Phylogeny of the Vitamin K 2,3-Epoxide Reductase (VKOR) Family and Evolutionary Relationship to the Disulfide Bond Formation Protein B (DsbB) Family

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Protein Sequences and Multiple Sequence Alignments
2.2. Phylogenetic Analyses
2.3. Assessment of Residue-Specific Evolutionary Conservation and Structural Correlates
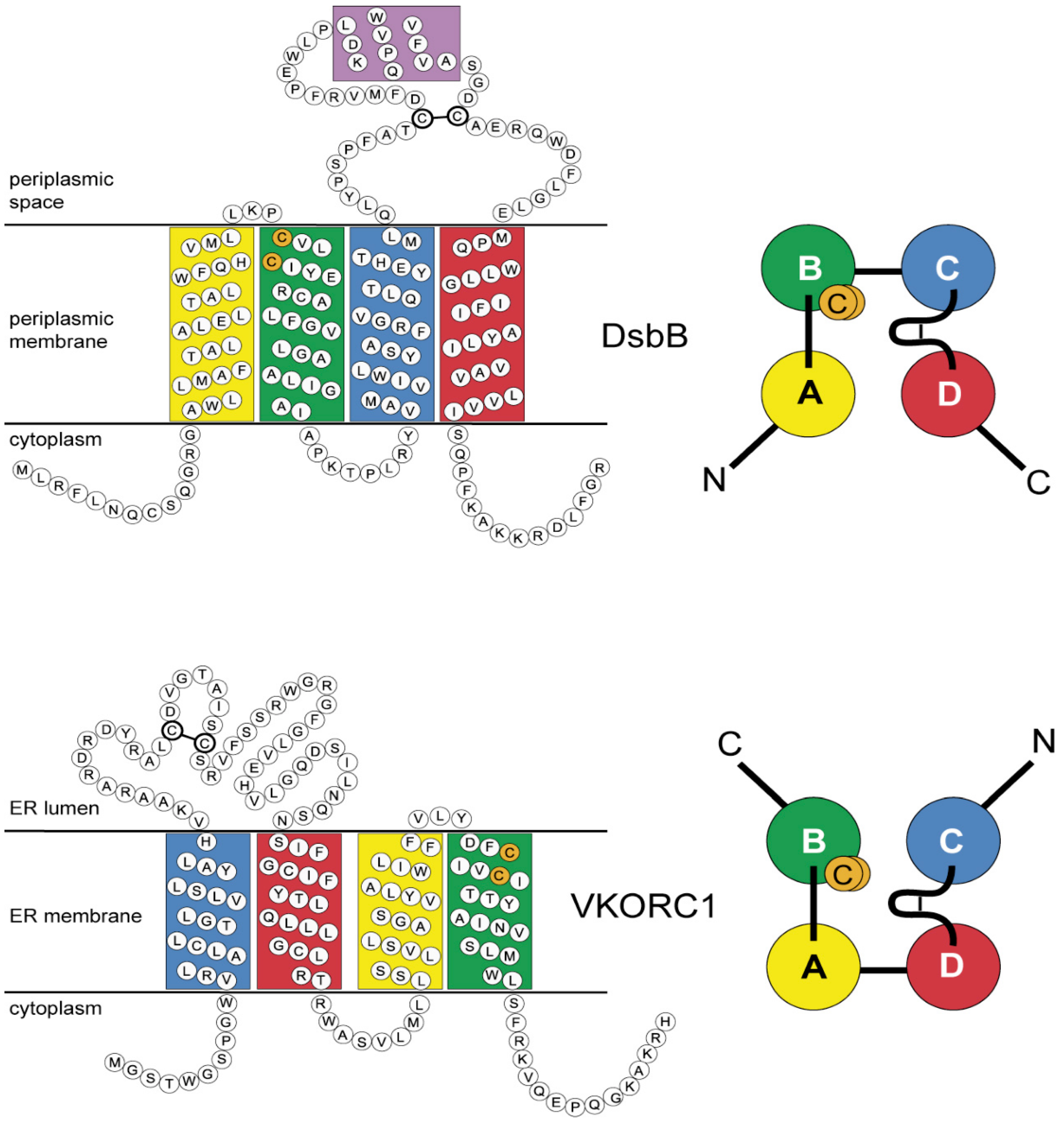
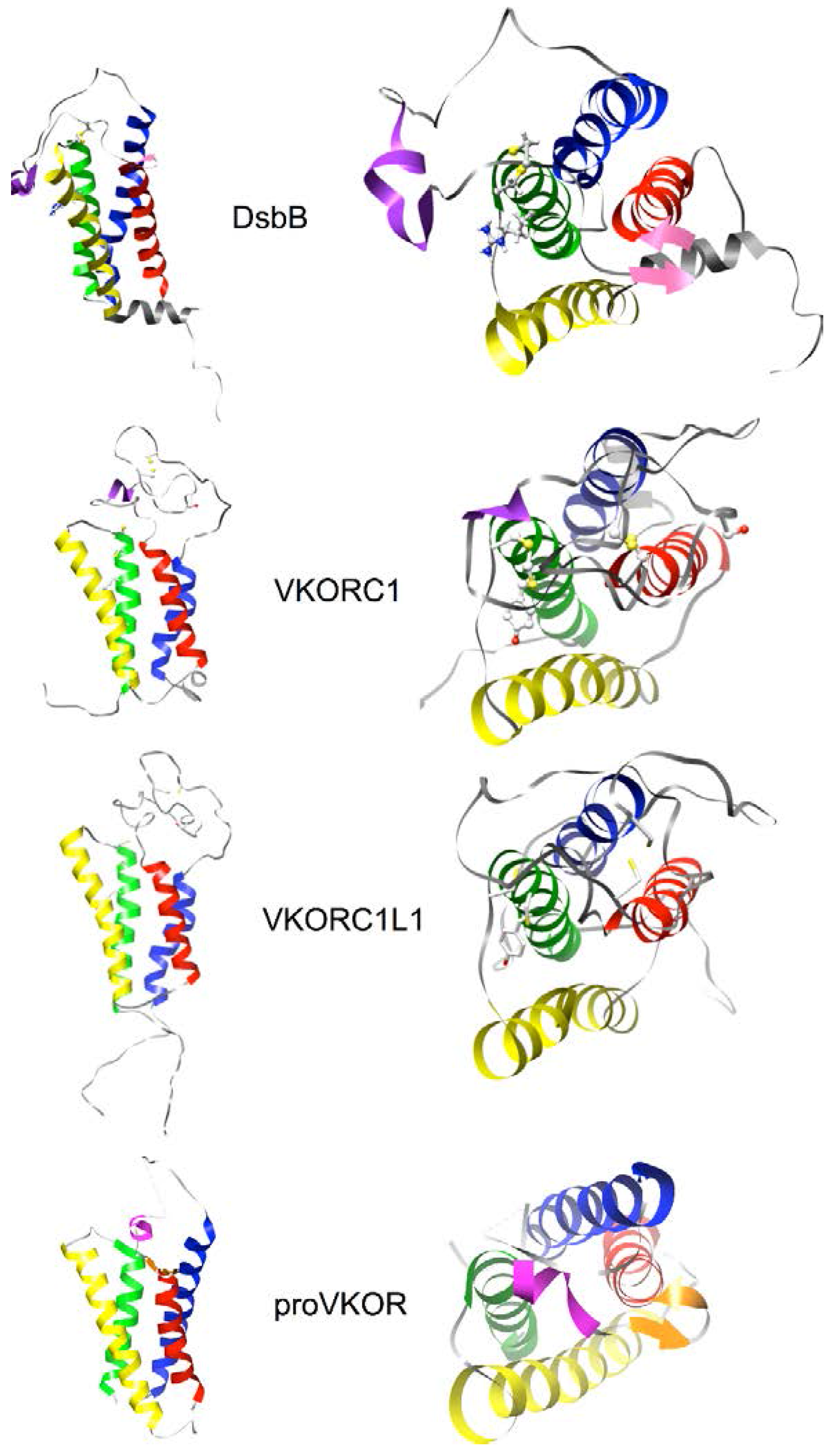
2.4. Homology Modeling of Human VKORC1 and VKORC1L1 Paralogs Using a Cyclic Permutation of E. coli DsbB as the Target Structure
3. Results
3.1. Multiple Sequence Alignments Reveal Greater Inhomogeneity in Indels for the VKOR Family Core Domain Relative to the DsbB Family
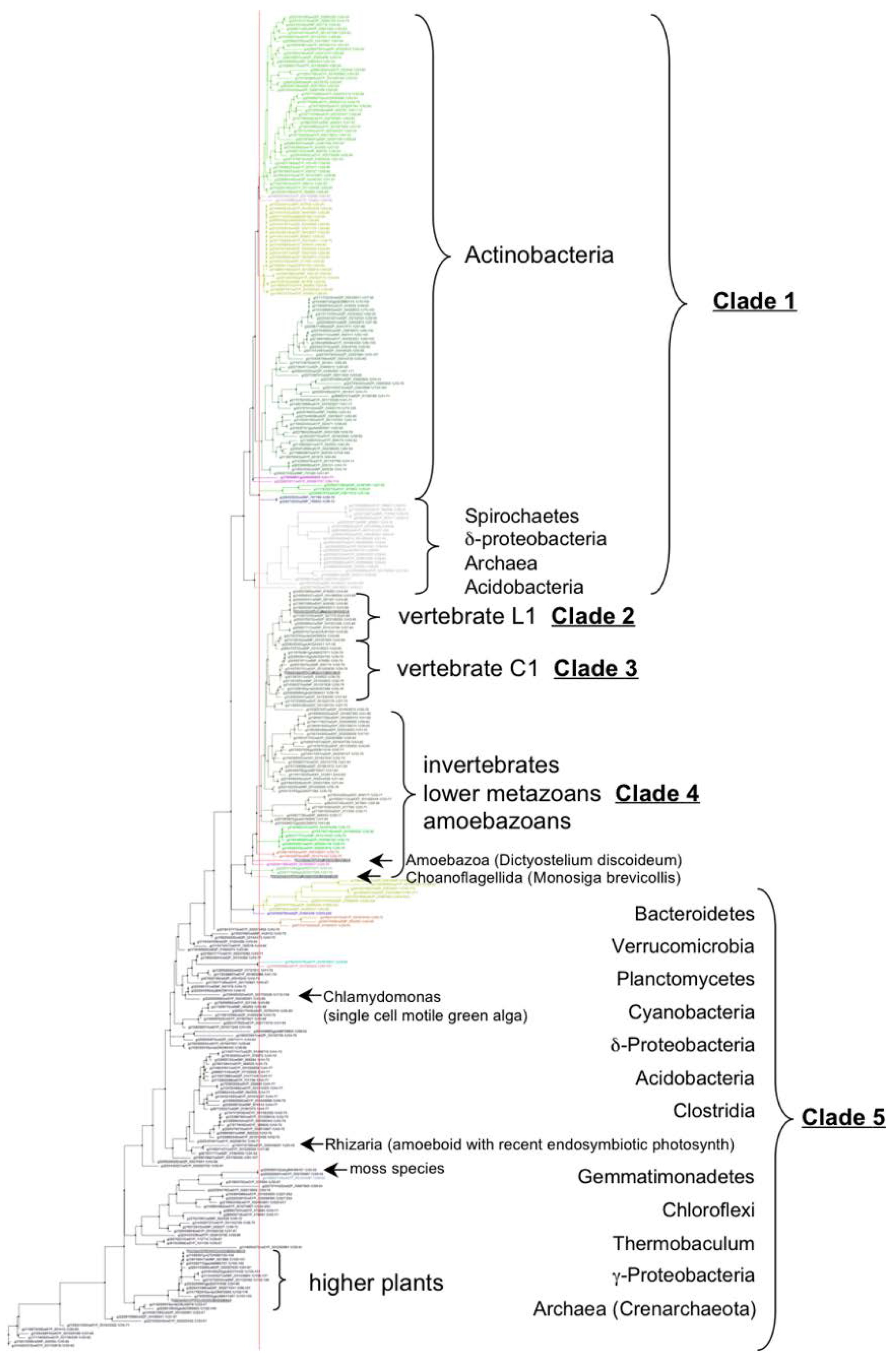
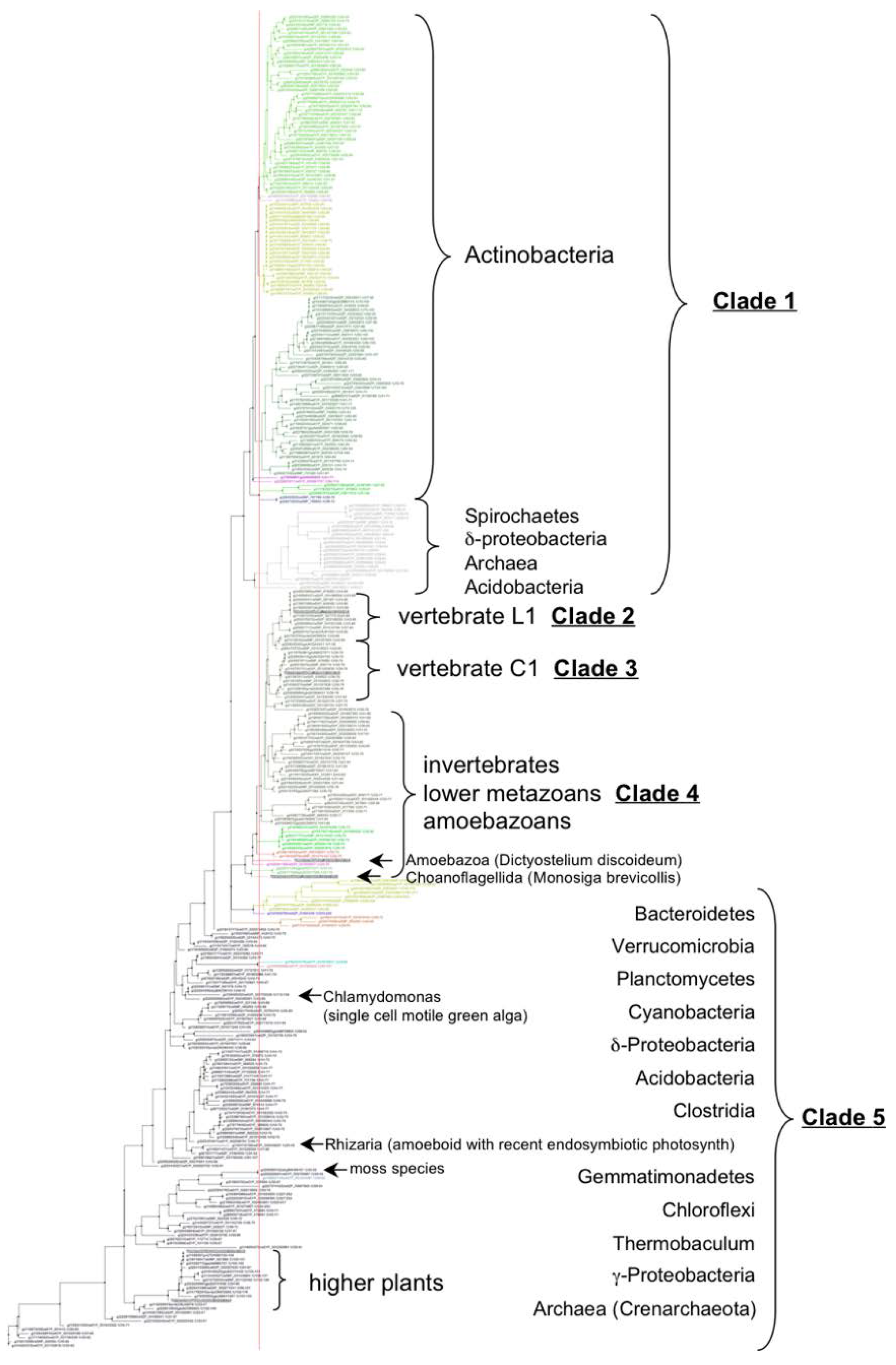
3.2. VKOR Family Phylogeny is Organized into Five Principal Clades

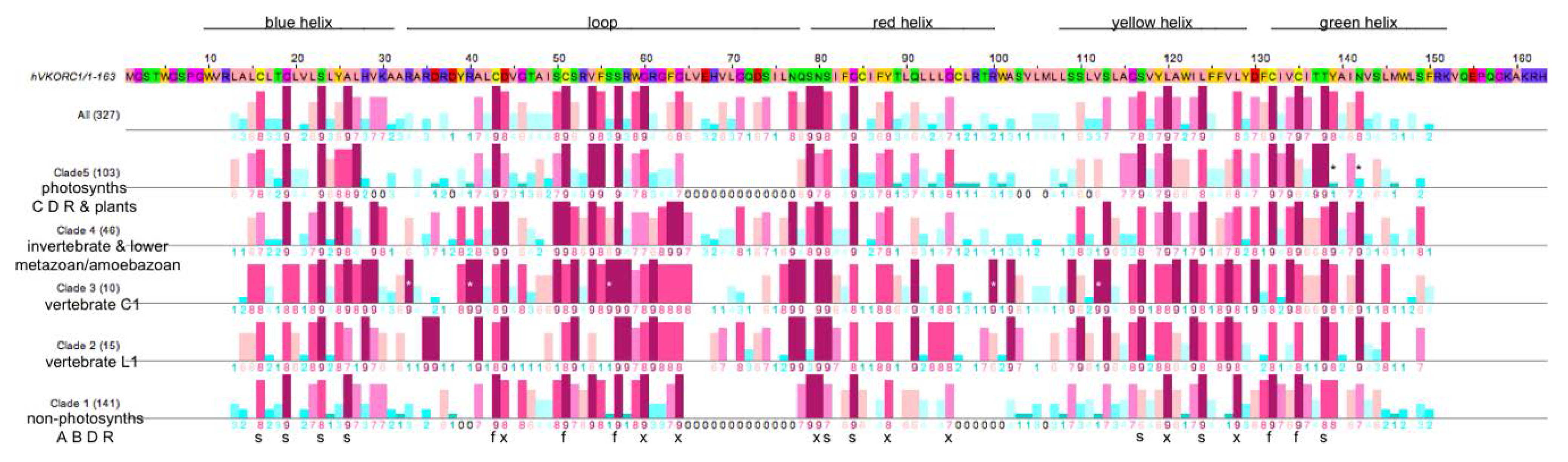
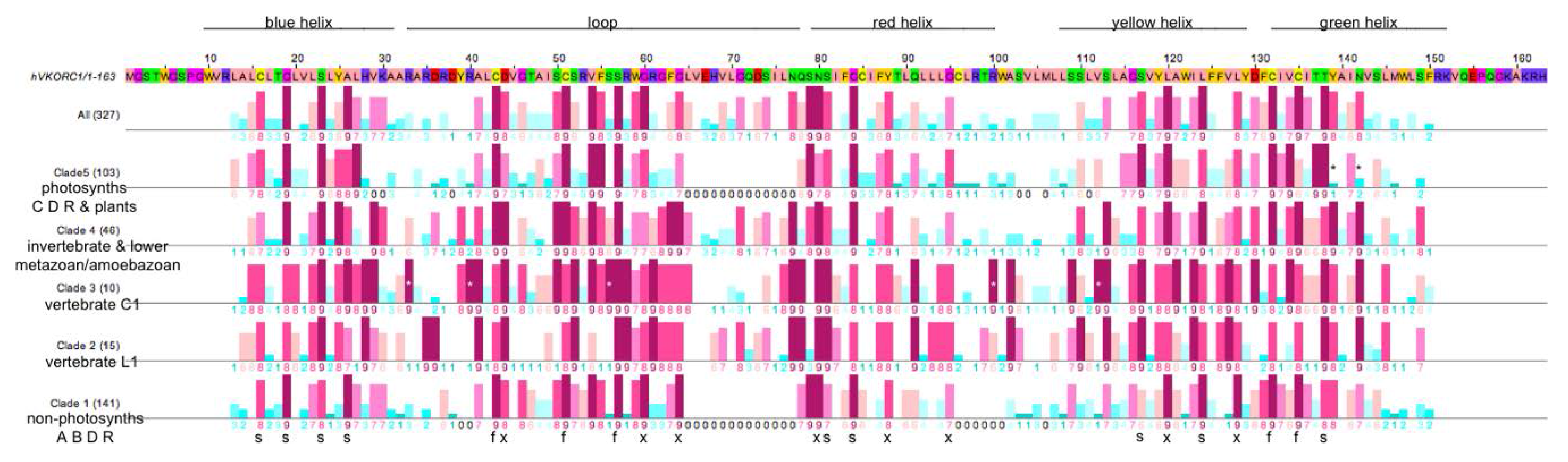
3.3. Similarities and Differences among Sequence Position-Specific Substitutions Reveal VKOR Family Clade-Specific Structural and Functional Residues
- (1)
- The five fully conserved VKOR signature residue positions (Figure 4, marked f) are functional, playing direct roles in vitamin K 2,3-epoxide to vitamin K quinone and vitamin K quinone to vitamin K quinol reduction (Cys43, Cys51, Ser57, Cys132, Cys135 according to human VKORC1 numbering);
- (2)
- Nine highly conserved positions (Figure 4, marked s) represent residues that form putative helix-helix structural contacts identified by visual inspection of the X-ray crystallographic structure for the prokaryotic VKOR homolog (PDB entry 3KP9, residue numbering corresponds to human VKORC1 sequence in Figure 4). On TMH1, Cys16, Gly19, Ser23 and Ala26 pack against TMH2, TMH2 and TMH4, TMH2, TMH2 and TMH3, respectively. On TMH2, Ser81 and Gly84 both pack against TMH1. On TMH3, Ser117 packs against TMH4, while Leu124 packs against only residues on the same helix (TMH3). On TMH4, Thr138 packs against TMH1;
- (3)
- Eight highly conserved positions (Figure 4, marked x) may be putative functional residues either essential for quinone substrate reduction or involved in substrate binding and specificity (Asp44, Gly80, Gly84, Asn80, Tyr88, Gly95, Leu120, Leu128).
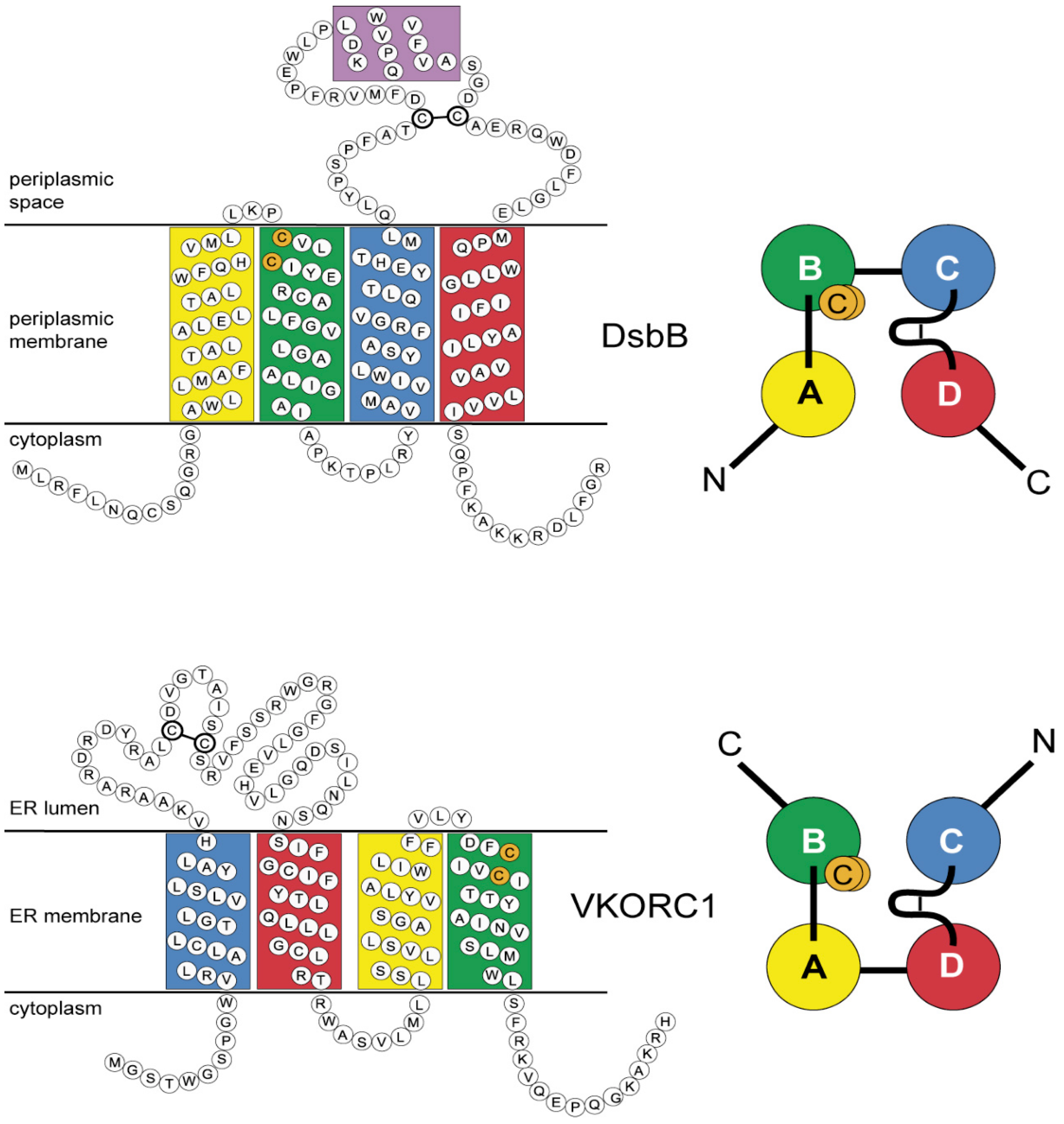
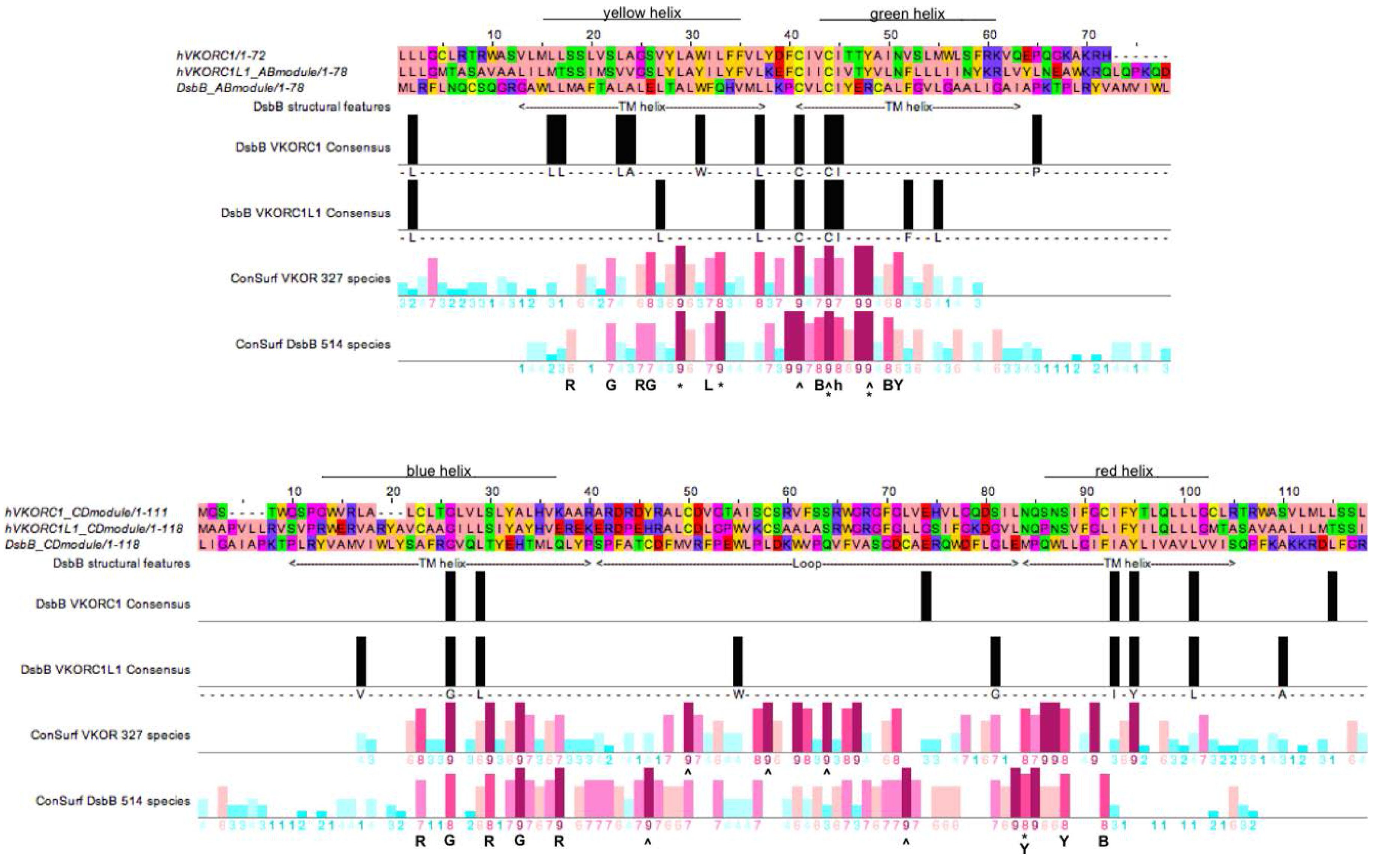
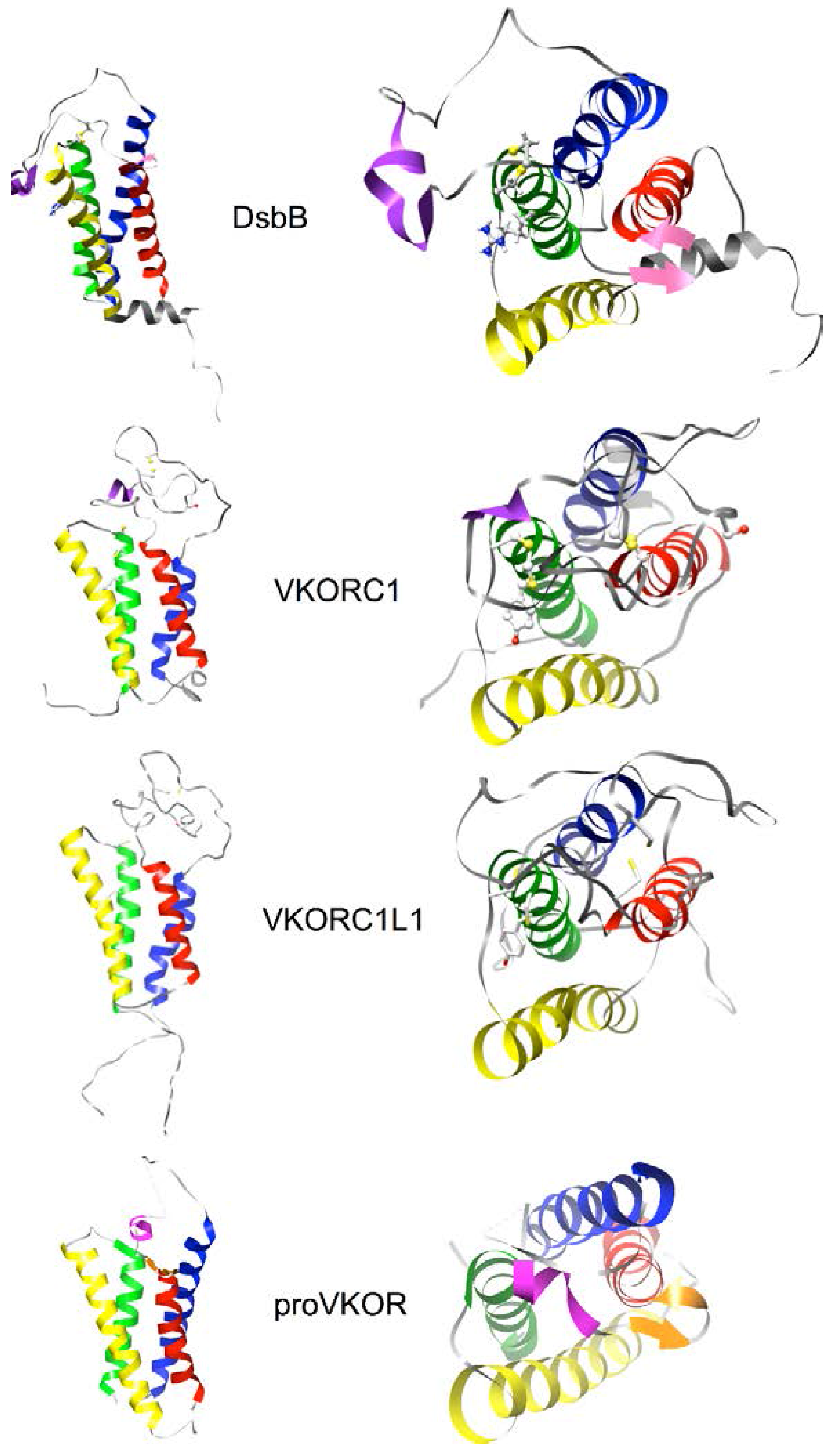
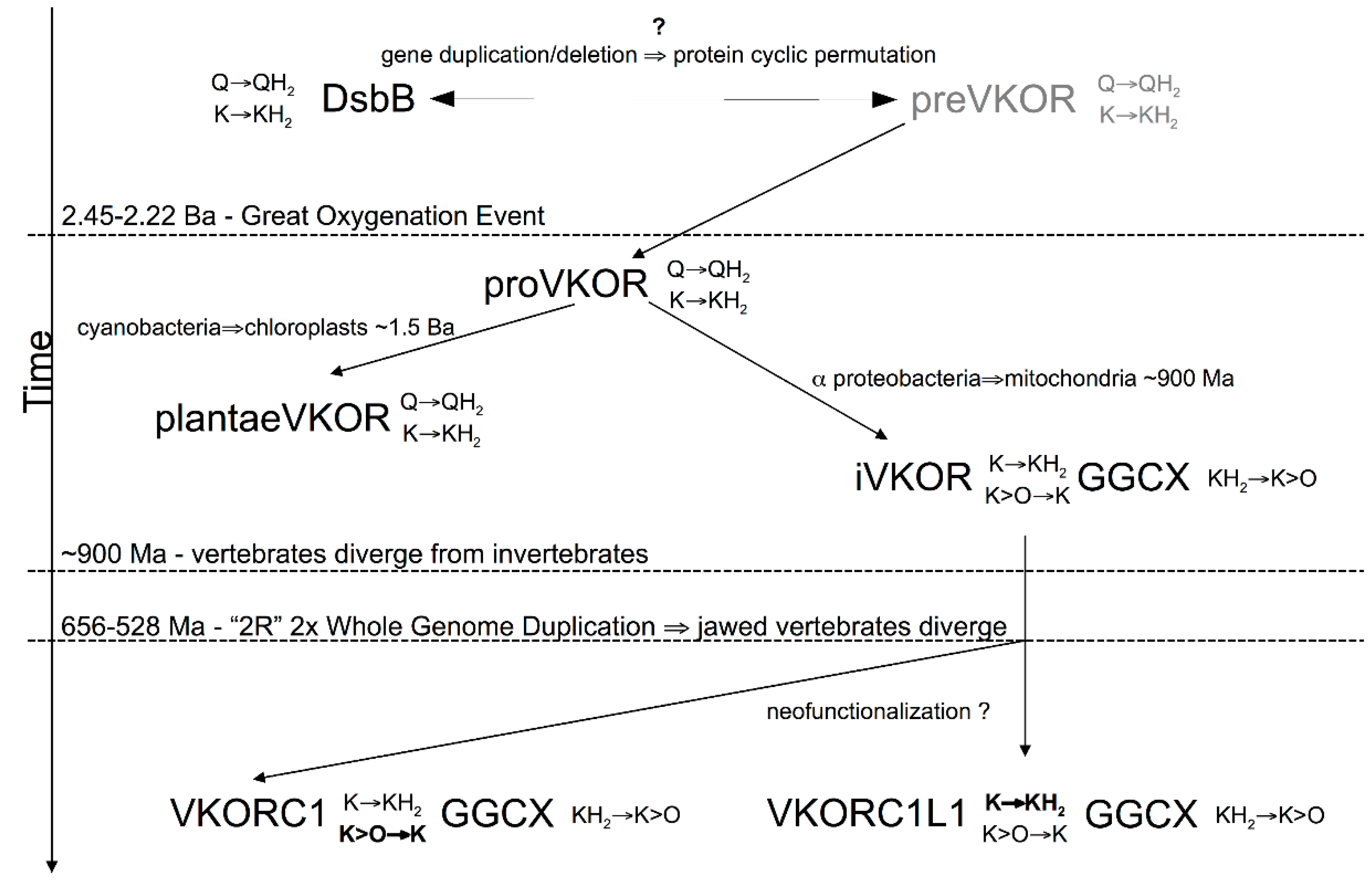
3.4. Evidence for an Evolutionary Relationship between VKOR and DsbB Families
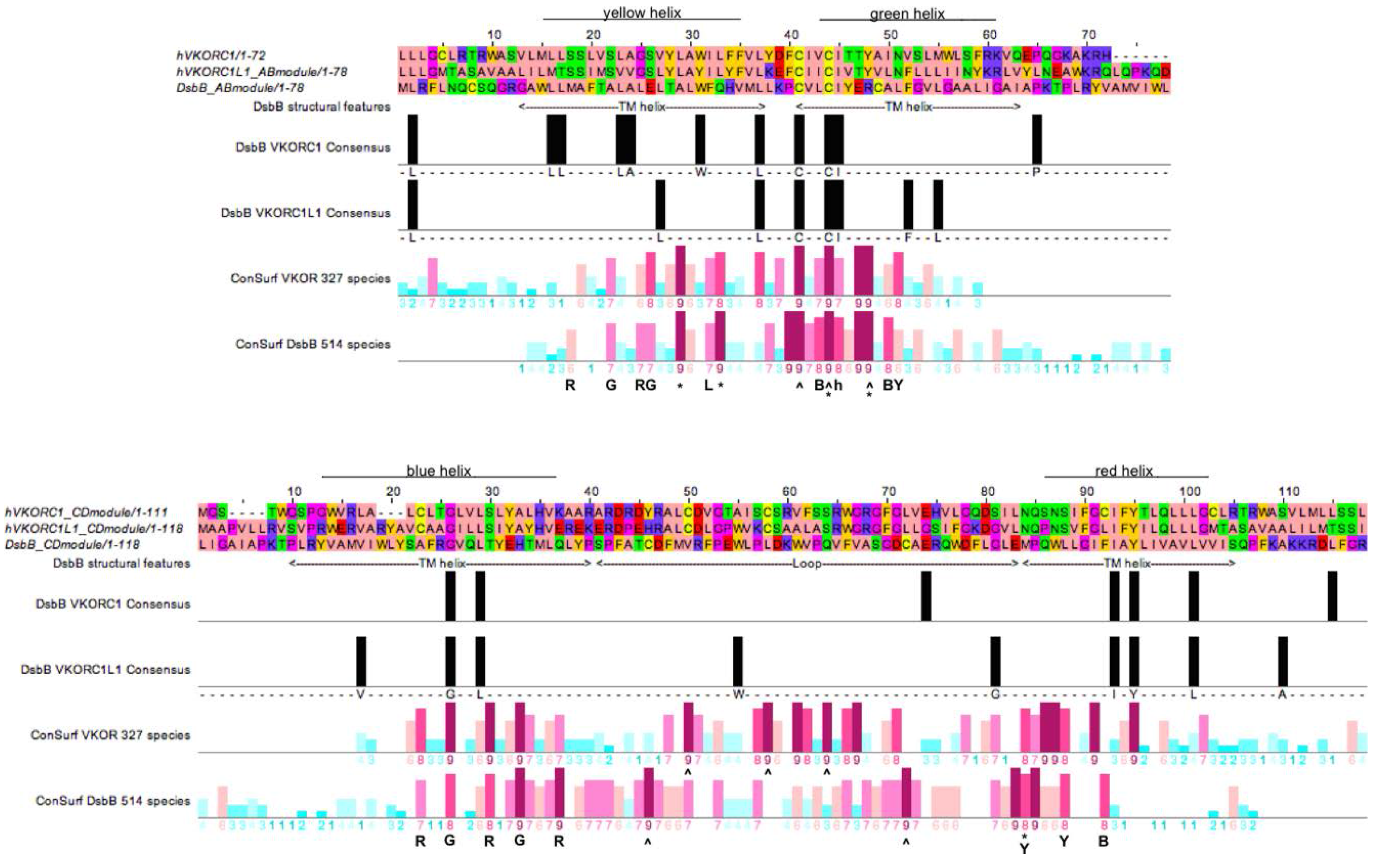
3.5. Identification of Transmembrane-Helical Bundle Protein Families Related by Cyclical Permuted Primary Sequences Can Extend Homology Modeling into the “Twilight Zone” (<30% Sequence Homology between Target and Template)
4. Discussion
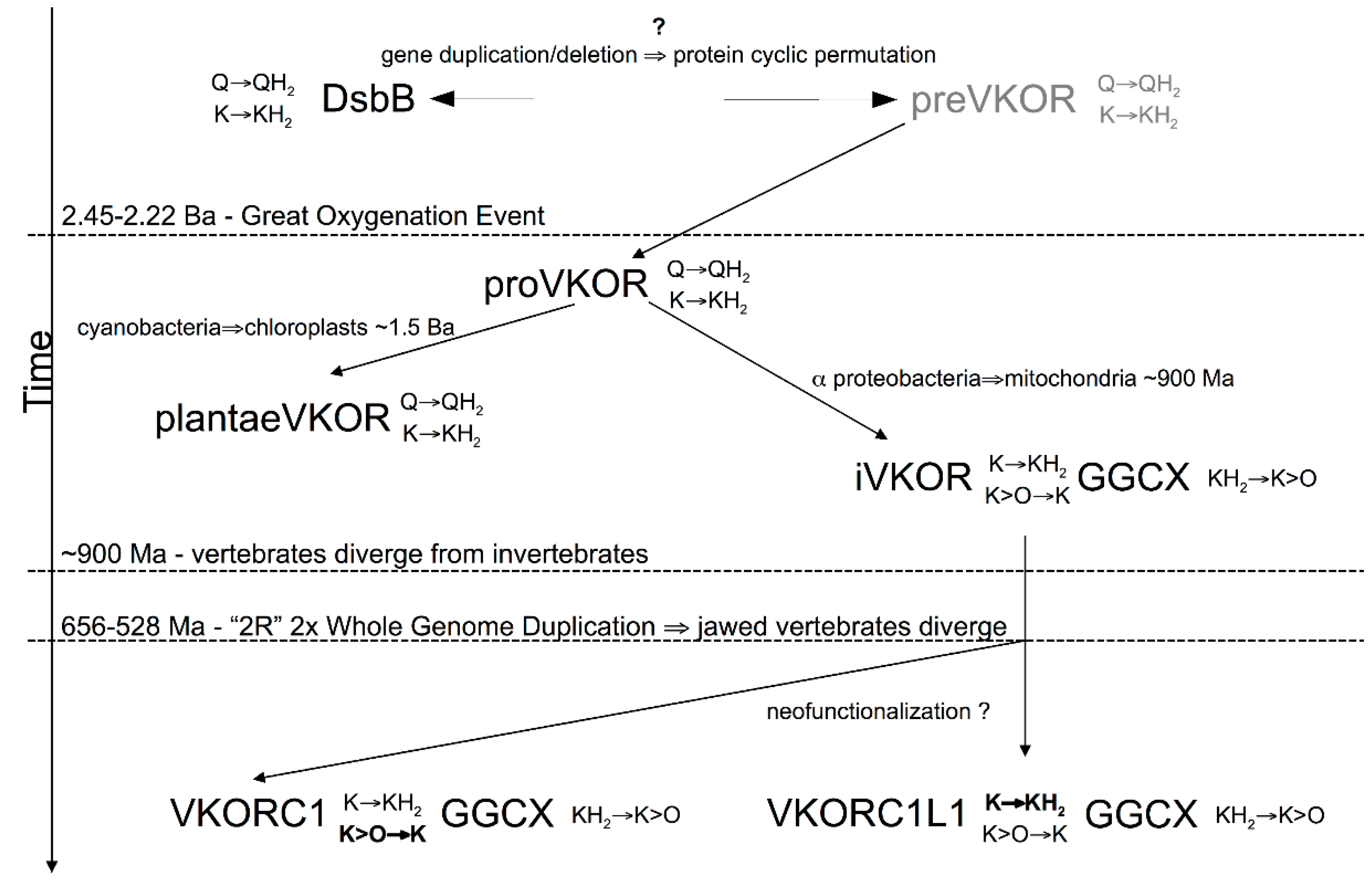
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Oldenburg, J.; Bevans, C.G.; Müller, C.R.; Watzka, M. Vitamin K epoxide reductase complex subunit 1 (VKORC1): The key protein of the vitamin K cycle. Antioxid. Redox Signal. 2006, 8, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Shearer, M.J.; Fu, X.; Booth, S.L. Vitamin K nutrition, metabolism, and requirements: Current concepts and future research. Adv. Nutr. 2012, 3, 182–195. [Google Scholar] [CrossRef] [PubMed]
- Shearer, M.J.; Newman, P. Recent trends in the metabolism and cell biology of vitamin K with special reference to vitamin K cycling and MK-4 biosynthesis. J. Lipid Res. 2014, 55, 345–362. [Google Scholar] [CrossRef] [PubMed]
- Spohn, G.; Kleinridders, A.; Wunderlich, F.T.; Watzka, M.; Zaucke, F.; Blumbach, K.; Geisen, C.; Seifried, E.; Müller, C.; Paulsson, M.; et al. VKORC1 deficiency in mice causes early postnatal lethality due to severe bleeding. Thromb. Haemost. 2009, 101, 1044–1050. [Google Scholar] [CrossRef] [PubMed]
- Dam, H. The antihaemorrhagic vitamin of the chick. Biochem. J. 1935, 29, 1273–1285. [Google Scholar] [PubMed]
- Watzka, M.; Geisen, C.; Bevans, C.G.; Sittinger, K.; Spohn, G.; Rost, S.; Seifried, E.; Müller, C.R.; Oldenburg, J. Thirteen novel VKORC1 mutations associated with oral anticoagulant resistance: Insights into improved patient diagnosis and treatment. J. Thromb. Haemost. 2011, 9, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Hammed, A.; Matagrin, B.; Spohn, G.; Prouillac, C.; Benoit, E.; Lattard, V. VKORC1L1, an enzyme rescuing the vitamin K 2,3-epoxide reductase activity in some extrahepatic tissues during anticoagulation therapy. J. Biol. Chem. 2013, 288, 28733–28742. [Google Scholar] [CrossRef] [PubMed]
- Caspers, M.; Czogalla, K.J.; Liphardt, K.; Müller, J.; Westhofen, P.; Watzka, M.; Oldenburg, J. Two enzymes catalyze vitamin K 2,3-epoxide reductase activity in mouse: VKORC1 is highly expressed in exocrine tissues while VKORC1L1 is highly expressed in brain. Thromb. Res. 2015, 135, 977–983. [Google Scholar] [CrossRef] [PubMed]
- Westhofen, P.; Watzka, M.; Marinova, M.; Hass, M.; Kirfel, G.; Müller, J.; Bevans, C.G.; Müller, C.R.; Oldenburg, J. Human vitamin K 2,3-epoxide reductase complex subunit 1-like 1 (VKORC1L1) mediates vitamin K-dependent intracellular antioxidant function. J. Biol. Chem. 2011, 286, 15085–15094. [Google Scholar] [CrossRef] [PubMed]
- Rost, S.; Fregin, A.; Ivaskevicius, V.; Conzelmann, E.; Hörtnagel, K.; Pelz, H.-J.; Lappegard, K.; Seifried, E.; Scharrer, I.; Tuddenham, E.G.D.; et al. Mutations in VKORC1 cause warfarin resistance and multiple coagulation factor deficiency type 2. Nature 2004, 427, 537–541. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Chang, C.-Y.; Jin, D.-Y.; Lin, P.-J.; Khvorova, A.; Stafford, D.W. Identification of the gene for vitamin K epoxide reductase. Nature 2004, 427, 541–544. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.-K.; Jin, D.-Y.; Stafford, D.W. Conserved loop cysteines of vitamin K epoxide reductase complex subunit 1-like 1 (VKORC1L1) are involved in its active site regeneration. J. Biol. Chem. 2014, 289, 9396–9407. [Google Scholar] [CrossRef] [PubMed]
- Tie, J.-K.; Jin, D.-Y.; Stafford, D.W. Mycobacterium tuberculosis vitamin K epoxide reductase homologue supports vitamin K-dependent carboxylation in mammalian cells. Antioxid. Redox Signal. 2012, 16, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Schulman, S.; Dutton, R.J.; Boyd, D.; Beckwith, J.; Rapoport, T.A. Structure of a bacterial homologue of vitamin K epoxide reductase. Nature 2010, 463, 507–512. [Google Scholar] [CrossRef] [PubMed]
- Furt, F.; van Oostende, C.; Widhalm, J.R.; Dale, M.A.; Wertz, J.; Basset, G.J.C. A bimodular oxidoreductase mediates the specific reduction of phylloquinone (vitamin K₁) in chloroplasts. Plant J. Cell Mol. Biol. 2010, 64, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.-M.; Yang, X.-J.; Du, J.-J.; Lu, Y.; Yu, Z.-B.; Feng, Y.-G.; Wang, X.-Y. Identification and characterization of SlVKOR, a disulfide bond formation protein from Solanum lycopersicum, and bioinformatic analysis of plant VKORs. Biochem. 2014, 79, 440–449. [Google Scholar] [CrossRef] [PubMed]
- Sevier, C.S.; Kadokura, H.; Tam, V.C.; Beckwith, J.; Fass, D.; Kaiser, C.A. The prokaryotic enzyme DsbB may share key structural features with eukaryotic disulfide bond forming oxidoreductases. Protein Sci. Publ. Protein Soc. 2005, 14, 1630–1642. [Google Scholar] [CrossRef] [PubMed]
- Sevier, C.S.; Kaiser, C.A. Conservation and diversity of the cellular disulfide bond formation pathways. Antioxid. Redox Signal. 2006, 8, 797–811. [Google Scholar] [CrossRef] [PubMed]
- Wajih, N.; Hutson, S.M.; Wallin, R. Disulfide-dependent protein folding is linked to operation of the vitamin K cycle in the endoplasmic reticulum: A protein disulfide isomerase-VKORC1 redox enzyme complex appears to be responsible for vitamin K1 2,3-epoxide reduction. J. Biol. Chem. 2007, 282, 2626–2635. [Google Scholar] [CrossRef] [PubMed]
- Rutkevich, L.A.; Williams, D.B. Vitamin K epoxide reductase contributes to protein disulfide formation and redox homeostasis within the endoplasmic reticulum. Mol. Biol. Cell 2012, 23, 2017–2027. [Google Scholar] [CrossRef] [PubMed]
- Oldenburg, J.; Müller, C.R.; Rost, S.; Watzka, M.; Bevans, C.G. Comparative genetics of warfarin resistance. Hamostaseologie 2014, 34, 143–159. [Google Scholar] [CrossRef] [PubMed]
- Goodstadt, L.; Ponting, C.P. Vitamin K epoxide reductase: homology, active site and catalytic mechanism. Trends Biochem. Sci. 2004, 29, 289–292. [Google Scholar] [CrossRef] [PubMed]
- Robertson, H.M. Genes encoding vitamin-K epoxide reductase are present in Drosophila and trypanosomatid protists. Genetics 2004, 168, 1077–1080. [Google Scholar] [CrossRef] [PubMed]
- Dutton, R.J.; Boyd, D.; Berkmen, M.; Beckwith, J. Bacterial species exhibit diversity in their mechanisms and capacity for protein disulfide bond formation. Proc. Natl. Acad. Sci. USA 2008, 105, 11933–11938. [Google Scholar] [CrossRef] [PubMed]
- Heras, B.; Shouldice, S.R.; Totsika, M.; Scanlon, M.J.; Schembri, M.A.; Martin, J.L. DSB proteins and bacterial pathogenicity. Nat. Rev. Microbiol. 2009, 7, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Bader, M.; Muse, W.; Ballou, D.P.; Gassner, C.; Bardwell, J.C. Oxidative protein folding is driven by the electron transport system. Cell 1999, 98, 217–227. [Google Scholar] [CrossRef]
- Reedstrom, C.K.; Suttie, J.W. Comparative distribution, metabolism, and utilization of phylloquinone and menaquinone-9 in rat liver. Biol. Med. 1995, 209, 403–409. [Google Scholar] [CrossRef]
- Collins, M.D.; Jones, D. Distribution of isoprenoid quinone structural types in bacteria and their taxonomic implication. Microbiol. Rev. 1981, 45, 316–354. [Google Scholar] [PubMed]
- Inaba, K.; Murakami, S.; Suzuki, M.; Nakagawa, A.; Yamashita, E.; Okada, K.; Ito, K. Crystal structure of the DsbB-DsbA complex reveals a mechanism of disulfide bond generation. Cell 2006, 127, 789–801. [Google Scholar] [CrossRef] [PubMed]
- Shixuan, L.; Wei, C.; Fowle Grider, R.; Guomin, S.; Weikai, L. Structures of an intramembrane vitamin K epoxide reductase homolog reveal control mechanisms for electron transfer. Nat. Commun. 2014, 5, 3110. [Google Scholar]
- Schwarz, R.; Seibel, P.N.; Rahmann, S.; Schoen, C.; Huenerberg, M.; Müller-Reible, C.; Dandekar, T.; Karchin, R.; Schultz, J.; Müller, T. Detecting species-site dependencies in large multiple sequence alignments. Nucleic Acids Res. 2009, 37, 5959–5968. [Google Scholar] [CrossRef] [PubMed]
- Geer, L.Y.; Domrachev, M.; Lipman, D.J.; Bryant, S.H. CDART: Protein homology by domain architecture. Genome Res. 2002, 12, 1619–1623. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Bateman, A.; Clements, J.; Coggill, P.; Eberhardt, R.Y.; Eddy, S.R.; Heger, A.; Hetherington, K.; Holm, L.; Mistry, J.; et al. Pfam: The protein families database. Nucleic Acids Res. 2014, 42, D222–D230. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [PubMed]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview Version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef] [PubMed]
- Milne, I.; Lindner, D.; Bayer, M.; Husmeier, D.; McGuire, G.; Marshall, D.F.; Wright, F. TOPALi v2: A rich graphical interface for evolutionary analyses of multiple alignments on HPC clusters and multi-core desktops. Bioinforma. Oxf. Engl. 2009, 25, 126–127. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinforma. Oxf. Engl. 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Minh, B.Q.; Nguyen, M.A.T.; von Haeseler, A. Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 2013, 30, 1188–1195. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL): An online tool for phylogenetic tree display and annotation. Bioinforma. Oxf. Engl. 2007, 23, 127–128. [Google Scholar] [CrossRef] [PubMed]
- Berezin, C.; Glaser, F.; Rosenberg, J.; Paz, I.; Pupko, T.; Fariselli, P.; Casadio, R.; Ben-Tal, N. ConSeq: The identification of functionally and structurally important residues in protein sequences. Bioinform. Oxf. Engl. 2004, 20, 1322–1324. [Google Scholar] [CrossRef] [PubMed]
- Ashkenazy, H.; Erez, E.; Martz, E.; Pupko, T.; Ben-Tal, N. ConSurf 2010: Calculating evolutionary conservation in sequence and structure of proteins and nucleic acids. Nucleic Acids Res. 2010, 38, W529–W533. [Google Scholar] [CrossRef] [PubMed]
- Lo, A.; Cheng, C.-W.; Chiu, Y.-Y.; Sung, T.-Y.; Hsu, W.-L. TMPad: An integrated structural database for helix-packing folds in transmembrane proteins. Nucleic Acids Res. 2011, 39, D347–D355. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Cierpicki, T.; Jimenez, R.H.F.; Lukasik, S.M.; Ellena, J.F.; Cafiso, D.S.; Kadokura, H.; Beckwith, J.; Bushweller, J.H. NMR solution structure of the integral membrane enzyme DsbB: Functional insights into DsbB-catalyzed disulfide bond formation. Mol. Cell 2008, 31, 896–908. [Google Scholar] [CrossRef] [PubMed]
- Sali, A.; Blundell, T.L. Comparative protein modelling by satisfaction of spatial restraints. J. Mol. Biol. 1993, 234, 779–815. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Cowtan, K. Coot: Model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 2126–2132. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef] [PubMed]
- Davis, I.W.; Leaver-Fay, A.; Chen, V.B.; Block, J.N.; Kapral, G.J.; Wang, X.; Murray, L.W.; Arendall, W.B.; Snoeyink, J.; Richardson, J.S.; et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007, 35, W375–W383. [Google Scholar] [CrossRef] [PubMed]
- Senes, A.; Gerstein, M.; Engelman, D.M. Statistical analysis of amino acid patterns in transmembrane helices: The GxxxG motif occurs frequently and in association with beta-branched residues at neighboring positions. J. Mol. Biol. 2000, 296, 921–936. [Google Scholar] [CrossRef] [PubMed]
- Stevens, T.J.; Arkin, I.T. Substitution rates in alpha-helical transmembrane proteins. Protein Sci. Publ. Protein Soc. 2001, 10, 2507–2517. [Google Scholar] [CrossRef]
- Raczko, A.M.; Bujnicki, J.M.; Pawlowski, M.; Godlewska, R.; Lewandowska, M.; Jagusztyn-Krynicka, E.K. Characterization of new DsbB-like thiol-oxidoreductases of Campylobacter jejuni and Helicobacter pylori and classification of the DsbB family based on phylogenomic, structural and functional criteria. Microbiol. Read. Engl. 2005, 151, 219–231. [Google Scholar] [CrossRef] [PubMed]
- Rishavy, M.A.; Usubalieva, A.; Hallgren, K.W.; Berkner, K.L. Novel insight into the mechanism of the vitamin K oxidoreductase (VKOR): Electron relay through Cys43 and Cys51 reduces VKOR to allow vitamin K reduction and facilitation of vitamin K-dependent protein carboxylation. J. Biol. Chem. 2011, 286, 7267–7278. [Google Scholar] [CrossRef] [PubMed]
- Doolittle, W.F. Phylogenetic classification and the universal tree. Science 1999, 284, 2124–2128. [Google Scholar] [CrossRef] [PubMed]
- Schaap, P. Guanylyl cyclases across the tree of life. Fron. Biosci. 2005, 10, 1485–1498. [Google Scholar] [CrossRef]
- Lake, J.A.; Skophammer, R.G.; Herbold, C.W.; Servin, J.A. Genome beginnings: Rooting the tree of life. Philos. Trans. R. Soc. Lond B. Biol. Sci. 2009, 364, 2177–2185. [Google Scholar] [CrossRef] [PubMed]
- Gribaldo, S.; Brochier, C. Phylogeny of prokaryotes: Does it exist and why should we care? Res. Microbiol. 2009, 23, 23. [Google Scholar] [CrossRef] [PubMed]
- Nakayama, T.; Archibald, J.M. Evolving a photosynthetic organelle. BMC Biol. 2012, 10, 35. [Google Scholar] [CrossRef] [PubMed]
- Nowack, E.C.M.; Melkonian, M.; Glöckner, G. Chromatophore genome sequence of Paulinella sheds light on acquisition of photosynthesis by eukaryotes. Curr. Biol. CB 2008, 18, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Marin, B.; Nowack, E.C.M.; Melkonian, M. A plastid in the making: Evidence for a second primary endosymbiosis. Protist 2005, 156, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.K.; Bhattacharyya-Pakrasi, M.; Pakrasi, H.B. Identification of an atypical membrane protein involved in the formation of protein disulfide bonds in oxygenic photosynthetic organisms. J. Biol. Chem. 2008, 283, 15762–15770. [Google Scholar] [CrossRef] [PubMed]
- Matagrin, B.; Hodroge, A.; Montagut-Romans, A.; Andru, J.; Fourel, I.; Besse, S.; Benoit, E.; Lattard, V. New insights into the catalytic mechanism of vitamin K epoxide reductase (VKORC1)—The catalytic properties of the major mutations of rVKORC1 explain the biological cost associated to mutations. FEBS Open Bio. 2013, 3, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Fasco, M.J.; Preusch, P.C.; Hildebrandt, E.; Suttie, J.W. Formation of hydroxyvitamin K by vitamin K epoxide reductase of warfarin-resistant rats. J. Biol. Chem. 1983, 258, 4372–4380. [Google Scholar] [PubMed]
- Rost, S.; Pelz, H.-J.; Menzel, S.; MacNicoll, A.D.; León, V.; Song, K.-J.; Jäkel, T.; Oldenburg, J.; Müller, C.R. Novel mutations in the VKORC1 gene of wild rats and mice—A response to 50 years of selection pressure by warfarin? BMC Genet. 2009, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Kapadia, M.; Rolston, K.V.I.; Han, X.Y. Invasive streptomyces infections: Six cases and literature review. Am. J. Clin. Pathol. 2007, 127, 619–624. [Google Scholar] [CrossRef] [PubMed]
- Berkner, K.L. Vitamin K-dependent carboxylation. Vitam. Horm. 2008, 78, 131–156. [Google Scholar] [PubMed]
- Note 1: Readers interested in viewing features summarized in Figure 5 for the all-atoms molecular models of hVKORC1 and hVKORC1L1 can download these from the Protein Model DataBase (https://bioinformatics.cineca.it/PMDB/main.php, PMDB identifiers PM0075969, PM007). The NMR structure for DsbB (PDB entry 2K74, model 1) can be downloaded from the RCSB Protein Data Bank (http://www.rcsb.org/pdb/home/home.do).
- White, S.H. Biophysical dissection of membrane proteins. Nature 2009, 459, 344–346. [Google Scholar] [CrossRef] [PubMed]
- Wallin, E.; von Heijne, G. Genome-wide analysis of integral membrane proteins from eubacterial, archaean, and eukaryotic organisms. Protein Sci. 1998, 7, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Lundstrom, K. Structural genomics and drug discovery. J. Cell Mol. Med. 2007, 11, 224–238. [Google Scholar] [CrossRef] [PubMed]
- Oberai, A.; Ihm, Y.; Kim, S.; Bowie, J.U. A limited universe of membrane protein families and folds. Protein Sci. 2006, 15, 1723–1734. [Google Scholar] [CrossRef] [PubMed]
- Vitkup, D.; Melamud, E.; Moult, J.; Sander, C. Completeness in structural genomics. Nat. Struct. Biol. 2001, 8, 559–566. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y. Protein structure prediction: When is it useful? Curr. Opin. Struct. Biol. 2009, 19, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Schwede, T.; Sali, A.; Honig, B.; Levitt, M.; Berman, H.M.; Jones, D.; Brenner, S.E.; Burley, S.K.; Das, R.; Dokholyan, N.V.; et al. Outcome of a workshop on applications of protein models in biomedical research. Structure 2009, 17, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Grosse-Kunstleve, R.W.; Read, R.J.; Richardson, J.S.; Richardson, D.C.; Terwilliger, T.C. Recent developments in phasing and structure refinement for macromolecular crystallography. Curr. Opin. Struct. Biol. 2009, 21, 21. [Google Scholar] [CrossRef] [PubMed]
- Barbar, E.; Lehoux, J.G.; Lavigne, P. Toward the NMR structure of StAR. Mol. Cell Endocrinol. 2009, 300, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Bax, A. Weak alignment offers new NMR opportunities to study protein structure and dynamics. Protein Sci. 2003, 12, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Rost, B. Twilight zone of protein sequence alignments. Protein Eng. 1999, 12, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Forrest, L.R.; Tang, C.L.; Honig, B. On the accuracy of homology modeling and sequence alignment methods applied to membrane proteins. Biophys. J. 2006, 91, 508–517. [Google Scholar] [CrossRef] [PubMed]
- Malojcić, G.; Owen, R.L.; Grimshaw, J.P.A.; Glockshuber, R. Preparation and structure of the charge-transfer intermediate of the transmembrane redox catalyst DsbB. FEBS Lett. 2008, 582, 3301–3307. [Google Scholar] [CrossRef] [PubMed]
- Inaba, K.; Murakami, S.; Nakagawa, A.; Iida, H.; Kinjo, M.; Ito, K.; Suzuki, M. Dynamic nature of disulphide bond formation catalysts revealed by crystal structures of DsbB. Embo J. 2009, 28, 779–791. [Google Scholar] [CrossRef] [PubMed]
- Vogel, C.; Morea, V. Duplication, divergence and formation of novel protein topologies. Bioessays 2006, 28, 973–978. [Google Scholar] [CrossRef] [PubMed]
- Grishin, N.V. Fold change in evolution of protein structures. J. Struct. Biol. 2001, 134, 167–185. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bevans, C.G.; Krettler, C.; Reinhart, C.; Watzka, M.; Oldenburg, J. Phylogeny of the Vitamin K 2,3-Epoxide Reductase (VKOR) Family and Evolutionary Relationship to the Disulfide Bond Formation Protein B (DsbB) Family. Nutrients 2015, 7, 6224-6249. https://doi.org/10.3390/nu7085281
Bevans CG, Krettler C, Reinhart C, Watzka M, Oldenburg J. Phylogeny of the Vitamin K 2,3-Epoxide Reductase (VKOR) Family and Evolutionary Relationship to the Disulfide Bond Formation Protein B (DsbB) Family. Nutrients. 2015; 7(8):6224-6249. https://doi.org/10.3390/nu7085281
Chicago/Turabian StyleBevans, Carville G., Christoph Krettler, Christoph Reinhart, Matthias Watzka, and Johannes Oldenburg. 2015. "Phylogeny of the Vitamin K 2,3-Epoxide Reductase (VKOR) Family and Evolutionary Relationship to the Disulfide Bond Formation Protein B (DsbB) Family" Nutrients 7, no. 8: 6224-6249. https://doi.org/10.3390/nu7085281
APA StyleBevans, C. G., Krettler, C., Reinhart, C., Watzka, M., & Oldenburg, J. (2015). Phylogeny of the Vitamin K 2,3-Epoxide Reductase (VKOR) Family and Evolutionary Relationship to the Disulfide Bond Formation Protein B (DsbB) Family. Nutrients, 7(8), 6224-6249. https://doi.org/10.3390/nu7085281