Tradeoff-in-the-Nephron: A Theory to Explain the Primacy of Phosphate in the Pathogenesis of Secondary Hyperparathyroidism

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Explications of Secondary Hyperparathyroidism: A Chronology
2.1. The Primacy of Phosphate Influx
2.2. The Original Tradeoff Hypothesis
2.3. Skeletal Resistance to PTH
2.4. Deficiency of 1,25-Dihyroxyvitamin D
2.5. Direct Stimulation of PTH Secretion by Circulating Phosphate
2.6. Impaired Suppression of the PTH Gene by Fibroblast Growth Factor 23 (FGF23)
2.7. Deficiency of 25-Hydroxyvitamin D (25D)
3. Tradeoff-in-the-Nephron
- (1)
- GFR[Ca]uf = ECa + TRCa. Division by GFR yields a formula for [Ca]uf:
- (2)
- [Ca]uf = ECa/GFR + TRCa/GFR. If creatinine clearance (Ccr) is assumed to equal GFR, then:
- (3)
- [Ca]uf = ECa/Ccr + TRCa/Ccr = [Ca]u[cr]s/[cr]u + TRCa/Ccr. It follows that:
- (4)
- TRCa/Ccr = [Ca]uf − ECa/Ccr = [Ca]uf − [Ca]u[cr]s/[cr]u [16].
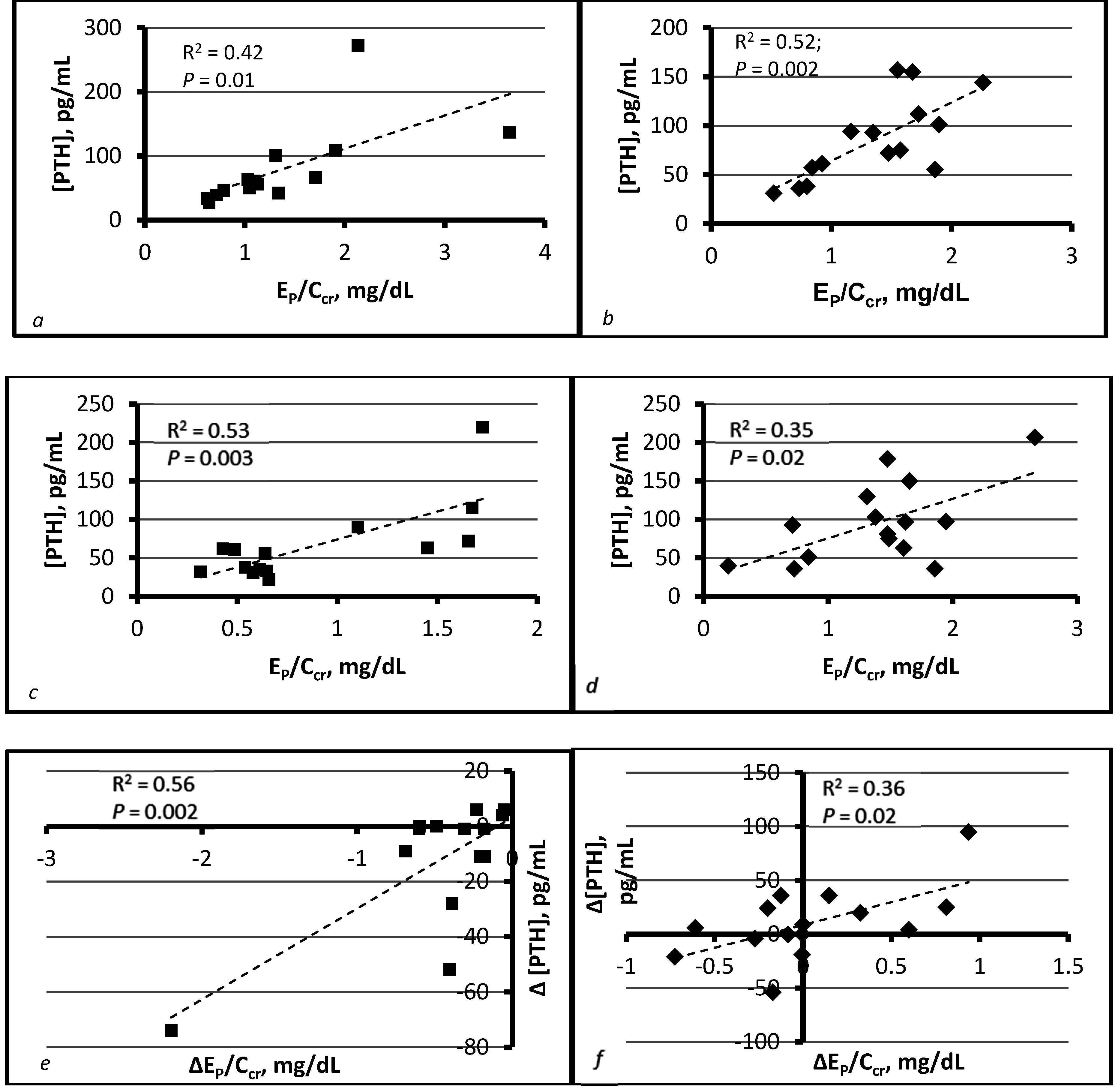
4. Compatibility of Tradeoff-in-the-Nephron with Existing Data
5. Therapeutic Implications of Tradeoff-in-the-Nephron
6. Conclusions
Acknowledgments
Conflicts of Interest
Abbreviations
| CKD | Chronic kidney disease |
| GFR | Glomerular filtration rate (volume/time) |
| Ccr | Creatinine clearance (volume/time) |
| PTH | Parathyroid hormone |
| PTE | Parathyroid extract |
| PHPT | Primary hyperparathyroidism |
| SHPT | Secondary hyperparathyroidism |
| CDN | Cortical distal nephron |
| [Ca]s | Serum calcium concentration, mass/volume |
| [Ca]i | Serum ionized calcium concentration, mass/volume |
| [Ca]uf | Serum ultrafilterable calcium concentration, mass/volume |
| [Ca]u | Urine calcium concentration, mass/volume |
| [Ca]CDN | Calcium concentration in the cortical distal nephron, mass/volume |
| ICa | Influx of calcium (into extracellular fluid or plasma), mass/time |
| ECa | Urinary excretion rate of calcium, mass/time |
| TRCa | Rate of tubular reabsorption of calcium, mass/time |
| ECa/Ccr | Amount of calcium excreted per volume of filtrate, mass/volume |
| TRCa/Ccr | Amount of calcium reabsorbed per volume of filtrate, mass/volume |
| [P]s | Serum phosphorus concentration, mass/volume |
| [P]p | Plasma phosphorus concentration, mass/volume |
| [P]u | Urine phosphorus concentration, mass/volume |
| [P]CDN | Phosphorus concentration in the cortical distal nephron, mass/volume |
| IP | Influx of phosphorus into extracellular fluid or plasma, mass/time |
| EP | Urinary excretion rate of phosphorus, mass/time |
| TRP | Tubular reabsorption rate of phosphorus, mass/time |
| EP/Ccr | Amount of phosphorus excreted per volume of filtrate, mass/volume |
| TRP/Ccr | Amount of phosphorus reabsorbed per volume of filtrate, mass/volume |
| 25D | 25-hydroxyvitamin D |
| 1,25D | 1,25-dihydroxyvitamin D |
| EDTA | Ethylenediaminetetraacetic acid |
| VDRA | Vitamin D receptor activator |
| mRNA | Messenger RNA |
| FGF23 | Fibroblast growth factor 23 |
| NHE3 | Sodium-hydrogen exchanger 3 |
References
- Pitts, T.O.; Piraino, B.H.; Mitro, R.; Chen, T.C.; Segre, G.V.; Greenberg, A.; Puschett, J.B. Hyperparathyroidism and 1,25-dihydroxyvitamin D deficiency in mild, moderate, and severe renal failure. J. Clin. Endocrinol. Metab. 1988, 67, 876–881. [Google Scholar] [CrossRef] [PubMed]
- Reichel, H.; Deibert, B.; Schmidt-Gayk, H.; Ritz, E. Calcium metabolism in early chronic renal failure: Implications for the pathogenesis of hyperparathyroidism. Nephrol. Dial. Transplant. 1991, 6, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Martinez, I.; Saracho, R.; Montenegro, J.; Llach, F. A deficit of calcitriol synthesis may not be the initial factor in the pathogenesis of secondary hyperparathyroidism. Nephrol. Dial. Transplant. 1996, 11, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.; Bakris, G.L.; Molitch, M.; Smulders, M.; Tian, J.; Williams, L.A.; Andress, D.L. Prevalence of abnormal serum vitamin D, PTH, calcium and phosphorus in patients with chronic kidney disease: Results of the study to evaluate early kidney disease. Kidney Int. 2007, 71, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Craver, L.; Marco, M.P.; Martinez, I.; Rue, M.; Borras, M.; Martin, M.L.; Sarro, F.; Valdivielso, J.M.; Fernandez, E. Mineral metabolism parameters throughout chronic kidney disease stages 1–5—Achievement of K/DOQI target ranges. Nephrol. Dial. Transplant. 2007, 22, 1171–1176. [Google Scholar] [CrossRef] [PubMed]
- Malluche, H.H.; Ritz, E.; Lange, H.P.; Kutschera, J.; Hodgson, M.; Seiffert, U.; Schoeppe, W. Bone histology in incipient and advanced renal failure. Kidney Int. 1976, 9, 355–362. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.M.; Long, J.; Montez-Rath, M.; Leonard, M.; Chertow, G.M. Hip fracture in patients with non-dialysis-requiring chronic kidney disease. J. Bone Miner. Res. 2016, 31, 1803–1809. [Google Scholar] [CrossRef] [PubMed]
- Ritz, E.; Stefanski, A.; Rambausek, M. The role of the parathyroid glands in the uremic syndrome. Am. J. Kidney Dis. 1995, 26, 808–813. [Google Scholar] [CrossRef]
- Goodman, W.G. The consequences of uncontrolled secondary hyperparathyroidism and its treatment in chronic kidney disease. Semin. Dial. 2004, 17, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Slatopolsky, E.; Caglar, S.; Pennell, J.P.; Taggart, D.D.; Canterbury, J.M.; Reiss, E.; Bricker, N.S. On the pathogenesis of hyperparathyroidism in chronic experimental renal insufficiency in the dog. J. Clin. Investig. 1971, 50, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Slatopolsky, E.; Caglar, S.; Gradowska, L.; Canterbury, J.; Reiss, E.; Bricker, N.S. On the prevention of secondary hyperparathyroidism in experimental chronic renal disease using “proportional reduction” of dietary phosphorus intake. Kidney Int. 1972, 2, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.A.; Canterbury, J.M.; Bourgoignie, J.J.; Veliz, G.; Gavellas, G.; Reiss, E.; Bricker, N.S. Reversal of hyperparathyroidism in response to dietary phosphorus restriction in the uremic dog. Kidney Int. 1979, 15, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Hilker, S.; Dusso, A.S.; Rapp, N.S.; Martin, K.J.; Slatopolsky, E. Phosphorus restriction reverses hyperparathyroidism in uremia independent of changes in calcium and calcitriol. Am. J. Physiol. 1990, 259, F432–F437. [Google Scholar] [PubMed]
- Denda, M.; Finch, J.; Slatopolsky, E. Phosphorus accelerates the development of parathyroid hyperplasia and secondary hyperparathyroidism in rats with renal failure. Am. J. Kidney Dis. 1996, 28, 596–602. [Google Scholar] [CrossRef]
- Boros, S.; Bindels, R.J.M.; Hoenderop, J.G.J. Active Ca2+ reabsorption in the connecting tubule. Pflug. Arch. 2009, 458, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Phelps, K.R.; Stote, K.S.; Mason, D. Tubular calcium reabsorption and other aspects of calcium homeostasis in primary and secondary hyperparathyroidism. Clin. Nephrol. 2014, 82, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Phelps, K.R.; Stote, K.S.; Mason, D. Use of sevelamer to examine the role of intraluminal phosphate in the pathogenesis of secondary hyperparathyroidism. Clin. Nephrol. 2014, 82, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Phelps, K.R.; Mason, D.L.; Stote, K.S. Phosphate homeostasis, parathyroid hormone, and fibroblast growth factor 23 in stages 3 and 4 chronic kidney disease. Clin. Nephrol. 2016, 85, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Phelps, K.R.; Mason, D.L. Parameters of phosphorus homeostasis at normal and reduced GFR: Theoretical considerations. Clin. Nephrol. 2015, 83, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Phelps, K.R.; Mason, D.L.; Stote, K.S. Parameters of phosphorus homeostasis at normal and reduced GFR: Empiric observations. Clin. Nephrol. 2015, 83, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Slatopolsky, E.; Robson, A.M.; Elkan, I.; Bricker, N.S. Control of phosphate excretion in uremic man. J. Clin. Investig. 1968, 47, 1865–1874. [Google Scholar] [CrossRef] [PubMed]
- Reiss, E.; Canterbury, J.M.; Bercovitz, M.A.; Kaplan, E.L. The role of phosphate in the secretion of parathyroid hormone in man. J. Clin. Investig. 1970, 49, 2146–2149. [Google Scholar] [CrossRef] [PubMed]
- Jowsey, J.; Reiss, E.; Canterbury, J.M. Long-term effects of high phosphate intake on parathyroid hormone levels and bone metabolism. Acta Orthop. Scand. 1974, 45, 801–808. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.A.; Canterbury, J.M.; Gavellas, G.; Jaffe, D.; Bourgoignie, J.J.; Reiss, E.; Bricker, N.S. Interrelations between phosphorus, calcium, parathyroid hormone and renal phosphate excretion in response to an oral phosphorus load in normal and uremic dogs. Kidney Int. 1978, 14, 207–214. [Google Scholar] [CrossRef] [PubMed]
- Bover, J.; Rodriguez, M.; Trinidad, P.; Jara, A.; Martinez, M.E.; Machado, L.; Llach, F.; Felsenfeld, A.J. Factors in the development of secondary hyperparathyroidism during graded renal failure in the rat. Kidney Int. 1994, 45, 953–961. [Google Scholar] [CrossRef] [PubMed]
- Krapf, R.; Glatz, M.; Hulter, H.N. Neutral phosphate administration generates and maintains renal metabolic alkalosis and hyperparathyroidism. Am. J. Physiol. 1995, 268, F802–F807. [Google Scholar] [PubMed]
- Estepa, J.C.; Aguilera-Tejero, E.; Lopez, I.; Almaden, Y.; Rodriguez, M.; Felsenfeld, A.J. Effect of phosphate on parathyroid hormone secretion in vivo. J. Bone Miner. Res. 1999, 14, 1848–1854. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, F.; Denda, M.; Finch, J.L.; Brown, A.L.; Slatopolsky, E. Hyperplasia of the parathyroid gland with secondary hyperparathyroidism. Kidney Int. 2002, 61, 1332–1338. [Google Scholar] [CrossRef] [PubMed]
- Ritter, C.S.; Martin, D.R.; Lu, Y.; Slatopolsky, E.; Brown, A.J. Reversal of secondary hyperparathyroidism by phosphate restriction restores parathyroid calcium-sensing receptor expression and function. J. Bone Miner. Res. 2002, 17, 2206–2213. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.R.; Ritter, C.S.; Slatopolsky, E.; Brown, A.J. Acute regulation of parathyroid hormone by dietary phosphate. Am. J. Physiol. 2005, 289, E729–E734. [Google Scholar] [CrossRef] [PubMed]
- Nagano, N.; Miyata, S.; Abe, M.; Kobayashi, N.; Wakita, S.; Yamashita, T.; Wada, M. Effect of manipulating serum phosphorus with phosphate binder on circulating PTH and FGF23 in renal failure rats. Kidney Int. 2006, 69, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Gutierrez, O.; Shah, A.; Castaldo, L.; Holmes, J.; Lee, H.; Wolf, M. Post-prandial mineral metabolism and secondary hyperparathyroidism in early chronic kidney disease. J. Am. Soc. Nephrol. 2008, 19, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Combe, C.; Aparicio, M. Phosphorus and protein restriction and parathyroid function in chronic renal failure. Kidney Int. 1994, 46, 1381–1386. [Google Scholar] [CrossRef] [PubMed]
- Combe, C.; Morel, D.; de Precigout, V.; Blanchetier, V.; Bouchet, J.L.; Potaux, L.; Fournier, A.; Aparicio, M. Long-term control of hyperparathyroidism in advanced renal failure by low-phosphorus low-protein diet supplemented with calcium (without changes in plasma calcitriol). Nephron 1995, 70, 287–295. [Google Scholar] [CrossRef] [PubMed]
- Portale, A.A.; Booth, B.E.; Halloran, B.P.; Morris, R.C., Jr. Effect of dietary phosphorus on circulating cconcentrations of 1,25-dihydroxyvitamin D and immunoreactive parathyroid hormone in children with moderate renal insufficiency. J. Clin. Investig. 1984, 73, 1580–1589. [Google Scholar] [CrossRef] [PubMed]
- Llach, F.; Massry, S.G. On the mechanism of secondary hyperparathyroidism in moderate renal insufficiency. J. Clin. Endocrinol. Metab. 1985, 61, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, R.B.; Cancela, A.L.E.; Graciolli, F.G.; Dos Reis, L.M.; Draibe, S.A.; Cuppari, L.; Carvalho, A.B.; Jorgetti, V.; Canziani, M.E.; Moyses, R.M.A. Early control of PTH and FGF23 in normophosphatemic CKD patients: A new target in CKD-MBD therapy? Clin. J. Am. Soc. Nephrol. 2010, 5, 286–291. [Google Scholar] [CrossRef] [PubMed]
- Wilson, L.; Felsenfeld, A.; Drezner, M.K.; Llach, F. Altered divalent metabolism in early renal failure. Role of 1,25(OH)2D. Kidney Int. 1985, 27, 565–573. [Google Scholar] [CrossRef] [PubMed]
- Rivkees, S.A.; El-Hajj-Fuleihan, G.; Brown, E.M.; Crawford, J.D. Tertiary hyperparathyroidism during high phosphate therapy of familial hypophosphatemic rickets. J. Clin. Endocrinol. Metab. 1992, 75, 1514–1518. [Google Scholar] [PubMed]
- Lucas, P.A.; Brown, R.C.; Woodhead, J.S.; Coles, G.A. 1,25-dihydroxycholecalciferol and parathyroid hormone in advanced chronic renal failure: Effects of simultaneous protein and phosphorus restriction. Clin. Nephrol. 1986, 25, 7–10. [Google Scholar] [PubMed]
- Rodriguez, M.; Martin-Malo, A.; Martinez, M.E.; Torres, A.; Felsenfeld, A.J.; Llach, F. Calcemic response to parathyroid hormone in renal failure: Role of phosphorus and its effect on calcitriol. Kidney Int. 1991, 40, 1055–1062. [Google Scholar] [CrossRef] [PubMed]
- Bricker, N.S. On the pathogenesis of the uremic state: An exposition of the “trade-off hypothesis”. N. Engl. J. Med. 1972, 286, 1093–1099. [Google Scholar] [PubMed]
- Adler, A.J.; Ferran, N.; Berlyne, G.M. Effect of inorganic phosphate on serum ionized calcium concentration: A reassessment of the “trade-off hypothesis”. Kidney Int. 1985, 28, 932–935. [Google Scholar] [CrossRef] [PubMed]
- Massry, S.G.; Coburn, J.W.; Lee, D.B.N.; Jowsey, J.; Kleeman, C.R. Skeletal resistance to parathyroid hormone in renal failure. Studies in 105 human subjects. Ann. Int. Med. 1973, 78, 357–364. [Google Scholar] [CrossRef] [PubMed]
- Llach, F.; Massry, S.G.; Singer, F.R.; Kurokawa, K.; Kaye, J.H.; Coburn, J.W. Skeletal resistance to endogenous parathyroid hormone in patients with early renal failure. A possible cause for secondary hyperparathyroidism. J. Clin. Endocrinol. Metab. 1975, 41, 339–345. [Google Scholar] [CrossRef] [PubMed]
- Massry, S.G.; Stein, R.; Garty, J.; Arieff, A.I.; Coburn, J.W.; Norman, A.W.; Friedler, R.M. Skeletal resistance to the calcemic action of parathyroid hormone in uremia: Role of 1,25(OH)2D. Kidney Int. 1976, 9, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Somerville, P.J.; Kaye, M. Resistance to parathyroid hormone in renal failure: Role of vitamin D metabolites. Kidney Int. 1978, 14, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Somerville, P.J.; Kaye, M. Evidence that resistance to the calcemic action of parathyroid hormone in rats with acute uremia is caused by phosphate retention. Kidney Int. 1979, 16, 552–560. [Google Scholar] [CrossRef] [PubMed]
- Somerville, P.J.; Kaye, M. Action of phosphorus on calcium release in isolated perfused rat tails. Kidney Int. 1982, 22, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Kaplan, M.A.; Canterbury, J.M.; Gavellas, G.; Jaffe, D.; Bourgoignie, J.J.; Reiss, E.; Bricker, N.S. The calcemic and phosphaturic effects of parathyroid hormone in the normal and uremic dog. Metabolism 1978, 27, 1785–1792. [Google Scholar] [CrossRef]
- Rodriguez, M.; Felsenfeld, A.J.; Llach, F. Calcemic response to parathyroid hormone in renal failure: Role of calcitriol and the effect of parathyroidectomy. Kidney Int. 1991, 40, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Zehnder, D.; Bland, R.; Walker, E.A.; Bradwell, A.R.; Howie, A.J.; Hewison, M.; Stewart, P.M. Expression of 25-hydroxyvitamin D3-1α hydroxylase in the human kidney. J. Am. Soc. Nephrol. 1999, 10, 2465–2473. [Google Scholar] [PubMed]
- Walling, M.W. Intestinal calcium and phosphate transport: Differential responses to vitamin D3 metabolites. Am. J. Physiol. 1977, 233, E488–E494. [Google Scholar] [PubMed]
- Silver, J.; Russell, J.; Sherwood, L.M. Regulation by vitamin D metabolites of messenger ribonucleic acid for preproparathyroid hormone in isolated bovine parathyroid cells. Proc. Natl. Acad. Sci. USA 1985, 82, 4270–4273. [Google Scholar] [CrossRef] [PubMed]
- Silver, J.; Naveh-Many, T.; Mayer, H.; Schmelzer, H.J.; Popovtzer, M.M. Regulation by vitamin D metabolites of parathyroid hormone gene transcription in vivo in the rat. J. Clin. Investig. 1986, 78, 1296–1301. [Google Scholar] [CrossRef] [PubMed]
- Sprague, S.M.; Coyne, D. Control of secondary hyperparathyroidism by vitamin D receptor agonists in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Slatopolsky, E.; Finch, J.; Denda, M.; Ritter, C.; Zhong, M.; Dusso, A.; MacDonald, P.N.; Brown, A.J. Phosphorus restriction prevents parathyroid gland growth. High phosphorus directly stimulates PTH secretion in vitro. J. Clin. Investig. 1996, 97, 2534–2540. [Google Scholar] [CrossRef] [PubMed]
- Almaden, Y.; Canalejo, A.; Hernandez, A.; Ballesteros, E.; Garcia-Navarro, S.; Torres, A.; Rodriguez, M. Direct effect of phosphorus on PTH secretion from whole rat parathyroid glands in vitro. J. Bone Miner. Res. 1996, 11, 970–976. [Google Scholar] [CrossRef] [PubMed]
- Almaden, Y.; Hernandez, A.; Torregrosa, V.; Canalejo, A.; Sabate, L.; Fernandez, C.L.; Campistol, J.M.; Torres, A.; Rodriguez, M. High phosphate level directly stimulates parathyroid hormone secretion and synthesis by human parathyroid hormone tissue in vitro. J. Am. Soc. Nephrol. 1998, 9, 1845–1852. [Google Scholar] [PubMed]
- Moallem, E.; Kilav, R.; Silver, J.; Naveh-Many, T. RNA-protein binding and post-transcriptional regulation of parathyroid hormone gene expression by calcium and phosphate. J. Biol. Chem. 1998, 273, 5253–5259. [Google Scholar] [CrossRef] [PubMed]
- Kates, D.M.; Sherrard, D.J.; Andress, D.L. Evidence that serum phosphate is independently associated with serum PTH in patients with chronic renal failure. Am. J. Kidney Dis. 1997, 30, 809–813. [Google Scholar] [CrossRef]
- Phelps, K.R.; Mason, D.L. Evidence that TmP/GFR can be estimated with the Walton-Bijvoet nomogram in chronic kidney disease. Clin. Nephrol. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Zhou, J.; Tang, W.; Jiang, X.; Rowe, D.W.; Quarles, L.D. Pathogenic role of FGF23 in Hyp mice. Am. J. Physiol. 2006, 291, E38–E49. [Google Scholar] [CrossRef] [PubMed]
- Zanchi, C.; Locatelli, M.; Benigni, A.; Corna, D.; Tomasoni, S.; Rottoli, D.; Gaspari, F.; Remuzzi, G.; Zoja, C. Renal expression of FGF23 in progressive renal disease of diabetes and the effect of ACE inhibitor. PLoS ONE 2013, 8, e70775. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Wahl, P.; Vargas, G.S.; Gutierrez, O.M.; Scialla, J.; Xie, H.; Appleby, D.; Nessel, L.; Bellovich, K.; Chen, J.; et al. Fibroblast growth factor 23 is elevated before parathyroid hormone and phosphate in chronic kidney disease. Kidney Int. 2011, 79, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Komaba, H.; Fukagawa, M. FGF23-parathyroid interaction: Implications in chronic kidney disease. Kidney Int. 2010, 77, 292–298. [Google Scholar] [CrossRef] [PubMed]
- Ben-Dov, I.Z.; Galitzer, H.; Lavi-Moshayoff, V.; Goetz, R.; Kuro-o, M.; Mohammadi, M.; Sirkis, R.; Naveh-Many, T.; Silver, J. The parathyroid is a target organ for FGF23 in rats. J. Clin. Investig. 2007, 117, 4003–4008. [Google Scholar] [CrossRef] [PubMed]
- Galitzer, H.; Ben-Dov, I.Z.; Silver, J.; Naveh-Many, T. Parathyroid cell resistance to fibroblast growth factor 23 in secondary hyperparathyroidism of chronic kidney disease. Kidney Int. 2010, 77, 211–218. [Google Scholar] [CrossRef] [PubMed]
- Canalejo, R.; Canalejo, A.; Martinez-Moreno, J.M.; Rodriguez-Ortiz, M.E.; Estepa, J.C.; Mendoza, F.J.; Munoz-Castaneda, J.R.; Shalhoub, V.; Almaden, Y.; Rodriguez, M. FGF23 fails to inhibit uremic parathyroid glands. J. Am. Soc. Nephrol. 2010, 21, 1125–1135. [Google Scholar] [CrossRef] [PubMed]
- Andrukhova, O.; Streicher, C.; Zeitz, U.; Erben, R.G. Fgf23 and parathyroid hormone signaling interact in kidney and bone. Mol. Cell. Endocrinol. 2016, 436, 224–239. [Google Scholar] [CrossRef] [PubMed]
- Chonchol, M.; Locatelli, F.; Abboud, H.E.; Charytan, C.; de Francisco, A.L.M.; Jolly, S.; Kaplan, M.; Roger, S.D.; Sarkar, S.; Albizem, M.B.; et al. A randomized, double-blind, placebo-controlled study to assess the efficacy and safety of cinacalcet HCl in participants with CKD not receiving dialysis. Am. J. Kidney Dis. 2009, 53, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Nagano, N.; Urakawa, I.; Yamazaki, Y.; Iijima, K.; Fujita, T.; Yamashita, T.; Fukumoto, S.; Shimada, T. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int. 2010, 78, 975–980. [Google Scholar] [CrossRef] [PubMed]
- Rosen, C.J. Vitamin D insufficiency. N. Engl. J. Med. 2011, 364, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. Vitamin D deficiency. N. Engl. J. Med. 2007, 357, 266–281. [Google Scholar] [CrossRef] [PubMed]
- Manson, J.E.; Brannon, P.M.; Rosen, C.J.; Taylor, C.L. Vitamin D deficiency—Is there really a pandemic? N. Engl. J. Med. 2016, 375, 1817–1820. [Google Scholar] [CrossRef] [PubMed]
- Hollis, B.W.; Wagner, C.L. Normal serum vitamin D levels. N. Engl. J. Med. 2005, 352, 515. [Google Scholar] [PubMed]
- Moe, S.M.; Saifullah, A.; LaClair, R.E.; Usman, S.A.; Yu, Z. A randomized trial of cholecalciferol versus doxercalciferol for lowering parathyroid hormone in chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2010, 5, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, J.A.; Law, J.; Coakley, K.E.; Zughaier, S.M.; Hao, L.; Salles, K.S.; Wasse, H.; Gutierrez, O.M.; Ziegler, T.R.; Tangpricha, V. High-dose cholecalciferol reduces parathyroid hormone in patients with early chronic kidney disease: A pilot, randomized, double-blind, placebo-controlled trial. Am. J. Clin. Nutr. 2012, 96, 672–679. [Google Scholar] [CrossRef] [PubMed]
- Nigwekar, S.U.; Bhan, I.; Thadhani, R. Ergocalciferol and cholecalciferol in CK.D. Am. J. Kidney Dis. 2012, 60, 139–156. [Google Scholar] [CrossRef] [PubMed]
- Sprague, S.M.; Silva, A.L.; Al-Saghir, F.; Damle, R.; Tabash, S.P.; Petkovich, M.; Messner, E.J.; White, J.A.; Melnick, J.Z.; Bishop, C.W. Modified-release calcifediol effectively controls secondary hyperparathyroidism associated with vitamin D insufficiency in chronic kidney disease. Am. J. Nephrol. 2014, 40, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Sprague, S.M.; Crawford, P.W.; Melnick, J.Z.; Strugnell, S.A.; Ali, S.; Mangoo-Karim, R.; Lee, S.; Petkovich, P.M.; Bishop, C.W. Use of extended-release calcifediol to treat secondary hyperparathyroidism in stages 3 and 4 chronic kidney disease. Am. J. Nephrol. 2016, 44, 316–325. [Google Scholar] [CrossRef] [PubMed]
- Wills, M.R.; Lewin, M.R. Plasma calcium fractions and the protein-binding of calcium in normal subjects and in patients with hypercalcaemia and hypocalcaemia. J. Clin. Pathol. 1971, 24, 856–866. [Google Scholar] [CrossRef] [PubMed]
- Bank, N.; Su, W.S.; Aynedjian, H.S. A micropuncture study of renal phosphate transport in rats with chronic renal failure and secondary hyperparathyroidism. J. Clin. Investig. 1978, 61, 884–894. [Google Scholar] [CrossRef] [PubMed]
- Tiselius, H.G. Estimated levels of supersaturation with calcium phosphate and calcium oxalate in the distal tubule. Urol. Res. 1997, 25, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Luptak, J.; Bek-Jensen, H.; Fornander, A.-M.; Hojgaard, I.; Nilsson, M.-A.; Tiselius, H.-G. Crystallization of calcium oxalate and calcium phosphate at supersaturation levels corresponding to those in different parts of the nephron. Scanning Microsp. 1994, 8, 47–62. [Google Scholar]
- Haut, L.L.; Alfrey, A.C.; Guggenheim, S.; Buddington, B.; Schrier, N. Renal toxicity of phosphate in rats. Kidney Int. 1980, 17, 722–731. [Google Scholar] [CrossRef] [PubMed]
- Markowitz, G.S.; Nasr, S.H.; Klein, P.; Anderson, H.; Stack, J.I.; Alterman, L.; Price, B.; Radhakrishnan, J.; D’Agati, V.D. Renal failure due to acute nephrocalcinosis following oral sodium phosphate bowel cleansing. Hum. Pathol. 2004, 35, 675–684. [Google Scholar] [CrossRef] [PubMed]
- Walser, M. Ion association. VI. Interactions between calcium, magnesium, inorganic phosphate, citrate, and protein in normal human plasma. J. Clin. Investig. 1961, 40, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Knox, F.G.; Osswald, H.; Marchand, G.R.; Spielman, W.S.; Haas, J.A.; Berndt, T.; Youngberg, S.P. Phosphate transport along the nephron. Am. J. Physiol. 1977, 233, F261–F268. [Google Scholar] [PubMed]
- Goodman, W.G.; Quarles, L.D. Development and progression of secondary hyperparathyroidism in chronic kidney disease: Lessons from molecular genetics. Kidney Int. 2008, 74, 276–288. [Google Scholar] [CrossRef] [PubMed]
- Martinez, I.; Saracho, R.; Montenegro, J.; Llach, F. The importance of dietary calcium and phosphorus in the secondary hyperparathyroidism of patients with early renal failure. Am. J. Kidney Dis. 1997, 29, 496–502. [Google Scholar] [CrossRef]
- Kovesdy, C.P.; Lu, J.L.; Malakauskas, S.M.; Andress, D.L.; Kalantar-Zadeh, K.; Ahmadzadeh, S. Paricalcitol versus ergocalciferol for secondary hyperparathyroidism in CKD stages 3 and 4: A randomized controlled trial. Am. J. Kidney Dis. 2012, 59, 58–66. [Google Scholar] [CrossRef] [PubMed]
- Uribarri, J.; Calvo, M.S. Hidden sources of phosphorus in the typical American diet: Does it matter in nephrology? Semin. Dial. 2003, 16, 186–188. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, C.; Sayre, S.S.; Leon, J.B.; Machekano, R.; Love, T.E.; Porter, D.; Marbury, M.; Sehgal, A. Effect of food additives on hyperphosphatemia among patients with end-stage renal disease. A randomized controlled trial. JAMA 2009, 301, 629–635. [Google Scholar] [CrossRef] [PubMed]
- Sprague, S.M.; Abboud, H.; Qiu, P.; Dauphin, M.; Zhang, P.; Finn, W. Lanthanum carbonate reduces phosphorus burden in patients with CKD stages 3 and 4: A randomized trial. Clin. J. Am. Soc. Nephrol. 2009, 4, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Block, G.A.; Wheeler, D.C.; Persky, M.S.; Kestenbaum, B.; Ketteler, M.; Spiegel, D.M.; Allison, M.A.; Asplin, J.; Smits, G.; Hoofnagle, A.N.; et al. Effects of phosphate binders in moderate CKD. J. Am. Soc. Nephrol. 2012, 23, 1407–1415. [Google Scholar] [CrossRef] [PubMed]
- Yokoyama, K.; Hirakata, H.; Akiba, T.; Fukagawa, M.; Nakayama, M.; Sawada, K.; Kumagai, Y.; Block, G.A. Ferric citrate hydrate for the treatment of hyperphosphatemia in nondialysis-dependent CKD. Clin. J. Am. Soc. Nephrol. 2014, 9, 543–552. [Google Scholar] [CrossRef] [PubMed]
- Block, G.A.; Fishbane, S.; Rodriguez, M.; Smits, G.; Shemesh, S.; Pergola, P.E.; Wolf, M.; Chertow, G.M. A 12-week, double-blind, placebo-controlled trial of ferric citrate for the treatment of iron deficiency anemia and reduction of serum phosphate in patients with CKD stages 3–5. Am. J. Kidney Dis. 2015, 65, 728–736. [Google Scholar] [CrossRef] [PubMed]
- Labonte, E.D.; Carreras, C.W.; Leadbetter, M.R.; Kozuka, K.; Kohler, J.; Koo-McCoy, S.; He, L.; Dy, E.; Black, D.; Zhong, Z.; et al. Gastrointestinal inhibition of sodium-hydrogen exchanger 3 reduces phosphorus absorption and protects against vascular calcification in CKD. J. Am. Soc. Nephrol. 2015, 26, 1138–1149. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Steffes, M.; Bostom, A.; Ix, J.H. Effect of niacin on FGF23 concentration in chronic kidney disease. Am. J. Nephrol. 2014, 39, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD-MBD Work Group. Clinical practice guidelines for the management of CKD-MBD. Kidney Int. 2009, 76, S1–S130. [Google Scholar]

© 2017 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phelps, K.R. Tradeoff-in-the-Nephron: A Theory to Explain the Primacy of Phosphate in the Pathogenesis of Secondary Hyperparathyroidism. Nutrients 2017, 9, 427. https://doi.org/10.3390/nu9050427
Phelps KR. Tradeoff-in-the-Nephron: A Theory to Explain the Primacy of Phosphate in the Pathogenesis of Secondary Hyperparathyroidism. Nutrients. 2017; 9(5):427. https://doi.org/10.3390/nu9050427
Chicago/Turabian StylePhelps, Kenneth R. 2017. "Tradeoff-in-the-Nephron: A Theory to Explain the Primacy of Phosphate in the Pathogenesis of Secondary Hyperparathyroidism" Nutrients 9, no. 5: 427. https://doi.org/10.3390/nu9050427
APA StylePhelps, K. R. (2017). Tradeoff-in-the-Nephron: A Theory to Explain the Primacy of Phosphate in the Pathogenesis of Secondary Hyperparathyroidism. Nutrients, 9(5), 427. https://doi.org/10.3390/nu9050427