Nutrients Turned into Toxins: Microbiota Modulation of Nutrient Properties in Chronic Kidney Disease

 and
and 
Abstract
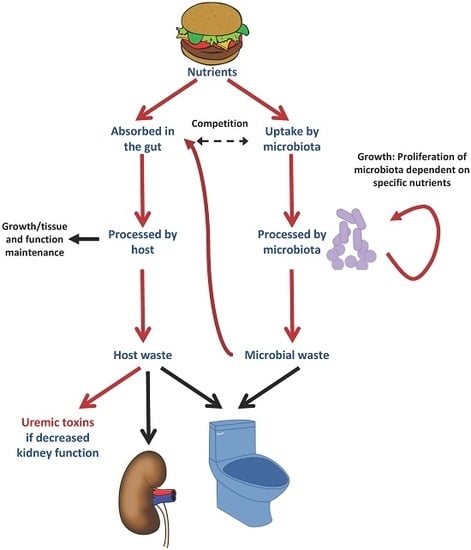
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. CKD and Uremic Toxins
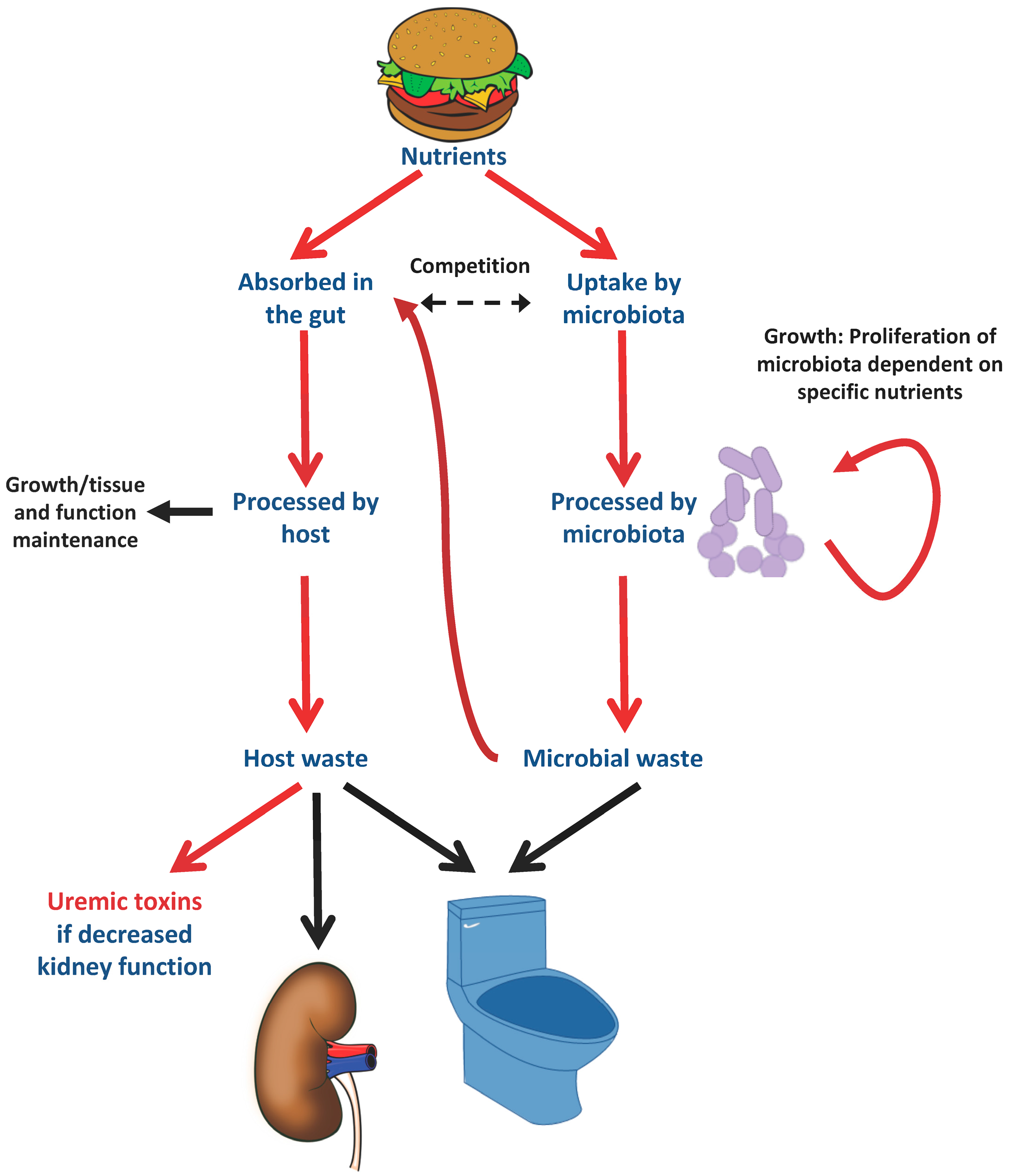
2. The Microbiota
3. Food-Derived Uremic Toxins
3.1. Phosphate
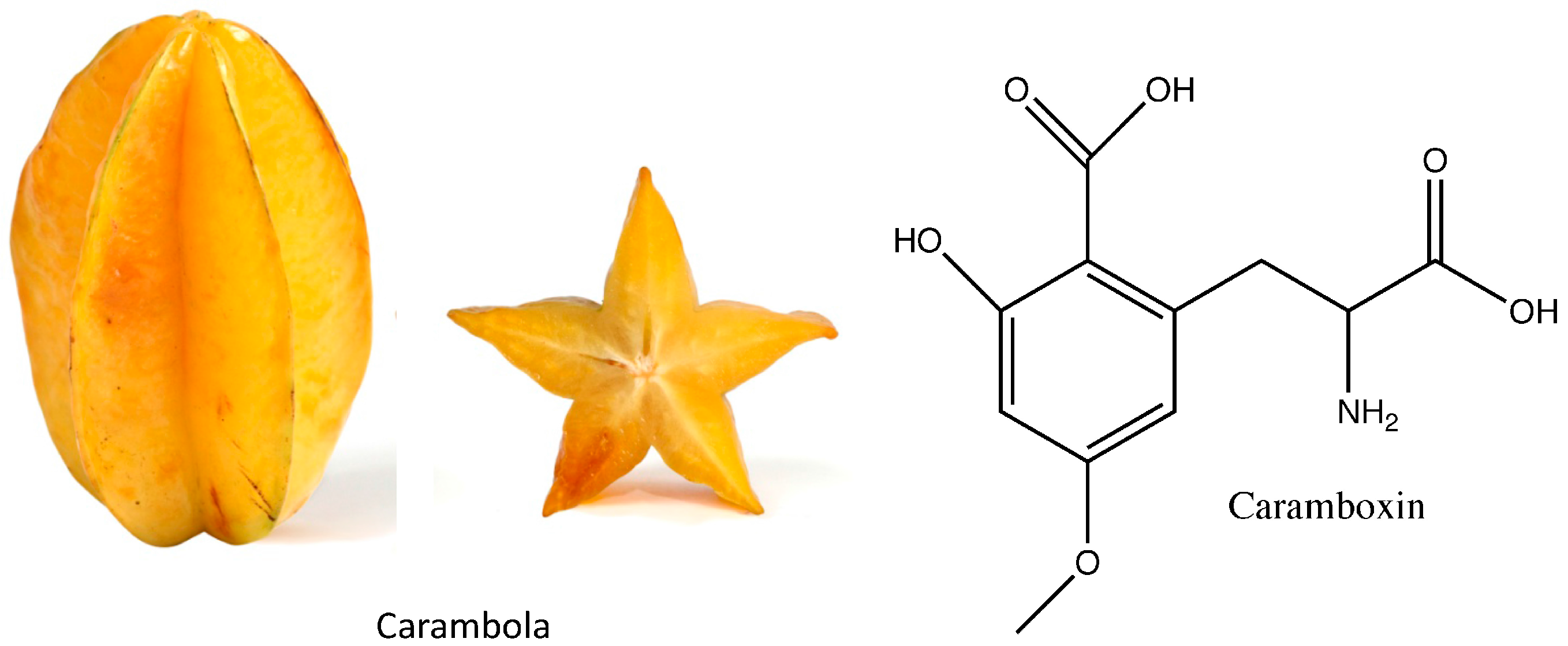
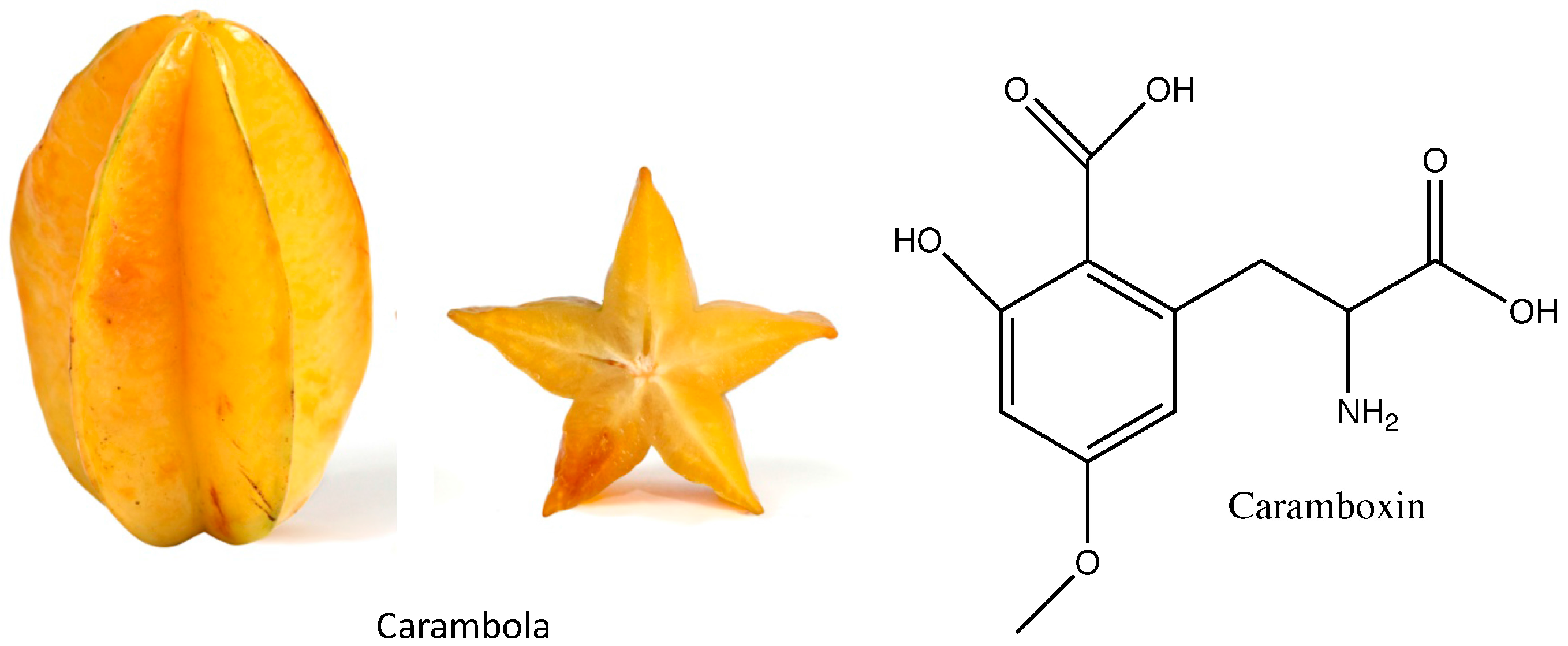
3.2. Caramboxin
3.3. Oxalate
4. Nutrients as Uremic Toxins Precursors via the Microbiota
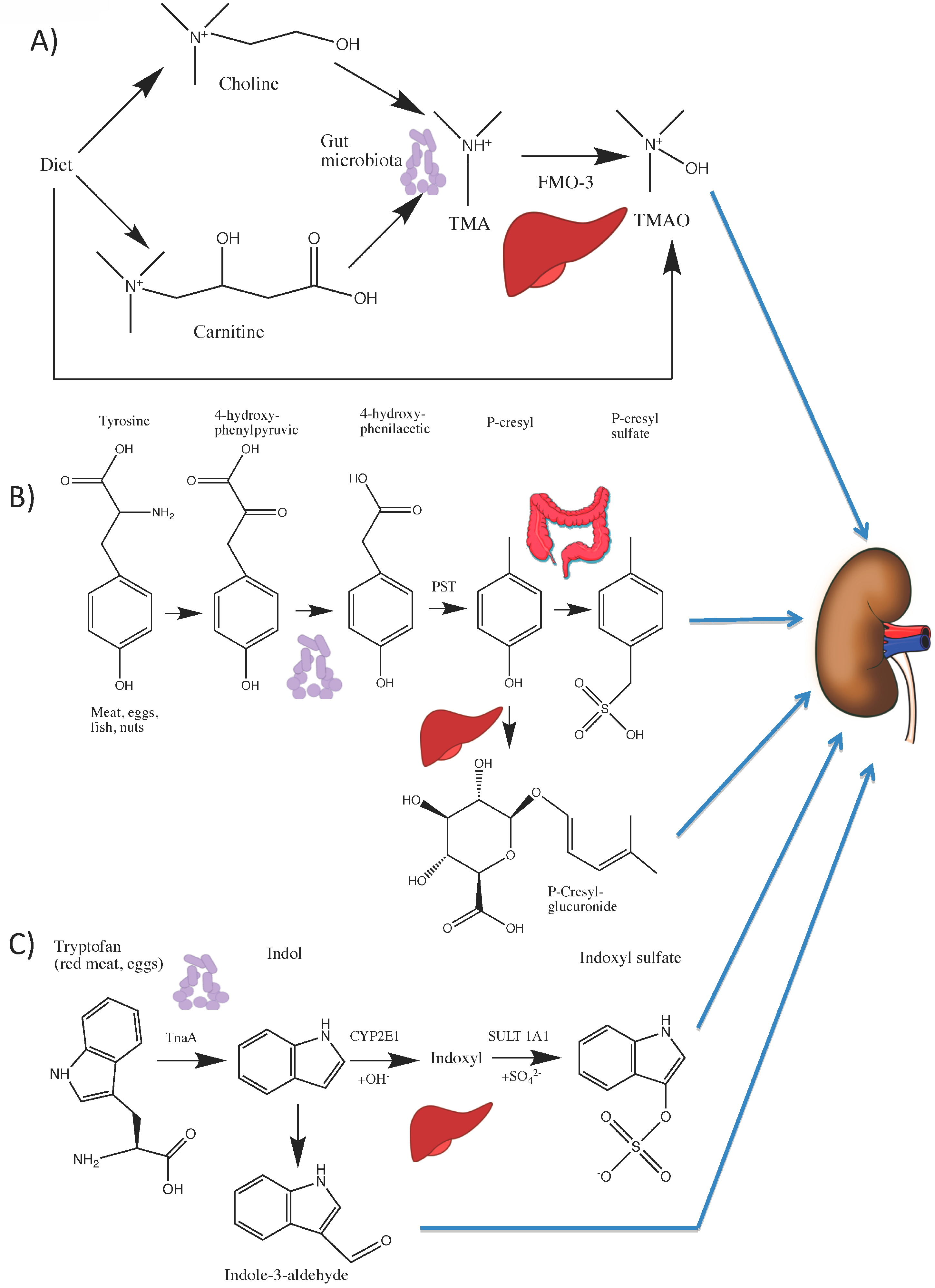
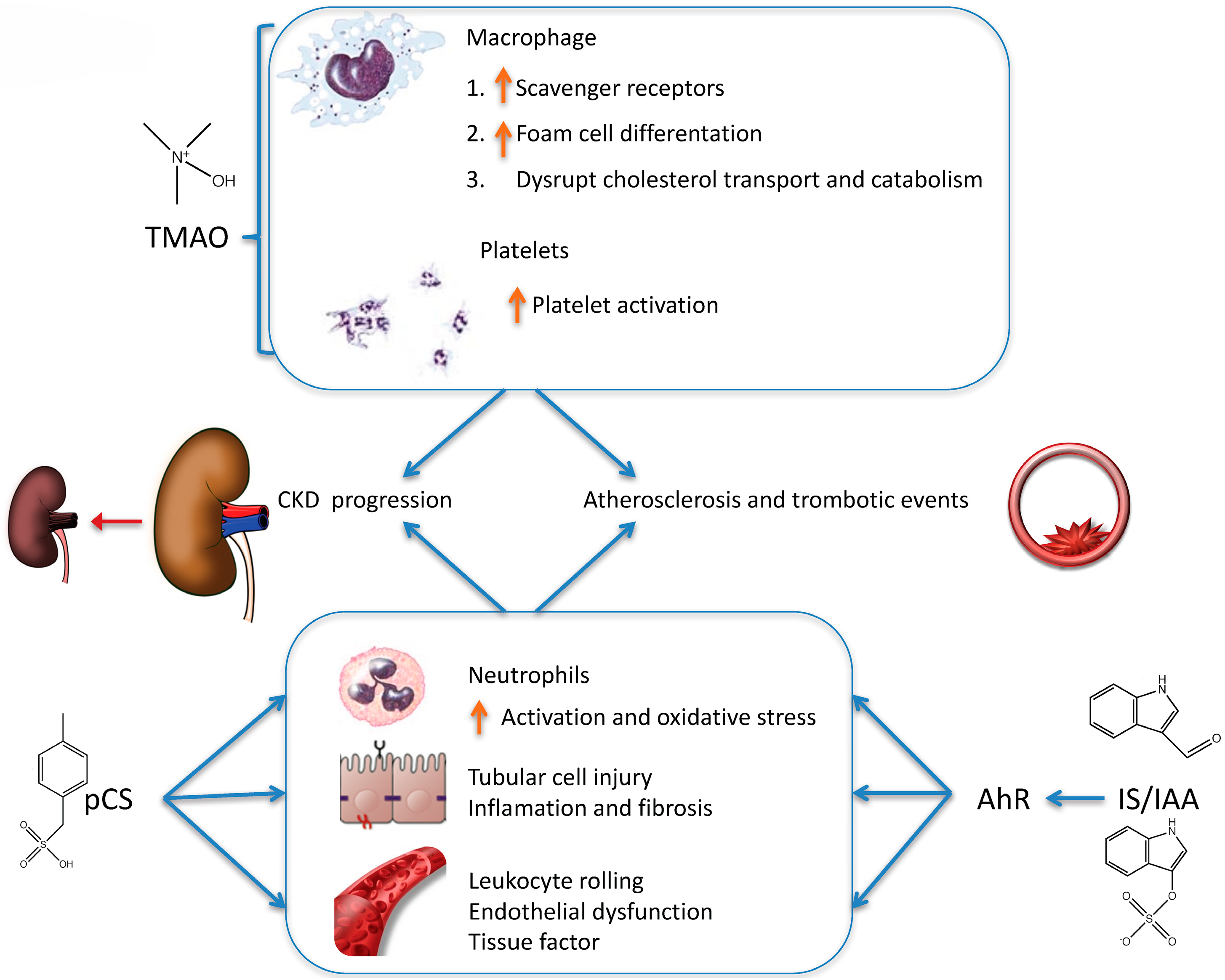
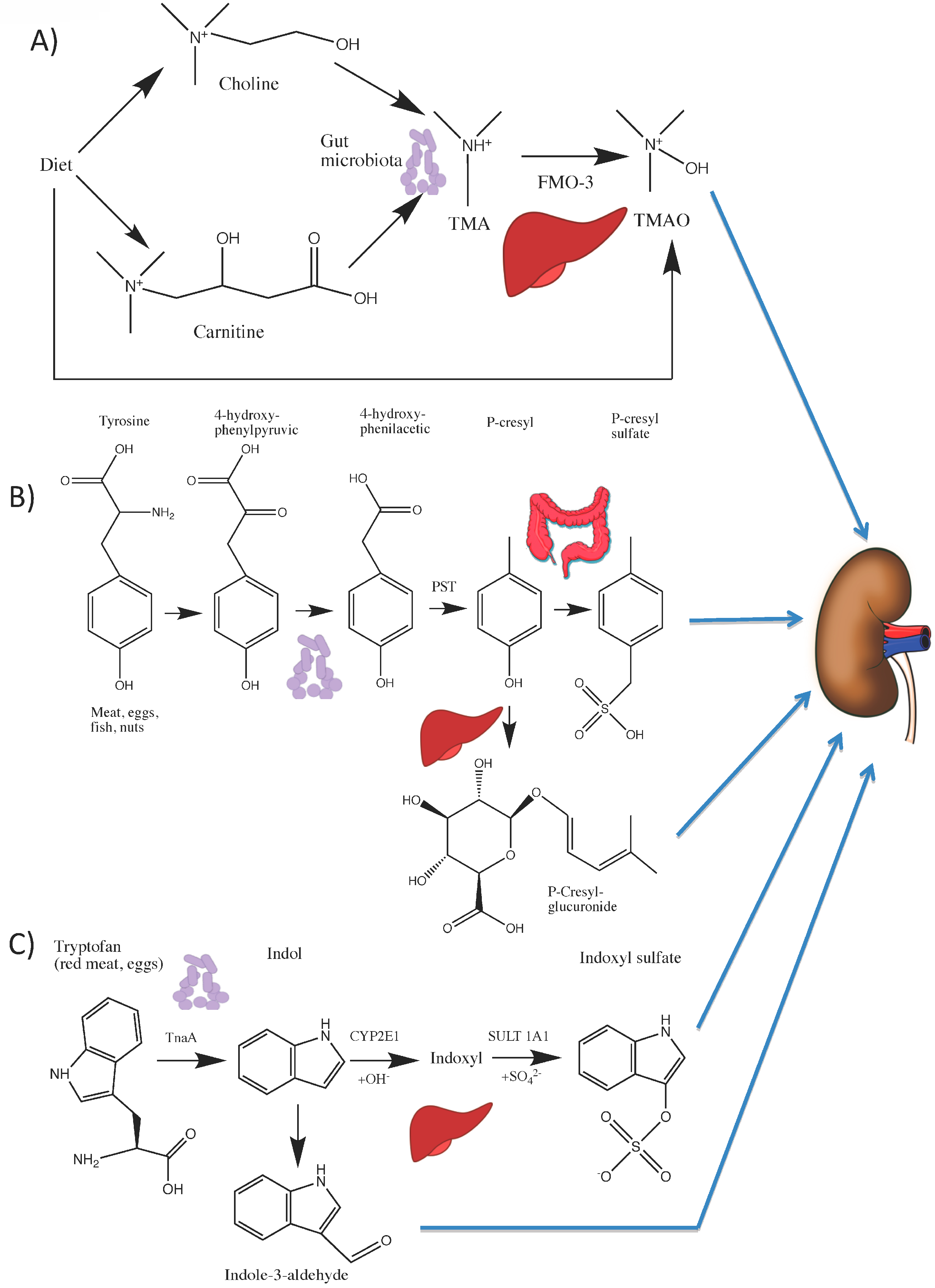
4.1. Choline and l-carnitine Are Metabolized to TMAO
4.2. Tryptophan Is Metabolized to Indoxyl Sulfate (IS) and Indole-3 Acetic Acid (IAA)
4.3. Tyrosine Is Metabolized to p-Cresyl Sulfate (pCS)
5. Potential Therapeutic Implications
Acknowledgments
Author Contributions
Conflicts of Interest
References
- United States Renal Data System, 2014 Annual Data Report: Epidemiology of Kidney Disease in the United States. Available online: https://iths.pure.elsevier.com/en/publications/us-renal-data-system-2014-annual-data-report-epidemiology-of-kidn (accessed on 20 April 2017).
- Brück, K.; Stel, V.S.; Gambaro, G.; Hallan, S.; Völzke, H.; Ärnlöv, J.; Kastarinen, M.; Guessous, I.; Vinhas, J.; Stengel, B.; et al. CKD Prevalence Varies across the European General Population. J. Am. Soc. Nephrol. JASN 2016, 27, 2135–2147. [Google Scholar] [CrossRef] [PubMed]
- Stevens, L.A.; Li, S.; Wang, C.; Huang, C.; Becker, B.N.; Bomback, A.S.; Brown, W.W.; Burrows, N.R.; Jurkovitz, C.T.; McFarlane, S.I.; et al. Prevalence of CKD and comorbid illness in elderly patients in the United States: Results from the Kidney Early Evaluation Program (KEEP). Am. J. Kidney Dis. 2010, 55, S23–S33. [Google Scholar] [CrossRef] [PubMed]
- GBD 2015 Mortality and Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1459–1544. [Google Scholar]
- Ortiz, A.; Covic, A.; Fliser, D.; Fouque, D.; Goldsmith, D.; Kanbay, M.; Mallamaci, F.; Massy, Z.A.; Rossignol, P.; Vanholder, R.; et al. Epidemiology, contributors to, and clinical trials of mortality risk in chronic kidney failure. Lancet 2014, 383, 1831–1843. [Google Scholar] [CrossRef]
- Vanholder, R.; Glorieux, G. The intestine and the kidneys: A bad marriage can be hazardous. Clin. Kidney J. 2015, 8, 168–179. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Fouque, D.; Glorieux, G.; Heine, G.H.; Kanbay, M.; Mallamaci, F.; Massy, Z.A.; Ortiz, A.; Rossignol, P.; Wiecek, A.; et al. Clinical management of the uraemic syndrome in chronic kidney disease. Lancet. Diabetes Endocrinol. 2016, 4, 360–373. [Google Scholar] [CrossRef]
- Vanholder, R.; De Smet, R.; Glorieux, G.; Argilés, A.; Baurmeister, U.; Brunet, P.; Clark, W.; Cohen, G.; De Deyn, P.P.; Deppisch, R.; et al. Review on uremic toxins: Classification, concentration, and interindividual variability. Kidney Int. 2003, 63, 1934–1943. [Google Scholar] [CrossRef] [PubMed]
- Duranton, F.; Cohen, G.; De Smet, R.; Rodriguez, M.; Jankowski, J.; Vanholder, R.; Argiles, A.; European Uremic Toxin Work Group. Normal and pathologic concentrations of uremic toxins. J. Am. Soc. Nephrol. JASN 2012, 23, 1258–1270. [Google Scholar] [CrossRef] [PubMed]
- Uremic Toxin–Data Base. Available online: http://www.uremic-toxins.org/DataBase.html (accessed on 20 October 2016).
- Bosch-Panadero, E.; Mas, S.; Sanchez-Ospina, D.; Camarero, V.; Pérez-Gómez, M.V.; Saez-Calero, I.; Abaigar, P.; Ortiz, A.; Egido, J.; González-Parra, E. The Choice of Hemodialysis Membrane Affects Bisphenol A Levels in Blood. J. Am. Soc. Nephrol. JASN 2016, 27, 1566–1574. [Google Scholar] [CrossRef] [PubMed]
- González-Parra, E.; Herrero, J.A.; Elewa, U.; Bosch, R.J.; Arduán, A.O.; Egido, J. Bisphenol a in chronic kidney disease. Int. J. Nephrol. 2013, 2013, 437857. [Google Scholar] [CrossRef] [PubMed]
- Lederberg, J.; McCray, A.T. ‘Ome Sweet’ Omics—A Genealogical Treasury of Words Genealogical Treasury of Words. Scientist 2001, 15, 8. [Google Scholar]
- Sabatino, A.; Regolisti, G.; Brusasco, I.; Cabassi, A.; Morabito, S.; Fiaccadori, E. Alterations of intestinal barrier and microbiota in chronic kidney disease. Nephrol. Dial. Transplant. 2015, 30, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Anand, P.K.; Malireddi, R.K.S.; Lukens, J.R.; Vogel, P.; Bertin, J.; Lamkanfi, M.; Kanneganti, T.-D. NLRP6 negatively regulates innate immunity and host defence against bacterial pathogens. Nature 2012, 488, 389–393. [Google Scholar] [CrossRef] [PubMed]
- Borges, N.A.; Barros, A.F.; Nakao, L.S.; Dolenga, C.J.; Fouque, D.; Mafra, D. Protein-Bound Uremic Toxins from Gut Microbiota and Inflammatory Markers in Chronic Kidney Disease. J. Ren. Nutr. 2016, 26, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.T.; Nieuwdorp, M.; Bäckhed, F. Microbial modulation of insulin sensitivity. Cell Metab. 2014, 20, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Integrative HMP (iHMP) Research Network Consortium. The Integrative Human Microbiome Project: Dynamic analysis of microbiome-host omics profiles during periods of human health and disease. Cell Host Microbe 2014, 16, 276–289. [Google Scholar]
- iHMP. Available online: http://hmp2.org (accessed on 20 April 2017).
- Johnson, A.M.F.; Olefsky, J.M. The origins and drivers of insulin resistance. Cell 2013, 152, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Cigarran Guldris, S.; González Parra, E.; Cases Amenós, A. Gut microbiota in chronic kidney disease. Nefrologia 2017, 37, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, N.D.; Wong, J.; Pahl, M.; Piceno, Y.M.; Yuan, J.; DeSantis, T.Z.; Ni, Z.; Nguyen, T.-H.; Andersen, G.L. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013, 83, 308–315. [Google Scholar] [CrossRef] [PubMed]
- Wong, J.; Piceno, Y.M.; Desantis, T.Z.; Pahl, M.; Andersen, G.L.; Vaziri, N.D. Expansion of urease- and uricase-containing, indole- and p-cresol-forming and contraction of short-chain fatty acid-producing intestinal microbiota in ESRD. Am. J. Nephrol. 2014, 39, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Parra, E.; Tuñón, J.; Egido, J.; Ortiz, A. Phosphate: A stealthier killer than previously thought? Cardiovasc. Pathol. 2012, 21, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Gutiérrez, O.M.; Wolf, M. A blueprint for randomized trials targeting phosphorus metabolism in chronic kidney disease. Kidney Int. 2009, 76, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.; Sacks, F.; Pfeffer, M.; Gao, Z.; Curhan, G.; Cholesterol and Recurrent Events Trial Investigators. Relation between serum phosphate level and cardiovascular event rate in people with coronary disease. Circulation 2005, 112, 2627–2633. [Google Scholar] [CrossRef] [PubMed]
- Dhingra, R.; Sullivan, L.M.; Fox, C.S.; Wang, T.J.; D’Agostino, R.B.; Gaziano, J.M.; Vasan, R.S. Relations of serum phosphorus and calcium levels to the incidence of cardiovascular disease in the community. Arch. Intern. Med. 2007, 167, 879–885. [Google Scholar] [CrossRef] [PubMed]
- Van Kuijk, J.-P.; Flu, W.-J.; Chonchol, M.; Valentijn, T.M.; Verhagen, H.J.M.; Bax, J.J.; Poldermans, D. Elevated preoperative phosphorus levels are an independent risk factor for cardiovascular mortality. Am. J. Nephrol. 2010, 32, 163–168. [Google Scholar] [CrossRef] [PubMed]
- Foley, R.N.; Collins, A.J.; Ishani, A.; Kalra, P.A. Calcium-phosphate levels and cardiovascular disease in community-dwelling adults: The Atherosclerosis Risk in Communities (ARIC) Study. Am. Heart J. 2008, 156, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Zoccali, C.; Ruggenenti, P.; Perna, A.; Leonardis, D.; Tripepi, R.; Tripepi, G.; Mallamaci, F.; Remuzzi, G.; REIN Study Group. Phosphate may promote CKD progression and attenuate renoprotective effect of ACE inhibition. J. Am. Soc. Nephrol. JASN 2011, 22, 1923–1930. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Gutiérrez, O.M.; Chang, Y.; Shah, A.; Tamez, H.; Smith, K.; Thadhani, R.; Wolf, M. Phosphorus binders and survival on hemodialysis. J. Am. Soc. Nephrol. JASN 2009, 20, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Hénaut, L.; Sanz, A.B.; Martin-Sanchez, D.; Carrasco, S.; Villa-Bellosta, R.; Aldamiz-Echevarria, G.; Massy, Z.A.; Sanchez-Nino, M.D.; Ortiz, A. TWEAK favors phosphate-induced calcification of vascular smooth muscle cells through canonical and non-canonical activation of NFκB. Cell Death Dis. 2016, 7, e2305. [Google Scholar] [CrossRef] [PubMed]
- Shuto, E.; Taketani, Y.; Tanaka, R.; Harada, N.; Isshiki, M.; Sato, M.; Nashiki, K.; Amo, K.; Yamamoto, H.; Higashi, Y.; et al. Dietary phosphorus acutely impairs endothelial function. J. Am. Soc. Nephrol. JASN 2009, 20, 1504–1512. [Google Scholar] [CrossRef] [PubMed]
- Saito, H.; Kusano, K.; Kinosaki, M.; Ito, H.; Hirata, M.; Segawa, H.; Miyamoto, K.-I.; Fukushima, N. Human fibroblast growth factor-23 mutants suppress Na+-dependent phosphate co-transport activity and 1alpha,25-dihydroxyvitamin D3 production. J. Biol. Chem. 2003, 278, 2206–2211. [Google Scholar] [CrossRef] [PubMed]
- Urakawa, I.; Yamazaki, Y.; Shimada, T.; Iijima, K.; Hasegawa, H.; Okawa, K.; Fujita, T.; Fukumoto, S.; Yamashita, T. Klotho converts canonical FGF receptor into a specific receptor for FGF23. Nature 2006, 444, 770–774. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Izquierdo, M.C.; Sanchez-Niño, M.D.; Suárez-Alvarez, B.; Lopez-Larrea, C.; Jakubowski, A.; Blanco, J.; Ramirez, R.; Selgas, R.; Ruiz-Ortega, M.; et al. The inflammatory cytokines TWEAK and TNFα reduce renal klotho expression through NFκB. J. Am. Soc. Nephrol. JASN 2011, 22, 1315–1325. [Google Scholar] [CrossRef] [PubMed]
- De Seigneux, S.; Courbebaisse, M.; Rutkowski, J.M.; Wilhelm-Bals, A.; Metzger, M.; Khodo, S.N.; Hasler, U.; Chehade, H.; Dizin, E.; Daryadel, A.; et al. Proteinuria Increases Plasma Phosphate by Altering Its Tubular Handling. J. Am. Soc. Nephrol. JASN 2015, 26, 1608–1618. [Google Scholar] [CrossRef] [PubMed]
- Morishita, K.; Shirai, A.; Kubota, M.; Katakura, Y.; Nabeshima, Y.; Takeshige, K.; Kamiya, T. The progression of aging in klotho mutant mice can be modified by dietary phosphorus and zinc. J. Nutr. 2001, 131, 3182–3188. [Google Scholar] [PubMed]
- Gutierrez, O.; Isakova, T.; Rhee, E.; Shah, A.; Holmes, J.; Collerone, G.; Jüppner, H.; Wolf, M. Fibroblast growth factor-23 mitigates hyperphosphatemia but accentuates calcitriol deficiency in chronic kidney disease. J. Am. Soc. Nephrol. JASN 2005, 16, 2205–2215. [Google Scholar] [CrossRef] [PubMed]
- Rossaint, J.; Oehmichen, J.; Van Aken, H.; Reuter, S.; Pavenstädt, H.J.; Meersch, M.; Unruh, M.; Zarbock, A. FGF23 signaling impairs neutrophil recruitment and host defense during CKD. J. Clin. Investig. 2016, 126, 962–974. [Google Scholar] [CrossRef] [PubMed]
- González-Parra, E.; Aceña, Á.; Lorenzo, Ó.; Tarín, N.; González-Casaus, M.L.; Cristóbal, C.; Huelmos, A.; Mahíllo-Fernández, I.; Pello, A.M.; Carda, R.; et al. Important abnormalities of bone mineral metabolism are present in patients with coronary artery disease with a mild decrease of the estimated glomerular filtration rate. J. Bone Miner. Metab. 2016, 34, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Tuñón, J.; Fernández-Fernández, B.; Carda, R.; Pello, A.M.; Cristóbal, C.; Tarín, N.; Aceña, Á.; González-Casaus, M.L.; Huelmos, A.; Alonso, J.; et al. Circulating fibroblast growth factor-23 plasma levels predict adverse cardiovascular outcomes in patients with diabetes mellitus with coronary artery disease. Diabetes Metab. Res. Rev. 2016, 32, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Aceña, Á.; Pello, A.M.; Carda, R.; Lorenzo, Ó.; Gonzalez-Casaus, M.L.; Blanco-Colio, L.M.; Martín-Ventura, J.L.; Palfy, J.; Orejas, M.; Rábago, R.; et al. Parathormone Levels Are Independently Associated with the Presence of Left Ventricular Hypertrophy in Patients with Coronary Artery Disease. J. Nutr. Health Aging 2016, 20, 659–664. [Google Scholar] [CrossRef] [PubMed]
- Tuñón, J.; Cristóbal, C.; Tarín, N.; Aceña, Á.; González-Casaus, M.L.; Huelmos, A.; Alonso, J.; Lorenzo, Ó.; González-Parra, E.; Mahíllo-Fernández, I.; et al. Coexistence of low vitamin D and high fibroblast growth factor-23 plasma levels predicts an adverse outcome in patients with coronary artery disease. PLoS ONE 2014, 9, e95402. [Google Scholar] [CrossRef] [PubMed]
- Leelarungrayub, J.; Yankai, A.; Pinkaew, D.; Puntumetakul, R.; Laskin, J.J.; Bloomer, R.J. A preliminary study on the effects of star fruit consumption on antioxidant and lipid status in elderly Thai individuals. Clin. Interv. Aging 2016, 11, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Leelarungrayub, J.; Laskin, J.J.; Bloomer, R.J.; Pinkaew, D. Consumption of star fruit juice on pro-inflammatory markers and walking distance in the community dwelling elderly. Arch. Gerontol. Geriatr. 2016, 64, 6–12. [Google Scholar] [CrossRef] [PubMed]
- Xie, Q.; Zhang, S.; Chen, C.; Li, J.; Wei, X.; Xu, X.; Xuan, F.; Chen, N.; Pham, T.; Qin, N.; et al. Protective Effect of 2-Dodecyl-6-Methoxycyclohexa-2, 5-Diene-1, 4-Dione, Isolated from Averrhoa carambola, L., Against Palmitic Acid-Induced Inflammation and Apoptosis in Min6 Cells by Inhibiting the TLR4-MyD88-NF-κB Signaling Pathway. Cell. Physiol. Biochem. 2016, 39, 1705–1715. [Google Scholar] [CrossRef] [PubMed]
- Muir, C.K.; Lam, C.K. Depressant action of Averrhoa carambola. Med. J. Malays. 1980, 34, 279–280. [Google Scholar]
- Garcia-Cairasco, N.; Moyses-Neto, M.; Del Vecchio, F.; Oliveira, J.A.C.; dos Santos, F.L.; Castro, O.W.; Arisi, G.M.; Dantas, M.; Carolino, R.O.G.; Coutinho-Netto, J.; et al. Elucidating the neurotoxicity of the star fruit. Angew. Chem. Int. Ed. 2013, 52, 13067–13070. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, E.S.M.; de Aguiar, A.S. Why eating star fruit is prohibited for patients with chronic kidney disease? J. Bras. Nefrol. 2015, 37, 241–247. [Google Scholar] [CrossRef] [PubMed]
- Abeysekera, R.A.; Wijetunge, S.; Nanayakkara, N.; Wazil, A.W.M.; Ratnatunga, N.V.I.; Jayalath, T.; Medagama, A. Star fruit toxicity: A cause of both acute kidney injury and chronic kidney disease: A report of two cases. BMC Res. Notes 2015, 8, 796. [Google Scholar] [CrossRef] [PubMed]
- Neto, M.M.; Silva, G.E.B.; Costa, R.S.; Vieira Neto, O.M.; Garcia-Cairasco, N.; Lopes, N.P.; Haendchen, P.F.C.; Silveira, C.; Mendes, A.R.; Filho, R.R.; et al. Star fruit: Simultaneous neurotoxic and nephrotoxic effects in people with previously normal renal function. NDT Plus 2009, 2, 485–488. [Google Scholar] [CrossRef] [PubMed]
- Neto, M.M.; da Costa, J.A.C.; Garcia-Cairasco, N.; Netto, J.C.; Nakagawa, B.; Dantas, M. Intoxication by star fruit (Averrhoa carambola) in 32 uraemic patients: Treatment and outcome. Nephrol. Dial. Transplant. 2003, 18, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.-C.; Lee, P.-T.; Lu, P.-J.; Chen, C.-L.; Chang, T.-Y.; Hsu, C.-Y.; Chung, H.-M.; Chou, K.-J. Mechanisms of star fruit-induced acute renal failure. Food Chem. Toxicol. 2008, 46, 1744–1752. [Google Scholar] [CrossRef] [PubMed]
- Husi, H.; Sanchez-Niño, M.D.; Delles, C.; Mullen, W.; Vlahou, A.; Ortiz, A.; Mischak, H. A combinatorial approach of Proteomics and Systems Biology in unravelling the mechanisms of acute kidney injury (AKI): Involvement of NMDA receptor GRIN1 in murine AKI. BMC Syst. Biol. 2013, 7, 110. [Google Scholar] [CrossRef] [PubMed]
- Mydlík, M.; Derzsiová, K. Oxalic Acid as a uremic toxin. J. Ren. Nutr. 2008, 18, 33–39. [Google Scholar] [CrossRef] [PubMed]
- Poesen, R.; Meijers, B.; Evenepoel, P. The colon: An overlooked site for therapeutics in dialysis patients. Semin. Dial. 2013, 26, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Getting, J.E.; Gregoire, J.R.; Phul, A.; Kasten, M.J. Oxalate nephropathy due to “juicing”: Case report and review. Am. J. Med. 2013, 126, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, S.H.; da Costa, K.-A. Choline: An essential nutrient for public health. Nutr. Rev. 2009, 67, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Hollenbeck, C.B. An introduction to the nutrition and metabolism of choline. Cent. Nerv. Syst. Agents Med. Chem. 2012, 12, 100–113. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine (US) Standing Committee on the Scientific Evaluation of Dietary Reference Intakes and its Panel on Folate, Other B Vitamins, and Choline. In Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline; National Academies Press: Washington, DC, USA, 1998.
- USDA Nutrients Database. Available online: https://ndb.nal.usda.gov/ndb/ (accessed on 22 April 2017).
- Rennick, B.; Acara, M.; Hysert, P.; Mookerjee, B. Choline loss during hemodialysis: Homeostatic control of plasma choline concentrations. Kidney Int. 1976, 10, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; West, A.A.; Caudill, M.A. Maternal choline supplementation: A nutritional approach for improving offspring health? Trends Endocrinol. Metab. TEM 2014, 25, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Mi, W.; van Wijk, N.; Cansev, M.; Sijben, J.W.C.; Kamphuis, P.J.G.H. Nutritional approaches in the risk reduction and management of Alzheimer’s disease. Nutrition 2013, 29, 1080–1089. [Google Scholar] [CrossRef] [PubMed]
- Conlay, L.A.; Wurtman, R.J.; Blusztajn, K.; Coviella, I.L.; Maher, T.J.; Evoniuk, G.E. Decreased plasma choline concentrations in marathon runners. N. Engl. J. Med. 1986, 315, 892. [Google Scholar] [PubMed]
- Buchman, A.L.; Awal, M.; Jenden, D.; Roch, M.; Kang, S.H. The effect of lecithin supplementation on plasma choline concentrations during a marathon. J. Am. Coll. Nutr. 2000, 19, 768–770. [Google Scholar] [CrossRef] [PubMed]
- Leermakers, E.T.M.; Moreira, E.M.; Kiefte-de Jong, J.C.; Darweesh, S.K.L.; Visser, T.; Voortman, T.; Bautista, P.K.; Chowdhury, R.; Gorman, D.; Bramer, W.M.; et al. Effects of choline on health across the life course: A systematic review. Nutr. Rev. 2015, 73, 500–522. [Google Scholar] [CrossRef] [PubMed]
- Boyd, W.D.; Graham-White, J.; Blackwood, G.; Glen, I.; McQueen, J. Clinical effects of choline in Alzheimer senile dementia. Lancet 1977, 2, 711. [Google Scholar] [CrossRef]
- Sanchez-Niño, M.D.; Ortiz, A. Differential effects of oral and intravenous l-carnitine on serum lipids: Is the microbiota the answer? Clin. Kidney J. 2014, 7, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Steiber, A.; Kerner, J.; Hoppel, C.L. Carnitine: A nutritional, biosynthetic, and functional perspective. Mol. Asp. Med. 2004, 25, 455–473. [Google Scholar] [CrossRef] [PubMed]
- Guarnieri, G. Carnitine in maintenance hemodialysis patients. J. Ren. Nutr. 2015, 25, 169–175. [Google Scholar] [CrossRef] [PubMed]
- Rospond, B.; Chłopicka, J. The biological function of l-carnitine and its content in the particular food examples. Prz. Lek. 2013, 70, 85–91. [Google Scholar] [PubMed]
- Evans, A. Dialysis-related carnitine disorder and levocarnitine pharmacology. Am. J. Kidney Dis. 2003, 41, S13–S26. [Google Scholar] [CrossRef]
- Evans, A.M.; Fornasini, G. Pharmacokinetics of L-Carnitine. Clin. Pharmacokinet. 2003, 42, 941–967. [Google Scholar] [CrossRef] [PubMed]
- Bain, M.A.; Milne, R.W.; Evans, A.M. Disposition and metabolite kinetics of oral l-carnitine in humans. J. Clin. Pharmacol. 2006, 46, 1163–1170. [Google Scholar] [CrossRef] [PubMed]
- Kerner, J.; Hoppel, C. Genetic disorders of carnitine metabolism and their nutritional management. Ann. Rev. Nutr. 1998, 18, 179–206. [Google Scholar] [CrossRef] [PubMed]
- Kalim, S.; Clish, C.B.; Wenger, J.; Elmariah, S.; Yeh, R.W.; Deferio, J.J.; Pierce, K.; Deik, A.; Gerszten, R.E.; Thadhani, R.; et al. A plasma long-chain acylcarnitine predicts cardiovascular mortality in incident dialysis patients. J. Am. Heart Assoc. 2013, 2, e000542. [Google Scholar] [CrossRef] [PubMed]
- Centers for Medicare & Medicaid Services (CMS); United States Department of Health and Human Services (HHS). Medicare program; end-stage renal disease quality incentive program. Final rule. Fed. Regist. 2011, 76, 627–646. [Google Scholar]
- Wasserstein, A.G. l-carnitine supplementation in dialysis: Treatment in quest of disease. Semin. Dial. 2013, 26, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Clinical practice guidelines for nutrition in chronic renal failure. K/DOQI, National Kidney Foundation. Am. J. Kidney Dis. 2000, 35, S1–S140.
- KDOQI Work Group. KDOQI Clinical Practice Guideline for Nutrition in Children with CKD: 2008 update. Executive summary. Am. J. Kidney Dis. 2009, 53, S11–S104. [Google Scholar]
- Kidney Disease: Improving Global Outcomes (KDIGO) Anemia Work Group. KDIGO Clinical Practice Guideline for Anemia in Chronic Kidney Disease. Kidney Int. Suppl. 2012, 2, 279. [Google Scholar]
- Kliger, A.S.; Foley, R.N.; Goldfarb, D.S.; Goldstein, S.L.; Johansen, K.; Singh, A.; Szczech, L. KDOQI US commentary on the 2012 KDIGO Clinical Practice Guideline for Anemia in CKD. Am. J. Kidney Dis. 2013, 62, 849–859. [Google Scholar] [CrossRef] [PubMed]
- Molyneux, R.; Seymour, A.-M.; Bhandari, S. Value of carnitine therapy in kidney dialysis patients and effects on cardiac function from human and animal studies. Curr. Drug Targets 2012, 13, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Abbate, M.; Tang, L.; Cai, G.; Gong, Z.; Wei, R.; Zhou, J.; Chen, X. l-Carnitine supplementation for adults with end-stage kidney disease requiring maintenance hemodialysis: A systematic review and meta-analysis. Am. J. Clin. Nutr. 2014, 99, 408–422. [Google Scholar] [CrossRef] [PubMed]
- Moraes, C.; Fouque, D.; Amaral, A.C.F.; Mafra, D. Trimethylamine N-Oxide From Gut Microbiota in Chronic Kidney Disease Patients: Focus on Diet. J. Ren. Nutr. 2015, 25, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of l-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.Q.; Mitchell, S.C.; Smith, R.L. Dietary precursors of trimethylamine in man: A pilot study. Food Chem. Toxicol. 1999, 37, 515–520. [Google Scholar] [CrossRef]
- Ufnal, M.; Zadlo, A.; Ostaszewski, R. TMAO: A small molecule of great expectations. Nutrition 2015, 31, 1317–1323. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, F.; Esteves, S.; Almeida, L.S.; Gaspar, A.; da Costa, C.D.; Janeiro, P.; Bandeira, A.; Martins, E.; Teles, E.L.; Garcia, P.; et al. Trimethylaminuria (fish odor syndrome): Genotype characterization among Portuguese patients. Gene 2013, 527, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Bain, M.A.; Faull, R.; Fornasini, G.; Milne, R.W.; Evans, A.M. Accumulation of trimethylamine and trimethylamine-N-oxide in end-stage renal disease patients undergoing haemodialysis. Nephrol. Dial. Transplant. 2006, 21, 1300–1304. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut microbiota-dependent trimethylamine N-oxide (TMAO) pathway contributes to both development of renal insufficiency and mortality risk in chronic kidney disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef] [PubMed]
- Posada-Ayala, M.; Zubiri, I.; Martin-Lorenzo, M.; Sanz-Maroto, A.; Molero, D.; Gonzalez-Calero, L.; Fernandez-Fernandez, B.; de la Cuesta, F.; Laborde, C.M.; Barderas, M.G.; et al. Identification of a urine metabolomic signature in patients with advanced-stage chronic kidney disease. Kidney Int. 2014, 85, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.A.; Corbin, K.D.; da Costa, K.-A.; Zhang, S.; Zhao, X.; Galanko, J.A.; Blevins, T.; Bennett, B.J.; O’Connor, A.; Zeisel, S.H. Effect of egg ingestion on trimethylamine-N-oxide production in humans: A randomized, controlled, dose-response study. Am. J. Clin. Nutr. 2014, 100, 778–786. [Google Scholar] [CrossRef] [PubMed]
- McEntyre, C.J.; Lever, M.; Chambers, S.T.; George, P.M.; Slow, S.; Elmslie, J.L.; Florkowski, C.M.; Lunt, H.; Krebs, J.D. Variation of betaine, N,N-dimethylglycine, choline, glycerophosphorylcholine, taurine and trimethylamine-N-oxide in the plasma and urine of overweight people with type 2 diabetes over a two-year period. Ann. Clin. Biochem. 2015, 52, 352–360. [Google Scholar] [CrossRef] [PubMed]
- Bain, M.A.; Faull, R.; Milne, R.W.; Evans, A.M. Oral l-carnitine: Metabolite formation and hemodialysis. Curr. Drug Metab. 2006, 7, 811–816. [Google Scholar] [CrossRef] [PubMed]
- Ozasa, H.; Shimizu, M.; Koizumi, A.; Wakabayashi, A.; Yamazaki, H. Trimethylamine generation in patients receiving hemodialysis treated with l-carnitine. Clin. Kidney J. 2014, 7, 329. [Google Scholar] [CrossRef] [PubMed]
- Kuhlmann, M.K.; Yoshino, M.; Levin, N.W. Differences in cardiovascular mortality rates among hemodialysis patients in the United States and Japan: The importance of background cardiovascular mortality. Hemodialysis international. Int. Symp. Home Hemodial. 2004, 8, 394–399. [Google Scholar] [CrossRef] [PubMed]
- Shafi, T.; Powe, N.R.; Meyer, T.W.; Hwang, S.; Hai, X.; Melamed, M.L.; Banerjee, T.; Coresh, J.; Hostetter, T.H. Trimethylamine N-Oxide and Cardiovascular Events in Hemodialysis Patients. J. Am. Soc. Nephrol. JASN 2017, 28, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef] [PubMed]
- Lew, Q.-L.J.; Jafar, T.H.; Koh, H.W.L.; Jin, A.; Chow, K.Y.; Yuan, J.-M.; Koh, W.-P. Red Meat Intake and Risk of ESRD. J. Am. Soc. Nephrol. JASN 2017, 28, 304–312. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; Dugar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.-M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Hazen, S.L. Microbiome, trimethylamine N-oxide, and cardiometabolic disease. Transl. Res. 2017, 179, 108–115. [Google Scholar] [CrossRef] [PubMed]
- Aron-Wisnewsky, J.; Clément, K. The gut microbiome, diet, and links to cardiometabolic and chronic disorders. Nat. Rev. Nephrol. 2016, 12, 169–181. [Google Scholar] [CrossRef] [PubMed]
- Senthong, V.; Li, X.S.; Hudec, T.; Coughlin, J.; Wu, Y.; Levison, B.; Wang, Z.; Hazen, S.L.; Tang, W.H.W. Plasma Trimethylamine N-Oxide, a Gut Microbe-Generated Phosphatidylcholine Metabolite, Is Associated With Atherosclerotic Burden. J. Am. Coll. Cardiol. 2016, 67, 2620–2628. [Google Scholar] [CrossRef] [PubMed]
- Obeid, R.; Awwad, H.M.; Rabagny, Y.; Graeber, S.; Herrmann, W.; Geisel, J. Plasma trimethylamine N-oxide concentration is associated with choline, phospholipids, and methyl metabolism. Am. J. Clin. Nutr. 2016, 103, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Wang, Z.; Fan, Y.; Levison, B.; Hazen, J.E.; Donahue, L.M.; Wu, Y.; Hazen, S.L. Prognostic value of elevated levels of intestinal microbe-generated metabolite trimethylamine-N-oxide in patients with heart failure: Refining the gut hypothesis. J. Am. Coll. Cardiol. 2014, 64, 1908–1914. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, T.; Heaney, L.M.; Bhandari, S.S.; Jones, D.J.L.; Ng, L.L. Trimethylamine N-oxide and prognosis in acute heart failure. Heart (Br. Card. Soc.) 2016, 102, 841–848. [Google Scholar]
- Ma, J.; Pazos, I.M.; Gai, F. Microscopic insights into the protein-stabilizing effect of trimethylamine N-oxide (TMAO). Proc. Natl. Acad. Sci. USA 2014, 111, 8476–8481. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Tamarappoo, B.K.; Verkman, A.S. Defective aquaporin-2 trafficking in nephrogenic diabetes insipidus and correction by chemical chaperones. J. Clin. Investig. 1998, 101, 2257–2267. [Google Scholar] [CrossRef] [PubMed]
- Tatzelt, J.; Prusiner, S.B.; Welch, W.J. Chemical chaperones interfere with the formation of scrapie prion protein. EMBO J. 1996, 15, 6363–6373. [Google Scholar] [PubMed]
- Hong, J.; Xiong, S. TMAO-Protein Preferential Interaction Profile Determines TMAO’s Conditional In Vivo Compatibility. Biophys. J. 2016, 111, 1866–1875. [Google Scholar] [CrossRef] [PubMed]
- Energy and protein requirements. Report of a joint FAO/WHO/UNU Expert Consultation. World Health Organ. Tech. Rep. Ser. 1985, 724, 1–206.
- Kalim, S.; Clish, C.B.; Deferio, J.J.; Ortiz, G.; Moffet, A.S.; Gerszten, R.E.; Thadhani, R.; Rhee, E.P. Cross-sectional examination of metabolites and metabolic phenotypes in uremia. BMC Nephrol. 2015, 16, 98. [Google Scholar] [CrossRef] [PubMed]
- Koenig, P.; Nagl, C.; Neurauter, G.; Schennach, H.; Brandacher, G.; Fuchs, D. Enhanced degradation of tryptophan in patients on hemodialysis. Clin. Nephrol. 2010, 74, 465–470. [Google Scholar] [CrossRef] [PubMed]
- Schefold, J.C.; Zeden, J.-P.; Fotopoulou, C.; von Haehling, S.; Pschowski, R.; Hasper, D.; Volk, H.-D.; Schuett, C.; Reinke, P. Increased indoleamine 2,3-dioxygenase (IDO) activity and elevated serum levels of tryptophan catabolites in patients with chronic kidney disease: A possible link between chronic inflammation and uraemic symptoms. Nephrol. Dial. Transplant. 2009, 24, 1901–1908. [Google Scholar] [CrossRef] [PubMed]
- Lovelace, M.D.; Varney, B.; Sundaram, G.; Franco, N.F.; Ng, M.L.; Pai, S.; Lim, C.K.; Guillemin, G.J.; Brew, B.J. Current Evidence for a Role of the Kynurenine Pathway of Tryptophan Metabolism in Multiple Sclerosis. Front. Immunol. 2016, 7, 246. [Google Scholar] [CrossRef] [PubMed]
- Ravindran, A.V.; da Silva, T.L. Complementary and alternative therapies as add-on to pharmacotherapy for mood and anxiety disorders: A systematic review. J. Affect. Disord. 2013, 150, 707–719. [Google Scholar] [CrossRef] [PubMed]
- Sallée, M.; Dou, L.; Cerini, C.; Poitevin, S.; Brunet, P.; Burtey, S. The aryl hydrocarbon receptor-activating effect of uremic toxins from tryptophan metabolism: A new concept to understand cardiovascular complications of chronic kidney disease. Toxins 2014, 6, 934–949. [Google Scholar] [CrossRef] [PubMed]
- Glassock, R.J. Uremic toxins: What are they? An integrated overview of pathobiology and classification. J. Ren. Nutr. 2008, 18, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Ellis, R.J.; Small, D.M.; Vesey, D.A.; Johnson, D.W.; Francis, R.; Vitetta, L.; Gobe, G.C.; Morais, C. Indoxyl sulphate and kidney disease: Causes, consequences and interventions. Nephrology 2016, 21, 170–177. [Google Scholar] [CrossRef] [PubMed]
- Wing, M.R.; Patel, S.S.; Ramezani, A.; Raj, D.S. Gut microbiome in chronic kidney disease. Exp. Physiol. 2016, 101, 471–477. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.S.; Davies, S.S. Microbial metabolism of dietary components to bioactive metabolites: Opportunities for new therapeutic interventions. Genome Med. 2016, 8, 46. [Google Scholar] [CrossRef] [PubMed]
- Chyan, Y.J.; Poeggeler, B.; Omar, R.A.; Chain, D.G.; Frangione, B.; Ghiso, J.; Pappolla, M.A. Potent neuroprotective properties against the Alzheimer beta-amyloid by an endogenous melatonin-related indole structure, indole-3-propionic acid. J. Biol. Chem. 1999, 274, 21937–21942. [Google Scholar] [CrossRef] [PubMed]
- Vanholder, R.; Schepers, E.; Pletinck, A.; Nagler, E.V.; Glorieux, G. The uremic toxicity of indoxyl sulfate and p-cresyl sulfate: A systematic review. J. Am. Soc. Nephrol. JASN 2014, 25, 1897–1907. [Google Scholar] [CrossRef] [PubMed]
- Barreto, F.C.; Barreto, D.V.; Liabeuf, S.; Meert, N.; Glorieux, G.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; European Uremic Toxin Work Group (EUTox). Serum indoxyl sulfate is associated with vascular disease and mortality in chronic kidney disease patients. Clin. J. Am. Soc. Nephrol. CJASN 2009, 4, 1551–1558. [Google Scholar] [CrossRef] [PubMed]
- Wu, I.-W.; Hsu, K.-H.; Lee, C.-C.; Sun, C.-Y.; Hsu, H.-J.; Tsai, C.-J.; Tzen, C.-Y.; Wang, Y.-C.; Lin, C.-Y.; Wu, M.-S. p-Cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrol. Dial. Transplant. 2011, 26, 938–947. [Google Scholar] [CrossRef] [PubMed]
- Mutsaers, H.A.M.; Stribos, E.G.D.; Glorieux, G.; Vanholder, R.; Olinga, P. Chronic Kidney Disease and Fibrosis: The Role of Uremic Retention Solutes. Front. Med. 2015, 2, 60. [Google Scholar] [CrossRef] [PubMed]
- Buckley, P.D.; Motion, R.L.; Blackwell, L.F.; Hill, J.P. pH effects on cytoplasmic aldehyde dehydrogenase from sheep liver. Adv. Exp. Med. Biol. 1991, 284, 31–41. [Google Scholar] [PubMed]
- Hung, S.-C.; Kuo, K.-L.; Huang, H.-L.; Lin, C.-C.; Tsai, T.-H.; Wang, C.-H.; Chen, J.-W.; Lin, S.-J.; Huang, P.-H.; Tarng, D.-C. Indoxyl sulfate suppresses endothelial progenitor cell-mediated neovascularization. Kidney Int. 2016, 89, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Shivanna, S.; Kolandaivelu, K.; Shashar, M.; Belghasim, M.; Al-Rabadi, L.; Balcells, M.; Zhang, A.; Weinberg, J.; Francis, J.; Pollastri, M.P.; et al. The Aryl Hydrocarbon Receptor is a Critical Regulator of Tissue Factor Stability and an Antithrombotic Target in Uremia. J. Am. Soc. Nephrol. JASN 2016, 27, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Chitalia, V.C.; Shivanna, S.; Martorell, J.; Balcells, M.; Bosch, I.; Kolandaivelu, K.; Edelman, E.R. Uremic serum and solutes increase post-vascular interventional thrombotic risk through altered stability of smooth muscle cell tissue factor. Circulation 2013, 127, 365–376. [Google Scholar] [CrossRef] [PubMed]
- Gondouin, B.; Cerini, C.; Dou, L.; Sallée, M.; Duval-Sabatier, A.; Pletinck, A.; Calaf, R.; Lacroix, R.; Jourde-Chiche, N.; Poitevin, S.; et al. Indolic uremic solutes increase tissue factor production in endothelial cells by the aryl hydrocarbon receptor pathway. Kidney Int. 2013, 84, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Yang, K.; Du, C.; Wang, X.; Li, F.; Xu, Y.; Wang, S.; Chen, S.; Chen, F.; Shen, M.; Chen, M.; et al. Uremic solute indoxyl sulfate-induced platelet hyperactivity contributes to CKD-associated thrombosis in mice. Blood 2017. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.-C.; Hsieh, M.-Y.; Hung, S.-C.; Kuo, K.-L.; Tsai, T.-H.; Lai, C.-L.; Chen, J.-W.; Lin, S.-J.; Huang, P.-H.; Tarng, D.-C. Serum Indoxyl Sulfate Associates with Postangioplasty Thrombosis of Dialysis Grafts. J. Am. Soc. Nephrol. JASN 2016, 27, 1254–1264. [Google Scholar] [CrossRef] [PubMed]
- Wallman, J.; Pettigrew, J.D. Conjugate and disjunctive saccades in two avian species with contrasting oculomotor strategies. J. Neurosci. 1985, 5, 1418–1428. [Google Scholar] [PubMed]
- Nangaku, M.; Mimura, I.; Yamaguchi, J.; Higashijima, Y.; Wada, T.; Tanaka, T. Role of uremic toxins in erythropoiesis-stimulating agent resistance in chronic kidney disease and dialysis patients. J. Ren. Nutr. 2015, 25, 160–163. [Google Scholar] [CrossRef] [PubMed]
- Nii-Kono, T.; Iwasaki, Y.; Uchida, M.; Fujieda, A.; Hosokawa, A.; Motojima, M.; Yamato, H.; Kurokawa, K.; Fukagawa, M. Indoxyl sulfate induces skeletal resistance to parathyroid hormone in cultured osteoblastic cells. Kidney Int. 2007, 71, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Soulage, C.O.; Koppe, L.; Fouque, D. Protein-bound uremic toxins … new targets to prevent insulin resistance and dysmetabolism in patients with chronic kidney disease. J. Ren. Nutr. 2013, 23, 464–466. [Google Scholar] [CrossRef] [PubMed]
- Sato, E.; Mori, T.; Mishima, E.; Suzuki, A.; Sugawara, S.; Kurasawa, N.; Saigusa, D.; Miura, D.; Morikawa-Ichinose, T.; Saito, R.; et al. Metabolic alterations by indoxyl sulfate in skeletal muscle induce uremic sarcopenia in chronic kidney disease. Sci. Rep. 2016, 6, 36618. [Google Scholar] [CrossRef] [PubMed]
- Dou, L.; Sallée, M.; Cerini, C.; Poitevin, S.; Gondouin, B.; Jourde-Chiche, N.; Fallague, K.; Brunet, P.; Calaf, R.; Dussol, B.; et al. The cardiovascular effect of the uremic solute indole-3 acetic acid. J. Am. Soc. Nephrol. JASN 2015, 26, 876–887. [Google Scholar] [CrossRef] [PubMed]
- Thurmond, J.B.; Freeman, G.B.; Soblosky, J.S.; Ieni, J.R.; Brown, J.W. Effects of dietary tyrosine on l-dopa- and amphetamine-induced changes in locomotor activity and neurochemistry in mice. Pharmacol. Biochem. Behav. 1990, 37, 259–266. [Google Scholar] [CrossRef]
- Fernstrom, J.D.; Fernstrom, M.H. Tyrosine, phenylalanine, and catecholamine synthesis and function in the brain. J. Nutr. 2007, 137, 1539S–1547S. [Google Scholar] [PubMed]
- Duranton, F.; Lundin, U.; Gayrard, N.; Mischak, H.; Aparicio, M.; Mourad, G.; Daurès, J.-P.; Weinberger, K.M.; Argilés, A. Plasma and urinary amino acid metabolomic profiling in patients with different levels of kidney function. Clin. J. Am. Soc. Nephrol. CJASN 2014, 9, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Bergström, J.; Alvestrand, A.; Fürst, P. Plasma and muscle free amino acids in maintenance hemodialysis patients without protein malnutrition. Kidney Int. 1990, 38, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Webster, D.; Wildgoose, J. Tyrosine supplementation for phenylketonuria. Cochrane Database Syst. Rev. 2013. [Google Scholar] [CrossRef]
- Jongkees, B.J.; Hommel, B.; Kühn, S.; Colzato, L.S. Effect of tyrosine supplementation on clinical and healthy populations under stress or cognitive demands—A review. J. Psychiatr. Res. 2015, 70, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Van de Rest, O.; van der Zwaluw, N.L.; de Groot, L.C.P.G.M. Literature review on the role of dietary protein and amino acids in cognitive functioning and cognitive decline. Amino Acids 2013, 45, 1035–1045. [Google Scholar] [CrossRef] [PubMed]
- Molnár, G.A.; Kun, S.; Sélley, E.; Kertész, M.; Szélig, L.; Csontos, C.; Böddi, K.; Bogár, L.; Miseta, A.; Wittmann, I. Role of Tyrosine Isomers in Acute and Chronic Diseases Leading to Oxidative Stress—A Review. Curr. Med. Chem. 2016, 23, 667–685. [Google Scholar] [CrossRef] [PubMed]
- Martinez, A.W.; Recht, N.S.; Hostetter, T.H.; Meyer, T.W. Removal of P-cresol sulfate by hemodialysis. J. Am. Soc. Nephrol. JASN 2005, 16, 3430–3436. [Google Scholar] [CrossRef] [PubMed]
- Gryp, T.; Vanholder, R.; Vaneechoutte, M.; Glorieux, G. p-Cresyl Sulfate. Toxins 2017, 9, 52. [Google Scholar] [CrossRef] [PubMed]
- Meert, N.; Schepers, E.; Glorieux, G.; Van Landschoot, M.; Goeman, J.L.; Waterloos, M.-A.; Dhondt, A.; Van der Eycken, J.; Vanholder, R. Novel method for simultaneous determination of p-cresylsulphate and p-cresylglucuronide: Clinical data and pathophysiological implications. Nephrol. Dial. Transplant. 2012, 27, 2388–2396. [Google Scholar] [CrossRef] [PubMed]
- Poveda, J.; Sanchez-Niño, M.D.; Glorieux, G.; Sanz, A.B.; Egido, J.; Vanholder, R.; Ortiz, A. P-Cresyl sulphate has pro-inflammatory and cytotoxic actions on human proximal tubular epithelial cells. Nephrol. Dial. Transplant. 2014, 29, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Pletinck, A.; Glorieux, G.; Schepers, E.; Cohen, G.; Gondouin, B.; Van Landschoot, M.; Eloot, S.; Rops, A.; Van de Voorde, J.; De Vriese, A.; et al. Protein-bound uremic toxins stimulate crosstalk between leukocytes and vessel wall. J. Am. Soc. Nephrol. JASN 2013, 24, 1981–1994. [Google Scholar] [CrossRef] [PubMed]
- Liabeuf, S.; Barreto, D.V.; Barreto, F.C.; Meert, N.; Glorieux, G.; Schepers, E.; Temmar, M.; Choukroun, G.; Vanholder, R.; Massy, Z.A.; et al. Free p-cresylsulphate is a predictor of mortality in patients at different stages of chronic kidney disease. Nephrol. Dial. Transplant. 2010, 25, 1183–1191. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, J.; Tanaka, T.; Inagi, R. Effect of AST-120 in Chronic Kidney Disease Treatment: Still a Controversy? Nephron 2017, 135, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Schulman, G.; Berl, T.; Beck, G.J.; Remuzzi, G.; Ritz, E.; Arita, K.; Kato, A.; Shimizu, M. Randomized Placebo-Controlled EPPIC Trials of AST-120 in CKD. J. Am. Soc. Nephrol. JASN 2015, 26, 1732–1746. [Google Scholar] [CrossRef] [PubMed]
- Emal, D.; Rampanelli, E.; Stroo, I.; Butter, L.M.; Teske, G.J.; Claessen, N.; Stokman, G.; Florquin, S.; Leemans, J.C.; Dessing, M.C. Depletion of Gut Microbiota Protects against Renal Ischemia-Reperfusion Injury. J. Am. Soc. Nephrol. JASN 2016. [Google Scholar] [CrossRef] [PubMed]
- Mishima, E.; Fukuda, S.; Shima, H.; Hirayama, A.; Akiyama, Y.; Takeuchi, Y.; Fukuda, N.N.; Suzuki, T.; Suzuki, C.; Yuri, A.; et al. Alteration of the Intestinal Environment by Lubiprostone Is Associated with Amelioration of Adenine-Induced CKD. J. Am. Soc. Nephrol. JASN 2015, 26, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Mafra, D.; Fouque, D. Gut microbiota and inflammation in chronic kidney disease patients. Clin. Kidney J. 2015, 8, 332–334. [Google Scholar] [CrossRef] [PubMed]
- Koppe, L.; Mafra, D.; Fouque, D. Probiotics and chronic kidney disease. Kidney Int. 2015, 88, 958–966. [Google Scholar] [CrossRef] [PubMed]
- Ramezani, A.; Massy, Z.A.; Meijers, B.; Evenepoel, P.; Vanholder, R.; Raj, D.S. Role of the Gut Microbiome in Uremia: A Potential Therapeutic Target. Am. J. Kidney Dis. 2016, 67, 483–498. [Google Scholar] [CrossRef] [PubMed]
- Meijers, B.K.I.; De Preter, V.; Verbeke, K.; Vanrenterghem, Y.; Evenepoel, P. p-Cresyl sulfate serum concentrations in haemodialysis patients are reduced by the prebiotic oligofructose-enriched inulin. Nephrol. Dial. Transplant. 2010, 25, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Rossi, M.; Johnson, D.W.; Morrison, M.; Pascoe, E.M.; Coombes, J.S.; Forbes, J.M.; Szeto, C.-C.; McWhinney, B.C.; Ungerer, J.P.J.; Campbell, K.L. Synbiotics Easing Renal Failure by Improving Gut Microbiology (SYNERGY): A Randomized Trial. Clin. J. Am. Soc. Nephrol. CJASN 2016, 11, 223–231. [Google Scholar] [CrossRef] [PubMed]
- Nakabayashi, I.; Nakamura, M.; Kawakami, K.; Ohta, T.; Kato, I.; Uchida, K.; Yoshida, M. Effects of synbiotic treatment on serum level of p-cresol in haemodialysis patients: A preliminary study. Nephrol. Dial. Transplant. 2011, 26, 1094–1098. [Google Scholar] [CrossRef] [PubMed]
- Tang, W.H.W.; Hazen, S.L. The contributory role of gut microbiota in cardiovascular disease. J. Clin. Investig. 2014, 124, 4204–4211. [Google Scholar] [CrossRef] [PubMed]
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandez-Prado, R.; Esteras, R.; Perez-Gomez, M.V.; Gracia-Iguacel, C.; Gonzalez-Parra, E.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Nutrients Turned into Toxins: Microbiota Modulation of Nutrient Properties in Chronic Kidney Disease. Nutrients 2017, 9, 489. https://doi.org/10.3390/nu9050489
Fernandez-Prado R, Esteras R, Perez-Gomez MV, Gracia-Iguacel C, Gonzalez-Parra E, Sanz AB, Ortiz A, Sanchez-Niño MD. Nutrients Turned into Toxins: Microbiota Modulation of Nutrient Properties in Chronic Kidney Disease. Nutrients. 2017; 9(5):489. https://doi.org/10.3390/nu9050489
Chicago/Turabian StyleFernandez-Prado, Raul, Raquel Esteras, Maria Vanessa Perez-Gomez, Carolina Gracia-Iguacel, Emilio Gonzalez-Parra, Ana B. Sanz, Alberto Ortiz, and Maria Dolores Sanchez-Niño. 2017. "Nutrients Turned into Toxins: Microbiota Modulation of Nutrient Properties in Chronic Kidney Disease" Nutrients 9, no. 5: 489. https://doi.org/10.3390/nu9050489
APA StyleFernandez-Prado, R., Esteras, R., Perez-Gomez, M. V., Gracia-Iguacel, C., Gonzalez-Parra, E., Sanz, A. B., Ortiz, A., & Sanchez-Niño, M. D. (2017). Nutrients Turned into Toxins: Microbiota Modulation of Nutrient Properties in Chronic Kidney Disease. Nutrients, 9(5), 489. https://doi.org/10.3390/nu9050489