Potential Links between Impaired One-Carbon Metabolism Due to Polymorphisms, Inadequate B-Vitamin Status, and the Development of Alzheimer’s Disease


Abstract
:1. Introduction
2. Evidence Linking B-Vitamins, Hcy and the Pathogenesis of AD
2.1. Observational Trials
2.2. Evidence from Supplementation with B-Vitamins
3. Role of Key Polymorphisms in the OCM
3.1. Overview of the Enzymes of the OCM
3.2. Polymorphisms in Key Enzymes of the OCM
3.2.1. Methylenetetrahydrofolate Reductase (MTHFR) Polymorphisms
3.2.2. Methionine Synthase (MS) Polymorphism
3.2.3. Methionine Synthase Reductase (MSR) Polymorphism
3.2.4. Cystathionine β-Synthase (CBS) Polymorphism
3.2.5. Serine Hydroxymethyltransferase (SHMT) Polymorphism
4. Proposed Mechanisms Linking Polymorphisms, Hcy, B-Vitamins and AD
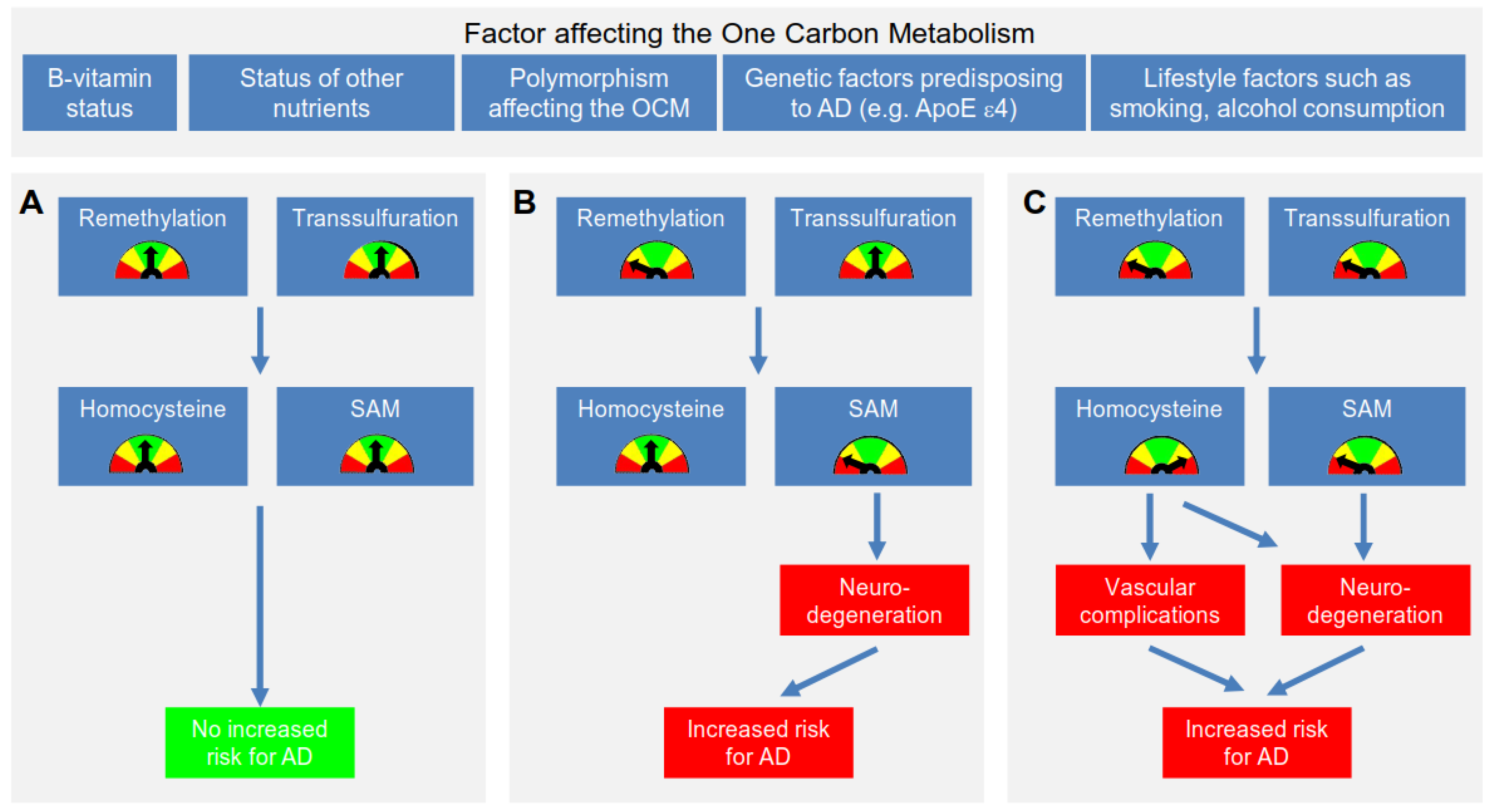
4.1. Proposed Mechanism Linking Hcy & AD
4.2. Further Mechanisms Linking an Impaired OCM to the Development of AD
5. Dietary Intake of B-Vitamins
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Duthey, B. Priority Medicines for Europe and the World Update Report-Background Paper "Alzheimer’s Disease and Other Dementias"; World Health Organization: Geneva, Switzerland, 2013. [Google Scholar]
- Kinsella, K.; He, W. An Aging World: 2008 International Population Reports; U.S. Department of Health and Human Services, National Institute of Health, National Institute of Aging: Washington, DC, USA, 2009.
- Alzheimer’s Association Website. Available online: Http://www.Alz.Org (accessed on 19 September 2016).
- Lindsley, C.W. Alzheimer’s disease: Development of disease-modifying treatments is the challenge for our generation. ACS Chem Neurosci 2012, 3, 804–805. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Association Website. Available online: Http://www.Alz.Org (accessed on 21 November 2014).
- Serrano-Pozo, A.; Frosch, M.P.; Masliah, E.; Hyman, B.T. Neuropathological alterations in Alzheimer’s disease. Cold Spring Harb. Perspect. Med. 2011, 1, a006189. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J. Resolving controversies on the path to Alzheimer’s therapeutics. Nat. Med. 2011, 17, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Moyer, M.W. Alzheimer’s researchers call for clinical revamp. Nat. Med. 2011, 17, 235. [Google Scholar] [CrossRef] [PubMed]
- Mohajeri, M.H.; Troesch, B.; Weber, P. Inadequate supply of vitamins and DHA in the elderly: Implications for brain aging and Alzheimer-type dementia. Nutrition 2015, 31, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Coppedè, F.; Tannorella, P.; Pezzini, I.; Migheli, F.; Ricci, G.; Caldarazzo lenco, E.; Piaceri, I.; Polini, A.; Nacmias, B.; Monzani, F.; et al. Folate, homocysteine, vitamin B12, and polymorphisms of genes participating in one-carbon metabolism in late-onset Alzheimer’s disease patients and healthy controls. Antioxid. Redox Signal. 2011, 17, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Migliore, L.; Coppedè, F. Genetics, environmental factors and the emerging role of epigenetics in neurodegenerative diseases. Mutat. Res. Fundam.Mol. Mech. Mutagen. 2009, 667, 82–97. [Google Scholar] [CrossRef] [PubMed]
- Finch, C.E. Evolution of the human lifespan and diseases of aging: Roles of infection, inflammation, and nutrition. Proc. Natl. Acad.Sci. USA 2010, 107, 1718–1724. [Google Scholar] [CrossRef] [PubMed]
- Egert, S.; Rimbach, G.; Huebbe, P. Apoe genotype: From geographic distribution to function and responsiveness to dietary factors. Proc. Nutr. Soc. 2012, 71, 410–424. [Google Scholar] [CrossRef] [PubMed]
- Ramassamy, C.; Averill, D.; Beffert, U.; Bastianetto, S.; Theroux, L.; Lussier-Cacan, S.; Cohn, J.S.; Christen, Y.; Davignon, J.; Quirion, R.; et al. Original contributions: Oxidative damage and protection by antioxidants in the frontal cortex of Alzheimer’s disease is related to the apolipoprotein e genotype. Free Radic. Biol. Med. 1999, 27, 544–553. [Google Scholar] [CrossRef]
- Selhub, J. Public health significance of elevated homocysteine. Food Nutr. Bull. 2008, 29, S116–S125. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.D.; Refsum, H. Homocysteine, B vitamins, and cognitive impairment. Annu. Rev. Nutr. 2016, 36, 211–239. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Selley, M.L. A metabolic link between S-adenosylhomocysteine and polyunsaturated fatty acid metabolism in Alzheimer’s disease. Neurobiol. Aging 2007, 28, 1834–1839. [Google Scholar] [CrossRef] [PubMed]
- Troesch, B.; Hoeft, B.; Weber, P.; Eggersdorfer, M. Understanding the genome: Implications for human nutrition? Vitam. Miner. 2014, 3. [Google Scholar] [CrossRef]
- Schwahn, B.; Rozen, R. Polymorphisms in the methylenetetrahydrofolate reductase gene: Clinical consequences. Am. J. Pharmacogenom. 2001, 1, 189–201. [Google Scholar] [CrossRef]
- Yamada, K.; Chen, Z.; Rozen, R.; Matthews, R.G. Effects of common polymorphisms on the properties of recombinant human methylenetetrahydrofolate reductase. Proc. Natl. Acad.Sci. USA 2001, 98, 14853–14858. [Google Scholar] [CrossRef] [PubMed]
- Guenther, B.D.; Sheppard, C.A.; Tran, P.; Rozen, R.; Matthews, R.G.; Ludwig, M.L. The structure and properties of methylenetetrahydrofolate reductase from Escherichia coli suggest how folate ameliorates human hyperhomocysteinemia. Nat. Struct. Mol. Biol. 1999, 6, 359–365. [Google Scholar]
- Weisberg, I.S.; Jacques, P.F.; Selhub, J.; Bostom, A.G.; Chen, Z.; Curtis Ellison, R.; Eckfeldt, J.H.; Rozen, R. The 1298a→c polymorphism in methylenetetrahydrofolate reductase (MTHFR): In vitro expression and association with homocysteine. Atherosclerosis 2001, 156, 409–415. [Google Scholar] [CrossRef]
- Weisberg, I.; Tran, P.; Christensen, B.; Sibani, S.; Rozen, R. A second genetic polymorphism in methylenetetrahydrofolate reductase (MTHFR) associated with decreased enzyme activity. Mol. Genet. Metab. 1998, 64, 169–172. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, D.; Campeau, E.; Goyette, P.; Adjalla, C.E.; Christensen, B.; Ross, M.; Eydoux, P.; Rosenblatt, D.S.; Rozen, R.; Gravel, R.A. Human methionine synthase: cDNA cloning and identification of mutations in patients of the cblg complementation group of folate/cobalamin disorders. Hum. Mol. Genet. 1996, 5, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.H.; Liu, M.-L.; Hwang, H.-Y.; Chen, L.-S.; Korenberg, J.; Shane, B. Human methionine synthase: cDNA cloning, gene localization, and expression. J. Biol. Chem. 1997, 272, 3628–3634. [Google Scholar] [CrossRef] [PubMed]
- Olteanu, H.; Munson, T.; Banerjee, R. Differences in the efficiency of reductive activation of methionine synthase and exogenous electron acceptors between the common polymorphic variants of human methionine synthase reductase. Biochemistry 2002, 41, 13378–13385. [Google Scholar] [CrossRef] [PubMed]
- Sebastio, G.; Sperandeo, M.P.; Panico, M.; de Franchis, R.; Kraus, J.P.; Andria, G. The molecular basis of homocystinuria due to cystathionine beta-synthase deficiency in italian families, and report of four novel mutations. Am. J. Hum. Genet. 1995, 56, 1324–1333. [Google Scholar] [PubMed]
- Nienaber-Rousseau, C.; Ellis, S.M.; Moss, S.J.; Melse-Boonstra, A.; Towers, G.W. Gene-environment and gene-gene interactions of specific MTHFR, MTR and CBS gene variants in relation to homocysteine in black south africans. Gene 2013, 530, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Lievers, K.J.; Kluijtmans, L.A.; Heil, S.G.; Boers, G.H.; Verhoef, P.; van Oppenraay-Emmerzaal, D.; den Heijer, M.; Trijbels, F.J.; Blom, H.J. A 31 bp VNTR in the cystathionine beta-synthase (CBS) gene is associated with reduced cbs activity and elevated post-load homocysteine levels. Eur. J. Hum. Genet. 2001, 9, 583–589. [Google Scholar] [CrossRef] [PubMed]
- Heil, S.G.; Van der Put, N.M.J.; Waas, E.T.; den Heijer, M.; Trijbels, F.J.M.; Blom, H.J. Is mutated serine hydroxymethyltransferase (SHMT) involved in the etiology of neural tube defects? Mol. Genet. Metabol. 2001, 73, 164–172. [Google Scholar] [CrossRef] [PubMed]
- Ho, R.C.M.; Cheung, M.W.L.; Fu, E.; Win, H.H.; Zaw, M.H.; Ng, A.; Mak, A. Is high homocysteine level a risk factor for cognitive decline in elderly? A systematic review, meta-analysis, and meta-regression. Am. J. Geriatr. Psychiatry 2011, 19, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Seshadri, S.; Beiser, A.; Selhub, J.; Jacques, P.F.; Rosenberg, I.H.; D’Agostino, R.B.; Wilson, P.W.F.; Wolf, P.A. Plasma homocysteine as a risk factor for dementia and Alzheimer’s disease. N. Engl. J. Med. 2002, 346, 476–483. [Google Scholar] [CrossRef] [PubMed]
- Schafer, J.H.; Glass, T.A.; Bolla, K.I.; Mintz, M.; Jedlicka, A.E.; Schwartz, B.S. Homocysteine and cognitive function in a population-based study of older adults. J. Am. Geriatr. Soc. 2005, 53, 381–388. [Google Scholar] [CrossRef] [PubMed]
- McCaddon, A.; Miller, J.W. Assessing the association between homocysteine and cognition: Reflections on bradford hill, meta-analyses, and causality. Nutr. Rev. 2015, 73, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.; Ji, H.F. Associations between homocysteine, folic acid, vitamin B12 and Alzheimer’s disease: Insights from meta-analyses. J. Alzheimers Dis. 2015, 46, 777–790. [Google Scholar] [CrossRef] [PubMed]
- Flicker, L.; Martins, R.N.; Thomas, J.; Acres, J.; Taddei, K.; Vasikaran, S.D.; Norman, P.; Jamrozik, K.; Almeida, O.P. B-vitamins reduce plasma levels of beta amyloid. Neurobiol. Aging 2008, 29, 303–305. [Google Scholar] [CrossRef] [PubMed]
- Durga, J.; van Boxtel, M.P.J.; Schouten, E.G.; Kok, F.J.; Jolles, J.; Katan, M.B.; Verhoef, P. Effect of 3-year folic acid supplementation on cognitive function in older adults in the facit trial: A randomised, double blind, controlled trial. Lancet 2007, 369, 208–216. [Google Scholar] [CrossRef]
- Smith, A.D.; Smith, S.M.; de Jager, C.A.; Whitbread, P.; Johnston, C.; Agacinski, G.; Oulhaj, A.; Bradley, K.M.; Jacoby, R.; Refsum, H. Homocysteine-lowering by B vitamins slows the rate of accelerated brain atrophy in mild cognitive impairment: A randomized controlled trial. PLoS ONE 2010, 5, e12244. [Google Scholar] [CrossRef] [PubMed]
- Douaud, G.; Refsum, H.; de Jager, C.A.; Jacoby, R.; Nichols, T.E.; Smith, S.M.; Smith, A.D. Preventing Alzheimer’s disease-related gray matter atrophy by B-vitamin treatment. Proc. Natl. Acad. Sci. USA 2013, 110, 9523–9528. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Halsey, J.; Bennett, D.; Lewington, S. Homocysteine and vascular disease: Review of published results of the homocysteine-lowering trials. J. Inherit. Metab. Dis. 2011, 34, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Bennett, D.; Parish, S.; Lewington, S.; Skeaff, M.; Eussen, S.J.; Lewerin, C.; Stott, D.J.; Armitage, J.; Hankey, G.J.; et al. Effects of homocysteine lowering with B vitamins on cognitive aging: Meta-analysis of 11 trials with cognitive data on 22,000 individuals. Am. J. Clin. Nutr. 2014, 100, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Fox, J.T.; Stover, P.J. Chapter 1 folate-mediated one-carbon metabolism. In Vitamins & Hormones; Gerald, L., Ed.; Academic Press: New York, NY, USA, 2008; Volume 79, pp. 1–44. [Google Scholar]
- Shane, B. Folate and vitamin B12 metabolism: Overview and interaction with riboflavin, vitamin B6, and polymorphisms. Food Nutr. Bull. 2008, 29, S5–S16. [Google Scholar] [CrossRef] [PubMed]
- Selhub, J. Homocysteine metabolism. Annu. Rev. Nutr. 1999, 19, 217–246. [Google Scholar] [CrossRef] [PubMed]
- Gregory, J.F., 3rd. Chemical and nutritional aspects of folate research: Analytical procedures, methods of folate synthesis, stability, and bioavailability of dietary folates. Adv. Food Nutr. Res. 1989, 33, 1–101. [Google Scholar] [PubMed]
- Malinow, M.R.; Nieto, F.J.; Kruger, W.D.; Duell, P.B.; Hess, D.L.; Gluckman, R.A.; Block, P.C.; Holzgang, C.R.; Anderson, P.H.; Seltzer, D.; et al. The effects of folic acid supplementation on plasma total homocysteine are modulated by multivitamin use and methylenetetrahydrofolate reductase genotypes. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 1157–1162. [Google Scholar] [CrossRef] [PubMed]
- Bailey, L.B.; Duhaney, R.L.; Maneval, D.R.; Kauwell, G.P.A.; Quinlivan, E.P.; Davis, S.R.; Cuadras, A.; Hutson, A.D.; Gregory, J.F. Vitamin B-12 status is inversely associated with plasma homocysteine in young women with C677T and/or a1298c methylenetetrahydrofolate reductase polymorphisms. J. Nutr. 2002, 132, 1872–1878. [Google Scholar] [PubMed]
- Gaughan, D.J.; Kluijtmans, L.A.J.; Barbaux, S.; McMaster, D.; Young, I.S.; Yarnell, J.W.G.; Evans, A.; Whitehead, A.S. The methionine synthase reductase (MTRR) A66G polymorphism is a novel genetic determinant of plasma homocysteine concentrations. Atherosclerosis 2001, 157, 451–456. [Google Scholar] [CrossRef]
- Ludwig, M.L.; Matthews, R.G. Structure-based perspectives on B12-dependent enzymes. Annu. Rev. Biochem. 1997, 66, 269–313. [Google Scholar] [CrossRef] [PubMed]
- Leclerc, D.; Wilson, A.; Dumas, R.; Gafuik, C.; Song, D.; Watkins, D.; Heng, H.H.Q.; Rommens, J.M.; Scherer, S.W.; Rosenblatt, D.S.; et al. Cloning and mapping of a cDNA for methionine synthase reductase, a flavoprotein defective in patients with homocystinuria. Proc. Natl. Acad. Sci. USA 1998, 95, 3059–3064. [Google Scholar] [CrossRef] [PubMed]
- Renwick, S.B.; Snell, K.; Baumann, U. The crystal structure of human cytosolic serine hydroxymethyltransferase: A target for cancer chemotherapy. Structure 1998, 6, 1105–1116. [Google Scholar] [CrossRef]
- Rosenblatt, D.S. Inherited disorders of folate transport and metabolism. In The Metabolic Basis of Inherited Disease; Seriver, A.L., Beaudet, W.S., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 1995; Volume 2, pp. 3111–3128. [Google Scholar]
- Pereira, A.C.; Schettert, I.T.; Morandini Filho, A.A.F.; Guerra-Shinohara, E.M.; Krieger, J.E. Methylenetetrahydrofolate reductase (MTHFR) C677T gene variant modulates the homocysteine folate correlation in a mild folate-deficient population. Clin. Chim. Acta 2004, 340, 99–105. [Google Scholar] [CrossRef] [PubMed]
- Meisel, C.; Cascorbi, I.; Gerloff, T.; Stangl, V.; Laule, M.; Müller, J.M.; Wernecke, K.D.; Baumann, G.; Roots, I.; Stangl, K. Identification of six methylenetetrahydrofolate reductase (MTHFR) genotypes resulting from common polymorphisms: Impact on plasma homocysteine levels and development of coronary artery disease. Atherosclerosis 2001, 154, 651–658. [Google Scholar] [CrossRef]
- Botto, L.D.; Yang, Q. 5,10-methylenetetrahydrofolate reductase gene variants and congenital anomalies: A huge review. Am. J. Epidemiol. 2000, 151, 862–877. [Google Scholar] [CrossRef] [PubMed]
- Wilcken, B.; Bamforth, F.; Li, Z.; Zhu, H.; Ritvanen, A.; Redlund, M.; Stoll, C.; Alembik, Y.; Dott, B.; Czeizel, A.; et al. Geographical and ethnic variation of the 677c>t allele of 5,10 methylenetetrahydrofolate reductase (MTHFR): Findings from over 7000 newborns from 16 areas world wide. J. Med. Genet. 2003, 40, 619–625. [Google Scholar] [CrossRef] [PubMed]
- Harmon, D.L.; Woodside, J.V.; Yarnell, J.W.G.; McMaster, D.; Young, I.S.; McCrum, E.E.; Gey, K.F.; Whitehead, A.S.; Evans, A.E. The Common ‘Thermolabile’ Variant of Methylene Tetrahydrofolate Reductase is a Major Determinant of Mild Hyperhomocysteinaemia. QJM 1996, 89, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.Y.; Bignell, M.; Yang, F.; Welge, B.G.; Graham, K.J.; Hanson, N.Q. Polygenic influence on plasma homocysteine: Association of two prevalent mutations, the 844INS68 of cystathionine β-synthase and A2756G of methionine synthase, with lowered plasma homocysteine levels. Atherosclerosis 2000, 149, 131–137. [Google Scholar] [CrossRef]
- Dekou, V.; Gudnason, V.; Hawe, E.; Miller, G.J.; Stansbie, D.; Humphries, S.E. Gene-environment and gene-gene interaction in the determination of plasma homocysteine levels in healthy middle-aged men. Thromb. Haemost. 2001, 85, 67–74. [Google Scholar] [PubMed]
- Kluijtmans, L.A.J.; Young, I.S.; Boreham, C.A.; Murray, L.; McMaster, D.; McNulty, H.; Strain, J.J.; McPartlin, J.; Scott, J.M.; Whitehead, A.S. Genetic and Nutritional Factors Contributing to Hyperhomocysteinemia in Young Adults. Blood 2003, 101, 2483–2488. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, S.; McConnell, J.; Little, J.; Sharp, L.; Piyathilake, C.J.; Powers, H.; Basten, G.; Duthie, S.J. Associations between two common variants C677T and A1298C in the methylenetetrahydrofolate reductase gene and measures of folate metabolism and DNA stability (strand breaks, misincorporated uracil, and DNA methylation status) in human lymphocytes in vivo. Cancer Epidemiol. Biomark. Prev. 2004, 13, 1436–1443. [Google Scholar]
- Devlin, A.M.; Clarke, R.; Birks, J.; Evans, J.G.; Halsted, C.H. Interactions among polymorphisms in folate-metabolizing genes and serum total homocysteine concentrations in a healthy elderly population. Am. J. Clin. Nutr. 2006, 83, 708–713. [Google Scholar] [PubMed]
- DeVos, L.; Chanson, A.; Liu, Z.; Ciappio, E.D.; Parnell, L.D.; Mason, J.B.; Tucker, K.L.; Crott, J.W. Associations between single nucleotide polymorphisms in folate uptake and metabolizing genes with blood folate, homocysteine, and DNA uracil concentrations. Am. J. Clin. Nutr. 2008, 88, 1149–1158. [Google Scholar] [PubMed]
- Kim, J.-M.; Stewart, R.; Kim, S.-W.; Yang, S.-J.; Shin, I.-S.; Shin, H.-Y.; Yoon, J.-S. Methylenetetrahydrofolate reductase gene and risk of Alzheimer’s disease in koreans. Int. J. Geriatr. Psychiatry 2008, 23, 454–459. [Google Scholar] [CrossRef] [PubMed]
- Yang, Q.-H.; Botto, L.D.; Gallagher, M.; Friedman, J.; Sanders, C.L.; Koontz, D.; Nikolova, S.; Erickson, J.D.; Steinberg, K. Prevalence and effects of gene-gene and gene-nutrient interactions on serum folate and serum total homocysteine concentrations in the united states: Findings from the third national health and nutrition examination survey DNA bank. Am. J. Clin. Nutr. 2008, 88, 232–246. [Google Scholar] [PubMed]
- Zappacosta, B.; Graziano, M.; Persichilli, S.; Di Castelnuovo, A.; Mastroiacovo, P.; Iacoviello, L. 5,10-methylenetetrahydrofolate reductase (MTHFR) C677T and A1298C polymorphisms: Genotype frequency and association with homocysteine and folate levels in middle-southern italian adults. Cell Biochem. Funct. 2014, 32, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Zhu, R.-X.; He, Z.-Y.; Liu, X.; Liu, H.-N. Association of MTHFR C677T with total homocysteine plasma levels and susceptibility to parkinson’s disease: A meta-analysis. Neurol. Sci. 2015, 36, 945–951. [Google Scholar] [CrossRef] [PubMed]
- Chango, A.; Potier De Courcy, G.; Boisson, F.; Guilland, J.C.; Barbe, F.; Perrin, M.O.; Christides, J.P.; Rabhi, K.; Pfister, M.; Galan, P.; et al. 5,10-methylenetetrahydrofolate reductase common mutations, folate status and plasma homocysteine in healthy french adults of the supplementation en vitamines et mineraux antioxydants (su.Vi.Max) cohort. Br. J. Nutr. 2000, 84, 891–896. [Google Scholar] [PubMed]
- Geisel, J.; Zimbelmann, I.; Schorr, H.; Knapp, J.P.; Bodis, M.; Hübner, U.; Herrmann, W. Genetic defects as important factors for moderate hyperhomocysteinemia. Clin. Chem. Lab. Med. 2001, 39, 698–704. [Google Scholar] [CrossRef] [PubMed]
- Jacques, P.F.; Bostom, A.G.; Williams, R.R.; Ellison, R.C.; Eckfeldt, J.H.; Rosenberg, I.H.; Selhub, J.; Rozen, R. Relation between folate status, a common mutation in methylenetetrahydrofolate reductase, and plasma homocysteine concentrations. Circulation 1996, 93, 7–9. [Google Scholar] [CrossRef] [PubMed]
- García-Minguillán, C.J.; Fernandez-Ballart, J.D.; Ceruelo, S.; Ríos, L.; Bueno, O.; Berrocal-Zaragoza, M.I.; Molloy, A.M.; Ueland, P.M.; Meyer, K.; Murphy, M.M. Riboflavin status modifies the effects of methylenetetrahydrofolate reductase (MTHFR) and methionine synthase reductase (mtrr) polymorphisms on homocysteine. Genes Nutr. 2014, 9, 435. [Google Scholar] [CrossRef] [PubMed]
- Guttormsen, A.B.; Ueland, P.M.; Nesthus, I.; Nyg, E.O.; Schneede, J.; Vollset, S.E.; Refsum, H. Determinants and vitamin responsiveness of intermediate hyperhomocysteinemia (> or = 40 micromol/liter). The hordaland homocysteine study. J. Clin. Investig. 1996, 98, 2174–2183. [Google Scholar] [CrossRef] [PubMed]
- Kauwell, G.P.; Wilsky, C.E.; Cerda, J.J.; Herrlinger-Garcia, K.; Hutson, A.D.; Theriaque, D.W.; Boddie, A.; Rampersaud, G.C.; Bailey, L.B. Methylenetetrahydrofolate reductase mutation (677c-->t) negatively influences plasma homocysteine response to marginal folate intake in elderly women. Metabolism 2000, 49, 1440–1443. [Google Scholar] [CrossRef] [PubMed]
- Hustad, S.; Ueland, P.M.; Vollset, S.E.; Zhang, Y.; Bjørke-Monsen, A.L.; Schneede, J. Riboflavin as a determinant of plasma total homocysteine: Effect modification by the methylenetetrahydrofolate reductase C677T polymorphism. Clin. Chem. 2000, 46, 1065–1071. [Google Scholar] [PubMed]
- McNulty, H.; Dowey, L.R.C.; Strain, J.J.; Dunne, A.; Ward, M.; Molloy, A.M.; McAnena, L.B.; Hughes, J.P.; Hannon-Fletcher, M.; Scott, J.M. Riboflavin lowers homocysteine in individuals homozygous for the MTHFR 677c→t polymorphism. Circulation 2006, 113, 74–80. [Google Scholar] [CrossRef] [PubMed]
- Jacques, P.F.; Kalmbach, R.; Bagley, P.J.; Russo, G.T.; Rogers, G.; Wilson, P.W.F.; Rosenberg, I.H.; Selhub, J. The relationship between riboflavin and plasma total homocysteine in the framingham offspring cohort is influenced by folate status and the C677T transition in the methylenetetrahydrofolate reductase gene. J. Nutr. 2002, 132, 283–288. [Google Scholar] [PubMed]
- Hustad, S.; Midttun, Ø.; Schneede, J.; Vollset, S.E.; Grotmol, T.; Ueland, P.M. The methylenetetrahydrofolate reductase 677c→t polymorphism as a modulator of a B vitamin network with major effects on homocysteine metabolism. Am. J. Hum. Genet. 2007, 80, 846–855. [Google Scholar] [CrossRef] [PubMed]
- D'Angelo, A.; Coppola, A.; Madonna, P.; Fermo, I.; Pagano, A.; Mazzola, G.; Galli, L.; Cerbone, A.M. The role of vitamin B12 in fasting hyperhomocysteinemia and its interaction with the homozygous C677T mutation of the methylenetetrahydrofolate reductase (MTHFR) gene a case-control study of patients with early-onset thrombotic events. Thromb. Haemost. 2000, 83, 563–570. [Google Scholar] [PubMed]
- Trimmer, E.E. Methylenetetrahydrofolate reductase: Biochemical characterization and medical significance. Curr. Pharm. Design 2013, 19, 2574–2593. [Google Scholar] [CrossRef]
- Friedman, G.; Goldschmidt, N.; Friedlander, Y.; Ben-Yehuda, A.; Selhub, J.; Babaey, S.; Mendel, M.; Kidron, M.; Bar-On, H. A common mutation A1298C in human methylenetetrahydrofolate reductase gene: Association with plasma total homocysteine and folate concentrations. J. Nutr. 1999, 129, 1656–1661. [Google Scholar] [PubMed]
- Vaughn, J.D.; Bailey, L.B.; Shelnutt, K.P.; Dunwoody, K.M.V.-C.; Maneval, D.R.; Davis, S.R.; Quinlivan, E.P.; Gregory, J.F.; Theriaque, D.W.; Kauwell, G.P.A. Methionine synthase reductase 66a→g polymorphism is associated with increased plasma homocysteine concentration when combined with the homozygous methylenetetrahydrofolate reductase 677c→t variant. J. Nutr. 2004, 134, 2985–2990. [Google Scholar] [PubMed]
- van der Put, N.M.; Gabreëls, F.; Stevens, E.M.; Smeitink, J.A.; Trijbels, F.J.; Eskes, T.K.; van den Heuvel, L.P.; Blom, H.J. A second common mutation in the methylenetetrahydrofolate reductase gene: An additional risk factor for neural-tube defects? Am. J. Hum. Genet. 1998, 62, 1044–1051. [Google Scholar] [CrossRef] [PubMed]
- Cabo, R.; Hernes, S.; Slettan, A.; Haugen, M.; Ye, S.; Blomhoff, R.; Mansoor, M.A. Effect of genetic polymorphisms involved in folate metabolism on the concentration of serum folate and plasma total homocysteine (p-thcy) in healthy subjects after short-term folic acid supplementation: A randomized, double blind, crossover study. Genes Nutr. 2015, 10, 7. [Google Scholar] [CrossRef] [PubMed]
- Morrison, K.; Edwards, Y.H.; Lynch, S.A.; Burn, J.; Hol, F.; Mariman, E. Methionine synthase and neural tube defects. J. Med. Genet. 1997, 34, 958. [Google Scholar] [CrossRef] [PubMed]
- van der Put, N.M.; van der Molen, E.F.; Kluijtmans, L.A.; Heil, S.G.; Trijbels, J.M.; Eskes, T.K.; Van Oppenraaij-Emmerzaal, D.; Banerjee, R.; Blom, H.J. Sequence analysis of the coding region of human methionine synthase: Relevance to hyperhomocysteinaemia in neural-tube defects and vascular disease. QJM 1997, 90, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Morita, H.; Kurihara, H.; Sugiyama, T.; Hamada, C.; Kurihara, Y.; Shindo, T.; Oh-hashi, Y.; Yazaki, Y. Polymorphism of the methionine synthase gene: Association with homocysteine metabolism and late-onset vascular diseases in the japanese population. Arterioscler. Thromb. Vasc. Biol. 1999, 19, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Jacques, P.F.; Bostom, A.G.; Selhub, J.; Rich, S.; Curtis Ellison, R.; Eckfeldt, J.H.; Gravel, R.A.; Rozen, R. Effects of polymorphisms of methionine synthase and methionine synthase reductase on total plasma homocysteine in the NHLBI family heart study. Atherosclerosis 2003, 166, 49–55. [Google Scholar] [CrossRef]
- Wang, X.L.; Duarte, N.; Cai, H.; Adachi, T.; Sim, A.S.; Cranney, G.; Wilcken, D.E.L. Relationship between total plasma homocysteine, polymorphisms of homocysteine metabolism related enzymes, risk factors and coronary artery disease in the australian hospital-based population. Atherosclerosis 1999, 146, 133–140. [Google Scholar] [CrossRef]
- Tsai, M.Y.; Welge, B.G.; Hanson, N.Q.; Bignell, M.K.; Vessey, J.; Schwichtenberg, K.; Yang, F.; Bullemer, F.E.; Rasmussen, R.; Graham, K.J. Genetic causes of mild hyperhomocysteinemia in patients with premature occlusive coronary artery diseases. Atherosclerosis 1999, 143, 163–170. [Google Scholar] [CrossRef]
- Ma, J.; Stampfer, M.J.; Christensen, B.; Giovannucci, E.; Hunter, D.J.; Chen, J.; Willett, W.C.; Selhub, J.; Hennekens, C.H.; Gravel, R.; et al. A polymorphism of the methionine synthase gene: Association with plasma folate, vitamin B12, homocyst(e)ine, and colorectal cancer risk. Cancer Epidemiol. Biomark. Prev. 1999, 8, 825–829. [Google Scholar]
- Hyndman, M.E.; Bridge, P.J.; Warnica, J.W.; Fick, G.; Parsons, H.G. Effect of heterozygosity for the methionine synthase 2756 a→g mutation on the risk for recurrent cardiovascular events. Am. J. Cardiol. 2000, 86, 1144–1146. [Google Scholar] [CrossRef]
- Harmon, D.L.; Shields, D.C.; Woodside, J.V.; McMaster, D.; Yarnell, J.W.G.; Young, I.S.; Peng, K.; Shane, B.; Evans, A.E.; Whitehead, A.S. Methionine synthase D919G polymorphism is a significant but modest determinant of circulating homocysteine concentrations. Genet. Epidemiol. 1999, 17, 298–309. [Google Scholar] [CrossRef]
- Chen, J.; Stampfer, M.J.; Ma, J.; Selhub, J.; Malinow, M.R.; Hennekens, C.H.; Hunter, D.J. Influence of a methionine synthase (D919G) polymorphism on plasma homocysteine and folate levels and relation to risk of myocardial infarction. Atherosclerosis 2001, 154, 667–672. [Google Scholar] [CrossRef]
- McCaddon, A.; Regland, B.; Hudson, P.; Davies, G. Functional vitamin B(12) deficiency and Alzheimer’s disease. Neurology 2002, 58, 1395–1399. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.; Platt, R.; Wu, Q.; Leclerc, D.; Christensen, B.; Yang, H.; Gravel, R.A.; Rozen, R. A common variant in methionine synthase reductase combined with low cobalamin (vitamin B12) increases risk for spina bifida. Mol. Genet. Metab. 1999, 67, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Rai, V.; Yadav, U.; Kumar, P.; Yadav, S.K. Analysis of methionine synthase reductase polymorphism (A66G) in indian muslim population. Indian J. Hum. Genet. 2013, 19, 183–187. [Google Scholar] [CrossRef] [PubMed]
- Rady, P.L.; Szucs, S.; Grady, J.; Hudnall, S.D.; Kellner, L.H.; Nitowsky, H.; Tyring, S.K.; Matalon, R.K. Genetic polymorphisms of methylenetetrahydrofolate reductase (MTHFR) and methionine synthase reductase (MTRR) in ethnic populations in texas; a report of a novel MTHFR polymorphic site, G1793A. Am. J. Med. Genet. 2002, 107, 162–168. [Google Scholar] [CrossRef] [PubMed]
- Brilakis, E.S.; Berger, P.B.; Ballman, K.V.; Rozen, R. Methylenetetrahydrofolate reductase (MTHFR) 677C>T and methionine synthase reductase (MTRR) 66A>G polymorphisms: Association with serum homocysteine and angiographic coronary artery disease in the era of flour products fortified with folic acid. Atherosclerosis 2003, 168, 315–322. [Google Scholar] [PubMed]
- Brown, C.A.; McKinney, K.Q.; Kaufman, J.S.; Gravel, R.A.; Rozen, R. A common polymorphism in methionine synthase reductase increases risk of premature coronary artery disease. J. Cardiovasc. Risk 2000, 7, 197–200. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, V.B.; Parle-McDermott, A.; Molloy, A.M.; Kirke, P.N.; Johnson, Z.; Conley, M.; Scott, J.M.; Mills, J.L. MTRR and MTHFR polymorphism: Link to down syndrome? Am. J. Med. Genet. 2002, 107, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Gaughan, D.J.; Kluijtmans, L.A.J.; Barbaux, S.; McMaster, D.; Young, I.S.; Yarnell, J.W.G.; Evans, A.; Whitehead, A.S. Corrigendum to “the methionine synthase reductase (MTRR) A66G polymorphism is a novel genetic determinant of plasma homocysteine concentrations” [ath 157 (2001) 451–456]. Atherosclerosis 2003, 167, 373. [Google Scholar] [CrossRef]
- De Bruijn, R.F.A.G.; Ikram, M.A. Cardiovascular risk factors and future risk of Alzheimer’s disease. BMC Med. 2014, 12, 130. [Google Scholar] [CrossRef] [PubMed]
- Kluijtmans, L.A.; van den Heuvel, L.P.; Boers, G.H.; Frosst, P.; Stevens, E.M.; van Oost, B.A.; den Heijer, M.; Trijbels, F.J.; Rozen, R.; Blom, H.J. Molecular genetic analysis in mild hyperhomocysteinemia: A common mutation in the methylenetetrahydrofolate reductase gene is a genetic risk factor for cardiovascular disease. Am. J. Hum. Genet. 1996, 58, 35–41. [Google Scholar] [PubMed]
- Gallagher, P.M.; Meleady, R.; Shields, D.C.; Tan, K.S.; McMaster, D.; Rozen, R.; Evans, A.; Graham, I.M.; Whitehead, A.S. Homocysteine and risk of premature coronary heart disease: Evidence for a common gene mutation. Circulation 1996, 94, 2154–2158. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.L.; Gu, Z.; Kozich, V.; Kraus, J.P.; Ramesh, V.; Shih, V.E. Molecular basis of cystathionine beta-synthase deficiency in pyridoxine responsive and nonresponsive homocystinuria. Hum. Mol. Genet. 1993, 2, 1857–1860. [Google Scholar] [CrossRef] [PubMed]
- Kraus, J.P. Komrower lecture. Molecular basis of phenotype expression in homocystinuria. J. Inherit. Metab. Dis. 1994, 17, 383–390. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, V.; Dekou, V.; Nicaud, V.; Chasse, J.F.; London, J.; Stansbie, D.; Humphries, S.E.; Gudnason, V. Linkage disequilibrium at the cystathionine beta synthase (CBS) locus and the association between genetic variation at the CBS locus and plasma levels of homocysteine. The ears II group. European atherosclerosis research study. Ann. Hum. Genet. 1998, 62, 481–490. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.Y.; Bignell, M.; Schwichtenberg, K.; Hanson, N.Q. High prevalence of a mutation in the cystathionine beta-synthase gene. Am. J. Hum. Genet. 1996, 59, 1262–1267. [Google Scholar] [PubMed]
- Kluijtmans, L.A.; Boers, G.H.; Trijbels, F.J.; van Lith-Zanders, H.M.; van den Heuvel, L.P.; Blom, H.J. A common 844INS68 insertion variant in the cystathionine beta-synthase gene. Biochem. Mol. Med. 1997, 62, 23–25. [Google Scholar] [CrossRef] [PubMed]
- Gan, Y.Y.; Chen, C.F. Novel alleles of 31-bp VNTR polymorphism in the human cystathionine beta-synthase (CBS) gene were detected in healthy asians. J. Genet. 2010, 89, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Vijaya Lakshmi, S.V.; Naushad, S.; Seshagiri Rao, D.; Kutala, V. Oxidative stress is associated with genetic polymorphisms in one-carbon metabolism in coronary artery disease. Cell Biochem. Biophys. 2013, 67, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Prince, J.A.; Feuk, L.; Sawyer, S.L.; Gottfries, J.; Ricksten, A.; Nagga, K.; Bogdanovic, N.; Blennow, K.; Brookes, A.J. Lack of replication of association findings in complex disease: An analysis of 15 polymorphisms in prior candidate genes for sporadic Alzheimer’s disease. Eur. J. Hum. Genet. 2001, 9, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Linnebank, M.; Linnebank, A.; Jeub, M.; Klockgether, T.; Wüllner, U.; Kölsch, H.; Heun, R.; Koch, H.G.; Suormala, T.; Fowler, B. Lack of genetic dispositions to hyperhomocysteinemia in Alzheimer’s disease. Am. J. Med. Genet. 2004, 131 A, 101–102. [Google Scholar] [CrossRef] [PubMed]
- Religa, D.; Styczynska, M.; Peplonska, B.; Gabryelewicz, T.; Pfeffer, A.; Chodakowska, M.; Luczywek, E.; Wasiak, B.; Stepien, K.; Golebiowski, M.; et al. Homocysteine, apolipoproteine E and methylenetetrahydrofolate reductase in Alzheimer’s disease and mild cognitive impairment. Dement. Geriatr. Cogn. Disord. 2003, 16, 64–70. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Smith, A.D.; Jobst, K.A.; Refsum, H.; Sutton, L.; Ueland, P.M. Folate, vitamin B12, and serum total homocysteine levels in confirmed Alzheimer’s disease. Arch. Neurol. 1998, 55, 1449–1455. [Google Scholar] [CrossRef] [PubMed]
- Chapman, J.; Wang, N.; Treves, T.A.; Korczyn, A.D.; Bornstein, N.M. Ace, MTHFR, factor v leiden, and apoe polymorphisms in patients with vascular and Alzheimer’s dementia. Stroke 1998, 29, 1401–1404. [Google Scholar] [CrossRef] [PubMed]
- Kida, T.; Kamino, K.; Yamamoto, M.; Kanayama, D.; Tanaka, T.; Kudo, T.; Takeda, M. C677T polymorphism of methylenetetrahydrofolate reductase gene affects plasma homocysteine level and is a genetic factor of late-onset Alzheimer’s disease. Psychogeriatrics 2004, 4, 4–10. [Google Scholar] [CrossRef]
- Brunelli, T.; Bagnoli, S.; Giusti, B.; Nacmias, B.; Pepe, G.; Sorbi, S.; Abbate, R. The C677T methylenetetrahydrofolate reductase mutation is not associated with Alzheimer’s disease. Neurosci. Lett. 2001, 315, 103–105. [Google Scholar] [CrossRef]
- Postiglione, A.; Milan, G.; Ruocco, A.; Gallotta, G.; Guiotto, G.; Di Minno, G. Plasma folate, vitamin B12, and total homocysteine and homozygosity for the C677T mutation of the 5,10-methylene tetrahydrofolate reductase gene in patients with Alzheimer’s dementia: A case-control study. Gerontology 2001, 47, 324–329. [Google Scholar] [CrossRef] [PubMed]
- Zuliani, G.; Ble, A.; Zanca, R.; Munari, M.R.; Zurlo, A.; Vavalle, C.; Atti, A.R.; Fellin, R. Genetic polymorphisms in older subjects with vascular or Alzheimer’s dementia. Acta Neurol. Scand. 2001, 103, 304–308. [Google Scholar] [CrossRef] [PubMed]
- Seripa, D.; Dal Forno, G.; Matera, M.G.; Gravina, C.; Margaglione, M.; Palermo, M.T.; Wekstein, D.R.; Antuono, P.; Davis, D.G.; Daniele, A.; et al. Methylenetetrahydrofolate reductase and angiotensin converting enzyme gene polymorphisms in two genetically and diagnostically distinct cohort of Alzheimer patients. Neurobiol. Aging 2003, 24, 933–939. [Google Scholar] [CrossRef]
- Bosco, P.; Gueant-Rodriguez, R.; Anello, G.; Romano, A.; Namour, B.; Spada, R.; Caraci, F.; Tringali, G.; Ferri, R.; Gueant, J. Association of IL-1 RN*2 allele and methionine synthase 2756 AA genotype with dementia severity of sporadic Alzheimer’s disease. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1036–1038. [Google Scholar] [CrossRef] [PubMed]
- Wehr, H.; Bednarska-Makaruk, M.; Łojkowska, W.; Graban, A.; Hoffman-Zacharska, D.; Kuczyńska-Zardzewiały, A.; Mrugała, J.; Rodo, M.; Bochyńska, A.; Sułek, A.; et al. Differences in risk factors for dementia with neurodegenerative traits and for vascular dementia. Dement. Geriatr. Cogn. Disord. 2006, 22, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Styczynska, M.; Strosznajder, J.B.; Religa, D.; Chodakowska-Zebrowska, M.; Pfeffer, A.; Gabryelewicz, T.; Czapski, G.A.; Kobrys, M.; Karciauskas, G.; Barcikowska, M. Association between genetic and environmental factors and the risk of Alzheimer’s disease. Folia Neuropathol. Assoc. Pol. Neuropathol. Med. Res. Centre Pol. Acad. Sci. 2008, 46, 249–254. [Google Scholar]
- Fernandez, L.L.; Scheibe, R.M. Is MTHFR polymorphism a risk factor for Alzheimer’s disease like apoe? Arquivos de Neuro-Psiquiatria 2005, 63, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.-Y.; Miao, L.; Li, Y.-S.; Hu, G.-Y. Meta-analysis of the methylenetetrahydrofolate reductase C677T polymorphism and susceptibility to Alzheimer’s disease. Neurosci. Res. 2010, 68, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Rai, V. Folate pathway gene methylenetetrahydrofolate reductase C677T polymorphism and Alzheimer’s disease risk in asian population. Indian J. Clin. Biochem. 2016, 31, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Keikhaee, M.; Hashemi, S.; Najmabadi, H.; Noroozian, M. C677T methylentetrahydrofulate reductase and angiotensin converting enzyme gene polymorphisms in patients with Alzheimer’s disease in iranian population. Neurochem. Res. 2006, 31, 1079–1083. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, V.C.; Ramos, F.J.D.C.; Freitas, E.M.; De Brito-Marques, P.R.; Cavalcanti, M.N.D.H.; D’Almeida, V.; Cabral-Filho, J.E.; Muniz, M.T.C. Alzheimer’s disease in brazilian elderly has a relation with homocysteine but not with MTHFR polymorphisms. Arquivos de Neuro-Psiquiatria 2006, 64, 941–945. [Google Scholar] [CrossRef] [PubMed]
- Bottiglieri, T.; Parnetti, L.; Arning, E.; Ortiz, T.; Amici, S.; Lanari, A.; Gallai, V. Plasma total homocysteine levels and the C677T mutation in the methylenetetrahydrofolate reductase (MTHFR) gene: A study in an italian population with dementia. Mech. Ageing Dev. 2001, 122, 2013–2023. [Google Scholar] [CrossRef]
- McGarel, C.; Pentieva, K.; Strain, J.J.; McNulty, H. Emerging roles for folate and related b-vitamins in brain health across the lifecycle. Proc. Nutr. Soc. 2015, 74, 46–55. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Nicolia, V.; Cavallaro, R.A.; Ricceri, L.; D’Anselmi, F.; Coluccia, P.; Calamandrei, G.; Scarpa, S. B-vitamin deprivation induces hyperhomocysteinemia and brain S-adenosylhomocysteine, depletes brain S-adenosylmethionine, and enhances ps1 and bace expression and amyloid-β deposition in mice. Mol. Cell. Neurosci. 2008, 37, 731–746. [Google Scholar] [CrossRef] [PubMed]
- Sai, X.; Kawamura, Y.; Kokame, K.; Yamaguchi, H.; Shiraishi, H.; Suzuki, R.; Suzuki, T.; Kawaichi, M.; Miyata, T.; Kitamura, T.; et al. Endoplasmic reticulum stress-inducible protein, herp, enhances presenilin-mediated generation of amyloid β-protein. J. Biol. Chem. 2002, 277, 12915–12920. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, J.M.; Portugal, G.S.; Kruger, W.D.; Wang, H.; Gould, T.J.; Praticò, D. Diet-induced hyperhomocysteinemia increases amyloid-β formation and deposition in a mouse model of Alzheimer’s disease. Curr. Alzheimer Res. 2010, 7, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Pacheco-Quinto, J.; Rodriguez de Turco, E.B.; DeRosa, S.; Howard, A.; Cruz-Sanchez, F.; Sambamurti, K.; Refolo, L.; Petanceska, S.; Pappolla, M.A. Hyperhomocysteinemic Alzheimer’s mouse model of amyloidosis shows increased brain amyloid β peptide levels. Neurobiol. Dis. 2006, 22, 651–656. [Google Scholar] [CrossRef] [PubMed]
- Sontag, E.; Nunbhakdi-Craig, V.; Sontag, J.-M.; Diaz-Arrastia, R.; Ogris, E.; Dayal, S.; Lentz, S.R.; Arning, E.; Bottiglieri, T. Protein phosphatase 2A methyltransferase links homocysteine metabolism with tau and amyloid precursor protein regulation. J. Neurosci. 2007, 27, 2751–2759. [Google Scholar] [CrossRef] [PubMed]
- Hooshmand, B.; Polvikoski, T.; Kivipelto, M.; Tanskanen, M.; Myllykangas, L.; Erkinjuntti, T.; Mäkelä, M.; Oinas, M.; Paetau, A.; Scheltens, P.; et al. Plasma homocysteine, Alzheimer and cerebrovascular pathology: A population-based autopsy study. Brain 2013, 136, 2707–2716. [Google Scholar] [CrossRef] [PubMed]
- Walsh, D.M.; Hartley, D.M.; Kusumoto, Y.; Fezoui, Y.; Condron, M.M.; Lomakin, A.; Benedek, G.B.; Selkoe, D.J.; Teplow, D.B. Amyloid beta-protein fibrillogenesis: Structure and biological activity of protofibrillar intermediates. J. Biol. Chem. 1999, 274, 25945–25952. [Google Scholar] [CrossRef] [PubMed]
- Hardy, J.; Allsop, D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. [Google Scholar] [CrossRef]
- Li, J.-G.; Praticò, D. High levels of homocysteine results in cerebral amyloid angiopathy in mice. J. Alzheimer’s Dis. 2015, 43, 29–35. [Google Scholar]
- Zhang, C.-E.; Wei, W.; Liu, Y.-H.; Peng, J.-H.; Tian, Q.; Liu, G.-P.; Zhang, Y.; Wang, J.-Z. Hyperhomocysteinemia increases β-amyloid by enhancing expression of γ-secretase and phosphorylation of amyloid precursor protein in rat brain. Am. J. Pathol. 2009, 174, 1481–1491. [Google Scholar] [CrossRef] [PubMed]
- Agnati, L.F.; Genedani, S.; Leo, G.; Forni, A.; Woods, A.S.; Filaferro, M.; Franco, R.; Fuxe, K. Aβ peptides as one of the crucial volume transmission signals in the trophic units and their interactions with homocysteine. Physiological implications and relevance for Alzheimer’s disease. J. Neural Transm. 2007, 114, 21–31. [Google Scholar] [CrossRef] [PubMed]
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Aβ oligomers induce neuronal oxidative stress through an N-methyl-d-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 2007, 282, 11590–11601. [Google Scholar] [CrossRef] [PubMed]
- Butterfield, D.A.; Boyd-Kimball, D. Amyloid β-peptide(1–42) contributes to the oxidative stress and neurodegeneration found in Alzheimer’s disease brain. Brain Pathol. 2004, 14, 426–432. [Google Scholar] [CrossRef] [PubMed]
- Troen, A.M.; Shea-Budgell, M.; Shukitt-Hale, B.; Smith, D.E.; Selhub, J.; Rosenberg, I.H. B-vitamin deficiency causes hyperhomocysteinemia and vascular cognitive impairment in mice. Proc. Natl. Acad. Sci. USA 2008, 105, 12474–12479. [Google Scholar] [CrossRef] [PubMed]
- Upchurch, G.R., Jr.; Welch, G.N.; Fabian, A.J.; Freedman, J.E.; Johnson, J.L.; Keaney, J.F., Jr.; Loscalzo, J. Homocyst(e)ine decreases bioavailable nitric oxide by a mechanism involving glutathione peroxidase. J. Biol. Chem. 1997, 272, 17012–17017. [Google Scholar] [CrossRef] [PubMed]
- Kruman, I.I.; Culmsee, C.; Chan, S.L.; Kruman, Y.; Guo, Z.; Penix, L.; Mattson, M.P. Homocysteine elicits a DNA damage response in neurons that promotes apoptosis and hypersensitivity to excitotoxicity. J. Neurosci. 2000, 20, 6920–6926. [Google Scholar] [PubMed]
- Grieve, A.; Butcher, S.P.; Griffiths, R. Synaptosomal plasma membrane transport of excitatory sulphur amino acid transmitter candidates: Kinetic characterisation and analysis of carrier specificity. J. Neurosci. Res. 1992, 32, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Farkas, M.; Keskitalo, S.; Smith, D.E.; Bain, N.; Semmler, A.; Ineichen, B.; Smulders, Y.; Blom, H.; Kulic, L.; Linnebank, M. Hyperhomocysteinemia in Alzheimer’s disease: The hen and the egg? J. Alzheimers Dis. 2013, 33, 1097–1104. [Google Scholar] [PubMed]
- Fuchs, D.; Jaeger, M.; Widner, B.; Wirleitner, B.; Artner-Dworzak, E.; Leblhuber, F. Is hyperhomocysteinemia due to the oxidative depletion of folate rather than to insufficient dietary intake? Clin. Chem. Lab. Med. 2001, 39, 691–694. [Google Scholar] [CrossRef] [PubMed]
- Widner, B.; Fuchs, D.; Leblhuber, F.; Sperner-Unterwege, B.; Reynolds, E.; Bottiglieri, T. Does disturbed homocysteine and folate metabolism in depression result from enhanced oxidative stress? J. Neurol. Neurosurg. Psychiatry 2001, 70, 419. [Google Scholar] [CrossRef] [PubMed]
- Mudd, S.H.; Poole, J.R. Labile methyl balances for normal humans on various dietary regimens. Metabolism 1975, 24, 721–735. [Google Scholar] [CrossRef]
- Obeid, R. The metabolic burden of methyl donor deficiency with focus on the betaine homocysteine methyltransferase pathway. Nutrients 2013, 5, 3481–3495. [Google Scholar] [CrossRef] [PubMed]
- McKeever, M.P.; Weir, D.G.; Molloy, A.; Scott, J.M. Betaine-homocysteine methyltransferase: Organ distribution in man, pig and rat and subcellular distribution in the rat. Clin. Sci. 1991, 81, 551–556. [Google Scholar] [CrossRef] [PubMed]
- Caudill, M.A.; Dellschaft, N.; Solis, C.; Hinkis, S.; Ivanov, A.A.; Nash-Barboza, S.; Randall, K.E.; Jackson, B.; Solomita, G.N.; Vermeylen, F. Choline intake, plasma riboflavin, and the phosphatidylethanolamine N-methyltransferase G5465A genotype predict plasma homocysteine in folate-deplete mexican-american men with the methylenetetrahydrofolate reductase 677TT genotype. J. Nutr. 2009, 139, 727–733. [Google Scholar] [CrossRef] [PubMed]
- Ortbauer, M.; Ripper, D.; Fuhrmann, T.; Lassi, M.; Auernigg-Haselmaier, S.; Stiegler, C.; König, J. Folate deficiency and over-supplementation causes impaired folate metabolism: Regulation and adaptation mechanisms in caenorhabditis elegans. Mol. Nutr. Food Res. 2016, 60, 949–956. [Google Scholar] [CrossRef] [PubMed]
- Field, M.S.; Kamynina, E.; Agunloye, O.C.; Liebenthal, R.P.; Lamarre, S.G.; Brosnan, M.E.; Brosnan, J.T.; Stover, P.J. Nuclear enrichment of folate cofactors and methylenetetrahydrofolate dehydrogenase 1 (MTHFD1) protect de novo thymidylate biosynthesis during folate deficiency. J. Biol. Chem. 2014, 289, 29642–29650. [Google Scholar] [CrossRef] [PubMed]
- Morrison, L.D.; Smith, D.D.; Kish, S.J. Brain S-adenosylmethionine levels are severely decreased in Alzheimer’s disease. J. Neurochem. 1996, 67, 1328–1331. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.-C.; Oelze, B.; Schumacher, A. Age-specific epigenetic drift in late-onset Alzheimer’s disease. PLoS ONE 2008, 3, e2698. [Google Scholar] [CrossRef] [PubMed]
- van Driel, L.M.J.W.; Eijkemans, M.J.C.; de Jonge, R.; de Vries, J.H.M.; van Meurs, J.B.J.; Steegers, E.A.P.; Steegers-Theunissen, R.P.M. Body mass index is an important determinant of methylation biomarkers in women of reproductive ages. J. Nutr. 2009, 139, 2315–2321. [Google Scholar] [CrossRef] [PubMed]
- Axelrod, J. Methylation reaction in the formation and metabolism of catecholamines and other biogenic amines. Pharmacol. Rev. 1966, 18, 95–113. [Google Scholar] [PubMed]
- Flynn, D.D.; Kloog, Y.; Potter, L.T.; Axelrod, J. Enzymatic methylation of the membrane-bound nicotinic acetylcholine receptor. J. Biol. Chem. 1982, 257, 9513–9517. [Google Scholar] [PubMed]
- Kim, S.; Lim, I.K.; Park, G.-H.; Paik, W.K. Biological methylation of myelin basic protein: Enzymology and biological significance. Int. J. Biochem. Cell Biol. 1997, 29, 743–751. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Hirata, F.; Axelrod, J. Regulation of the beta-adrenergic receptor by methylation of membrane phospholipids. Adv. Cyclic Nucleotide Res. 1981, 14, 83–91. [Google Scholar] [PubMed]
- Sontag, J.-M.; Nunbhakdi-Craig, V.; Montgomery, L.; Arning, E.; Bottiglieri, T.; Sontag, E. Folate deficiency induces in vitro and mouse brain region-specific downregulation of leucine carboxyl methyltransferase-1 and protein phosphatase 2A Bα subunit expression that correlate with enhanced tau phosphorylation. J. Neurosci. 2008, 28, 11477–11487. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A.; Cavallaro, R.A.; Zampelli, A.; D'Anselmi, F.; Piscopo, P.; Confaloni, A.; Scarpa, S. Gamma-secretase is differentially modulated by alterations of homocysteine cycle in neuroblastoma and glioblastoma cells. J. Alzheimers Dis. 2007, 11, 275–290. [Google Scholar] [PubMed]
- Fuso, A.; Nicolia, V.; Pasqualato, A.; Fiorenza, M.T.; Cavallaro, R.A.; Scarpa, S. Changes in presenilin 1 gene methylation pattern in diet-induced b vitamin deficiency. Neurobiol. Aging 2011, 32, 187–199. [Google Scholar] [CrossRef] [PubMed]
- Fuso, A. The ‘golden age’ of DNA methylation in neurodegenerative diseases. Clin. Chem. Lab. Med. 2013, 51, 523–534. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Liu, H.; Yu, M.; Zhang, X.; Zhang, M.; Wilson, J.X.; Huang, G. Folic acid administration inhibits amyloid β-peptide accumulation in app/ps1 transgenic mice. J. Nutr. Biochem. 2015, 26, 883–891. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.J.; Green, P.; Mann, J.J.; Rapoport, S.I.; Sublette, M.E. Pathways of polyunsaturated fatty acid utilization: Implications for brain function in neuropsychiatric health and disease. Brain Res. 2015, 1597, 220–246. [Google Scholar] [CrossRef] [PubMed]
- Watkins, S.M.; Zhu, X.; Zeisel, S.H. Phosphatidylethanolamine-n-methyltransferase activity and dietary choline regulate liver-plasma lipid flux and essential fatty acid metabolism in mice. J. Nutr. 2003, 133, 3386–3391. [Google Scholar] [PubMed]
- Vance, D.E.; Walkey, C.J.; Cui, Z. Phosphatidylethanolamine N-methyltransferase from liver. Biochim. Biophys. Acta 1997, 1348, 142–150. [Google Scholar] [CrossRef]
- Astarita, G.; Jung, K.-M.; Berchtold, N.C.; Nguyen, V.Q.; Gillen, D.L.; Head, E.; Cotman, C.W.; Piomelli, D. Deficient liver biosynthesis of docosahexaenoic acid correlates with cognitive impairment in Alzheimer’s disease. PLoS ONE 2010, 5, e12538. [Google Scholar] [CrossRef] [PubMed]
- Beyer, K.; Lao, J.I.; Latorre, P.; Riutort, N.; Matute, B.; Fernandez-Figueras, M.T.; Mate, J.L.; Ariza, A. Methionine synthase polymorphism is a risk factor for Alzheimer’s disease. Neuroreport 2003, 14, 1391–1394. [Google Scholar] [CrossRef] [PubMed]
- Dorszewska, J.; Florczak, J.; Rozycka, A.; Kempisty, B.; Jaroszewska-Kolecka, J.; Chojnacka, K.; Trzeciak, W.H.; Kozubski, W. Oxidative DNA damage and level of thiols as related to polymorphisms of MTHFR, mtr, mthfd1 in Alzheimer’s and Parkinson’s diseases. Acta Neurobiol. Exp. 2007, 67, 113–129. [Google Scholar]
- Shea, T.B.; Rogers, E.; Ashline, D.; Ortiz, D.; Sheu, M.-S. Original contribution: Apolipoprotein e deficiency promotes increased oxidative stress and compensatory increases in antioxidants in brain tissue. Free Radic. Biol. Med. 2002, 33, 1115–1120. [Google Scholar] [CrossRef]
- Shea, T.B.; Rogers, E. Research report: Folate quenches oxidative damage in brains of apolipoprotein E-deficient mice: Augmentation by vitamin E. Mol. Brain Res. 2002, 108, 1–6. [Google Scholar] [CrossRef]
- Huang, R.-F.S.; Hsu, Y.-C.; Lin, H.-L.; Yang, F.L. Folate depletion and elevated plasma homocysteine promote oxidative stress in rat livers. J. Nutr. 2001, 131, 33–38. [Google Scholar] [PubMed]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative flux analysis reveals folate-dependent nadph production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Wakutani, Y.; Kowa, H.; Kusumi, M.; Nakaso, K.; Yasui, K.I.; Isoe-Wada, K.; Yano, H.; Urakami, K.; Takeshima, T.; Nakashima, K. A haplotype of the methylenetetrahydrofolate reductase gene is protective against late-onset Alzheimer’s disease. Neurobiol. Aging 2004, 25, 291–294. [Google Scholar] [CrossRef]
- Kruman, II; Kumaravel, T.S.; Lohani, A.; Pedersen, W.A.; Cutler, R.G.; Kruman, Y.; Haughey, N.; Lee, J.; Evans, M.; Mattson, M.P. Folic acid deficiency and homocysteine impair DNA repair in hippocampal neurons and sensitize them to amyloid toxicity in experimental models of Alzheimer’s disease. J. Neurosci. 2002, 22, 1752–1762. [Google Scholar] [PubMed]
- Taoka, S.; Ohja, S.; Shan, X.; Kruger, W.D.; Banerjee, R. Evidence for heme-mediated redox regulation of human cystathionine β-synthase activity. J. Biol. Chem. 1998, 273, 25179–25184. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, C.M.; Sternberg, M.R.; Fazili, Z.; Lacher, D.A.; Zhang, M.; Johnson, C.L.; Hamner, H.C.; Bailey, R.L.; Rader, J.I.; Yamini, S.; et al. Folate status and concentrations of serum folate forms in the us population: National health and nutrition examination survey 2011–2. Br. J. Nutr. 2015, 113, 1965–1977. [Google Scholar] [CrossRef] [PubMed]
- Regland, B.; Blennow, K.; Germgård, T.; Koch-Schmidt, A.C.; Gottfries, C.G. The role of the polymorphic genes apolipoprotein E and methylene-tetrahydrofolate reductase in the development of dementia of the Alzheimer type. Dement. Geriatr. Cogn. Disord. 1999, 10, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Bailey, R.L.; Carmel, R.; Green, R.; Pfeiffer, C.M.; Cogswell, M.E.; Osterloh, J.D.; Sempos, C.T.; Yetley, E.A. Monitoring of vitamin B-12 nutritional status in the united states by using plasma methylmalonic acid and serum vitamin B-12. Am. J. Clin. Nutr. 2011, 94, 552–561. [Google Scholar] [CrossRef] [PubMed]
- Bailey, R.L.; Dodd, K.W.; Gahche, J.J.; Dwyer, J.T.; McDowell, M.A.; Yetley, E.A.; Sempos, C.A.; Burt, V.L.; Radimer, K.L.; Picciano, M.F. Total folate and folic acid intake from foods and dietary supplements in the united states: 2003–2006. Am. J. Clin. Nutr. 2010, 91, 231–237. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Reider, C.; Brooks, J.R.; Fulgoni, V.L. Comparison of prevalence of inadequate nutrient intake based on body weight status of adults in the united states: An analysis of nhanes 2001–2008. J. Am. Coll. Nutr. 2015, 34, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Roman Vinas, B.; Ribas Barba, L.; Ngo, J.; Gurinovic, M.; Novakovic, R.; Cavelaars, A.; de Groot, L.C.; Van’t Veer, P.; Matthys, C.; Serra Majem, L. Projected prevalence of inadequate nutrient intakes in Europe. Ann. Nutr. Metab. 2011, 59, 84–95. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, S.M.; Gibney, M.J.; Nugent, A.P.; McNulty, H.; Molloy, A.M.; Scott, J.M.; Flynn, A.; Strain, J.J.; Ward, M.; Walton, J.; et al. Impact of voluntary fortification and supplement use on dietary intakes and biomarker status of folate and vitamin B-12 in irish adults. Am. J. Clin. Nutr. 2015, 101, 1163–1172. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, W.; Obeid, R. Cobalamin deficiency. In Water Soluble Vitamins; Stanger, O., Ed.; Springer: Dordrecht, The Netherlands, 2012; Volume 56, pp. 301–322. [Google Scholar]
- Carmel, R. Current concepts in cobalamin deficiency. Annu. Rev. Med. 2000, 51, 357–375. [Google Scholar] [CrossRef] [PubMed]
- Carmel, R. Efficacy and safety of fortification and supplementation with vitamin B12: Biochemical and physiological effects. Food Nutr. Bull. 2008, 29, S177–S187. [Google Scholar] [CrossRef] [PubMed]
- Carmel, R. Nutritional anemias and the elderly. Semin. Hematol. 2008, 45, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Oulhaj, A.; Jerneren, F.; Refsum, H.; Smith, A.D.; de Jager, C.A. Omega-3 fatty acid status enhances the prevention of cognitive decline by B vitamins in mild cognitive impairment. J. Alzheimers Dis. 2016, 50, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Stark, K.D.; Van Elswyk, M.E.; Higgins, M.R.; Weatherford, C.A.; Salem, N., Jr. Global survey of the omega-3 fatty acids, docosahexaenoic acid and eicosapentaenoic acid in the blood stream of healthy adults. Prog. Lipid Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, K.; Gustafson, L.; Hultberg, B. Improvement of cognitive functions after cobalamin/folate supplementation in elderly patients with dementia and elevated plasma homocysteine. Int. J. Geriatr. Psychiatry 2001, 16, 609–614. [Google Scholar] [CrossRef] [PubMed]
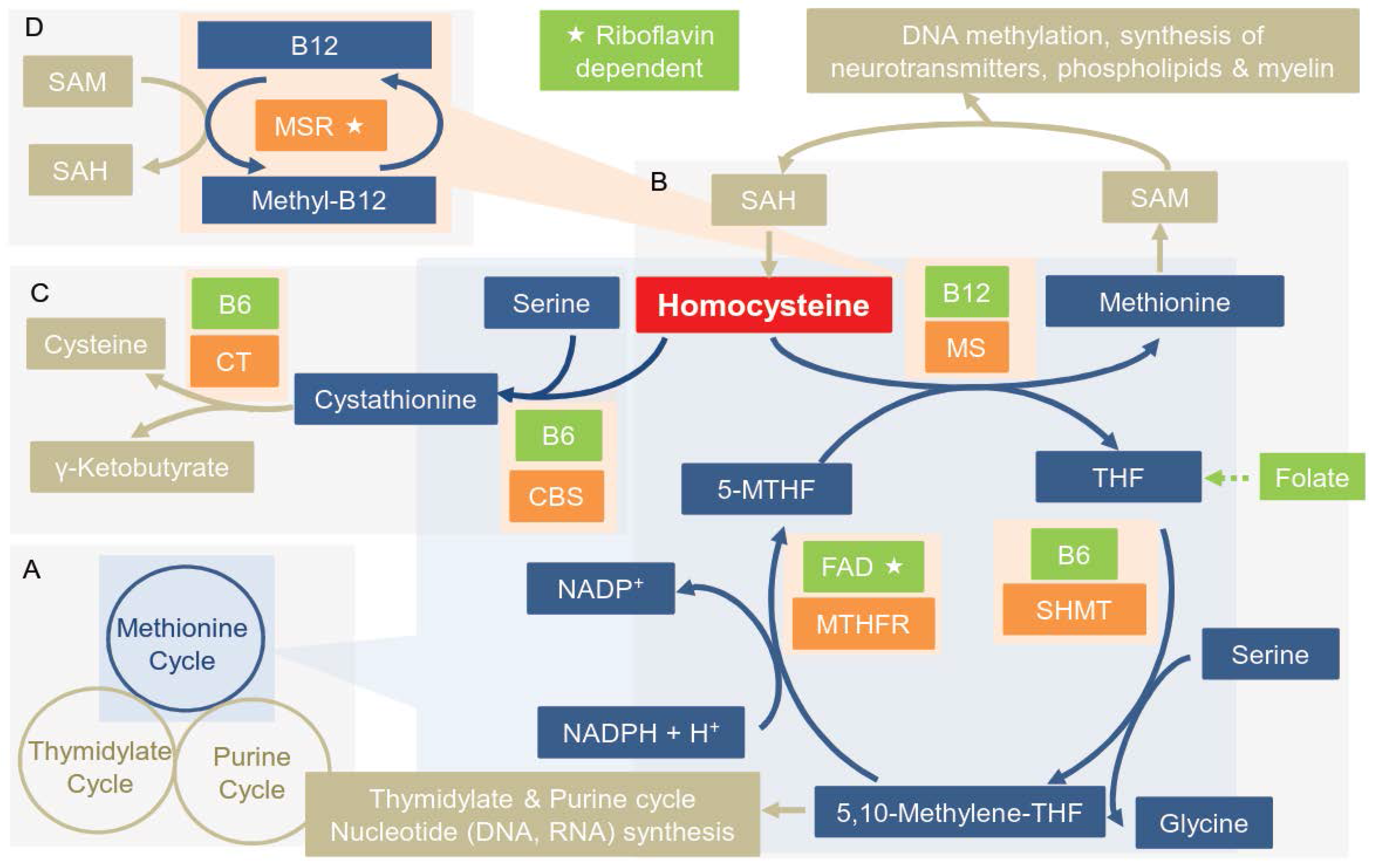

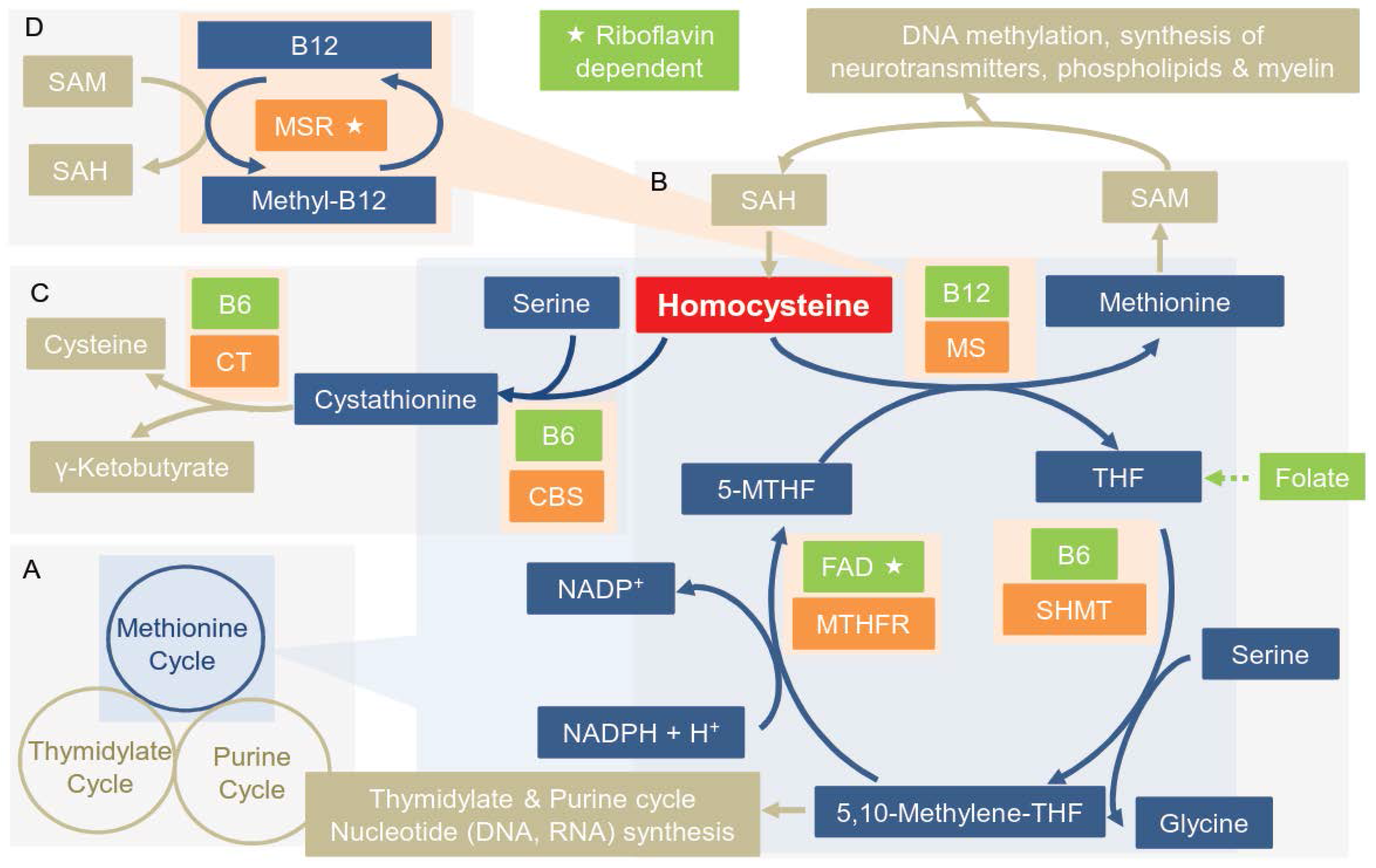{kind=link}
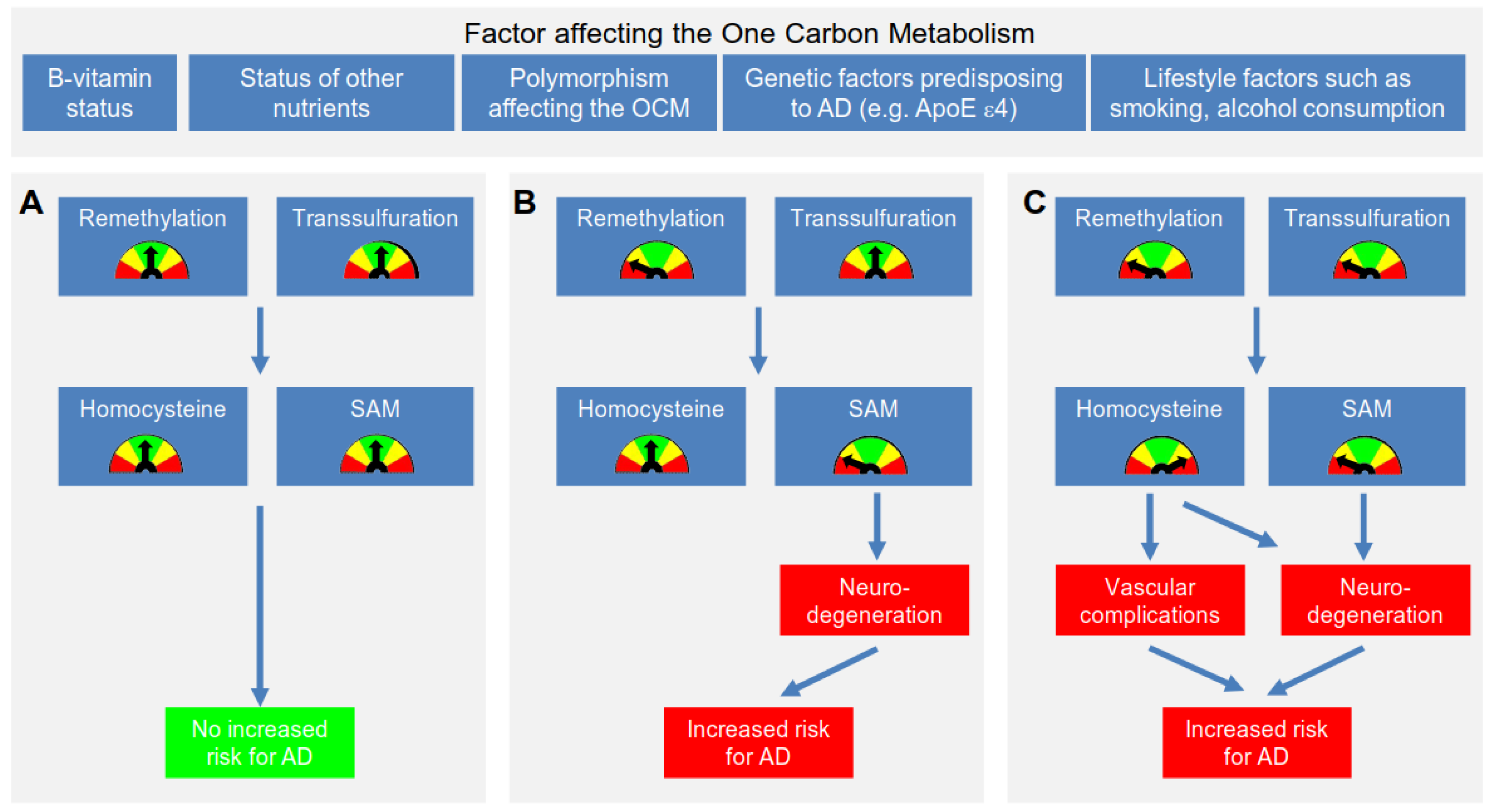{kind=link}
| Enzyme | Polymorphism | Reference |
|---|---|---|
| MTHFR | C677T | Schwahn and Rozen 2001 [20], Yamada et al., 2001 [21], Guenther et al., 1999 [22] |
| A1298C | Weisberg et al., 2001 [23] | |
| T1317C | Weisberg et al., 1998 [24] | |
| MS | A2756G | Leclerc et al., 1996 [25], Chen et al., 1997 [26] |
| MSR | A66G | Olteanu et al., 2002 [27] |
| C524T | Olteanu et al., 2002 [27] | |
| CBS | 68 bp insertion at exon 8 | Sebastio et al., 1995 [28] |
| G9276A | Nienaber-Rousseau et al., 2013 [29] | |
| 31 bp variable number of tandem repeats | Lievers et al., 2001 [30] | |
| SHMT | C1420T | Heil et al., 2001 [31] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Troesch, B.; Weber, P.; Mohajeri, M.H. Potential Links between Impaired One-Carbon Metabolism Due to Polymorphisms, Inadequate B-Vitamin Status, and the Development of Alzheimer’s Disease. Nutrients 2016, 8, 803. https://doi.org/10.3390/nu8120803
Troesch B, Weber P, Mohajeri MH. Potential Links between Impaired One-Carbon Metabolism Due to Polymorphisms, Inadequate B-Vitamin Status, and the Development of Alzheimer’s Disease. Nutrients. 2016; 8(12):803. https://doi.org/10.3390/nu8120803
Chicago/Turabian StyleTroesch, Barbara, Peter Weber, and M. Hasan Mohajeri. 2016. "Potential Links between Impaired One-Carbon Metabolism Due to Polymorphisms, Inadequate B-Vitamin Status, and the Development of Alzheimer’s Disease" Nutrients 8, no. 12: 803. https://doi.org/10.3390/nu8120803
APA StyleTroesch, B., Weber, P., & Mohajeri, M. H. (2016). Potential Links between Impaired One-Carbon Metabolism Due to Polymorphisms, Inadequate B-Vitamin Status, and the Development of Alzheimer’s Disease. Nutrients, 8(12), 803. https://doi.org/10.3390/nu8120803