Vitamin C, Aging and Alzheimer’s Disease

Abstract
:1. Ascorbic Acid and Its Relevance to the Aging Process
2. Ascorbic Acid, Epigenetic Modulation and Nutrigenomics
3. Ascorbic Acid and the Aging Process: In Vitro Models
Ascorbic Acid and the Aging Process: In Vivo Evidence
4. Ascorbic Acid and the Aging Process: Oxidative Stress and Antioxidant Defense
5. Evidence for Ascorbic Acid in Brain Aging
6. Ascorbic Acid and Its Relevance to Alzheimer’s Disease
6.1. Ascorbic Acid and Oxidative Stress in Alzheimer’s Disease
6.2. Metals, Oxidative Stress and Ascorbic Acid in Alzheimer’s Disease: The AA Oxidative Balance in the Brain
6.3. Ascorbic Acid and Neuroinflammation in Alzheimer’s Disease
6.4. Ascorbic Acid and Amyloid Plaque Accumulation in Alzheimer’s Disease
6.5. Acid Ascorbic and Vascular Disease Associated with Alzheimer’s Disease
7. From Bench to Bedside
8. Conclusions
Author Contributions
Conflicts of Interest
References
- Bloom, D.E. 7 billion and counting. Science 2011, 333, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Cannizzo, E.S.; Clement, C.C.; Sahu, R.; Follo, C.; Santambrogio, L. Oxidative stress, inflamm-aging and immunosenescence. J. Proteom. 2011, 74, 2313–2323. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. Ser. A Biol. Sci. Med. Sci. 2014, 69 (Suppl. 1), S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Vasto, S.; Candore, G.; Balistreri, C.R.; Caruso, M.; Colonna-Romano, G.; Grimaldi, M.P.; Listi, F.; Nuzzo, D.; Lio, D.; Caruso, C. Inflammatory networks in ageing, age-related diseases and longevity. Mech. Ageing Dev. 2007, 128, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Cevenini, E.; Monti, D.; Franceschi, C. Inflamm-ageing. Curr. Opin. Clin. Nutr. Metab. Care 2013, 16, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Balardy, L.; Moulis, G.; Gaudin, C.; Peyrot, C.; Vellas, B.; Cesari, M.; Nourhashemi, F. Proinflammatory cytokines, aging, and age-related diseases. J. Am. Med. Dir. Assoc. 2013, 14, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging. An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Szarc vel Szic, K.; Declerck, K.; Vidakovic, M.; Vanden Berghe, W. From inflammaging to healthy aging by dietary lifestyle choices: Is epigenetics the key to personalized nutrition? Clin. Epigenetics 2015, 7, 33. [Google Scholar] [CrossRef] [PubMed]
- Santoro, A.; Pini, E.; Scurti, M.; Palmas, G.; Berendsen, A.; Brzozowska, A.; Pietruszka, B.; Szczecinska, A.; Cano, N.; Meunier, N.; et al. Combating inflammaging through a Mediterranean whole diet approach: The nu-age project’s conceptual framework and design. Mech. Ageing Dev. 2014, 136, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Berendsen, A.; Santoro, A.; Pini, E.; Cevenini, E.; Ostan, R.; Pietruszka, B.; Rolf, K.; Cano, N.; Caille, A.; Lyon-Belgy, N.; et al. Reprint of: A parallel randomized trial on the effect of a healthful diet on inflammageing and its consequences in European elderly people: Design of the nu-age dietary intervention study. Mech. Ageing Dev. 2014, 136, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Neufcourt, L.; Assmann, K.E.; Fezeu, L.K.; Touvier, M.; Graffouillere, L.; Shivappa, N.; Hebert, J.R.; Wirth, M.D.; Hercberg, S.; Galan, P.; et al. Prospective association between the dietary inflammatory index and metabolic syndrome: Findings from the su.Vi.Max study. Nutr. Metab. Cardiovasc. Dis. NMCD 2015, 25, 988–996. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Katz, A.; Wang, Y.; Eck, P.; Kwon, O.; Lee, J.H.; Chen, S.; Corpe, C.; Dutta, A.; Dutta, S.K.; et al. Vitamin C as an antioxidant: Evaluation of its role in disease prevention. J. Am. Coll. Nutr. 2003, 22, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Naidu, K.A. Vitamin C in human health and disease is still a mystery? An overview. Nutr. J. 2003, 2, 7. [Google Scholar] [CrossRef] [PubMed]
- Michels, A.J.; Joisher, N.; Hagen, T.M. Age-related decline of sodium-dependent ascorbic acid transport in isolated rat hepatocytes. Arch. Biochem. Biophys. 2003, 410, 112–120. [Google Scholar] [CrossRef]
- Grosso, G.; Bei, R.; Mistretta, A.; Marventano, S.; Calabrese, G.; Masuelli, L.; Giganti, M.G.; Modesti, A.; Galvano, F.; Gazzolo, D. Effects of vitamin c on health: A review of evidence. Front. Biosci. 2013, 18, 1017–1029. [Google Scholar]
- Duarte, T.L.; Lunec, J. Review: When is an antioxidant not an antioxidant? A review of novel actions and reactions of vitamin C. Free Radic. Res. 2005, 39, 671–686. [Google Scholar] [CrossRef] [PubMed]
- Regine, H.; Gabriele, W.-F.; Ernst, R.W. Antioxidants and endothelial nitric oxide synthesis. Eur. J. Clin. Pharmacol. 2006, 62 (Suppl. 1), 21–28. [Google Scholar]
- Heller, R.; Unbehaun, A.; Schellenberg, B.; Mayer, B.; Werner-Felmayer, G.; Werner, E.R. l-ascorbic acid potentiates endothelial nitric oxide synthesis via a chemical stabilization of tetrahydrobiopterin. J. Biol. Chem. 2001, 276, 40–47. [Google Scholar] [CrossRef] [PubMed]
- Ladurner, A.; Schmitt, C.A.; Schachner, D.; Atanasov, A.G.; Werner, E.R.; Dirsch, V.M.; Heiss, E.H. Ascorbate stimulates endothelial nitric oxide synthase enzyme activity by rapid modulation of its phosphorylation status. Free Radic. Biol. Med. 2012, 52, 2082–2090. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Vitamin C and genomic stability. Mutat. Res. 2001, 475, 29–35. [Google Scholar] [CrossRef]
- Camarena, V.; Wang, G. The epigenetic role of vitamin C in health and disease. Cell. Mol. Life Sci. CMLS 2016, 73, 1645–1658. [Google Scholar] [CrossRef] [PubMed]
- Young, J.I.; Zuchner, S.; Wang, G. Regulation of the epigenome by vitamin C. Annu. Rev. Nutr. 2015, 35, 545–564. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Mao, S.Q.; Zhao, B.; Chong, Z.; Yang, Y.; Zhao, C.; Zhang, D.; Huang, H.; Gao, J.; Li, Z.; et al. Ascorbic acid enhances Tet-mediated 5-methylcytosine oxidation and promotes DNA demethylation in mammals. J. Am. Chem. Soc. 2013, 135, 10396–10403. [Google Scholar] [CrossRef] [PubMed]
- Cahill, L.E.; El-Sohemy, A. Vitamin c transporter gene polymorphisms, dietary vitamin C and serum ascorbic acid. J. Nutr. Nutr. 2009, 2, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Langlois, M.R.; Martin, M.E.; Boelaert, J.R.; Beaumont, C.; Taes, Y.E.; De Buyzere, M.L.; Bernard, D.R.; Neels, H.M.; Delanghe, J.R. The haptoglobin 2-2 phenotype affects serum markers of iron status in healthy males. Clin. Chem. 2000, 46, 1619–1625. [Google Scholar] [PubMed]
- Horska, A.; Mislanova, C.; Bonassi, S.; Ceppi, M.; Volkovova, K.; Dusinska, M. Vitamin C levels in blood are influenced by polymorphisms in glutathione S-transferases. Eur. J. Nutr. 2011, 50, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, B. New criteria for supplementation of selected micronutrients in the era of nutrigenetics and nutrigenomics. Int. J. Food Sci. Nutr. 2014, 65, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Blaschke, K.; Ebata, K.T.; Karimi, M.M.; Zepeda-Martinez, J.A.; Goyal, P.; Mahapatra, S.; Tam, A.; Laird, D.J.; Hirst, M.; Rao, A.; et al. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst-like state in ES cells. Nature 2013, 500, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, J.S.; Goodwin, J.M.; Garry, P.J. Association between nutritional status and cognitive functioning in a healthy elderly population. JAMA 1983, 249, 2917–2921. [Google Scholar] [CrossRef] [PubMed]
- Lebel, M.; Massip, L.; Garand, C.; Thorin, E. Ascorbate improves metabolic abnormalities in Wrn mutant mice but not the free radical scavenger catechin. Ann. N. Y. Acad. Sci. 2010, 1197, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Aumailley, L.; Warren, A.; Garand, C.; Dubois, M.J.; Paquet, E.R.; Le Couteur, D.G.; Marette, A.; Cogger, V.C.; Lebel, M. Vitamin C modulates the metabolic and cytokine profiles, alleviates hepatic endoplasmic reticulum stress, and increases the life span of gulo−/− mice. Aging 2016, 8, 458–483. [Google Scholar] [CrossRef] [PubMed]
- Dallaire, A.; Proulx, S.; Simard, M.J.; Lebel, M. Expression profile of Caenorhabditis elegans mutant for the Werner syndrome gene ortholog reveals the impact of vitamin C on development to increase life span. BMC Genom. 2014, 15, 940. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, W.; Chang, L.; Han, Y.; Sun, L.; Gong, X.; Tang, H.; Liu, Z.; Deng, H.; Ye, Y.; et al. Vitamin C alleviates aging defects in a stem cell model for Werner syndrome. Protein Cell 2016, 7, 478–488. [Google Scholar] [CrossRef] [PubMed]
- Massip, L.; Garand, C.; Paquet, E.R.; Cogger, V.C.; O’Reilly, J.N.; Tworek, L.; Hatherell, A.; Taylor, C.G.; Thorin, E.; Zahradka, P.; et al. Vitamin C restores healthy aging in a mouse model for Werner syndrome. FASEB J. 2010, 24, 158–172. [Google Scholar] [CrossRef] [PubMed]
- Pangrazzi, L.; Meryk, A.; Naismith, E.; Koziel, R.; Lair, J.; Krismer, M.; Trieb, K.; Grubeck-Loebenstein, B. “Inflamm-aging” influences immune cell survival factors in human bone marrow. Eur. J. Immunol. 2016, 47. [Google Scholar] [CrossRef] [PubMed]
- Rahman, F.; Al Frouh, F.; Bordignon, B.; Fraterno, M.; Landrier, J.F.; Peiretti, F.; Fontes, M. Ascorbic acid is a dose-dependent inhibitor of adipocyte differentiation, probably by reducing camp pool. Front. Cell Dev. Biol. 2014, 2, 29. [Google Scholar] [CrossRef] [PubMed]
- Rahman, F.; Bordignon, B.; Culerrier, R.; Peiretti, F.; Spicuglia, S.; Djabali, M.; Landrier, J.F.; Fontes, M. Ascorbic acid drives the differentiation of mesoderm-derived embryonic stem cells. Involvement of p38 MAPK/CREB and SVCT2 transporter. Mol. Nutr. Food Res. 2016, 61. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.G.; Cho, G.W. Endogenous ROS levels are increased in replicative senescence in human bone marrow mesenchymal stromal cells. Biochem. Biophys. Res. Commun. 2015, 460, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Aumailley, L.; Dubois, M.J.; Garand, C.; Marette, A.; Lebel, M. Impact of vitamin C on the cardiometabolic and inflammatory profiles of mice lacking a functional Werner syndrome protein helicase. Exp. Gerontol. 2015, 72, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Uchio, R.; Hirose, Y.; Murosaki, S.; Yamamoto, Y.; Ishigami, A. High dietary intake of vitamin c suppresses age-related thymic atrophy and contributes to the maintenance of immune cells in vitamin C-deficient senescence marker protein-30 knockout mice. Br. J. Nutr. 2015, 113, 603–609. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Amano, A.; Kishimoto, Y.; Takahashi, K.; Handa, S.; Maruyama, N.; Ishigami, A. Ascorbic acid prevents protein oxidation in livers of senescence marker protein-30/gluconolactonase knockout mice. Geriatr. Gerontol. Int. 2014, 14, 989–995. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Lim, S.M.; Yoo, J.A.; Woo, M.J.; Cho, K.H. Consumption of high-dose vitamin C (1250 mg per day) enhances functional and structural properties of serum lipoprotein to improve anti-oxidant, anti-atherosclerotic, and anti-aging effects via regulation of anti-inflammatory microrna. Food Funct. 2015, 6, 3604–3612. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; May, J.M. Vitamin c function in the brain: Vital role of the ascorbate transporter SVCT2. Free Radic. Biol. Med. 2009, 46, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.; Sui, B.; Hu, C.; Jin, Y. Vitamin c promotes in vitro proliferation of bone marrow mesenchymal stem cells derived from aging mice. J. Sourn Med. Univ. 2015, 35, 1689–1693. [Google Scholar]
- Chen, B.Y.; Wang, X.; Chen, L.W.; Luo, Z.J. Molecular targeting regulation of proliferation and differentiation of the bone marrow-derived mesenchymal stem cells or mesenchymal stromal cells. Curr. Drug Targets 2012, 13, 561–571. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Hekimi, S. A mitochondrial superoxide signal triggers increased longevity in Caenorhabditis elegans. PLoS Biol. 2010, 8, e1000556. [Google Scholar] [CrossRef] [PubMed]
- Van Raamsdonk, J.M.; Hekimi, S. Superoxide dismutase is dispensable for normal animal lifespan. Proc. Natl. Acad. Sci. USA 2012, 109, 5785–5790. [Google Scholar] [CrossRef] [PubMed]
- Desjardins, D.; Cacho-Valadez, B.; Liu, J.L.; Wang, Y.; Yee, C.; Bernard, K.; Khaki, A.; Breton, L.; Hekimi, S. Antioxidants reveal an inverted U-shaped dose-response relationship between reactive oxygen species levels and the rate of aging in Caenorhabditis elegans. Aging Cell 2017, 16, 104–112. [Google Scholar] [CrossRef] [PubMed]
- Riscuta, G. Nutrigenomics at the interface of aging, lifespan, and cancer prevention. J. Nutr. 2016, 146, 1931–1939. [Google Scholar] [CrossRef] [PubMed]
- Soysal, P.; Isik, A.T.; Carvalho, A.F.; Fernandes, B.S.; Solmi, M.; Schofield, P.; Veronese, N.; Stubbs, B. Oxidative stress and frailty: A systematic review and synthesis of the best evidence. Maturitas 2017, 99, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Leelarungrayub, J.; Laskin, J.J.; Bloomer, R.J.; Pinkaew, D. Consumption of star fruit juice on pro-inflammatory markers and walking distance in the community dwelling elderly. Arch. Gerontol. Geriatr. 2016, 64, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Leelarungrayub, J.; Yankai, A.; Pinkaew, D.; Puntumetakul, R.; Laskin, J.J.; Bloomer, R.J. A preliminary study on the effects of star fruit consumption on antioxidant and lipid status in elderly Thai individuals. Clin. Interv. Aging 2016, 11, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Nualart, F.; Mack, L.; Garcia, A.; Cisternas, P.; Bongarzone, E.R.; Heitzer, M.; Jara, N.; Martinez, F.; Ferrada, L.; Espinoza, F.; et al. Vitamin C transporters, recycling and the bystander effect in the nervous system: SVCT2 versus gluts. J. Stem Cell Res. Ther. 2014, 4, 209. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.E. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000, 23, 209–216. [Google Scholar] [CrossRef]
- Lee, J.Y.; Chang, M.Y.; Park, C.H.; Kim, H.Y.; Kim, J.H.; Son, H.; Lee, Y.S.; Lee, S.H. Ascorbate-induced differentiation of embryonic cortical precursors into neurons and astrocytes. J. Neurosci. Res. 2003, 73, 156–165. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Li, L.; Weeber, E.J.; May, J.M. Ascorbate transport by primary cultured neurons and its role in neuronal function and protection against excitotoxicity. J. Neurosci. Res. 2007, 85, 1046–1056. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; Bowman, G.L.; Polidori, M.C. Ascorbic acid and the brain: Rationale for the use against cognitive decline. Nutrients 2014, 6, 1752–1781. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias-Pinto, A.; Acuna, A.I.; Beltran, F.A.; Torres-Diaz, L.; Castro, M.A. Old things new view: Ascorbic acid protects the brain in neurodegenerative disorders. Int. J. Mol. Sci. 2015, 16, 28194–28217. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.; Wang, X. Antioxidant therapies for Alzheimer’s disease. Oxid. Med. Cell. Longev. 2012, 2012, 472932. [Google Scholar] [CrossRef] [PubMed]
- Ientile, L.; De Pasquale, R.; Monacelli, F.; Odetti, P.; Traverso, N.; Cammarata, S.; Tabaton, M.; Dijk, B. Survival rate in patients affected by dementia followed by memory clinics (UVA) in Italy. J. Alzheimers Dis. 2013, 36, 303–309. [Google Scholar] [PubMed]
- Yao, Y.; Chinnici, C.; Tang, H.; Trojanowski, J.Q.; Lee, V.M.; Pratico, D. Brain inflammation and oxidative stress in a transgenic mouse model of Alzheimer-like brain amyloidosis. J. Neuroinflamm. 2004, 1, 21. [Google Scholar] [CrossRef] [PubMed]
- Choudhry, F.; Howlett, D.R.; Richardson, J.C.; Francis, P.T.; Williams, R.J. Pro-oxidant diet enhances beta/gamma secretase-mediated APP processing in APP/PS1 transgenic mice. Neurobiol. Aging 2012, 33, 960–968. [Google Scholar] [CrossRef] [PubMed]
- McGeer, P.L.; McGeer, E.G. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: Implications for therapy. Acta Neuropathol. 2013, 126, 479–497. [Google Scholar] [CrossRef] [PubMed]
- Apelt, J.; Bigl, M.; Wunderlich, P.; Schliebs, R. Aging-related increase in oxidative stress correlates with developmental pattern of beta-secretase activity and beta-amyloid plaque formation in transgenic Tg2576 mice with Alzheimer-like pathology. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2004, 22, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Sanmartin, C.D.; Adasme, T.; Hidalgo, C.; Paula-Lima, A.C. The antioxidant N-acetylcysteine prevents the mitochondrial fragmentation induced by soluble amyloid-beta peptide oligomers. Neuro-Degener. Dis. 2012, 10, 34–37. [Google Scholar] [CrossRef] [PubMed]
- Ma, T.; Hoeffer, C.A.; Wong, H.; Massaad, C.A.; Zhou, P.; Iadecola, C.; Murphy, M.P.; Pautler, R.G.; Klann, E. Amyloid beta-induced impairments in hippocampal synaptic plasticity are rescued by decreasing mitochondrial superoxide. J. Neurosci. Off. J. Soc. Neurosci. 2011, 31, 5589–5595. [Google Scholar] [CrossRef] [PubMed]
- Bloom, G.S. Amyloid-beta and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Moreira, P.I.; Santos, M.S.; Oliveira, C.R. Alzheimer’s disease: A lesson from mitochondrial dysfunction. Antioxid. Redox Signal. 2007, 9, 1621–1630. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; Shimizu, T. Cytoplasmic superoxide radical: A possible contributing factor to intracellular abeta oligomerization in Alzheimer disease. Commun. Integr. Biol. 2012, 5, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I.; Masters, C.L.; Tanzi, R.E. Copper, beta-amyloid, and Alzheimer’s disease: Tapping a sensitive connection. Proc. Natl. Acad. Sci. USA 2003, 100, 11193–11194. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I.; Curtain, C.C. Twenty years of metallo-neurobiology: Where to now? Eur. Biophys. J. EBJ 2008, 37, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Jomova, K.; Vondrakova, D.; Lawson, M.; Valko, M. Metals, oxidative stress and neurodegenerative disorders. Mol. Cell. Biochem. 2010, 345, 91–104. [Google Scholar] [CrossRef] [PubMed]
- Monacelli, F.; Borghi, R.; Pacini, D.; Serrati, C.; Traverso, N.; Odetti, P. Pentosidine determination in CSF: A potential biomarker of Alzheimer’s disease? Clin. Chem. Lab. Med. 2014, 52, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Reed, T.; Newman, S.F.; Sultana, R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic. Biol. Med. 2007, 43, 658–677. [Google Scholar] [CrossRef] [PubMed]
- Polidori, M.C.; Mecocci, P. Plasma susceptibility to free radical-induced antioxidant consumption and lipid peroxidation is increased in very old subjects with Alzheimer disease. J. Alzheimers Dis. 2002, 4, 517–522. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Cisse, S.; Stopa, E.G. Expression of heme oxygenase-1 in the senescent and Alzheimer-diseased brain. Ann. Neurol. 1995, 37, 758–768. [Google Scholar] [CrossRef] [PubMed]
- Ischiropoulos, H. Biological tyrosine nitration: A pathophysiological function of nitric oxide and reactive oxygen species. Arch. Biochem. Biophys. 1998, 356, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Krauss, A.; Ferrada, L.; Astuya, A.; Salazar, K.; Cisternas, P.; Martinez, F.; Ramirez, E.; Nualart, F. Dehydroascorbic acid promotes cell death in neurons under oxidative stress: A protective role for astrocytes. Mol. Neurobiol. 2016, 53, 5847–5863. [Google Scholar] [CrossRef] [PubMed]
- May, J.M. Vitamin c transport and its role in the central nervous system. Sub-Cell. Biochem. 2012, 56, 85–103. [Google Scholar]
- Sultana, R.; Mecocci, P.; Mangialasche, F.; Cecchetti, R.; Baglioni, M.; Butterfield, D.A. Increased protein and lipid oxidative damage in mitochondria isolated from lymphocytes from patients with Alzheimer’s disease: Insights into the role of oxidative stress in Alzheimer’s disease and initial investigations into a potential biomarker for this dementing disorder. J. Alzheimers Dis. 2011, 24, 77–84. [Google Scholar] [PubMed]
- Dixit, S.; Bernardo, A.; Walker, J.M.; Kennard, J.A.; Kim, G.Y.; Kessler, E.S.; Harrison, F.E. Vitamin C deficiency in the brain impairs cognition, increases amyloid accumulation and deposition, and oxidative stress in APP/PSEN1 and normally aging mice. ACS Chem. Neurosci. 2015, 6, 570–581. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, S.; Yoo, B.; McElheny, D.; Tay, W.; Ishii, Y. Capturing a reactive state of amyloid aggregates: Nmr-based characterization of copper-bound Alzheimer disease amyloid beta-fibrils in a redox cycle. J. Biol. Chem. 2014, 289, 9998–10010. [Google Scholar] [CrossRef] [PubMed]
- Berger, T.M.; Polidori, M.C.; Dabbagh, A.; Evans, P.J.; Halliwell, B.; Morrow, J.D.; Roberts, L.J., II; Frei, B. Antioxidant activity of vitamin C in iron-overloaded human plasma. J. Biol. Chem. 1997, 272, 15656–15660. [Google Scholar] [CrossRef] [PubMed]
- Sil, S.; Ghosh, T.; Gupta, P.; Ghosh, R.; Kabir, S.N.; Roy, A. Dual role of vitamin C on the neuroinflammation mediated neurodegeneration and memory impairments in colchicine induced rat model of Alzheimer disease. J. Mol. Neurosci. MN 2016, 60, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; Hosseini, A.H.; McDonald, M.P.; May, J.M. Vitamin C reduces spatial learning deficits in middle-aged and very old APP/PSEN1 transgenic and wild-type mice. Pharmacol. Biochem. Behav. 2009, 93, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; May, J.M. Ascorbic acid protects SH-SY5Y neuroblastoma cells from apoptosis and death induced by beta-amyloid. Brain Res. 2006, 1097, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Shah, S.A.; Badshah, H.; Kim, M.J.; Ali, T.; Yoon, G.H.; Kim, T.H.; Abid, N.B.; Rehman, S.U.; Khan, S.; et al. Neuroprotection by vitamin c against ethanol-induced neuroinflammation associated neurodegeneration in the developing rat brain. CNS Neurol. Disord. Drug Targets 2016, 15, 360–370. [Google Scholar] [CrossRef] [PubMed]
- Ballaz, S.; Morales, I.; Rodriguez, M.; Obeso, J.A. Ascorbate prevents cell death from prolonged exposure to glutamate in an in vitro model of human dopaminergic neurons. J. Neurosci. Res. 2013, 91, 1609–1617. [Google Scholar] [CrossRef] [PubMed]
- Chan, S.; Kantham, S.; Rao, V.M.; Palanivelu, M.K.; Pham, H.L.; Shaw, P.N.; McGeary, R.P.; Ross, B.P. Metal chelation, radical scavenging and inhibition of abeta(4)(2) fibrillation by food constituents in relation to Alzheimer’s disease. Food Chem. 2016, 199, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Polidori, M.C.; Pientka, L.; Mecocci, P. A review of the major vascular risk factors related to Alzheimer’s disease. J. Alzheimers Dis. 2012, 32, 521–530. [Google Scholar] [PubMed]
- Murakami, K.; Murata, N.; Ozawa, Y.; Kinoshita, N.; Irie, K.; Shirasawa, T.; Shimizu, T. Vitamin c restores behavioral deficits and amyloid-beta oligomerization without affecting plaque formation in a mouse model of Alzheimer’s disease. J. Alzheimers Dis. 2011, 26, 7–18. [Google Scholar] [PubMed]
- Hauss-Wegrzyniak, B.; Wenk, G.L. Beta-amyloid deposition in the brains of rats chronically infused with thiorphan or lipopolysaccharide: The role of ascorbic acid in the vehicle. Neurosci. Lett. 2002, 322, 75–78. [Google Scholar] [CrossRef]
- Deane, R.; Bell, R.D.; Sagare, A.; Zlokovic, B.V. Clearance of amyloid-beta peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 16–30. [Google Scholar] [CrossRef] [PubMed]
- Dede, D.S.; Yavuz, B.; Yavuz, B.B.; Cankurtaran, M.; Halil, M.; Ulger, Z.; Cankurtaran, E.S.; Aytemir, K.; Kabakci, G.; Ariogul, S. Assessment of endothelial function in Alzheimer’s disease: Is Alzheimer’s disease a vascular disease? J. Am. Geriatr. Soc. 2007, 55, 1613–1617. [Google Scholar] [CrossRef] [PubMed]
- Lam, V.; Hackett, M.; Takechi, R. Antioxidants and dementia risk: Consideration through a cerebrovascular perspective. Nutrients 2016, 8, 828. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Mukherjee, A.; Swarnakar, S.; Das, N. Nanocapsulated ascorbic acid in combating cerebral ischemia reperfusion-induced oxidative injury in rat brain. Curr. Alzheimer Res. 2016, 13, 1363–1373. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Kussmaul, L.; Gutterer, J.M.; Hirrlinger, J.; Hamprecht, B. The glutathione system of peroxide detoxification is less efficient in neurons than in astroglial cells. J. Neurochem. 1999, 72, 2523–2530. [Google Scholar] [CrossRef] [PubMed]
- Rohl, C.; Armbrust, E.; Herbst, E.; Jess, A.; Gulden, M.; Maser, E.; Rimbach, G.; Bosch-Saadatmandi, C. Mechanisms involved in the modulation of astroglial resistance to oxidative stress induced by activated microglia: Antioxidative systems, peroxide elimination, radical generation, lipid peroxidation. Neurotox. Res. 2010, 17, 317–331. [Google Scholar] [CrossRef] [PubMed]
- Heo, J.H.; Hyon, L.; Lee, K.M. The possible role of antioxidant vitamin C in Alzheimer’s disease treatment and prevention. Am. J. Alzheimers Dis. Other Dement. 2013, 28, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Rosales-Corral, S.; Tan, D.X.; Reiter, R.J.; Valdivia-Velazquez, M.; Martinez-Barboza, G.; Acosta-Martinez, J.P.; Ortiz, G.G. Orally administered melatonin reduces oxidative stress and proinflammatory cytokines induced by amyloid-beta peptide in rat brain: A comparative, in vivo study versus vitamin C and E. J. Pineal Res. 2003, 35, 80–84. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, D.; Parle, M.; Kulkarni, S.K. Comparative brain cholinesterase-inhibiting activity of Glycyrrhiza glabra, Myristica fragrans, ascorbic acid, and metrifonate in mice. J. Med. Food 2006, 9, 281–283. [Google Scholar] [CrossRef] [PubMed]
- Kuo, C.H.; Hata, F.; Yoshida, H.; Yamatodani, A.; Wada, H. Effect of ascorbic acid on release of acetylcholine from synaptic vesicles prepared from different species of animals and release of noradrenaline from synaptic vesicles of rat brain. Life Sci. 1979, 24, 911–915. [Google Scholar] [CrossRef]
- Cheng, F.; Cappai, R.; Ciccotosto, G.D.; Svensson, G.; Multhaup, G.; Fransson, L.A.; Mani, K. Suppression of amyloid beta a11 antibody immunoreactivity by vitamin C: Possible role of heparan sulfate oligosaccharides derived from glypican-1 by ascorbate-induced, nitric oxide (no)-catalyzed degradation. J. Biol. Chem. 2011, 286, 27559–27572. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.; Li, X.; Liu, L.; Yagnik, G.B.; Zhou, F. Reaction rates and mechanism of the ascorbic acid oxidation by molecular oxygen facilitated by cu(II)-containing amyloid-beta complexes and aggregates. J. Phys. Chem. B 2010, 114, 4896–4903. [Google Scholar] [CrossRef] [PubMed]
- Chambial, S.; Dwivedi, S.; Shukla, K.K.; John, P.J.; Sharma, P. Vitamin c in disease prevention and cure: An overview. Indian J. Clin. Biochem. IJCB 2013, 28, 314–328. [Google Scholar] [CrossRef] [PubMed]
- Rahal, A.; Kumar, A.; Singh, V.; Yadav, B.; Tiwari, R.; Chakraborty, S.; Dhama, K. Oxidative stress, prooxidants, and antioxidants: The interplay. BioMed Res. Int. 2014, 2014, 761264. [Google Scholar] [CrossRef] [PubMed]
- Cheng, R.; Lin, B.; Lee, K.W.; Ortwerth, B.J. Similarity of the yellow chromophores isolated from human cataracts with those from ascorbic acid-modified calf lens proteins: Evidence for ascorbic acid glycation during cataract formation. Biochim. Biophys. Acta 2001, 1537, 14–26. [Google Scholar] [CrossRef]
- Carr, A.; Frei, B. Does vitamin c act as a pro-oxidant under physiological conditions? FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 1999, 13, 1007–1024. [Google Scholar]
- Valko, M.; Morris, H.; Cronin, M.T. Metals, toxicity and oxidative stress. Curr. Med. Chem. 2005, 12, 1161–1208. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S.I.; Vitek, M.P.; Mason, R.P. Cupric-amyloid beta peptide complex stimulates oxidation of ascorbate and generation of hydroxyl radical. Free Radic. Biol. Med. 2004, 36, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Shearer, J.; Szalai, V.A. The amyloid-beta peptide of Alzheimer’s disease binds Cu(I) in a linear Bis-His coordination environment: Insight into a possible neuroprotective mechanism for the amyloid-beta peptide. J. Am. Chem. Soc. 2008, 130, 17826–17835. [Google Scholar] [CrossRef] [PubMed]
- Nadal, R.C.; Rigby, S.E.; Viles, J.H. Amyloid beta-Cu2+ complexes in both monomeric and fibrillar forms do not generate H2O2 catalytically but quench hydroxyl radicals. Biochemistry 2008, 47, 11653–11664. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.N.; Lai, C.C.; Chiu, C.T.; Lin, J.J.; Wang, J.Y. l-ascorbate attenuates the endotoxin-induced production of inflammatory mediators by inhibiting MAPK activation and NF-kappaB translocation in cortical neurons/glia Cocultures. PloS ONE 2014, 9, e97276. [Google Scholar]
- Kook, S.Y.; Lee, K.M.; Kim, Y.; Cha, M.Y.; Kang, S.; Baik, S.H.; Lee, H.; Park, R.; Mook-Jung, I. High-dose of vitamin c supplementation reduces amyloid plaque burden and ameliorates pathological changes in the brain of 5XFAD mice. Cell Death Dis. 2014, 5, e1083. [Google Scholar] [CrossRef] [PubMed]
- Polidori, M.C.; Ruggiero, C.; Croce, M.F.; Raichi, T.; Mangialasche, F.; Cecchetti, R.; Pelini, L.; Paolacci, L.; Ercolani, S.; Mecocci, P. Association of increased carotid intima-media thickness and lower plasma levels of vitamin C and vitamin E in old age subjects: Implications for Alzheimer’s disease. J. Neural Transm. 2015, 122, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Bomboi, G.; Castello, L.; Cosentino, F.; Giubilei, F.; Orzi, F.; Volpe, M. Alzheimer’s disease and endothelial dysfunction. Neurol. Sci. Off. J. Ital. Neurol. Soc. Ital. Soc. Clin. Neurophysiol. 2010, 31, 1–8. [Google Scholar] [CrossRef] [PubMed]
- May, J.M.; Qu, Z.C. Ascorbic acid prevents increased endothelial permeability caused by oxidized low density lipoprotein. Free Radic. Res. 2010, 44, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- May, J.M.; Harrison, F.E. Role of vitamin C in the function of the vascular endothelium. Antioxid. Redox Signal. 2013, 19, 2068–2083. [Google Scholar] [CrossRef] [PubMed]
- Polidori, M.C.; Pientka, L. Bridging the pathophysiology of Alzheimer’s disease with vascular pathology: The feed-back, the feed-forward, and oxidative stress. J. Alzheimers Dis. 2012, 28, 1–9. [Google Scholar] [PubMed]
- Miller, M.C.; Tavares, R.; Johanson, C.E.; Hovanesian, V.; Donahue, J.E.; Gonzalez, L.; Silverberg, G.D.; Stopa, E.G. Hippocampal rage immunoreactivity in early and advanced Alzheimer’s disease. Brain Res. 2008, 1230, 273–280. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.L.; Huang, Y.H.; Shen, Y.C.; Huang, H.C.; Liu, P.H. Ascorbic acid prevents blood-brain barrier disruption and sensory deficit caused by sustained compression of primary somatosensory cortex. J. Cereb. Blood Flow Metab. Off. J. Int. Soc. Cereb. Blood Flow Metab. 2010, 30, 1121–1136. [Google Scholar] [CrossRef] [PubMed]
- May, J.M.; Qu, Z.C. Nitric oxide mediates tightening of the endothelial barrier by ascorbic acid. Biochem. Biophys. Res. Commun. 2011, 404, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Scuteri, A.; Tesauro, M.; Appolloni, S.; Preziosi, F.; Brancati, A.M.; Volpe, M. Arterial stiffness as an independent predictor of longitudinal changes in cognitive function in the older individual. J. Hypertens. 2007, 25, 1035–1040. [Google Scholar] [CrossRef] [PubMed]
- Ellingsen, I.; Seljeflot, I.; Arnesen, H.; Tonstad, S. Vitamin C consumption is associated with less progression in carotid intima media thickness in elderly men: A 3-year intervention study. Nutr. Metab. Cardiovasc. Dis. NMCD 2009, 19, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Donahue, J.E.; Flaherty, S.L.; Johanson, C.E.; Duncan, J.A., III; Silverberg, G.D.; Miller, M.C.; Tavares, R.; Yang, W.; Wu, Q.; Sabo, E.; et al. Rage, lrp-1, and amyloid-beta protein in Alzheimer’s disease. Acta Neuropathol. 2006, 112, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Pleiner, J.; Schaller, G.; Mittermayer, F.; Marsik, C.; MacAllister, R.J.; Kapiotis, S.; Ziegler, S.; Ferlitsch, A.; Wolzt, M. Intra-arterial vitamin C prevents endothelial dysfunction caused by ischemia-reperfusion. Atherosclerosis 2008, 197, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Scorza, G.; Pietraforte, D.; Minetti, M. Role of ascorbate and protein thiols in the release of nitric oxide from S-nitroso-albumin and S-nitroso-glutathione in human plasma. Free Radic. Biol. Med. 1997, 22, 633–642. [Google Scholar] [CrossRef]
- Allahtavakoli, M.; Amin, F.; Esmaeeli-Nadimi, A.; Shamsizadeh, A.; Kazemi-Arababadi, M.; Kennedy, D. Ascorbic acid reduces the adverse effects of delayed administration of tissue plasminogen activator in a rat stroke model. Basic Clin. Pharmacol. Toxicol. 2015, 117, 335–339. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Park, J.; Kim, J.H.; Choi, J.Y.; Kim, J.Y.; Lee, K.M.; Lee, J.E. Dehydroascorbic acid attenuates ischemic brain edema and neurotoxicity in cerebral ischemia: An in vivo study. Exp. Neurobiol. 2015, 24, 41–54. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.L.; Bayraktutan, U. Antioxidants attenuate hyperglycaemia-mediated brain endothelial cell dysfunction and blood-brain barrier hyperpermeability. Diabetes Obes. Metab. 2009, 11, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Bowman, G.L. Ascorbic acid, cognitive function, and Alzheimer’s disease: A current review and future direction. BioFactors 2012, 38, 114–122. [Google Scholar] [CrossRef] [PubMed]
- Bowman, G.L.; Dodge, H.; Frei, B.; Calabrese, C.; Oken, B.S.; Kaye, J.A.; Quinn, J.F. Ascorbic acid and rates of cognitive decline in Alzheimer’s disease. J. Alzheimers Dis. 2009, 16, 93–98. [Google Scholar] [CrossRef] [PubMed]
- Bowman, G.L.; Kaye, J.A.; Moore, M.; Waichunas, D.; Carlson, N.E.; Quinn, J.F. Blood-brain barrier impairment in alzheimer disease: Stability and functional significance. Neurology 2007, 68, 1809–1814. [Google Scholar] [CrossRef] [PubMed]
- Spector, R.; Johanson, C.E. Sustained choroid plexus function in human elderly and Alzheimer’s disease patients. Fluids Barriers CNS 2013, 10, 28. [Google Scholar] [CrossRef] [PubMed]
- Charlton, K.E.; Rabinowitz, T.L.; Geffen, L.N.; Dhansay, M.A. Lowered plasma vitamin C, but not vitamin E, concentrations in dementia patients. J. Nutr. Health Aging 2004, 8, 99–107. [Google Scholar] [PubMed]
- Rinaldi, P.; Polidori, M.C.; Metastasio, A.; Mariani, E.; Mattioli, P.; Cherubini, A.; Catani, M.; Cecchetti, R.; Senin, U.; Mecocci, P. Plasma antioxidants are similarly depleted in mild cognitive impairment and in Alzheimer’s disease. Neurobiol. Aging 2003, 24, 915–919. [Google Scholar] [CrossRef]
- Morris, M.C.; Beckett, L.A.; Scherr, P.A.; Hebert, L.E.; Bennett, D.A.; Field, T.S.; Evans, D.A. Vitamin E and vitamin C supplement use and risk of incident Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1998, 12, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Zandi, P.P.; Anthony, J.C.; Khachaturian, A.S.; Stone, S.V.; Gustafson, D.; Tschanz, J.T.; Norton, M.C.; Welsh-Bohmer, K.A.; Breitner, J.C.; Cache County Study, G. Reduced risk of Alzheimer disease in users of antioxidant vitamin supplements: The cache county study. Arch. Neurol. 2004, 61, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Devore, E.E.; Kang, J.H.; Stampfer, M.J.; Grodstein, F. The association of antioxidants and cognition in the nurses’ health study. Am. J. Epidemiol. 2013, 177, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Engelhart, M.J.; Geerlings, M.I.; Ruitenberg, A.; van Swieten, J.C.; Hofman, A.; Witteman, J.C.; Breteler, M.M. Dietary intake of antioxidants and risk of Alzheimer disease. JAMA 2002, 287, 3223–3229. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.; Suh, J.; Moore, M.M.; Kaye, J.; Frei, B. Antioxidants in Alzheimer’s disease-vitamin c delivery to a demanding brain. J. Alzheimers Dis. 2003, 5, 309–313. [Google Scholar] [CrossRef] [PubMed]
- Fillenbaum, G.G.; Kuchibhatla, M.N.; Hanlon, J.T.; Artz, M.B.; Pieper, C.F.; Schmader, K.E.; Dysken, M.W.; Gray, S.L. Dementia and Alzheimer’s disease in community-dwelling elders taking vitamin C and/or vitamin E. Ann. Pharmacother. 2005, 39, 2009–2014. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E. A critical review of vitamin C for the prevention of age-related cognitive decline and Alzheimer’s disease. J. Alzheimers Dis. 2012, 29, 711–726. [Google Scholar] [PubMed]
- Emir, U.E.; Raatz, S.; McPherson, S.; Hodges, J.S.; Torkelson, C.; Tawfik, P.; White, T.; Terpstra, M. Noninvasive quantification of ascorbate and glutathione concentration in the elderly human brain. NMR Biomed. 2011, 24, 888–894. [Google Scholar] [CrossRef] [PubMed]
- Minor, E.A.; Court, B.L.; Young, J.I.; Wang, G. Ascorbate induces ten-eleven translocation (Tet) methylcytosine dioxygenase-mediated generation of 5-hydroxymethylcytosine. J. Biol. Chem. 2013, 288, 13669–13674. [Google Scholar] [CrossRef] [PubMed]
- Smuda, M.; Glomb, M.A. Maillard degradation pathways of vitamin C. Angew. Chem. 2013, 52, 4887–4891. [Google Scholar] [CrossRef] [PubMed]
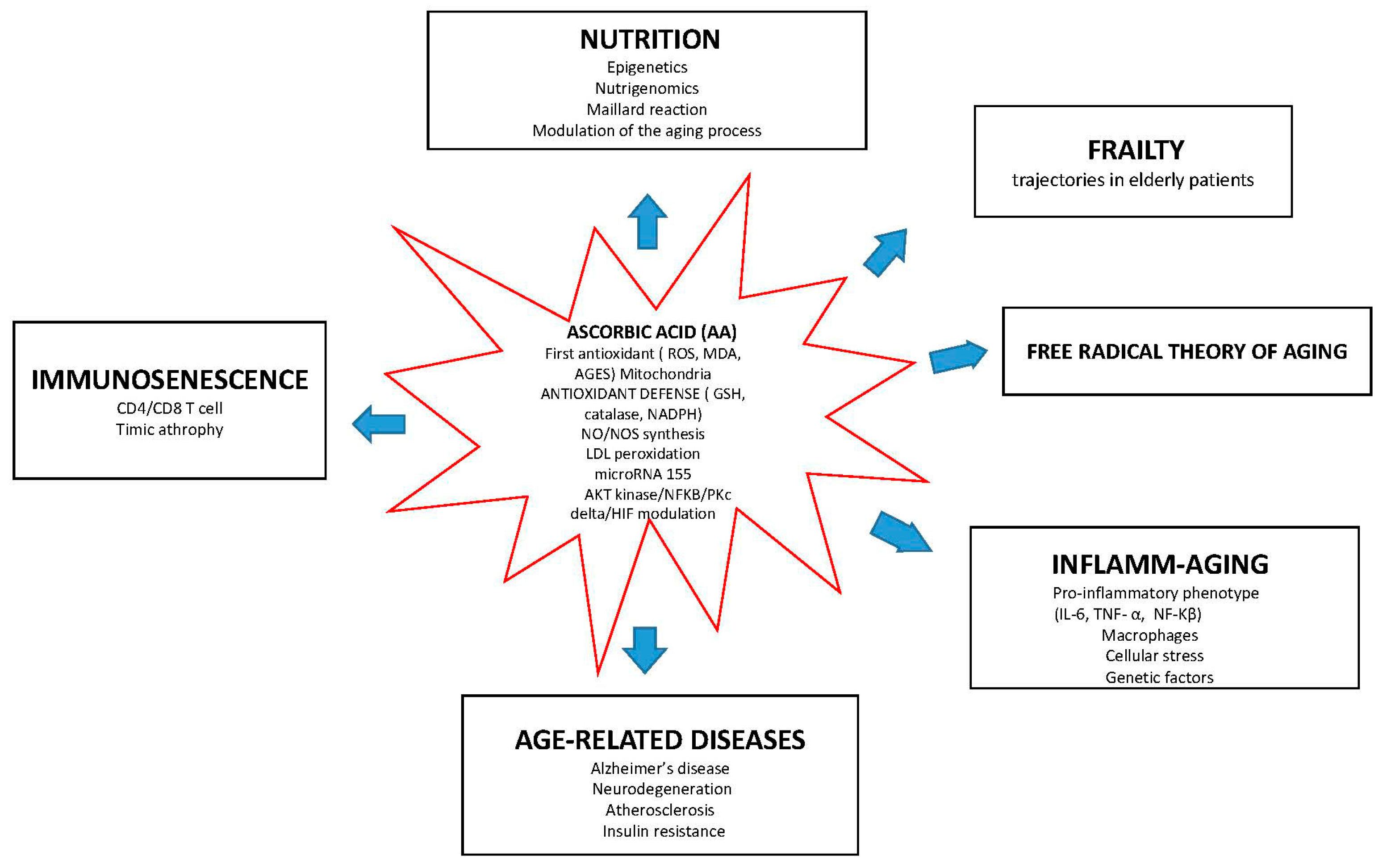

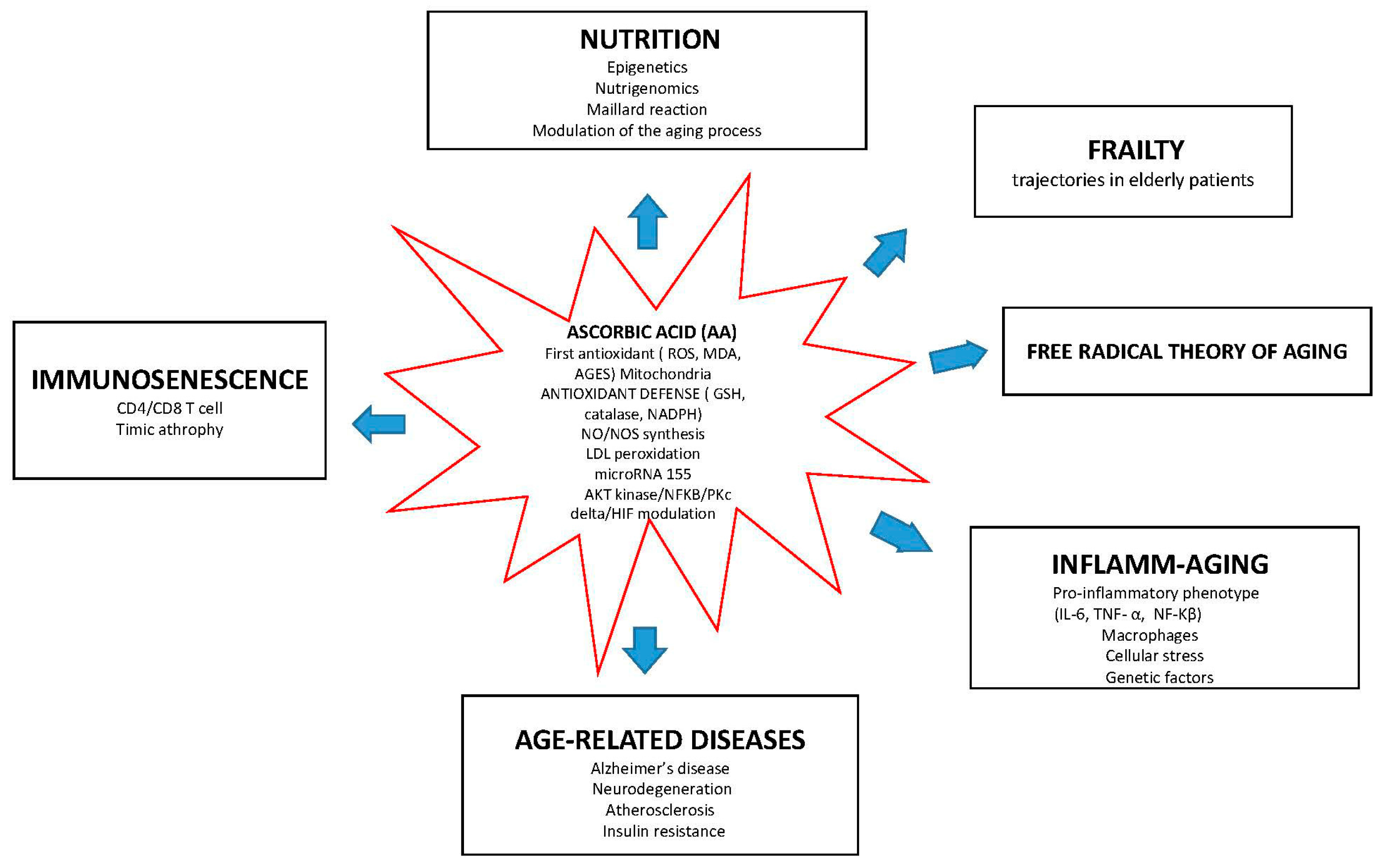{kind=link}
| Species | Model | Design and Methods | Conclusion | References |
|---|---|---|---|---|
| Human | ESCs | Methylation | Epigenetic regulation of Tet activity and DNA methylation | Blaschke, K. 2013 [28] |
| Mouse | Embryonic fibroblasts cultured | Expression of Tet genes, GSH antioxidant activity | Epigenetic modulation of genome activity and stability | Minor, E.A. [29] |
| Human | Umbilical cord vein endothelial cells (HUVEC) | Measurement of citrulline synthesis, determination of cGMP, eNOS activity, GTP Cyclohydrolase I | Anti-oxidative pathways (protection of tetrahydrobiopterin Endothelial integrity (cellular NO synthesis)) | Heller, R. 2001 [18] |
| In Vitro | EA. hy926 | Determination of BH4 levels, H2O2, expression of PP2Ac | Endothelial integrity (eNOS activity/eNOS phosphorylation) | Ladurner, A. 2012 [19] |
| Mouse | WrnΔhel/Δhel | Measurement ROS and oxidative DNA damage | Extended life span, improvement of inflammation, metabolic profile, lipid profile | Lebel, M. 2010 [30] |
| Mouse | Gulo−/− | Measurement cytokines and metabolites | Extended life span. Model of rejuvenation | Aumailley, L. 2016 [31] |
| Insect | Wrn-1(gk99) mutant | Gene expression and regulation | Extended mean life span of C. elegans (regulatory genes of lipid metabolism, ketones, organic acids, carboxylic acids. Locomotion and developmental anatomical structure) | Dallaire, A. 2014 [32] |
| In Vitro | WS MSC model | Oxidative stress levels, IL-6 anIL-8 | Model of rejuvenation | Li, Y. 2016 [33] |
| Mouse | WrnΔhel/Δhel | Measurements of ROS and oxidative DNA damage, GSH, ATP, protein analysis, lactonase activity | Beneficial effects on oxidative pathways, genome stability | Massip, L. 2010 [34] |
| Human and Mouse | PBMCs and BMMC and BM from SOD1−/− | PBMC: IFN-γ ± NAC Human and mouse BM: BFA ± NAC | Beneficial effects on immunosenescence through inflamm-aging | Pangrazzi, L. 2016 [35] |
| Mouse and In Vitro | 3T3-L1 cells, OP9 cells | Adipocytes differentiation | Adipocyte differentiation: implications for the aging process | Rahaman, F. 2014 [36] |
| Mouse | Embryonic stem cell line CGR8 | Stem cells differentiation | AA-dependent differentiation (p38 MAPK/CREB pathway). Epigenetic regulation | Rahman, F. 2016 [37] |
| Human In Vitro | hBM-MSCs | Osteocyte and adipocyte differentiation | Beneficial effects on cells differentiation mediated by anti-oxidation | Jeong, S.G. 2015 [38] |
| Mouse | WrnΔhel/Δhel | Metabolite, cytokine and chemokine measurements | Potential predictive cardiometabolic biomarkers in patients with WS. | Aumailley, L. 2015 [39] |
| Mouse | SMP30KO | Immunosenescence and aging | Beneficial effect on the maintenance of immune cells (thymic atrophy) | Uchio, R. 2015 [40] |
| Mouse | SMP30/GNL KO | Model of senescence | Beneficial effects on liver protein oxidation in vivo | Sato, Y. 2014 [41] |
| In Vitro | Cortical precursor cells | Survival, proliferation, and differentiation of AA-treated CNS precursor cells | Brain development: the generation of neurons and glia | Lee, J.Y. 2003 [42] |
| Mouse | Hippocampal and cortical neurons from mice lacking one allele of the SVCT2 | Combined treatment of AA and GSH | Beneficial effects on neuronal development, functional maturation, and antioxidant responses | Qiu, S. 2007 [43] |
| Species | Model | Design and Methods | Conclusion | References |
|---|---|---|---|---|
| Mouse | TASTPM | Evaluation of carbonyls, glutathione, Αβ, APP | Decreased oxidative stress markers, Nrf2, GSH, APP, soluble Aβ1-42. No increase of BACE 1, PS1and AB plaque | Choundhry, F. 2012 [58] |
| Mouse | Model with human APP695 and double mutation (K670N, M671L) | Evaluation of Aβ, BACE1, antioxidant system and IL-1β | Increased antioxidant system, reduced activity of BACE, IL-1β and NO levels, Aβ deposition | Apelt, J. 2004 [60] |
| Mouse | APP/PS1 transgenic | ROS scavengers and inhibitors effects on Aβ-induced impairments in LTP | Reversal of Aβ- deposition by mitochondria-targeted ROS scavenging | Ma, T. 2011 [62] |
| Mouse | HAPP/Sod1−/− | Anti-Aβ1-16 antibody | Inhibition of amyloid plaques (Aβ hexamers /BACE1 modulation) | Murakami, K. 2012 [65] |
| Mouse | Tg2576 | Aβ levels brain deposition | Suppressed brain inflammatory and oxidative stress responses in mice, significant reduction of soluble and insoluble Aβ1-40 and Aβ1-42 | Yao, Y. 2004 [57] |
| Mouse | APPS we/PSEN1ΔE9 | MDA, Aβ levels, AChE activity. Learning and memory | Improvement of learning and memory Beneficial effects against MDA, and Beneficial effects on AChE function | Harrison, F.E. 2009 [50] |
| Mouse | APPSWE/PSEN1ΔE9 mice, SVCT2+/− | Behavioural test, GSH, MDA, isoprostanes | Decreased Aβ deposition (senile plaque formation and accumulation) | Dixit, S. 2015 [81] |
| Rat | F-344 | Aβ deposition | Decreased-amyloid immunoreactive fibrils | Hauss-Wegrzyniak, B. 2002 [82] |
| Rat and Mouse | Charles-Foster, Swiss Albino mouse | Cognitive test, cytokines, ROS Cytotoxic Activity Assay | Enhancement of anti-oxidative pathway | Sil, S. 2016 [83] |
| Mouse | Gulo−/−5XFAD | Identification modification of cerebral capillaries | Reduction of Aβ accumulation | Kook, S.Y. 2014 [84] |
| In Vitro | neuroblastoma cell line SH-SY5Y | Apoptosis (phosphatidylserine, TUNEL assay, caspase-3 activity) | Prevention of toxicity induced by Aβ | Huang, J. 2006 [85] |
| Mouse | Tg2576, 3xTg-AD | Aβ staining, investigation APP and HS oligosaccharides | Modulation of Aβ fibrillogenesis | Cheng, F. 2011 [86] |
| Mouse | AD model | Fibrillogenesis: senile plaques | Modulation of synaptophysin and the phosphorylation of tau at Ser396 | Murakami, K. 2011 [87] |
| Rat | Wistar | Lipoperoxidation, oxidation, Inflammation, nitrites | Reduction of pro-inflammatory cytokine Inhibition of Aβ deposition | Rosales-Corral, S. 2003 [88] |
| Human In Vitro | NT2 undifferentiated cells | Measurement levels of Aβx-40 and Aβx-42, HNE, expression of BACE-1. Evaluation apoptotic cell death induced by HNE | Increased anti-oxidative pathways against SAPK pathways and BACE-1 that regulate AβPP processing | Tamagno, E. 2005 [74] |
| Human In Vitro | Neuroblastoma cell line SH-SY5Y | Glutathione, superoxide dismutase, and catalase | Neuroprotection anti-oxidative pathways Improvement of antioxidant defense system | Ballaz, S. 2013 [80] |
| Rat | PND7 | Induction of ROS, apoptotic markers. Quantification of Bax/Bcl-2 ratio, cytochrome c and caspases | Reduction of oxidation, neuroinflammation (both activated microglia and astrocytes). reduced ethanol-induced activation of PARP-1 and neurodegeneration | Ahmad, A. 2016 [89] |
| In Vitro | EA. hy926 cells | Measurement intracellular ascorbate and GSH | Endothelial integrity (NO: eNOS/guanylate cyclase pathway) | May, J.M. 2011 [90] |
| In Vitro | EA. hy926 cells | Quantification LDL-enriched lipoproteins, GSH, and lipid peroxidation | Endothelial integrity | May, J.M. 2010 [91] |
| Rat | Cortical neuron/glia co-cultures of neonatal | Measuring nitrites IL-6 and MIP-2, LDH. p38 and ERK MAPKs | Suppression of the LPS-stimulated production of inflammatory mediators | Huang, Y.N. 2014 [92] |
| Rat | Sprague–Dawley | Behavioural test BBB components | Modulation of cortical compression and/or BBB dysfunction | Lin, J.L. 2010 [12] |
| Rat | MCAO | Measurement of infarct and edema brain, measurement of serum MMP-9 levels, behavioural testing | decreased MMP-9 levels, Improvement of the vascular insult (BBB disruption and brain edema) | Allahtavakoli, M. 2015 [93] |
| Rat | Brains | Assessment the role of nanocapsulated ascorbic acid (NAA) | NAA exerted protection to brain mitochondria by preventing oxidative damage in ROS mediated CIR injury | Sarkar, S. 2016 [94] |
| Rat | Hippocampal neurons | Incubation with Aβ Os or 4-CMC ± NAC | NAC prevention of Aβ O-induced mitochondrial Fragmentation by anti-oxidative pathways | Sanmartin, C.D. 2012 [61] |
| Rat | Cortical neurons Neuroblastoma cells | Oxidative stress and DHA uptake, analysis of GLUTs | Improvement of anti-oxidative defense of neurons | García-Krauss, A. 2016 [75] |
| Rat | Primary neurons | Incubation with H2O2, ratio GSH/GSSG | Increased glutathione system of peroxide detoxification | Dringen, R. 1999 [78] |
| Rat | Astroglial cells | Treatment with H2O2 or hydro peroxide, NO release, Lipid Peroxidation, ROS | Reduction of neuroinflammation (microglial-astroglial cells) | Röhl, C. 2010 [79] |
| Rat | SD | Induction of transient focal cerebral ischemia, treatment with DHA | DHA reduced brain edema and vascular permeability formation following cerebral ischemia | Song, J. 2015 [95] |
| Human | Endothelial cell (HBMEC) and astrocyte co-colture | BBB after hyperglycaemic insult | Improvement of BBB permeability by reducing oxidative stress associated with glucose normalization | Allen, C.L. 2009 [96] |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Monacelli, F.; Acquarone, E.; Giannotti, C.; Borghi, R.; Nencioni, A. Vitamin C, Aging and Alzheimer’s Disease. Nutrients 2017, 9, 670. https://doi.org/10.3390/nu9070670
Monacelli F, Acquarone E, Giannotti C, Borghi R, Nencioni A. Vitamin C, Aging and Alzheimer’s Disease. Nutrients. 2017; 9(7):670. https://doi.org/10.3390/nu9070670
Chicago/Turabian StyleMonacelli, Fiammetta, Erica Acquarone, Chiara Giannotti, Roberta Borghi, and Alessio Nencioni. 2017. "Vitamin C, Aging and Alzheimer’s Disease" Nutrients 9, no. 7: 670. https://doi.org/10.3390/nu9070670
APA StyleMonacelli, F., Acquarone, E., Giannotti, C., Borghi, R., & Nencioni, A. (2017). Vitamin C, Aging and Alzheimer’s Disease. Nutrients, 9(7), 670. https://doi.org/10.3390/nu9070670