Pharmacother. 2016, 83, 302–313. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Li, Y.; Yu, H.; Gao, C.; Liu, L.; Chen, S.; Yao, P. Quercetin prevents ethanol-induced iron overload by regulating hepcidin through the BMP6/SMAD4 signaling pathway. J. Nutr. Biochem. 2014, 25, 675–682. [Google Scholar] [CrossRef] [PubMed]
- Mu, M.; An, P.; Wu, Q.; Shen, X.; Shao, D.; Wang, H.; Wang, F. The dietary flavonoid myricetin regulates iron homeostasis by suppressing hepcidin expression. J. Nutr. Biochem. 2016, 30, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Zhen, A.W.; Nguyen, N.H.; Gibert, Y.; Motola, S.; Buckett, P.; Wessling-Resnick, M.; Fraenkel, P.G. The small molecule, genistein, increases hepcidin expression in human hepatocytes. Hepatology 2013, 58, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- Moayedi Esfahani, B.A.; Reisi, N.; Mirmoghtadaei, M. Evaluating the safety and efficacy of silymarin in β-thalassemia patients: A review. Hemoglobin 2015, 39, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; He, H.; Zhang, Z.; Liao, Z.; Yin, D.; Liu, D.; He, M. Long-term sodium ferulate supplementation scavenges oxygen radicals and reverses liver damage induced by iron overloading. Molecules 2016, 21, 1219. [Google Scholar] [CrossRef] [PubMed]
- Das, S.K.; Wang, W.; Zhabyeyev, P.; Basu, R.; McLean, B.; Fan, D.; Dyck, J.R. Iron-overload injury and cardiomyopathy in acquired and genetic models is attenuated by resveratrol therapy. Sci. Rep. 2014, 5, 18132. [Google Scholar] [CrossRef] [PubMed]
- Skarpańska-Stejnborn, A.; Basta, P.; Sadowska, J.; Pilaczyńska-Szczeńniak, Ł. Effect of supplementation with chokeberry juice on the inflammatory status and markers of iron metabolism in rowers. J. Int. Soc. Sports Nutr. 2014, 11, 48. [Google Scholar] [CrossRef] [PubMed]
- García-Casal, M.N.; Layrisse, M.; Solano, L.; Barón, M.A.; Arguello, F.; Llovera, D.; Ramírez, J.; Leets, I.; Tropper, E. Vitamin A and β-carotene can improve nonheme iron absorption from rice, wheat and corn by humans. J. Nutr. 1997, 128, 646–650. [Google Scholar]
- Citelli, M.; Bittencourt, L.L.; Da Silva, S.V.; Pierucci, A.P.; Pedrosa, C. Vitamin A modulates the expression of genes involved in iron bioavailability. Biol. Trace Elem. Res. 2012, 149, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Katz, O.; Reifen, R.; Lerner, A. β-Carotene can reverse dysregulation of iron protein in an in vitro model of inflammation. Immunol. Res. 2015, 61, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Chiu, P.F.; Ko, S.Y.; Chang, C.C. Vitamin C affects the expression of hepcidin and erythropoietin receptor in HepG2 cells. J. Ren. Nutr. 2012, 22, 373–376. [Google Scholar] [CrossRef] [PubMed]
- Fustinoni-Reis, A.M.; Arruda, S.F.; Dourado, L.P.; da Cunha, M.S.; Siqueira, E. Tucum-Do-Cerrado (Bactrissetosa Mart.) consumption modulates iron homeostasis and prevents iron-induced oxidative stress in the rat liver. Nutrients 2016, 8, 38. [Google Scholar] [CrossRef] [PubMed]
- Perez, C.A.; Wei, Y.; Guo, M. Iron-binding and anti-Fenton properties of baicalein and baicalin. J. Inorg. Biochem. 2009, 103, 326–332. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.; An, P.; Zhang, Z.; Zhang, F.; Yu, Y.; Wu, Q.; Wang, F. Screening identifies the Chinese medicinal plant Caulis Spatholobi as an effective HAMP expression inhibitor. J. Nutr. 2013, 143, 1061–1066. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Gu, J.Y.; Chen, Z.S.; Xing, K.C.; Sun, B. Astragalus polysaccharide suppresses palmitate-induced apoptosis in human cardiac myocytes: The role of Nrf1 and antioxidant response. Int. J. Clin. Exp. Pathol. 2015, 8, 2515. [Google Scholar] [PubMed]
- Ren, F.; Qian, X.H.; Qian, X.L. Astragalus polysaccharide upregulateshepcidin and reduces iron overload in mice via activation of p38 mitogen-activated protein kinase. Biochem. Biophys. Res. Commun. 2016, 472, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, C.; Xu, N.W.; Gao, G.M.; Ni, S.; Miao, K.S.; Li, C.K.; Xie, H.G. Polysaccharide from Angelica sinensis protects chondrocytes from H2O2-induced apoptosis through its antioxidant effects in vitro. Int. J. Biol. Macromol. 2016, 87, 322–328. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Zhou, J.; Li, Q.; Liu, Y.; Wang, K.; Zhang, Y. The effects of polysaccharides from the root of Angelica sinensis on tumor growth and iron metabolism in H22-bearing mice. Food Funct. 2016, 7, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Scherbinina, S.P.; Levina, A.A.; Lisovskaya, I.L.; Ataullakhanov, F.I. Effect of exogenous antioxidants on erythrocyte redox status and hepcidin content in disorders of iron metabolism regulation. Biochemistry 2012, 6, 338–342. [Google Scholar] [CrossRef]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef]
- Vauzour, D.; Rodriguez-Mateos, A.; Corona, G.; Oruna-Concha, M.J.; Spencer, J.P. Polyphenols and human health: prevention of disease and mechanisms of action. Nutrients 2010, 2, 1106–1131. [Google Scholar] [CrossRef] [PubMed]
- Pandey, K.B.; Rizvi, S.I. Plant polyphenols as dietary antioxidants in human health and disease. Oxid. Med. Cell. Longev. 2009, 2, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Niu, Q.; Mu, L.; Li, S.; Xu, S.; Ma, R.; Guo, S. Proanthocyanidin protects human embryo hepatocytes from fluoride-induced oxidative stress by regulating iron metabolism. Biol. Trace Elem. Res. 2016, 169, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Cao, G.; Sofic, E.; Prior, R.L. Antioxidant and prooxidant behavior of flavonoids: Structure-activity relationships. Free Radic. Biol. Med. 1997, 22, 749–760. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, Y.; Liu, J.; Wang, K.; Guo, X.; Ji, B.; Zhou, F. Protective Effects of genistein and puerarin against chronic alcohol-induced liver injury in mice via antioxidant, anti-inflammatory, and anti-apoptotic mechanisms. J. Agric. Food Chem. 2016, 64, 7291–7297. [Google Scholar] [CrossRef] [PubMed]
- Rathee, P.; Chaudhary, H.; Rathee, S.; Rathee, D.; Kumar, V.; Kohli, K. Mechanism of action of flavonoids as anti-inflammatory agents: A review. Inflamm. Allergy Drug Targets 2009, 8, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Bayele, H.K.; Balesaria, S.; Srai, S.K. Phytoestrogens modulate hepcidin expression by Nrf2: Implications for dietary control of iron absorption. Free Radic. Biol. Med. 2015, 89, 1192–1202. [Google Scholar] [CrossRef] [PubMed]
- Oboh, G.; Ademosun, A.O.; Opeyemi, O.B. Quercetin and its role in chronic diseases. Adv. Exp. Med. Biol. 2016, 929, 377–387. [Google Scholar] [PubMed]
- Kumar, N.; Pruthi, V. Potential applications of ferulic acid from natural sources. Biotechnol. Rep. 2014, 4, 86–93. [Google Scholar] [CrossRef] [PubMed]
- Bradamante, S.; Barenghi, L.; Villa, A. Cardiovascular protective effects of resveratrol. Cardiovasc. Drug Rev. 2004, 22, 169–188. [Google Scholar] [CrossRef] [PubMed]
- Tang, P.C.T.; Ng, Y.F.; Ho, S.; Gyda, M.; Chan, S.W. Resveratrol and cardiovascular health—Promising therapeutic or hopeless illusion? Pharmacol. Res. 2014, 90, 88–115. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Gu, L.; Prior, R.L.; McKay, S. Characterization of anthocyanins and proanthocyanidins in some cultivars of Ribes, Aronia and Sambucus and their antioxidant capacity. J. Agric. Food Chem. 2004, 52, 7846–7856. [Google Scholar] [CrossRef] [PubMed]
- Taheri, R.; Connolly, B.A.; Brand, M.H.; Bolling, B.W. Underutilized chokeberry (Aroniamelanocarpa, Aroniaarbutifolia, Aroniaprunifolia) accessions are rich sources of anthocyanins, flavonoids, hydroxycinnamic acids, and proanthocyanidins. J. Agric. Food Chem. 2013, 61, 8581–8588. [Google Scholar] [CrossRef] [PubMed]
- McDowell, L.R.; Wilkinson, N.; Madison, R.; Felix, T.L. Vitamins and minerals functioning as antioxidants with supplementation considerations. In Florida Ruminant Nutrition Symposium; Best Western Gateway Grand: Gainesville, FL, USA, 2007; pp. 30–31. [Google Scholar]
- Kennedy, T.A.; Liebler, D.C. Peroxyl radical scavenging by beta-carotene in lipid bilayers. Effect of oxygen partial pressure. J. Biol. Chem. 1992, 267, 4658–4663. [Google Scholar] [PubMed]
- Ramalho, A.; Padilha, P.; Saunders, C. Critical analysis of Brazilian studies about vitamin A deficiency in maternal child group. Rev. Paul. Pediatr. 2008, 26, 392–399. [Google Scholar] [CrossRef]
- Mejía, L.A.; Chew, F. Hematological effect of supplementing anemic children with vitamin A alone and in combinations with iron. Am. J. Clin. Nutr. 1998, 48, 595–600. [Google Scholar]
- Bloem, M.W.; Wedel, M.; van Agtamal, E.J.; Speek, A.J.; Saowakontha, S.; Schreurs, W.H. Vitamin A intervention: Short-term effects of a single, oral, massive dose on iron metabolism. Am. J. Clin. Nutr. 1990, 51, 76–79. [Google Scholar] [PubMed]
- Kelleher, S.L.; Lönnerdal, B. Low vitamin A intake affects milk iron and iron transporters in rat mammary gland and liver. J. Nutr. 2005, 135, 27–32. [Google Scholar] [PubMed]
- Imam, M.U.; Ismail, M.; Ooi, D.J.; Azmi, N.H.; Sarega, N.; Chan, K.W.; Bhanger, M.I. Are bioactive-rich fractions functionally richer? Crit. Rev. Biotechnol. 2016, 36, 585–593. [Google Scholar] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antioxidant | Mechanism of Fe Regulation | Reference(s) |
|---|---|---|
| Citrus flavonoid-rich extracts of orange and bergamot juice | Chelation of iron in iron-overloaded human lung epithelial cells (A549), induction of catalase enzyme, and attenuation of reactive oxygen species (ROS) and membrane lipid peroxidation. | [65] |
| Epigallocatechin-3-gallate | Chelation of iron, reduced basolateral iron export in Caco-2 cells, and activation of nuclear factor erythroid 2-related factor 2 (Nrf2), a master transcriptional regulator of antioxidant genes in human mesenchymal stem cells (hMSCs). | [66,67] |
| Grape seed extract and anthocyanins | Grape seed extract induced chelation of iron and reduced basolateral iron export in Caco-2 cells. Anthocyanins induced attenuation of sodium fluoride-induced oxidative damage to human embryo hepatic cells via decreased iron content and increased antioxidants including glutathione peroxidase (GPx), superoxide dismutase (SOD), and total antioxidant capacity, mediated via decreased hepcidin and increased ferroportin expression. | [66,68] |
| Curcumin | Decreased iron levels in the bone marrow, spleen and liver, attenuated lipopolysaccharide (LPS)-induced oxidative stress-related inflammation, activated hepatic iron-responsive element-binding protein (IRP) and transferrin receptor 1 (TfR1), and repressed hepatic ferritin and hepcidin synthesis. | [69] |
| Quercetin | Attenuation of hepatic iron deposition in mice exposed to ethanol or excess iron, induction of bone morphogentic protein 6 (BMP6), intranuclear suppressor of mother of mothers against decapentaplegic homolog 4 (SMAD4), SMAD4 binding to hepcidin antimicrobial peptide (HAMP) promoter and hepcidin expression. | [70] |
| Black soybean seed coat anthocyanins | Reduced hepatic hepcidin expression, decreased splenic iron and increased serum iron, mediated via reduced SMAD1/5/8 phosphorylation. | [71] |
| Myrecitin | Reduced hepatic hepcidin expression, reduced hepcidin promoter activity, and reduced SMAD1/5/8 phosphorylation in HepG2 cells. Reduced hepatic hepcidin expression, decreased splenic iron levels, and increased serum iron levels in mice. | [72] |
| Genistein | Increased hepcidin expression and promoter activity in zebrafish and human hepatocytes in a signal transducer and activator of transcription 3- (STAT3-) dependent and SMAD4-dependent manner. | [73] |
| Silymarin | Iron chelation. | [74] |
| Ferulic acid | Attenuates iron-induced oxidative stress by reducing liver injury, apoptotic changes and ROS production; increases hepatic antioxidants and mitochondrial membrane potential; and reverses mitochondrial swelling. | [75] |
| Resveratrol | Attenuation of iron-induced cardiac iron overload, oxidative stress, altered Ca2+ homeostasis and myocardial fibrosis; increased cardiac nuclear and acetylated Forkhead box protein O1 (FOXO1) levels; Decreased sirtuin 1 (SIRT1) and sarco/endoplasmic reticulum Ca2+-ATPase 2a (SERCA2a) levels. | [76] |
| Chokeberries | Reduced inflammatory markers; increased total antioxidant status and serum iron levels. | [77] |
| Vitamin A and beta-carotene | Increased expression of TfR and hepcidin; increased intestinal iron absorption; reduced ferroportin expression; reduced inflammatory signaling; increased intracellular ferritin levels; release of intracellular trapped iron. | [78,79,80] |
| Vitamin C | Reduction of Fe3+ to Fe2+; inhibition of hepcidin expression. | [81] |
| Tucum-Do-Cerrado (Bactris setosaMart.) | Attenuation of iron-induced increases in serum and tissue iron levels, transferrin (Tf) saturation, and lipid oxidation via increasing expression of hepatic HAMP, ferritin, heme oxygenase 1 (Hmox1), NADPH dehydrogenase quinone 1 (Nqo1), and Nrf2 and BMP6, and intestinal Nrf2; increased antioxidant enzymes including catalase, glutathione reductase, and GPx. | [82] |
| Baicalein (Scutellaria baicalensis) | Iron chelation; inhibition of iron-mediated Fenton reaction under physiological conditions in vitro. | [83] |
| Caulis spatholobi | Inhibition of hepcidin, BMP6, and SMAD1/5/8 expression in Huh7 cells; reduced hepatic iron levels; increased serum iron levels in mice. | [84] |
| Astragalus polysaccharide | Attenuation of ROS and Nrf1 accumulation in human cardiac myocytes (HCMs); increased hepcidin expression via the activation of p38 mitogen-activated protein kinase (MAPK) and release of interleukin 6 (IL-6). | [85,86] |
| Angelica sinensis polysaccharide (ASP) | Increased chondrocyte cell viability, and increased SOD and catalase levels; reduced malondialdehyde production, apoptosis, and inflammatory cytokines; reduced levels of serum hepcidin, IL-6, ferritin, Tf, TfR1, and TfR2 in H22-bearing mice. | [87,88] |
| Mexidol | Reversal of oxidative hemolysis and increased serum hepcidin levels in hemochromatosis patients. | [89] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Imam, M.U.; Zhang, S.; Ma, J.; Wang, H.; Wang, F. Antioxidants Mediate Both Iron Homeostasis and Oxidative Stress. Nutrients 2017, 9, 671. https://doi.org/10.3390/nu9070671
Imam MU, Zhang S, Ma J, Wang H, Wang F. Antioxidants Mediate Both Iron Homeostasis and Oxidative Stress. Nutrients. 2017; 9(7):671. https://doi.org/10.3390/nu9070671
Chicago/Turabian StyleImam, Mustapha Umar, Shenshen Zhang, Jifei Ma, Hao Wang, and Fudi Wang. 2017. "Antioxidants Mediate Both Iron Homeostasis and Oxidative Stress" Nutrients 9, no. 7: 671. https://doi.org/10.3390/nu9070671
APA StyleImam, M. U., Zhang, S., Ma, J., Wang, H., & Wang, F. (2017). Antioxidants Mediate Both Iron Homeostasis and Oxidative Stress. Nutrients, 9(7), 671. https://doi.org/10.3390/nu9070671