Pyrosequencing Analysis Reveals Changes in Intestinal Microbiota of Healthy Adults Who Received a Daily Dose of Immunomodulatory Probiotic Strains

 ,
,  , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethical Statement
2.2. Subjects and Experimental Design
2.3. Fecal Samples, DNA Extraction and 454 Pyrosequencing
2.4. Taxonomic Analysis
2.5. Statistical Analysis
3. Results
3.1. Subject Information and Sequencing Coverage

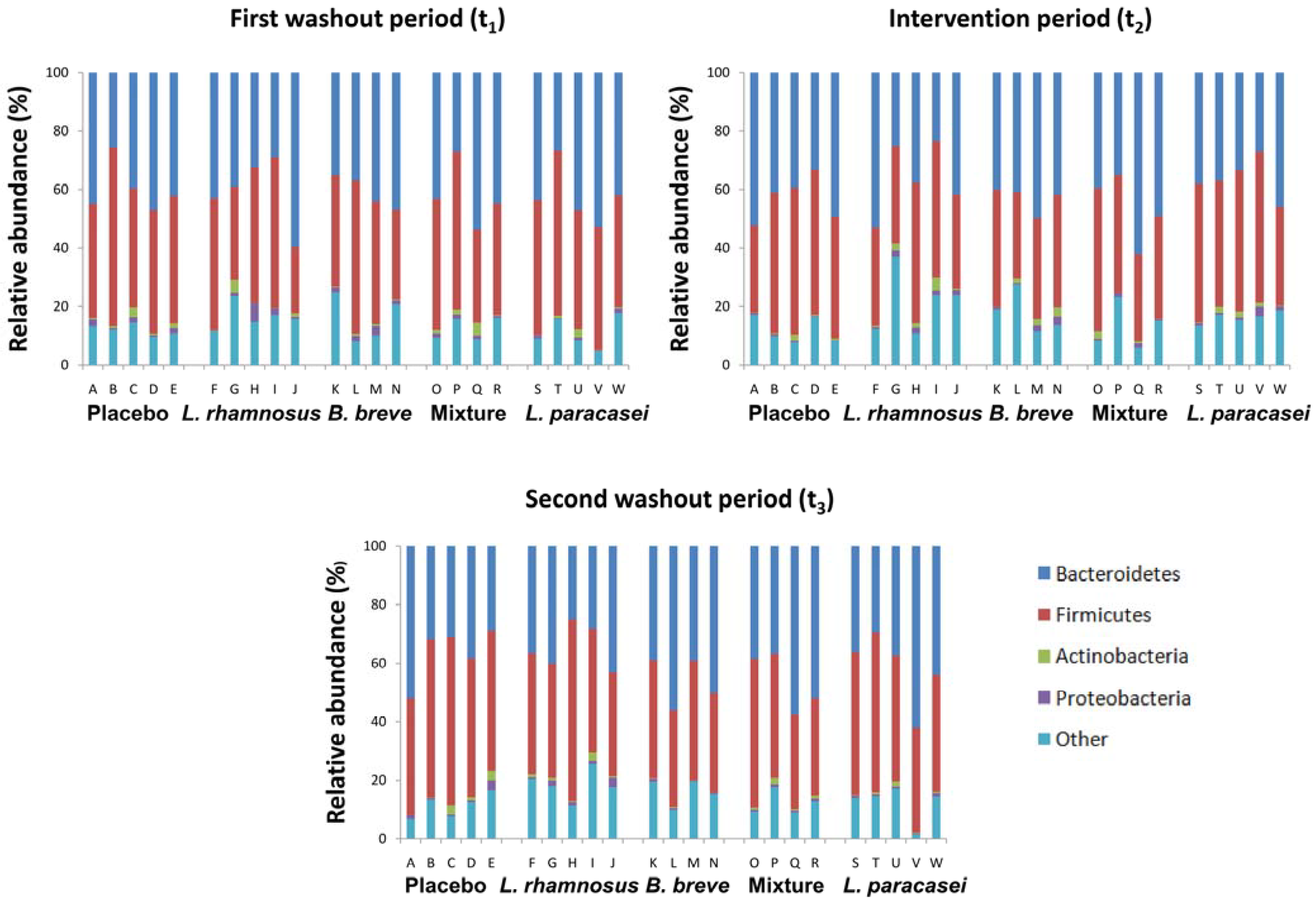{kind=link}
{kind=link}
{kind=link}
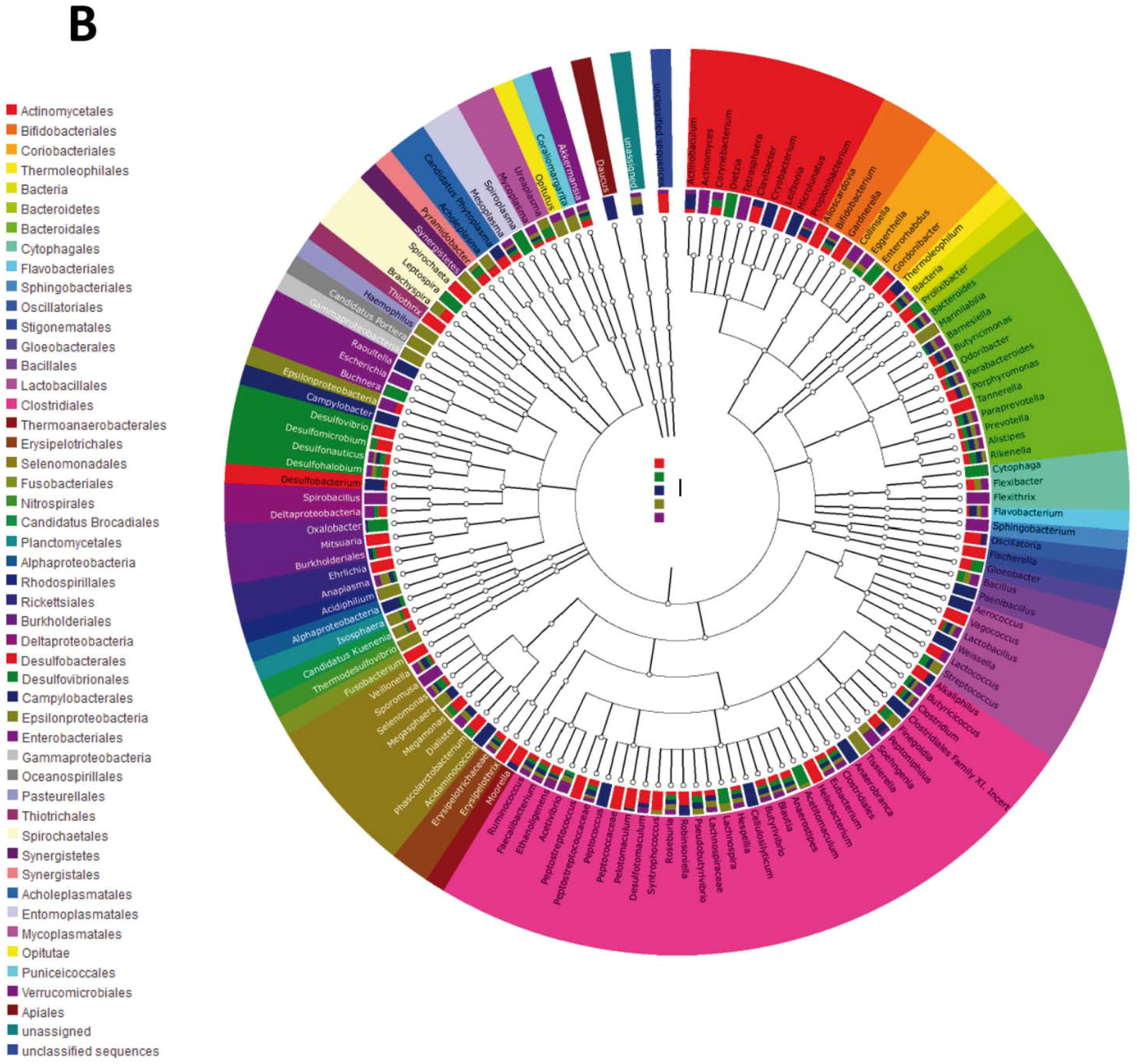{kind=link}
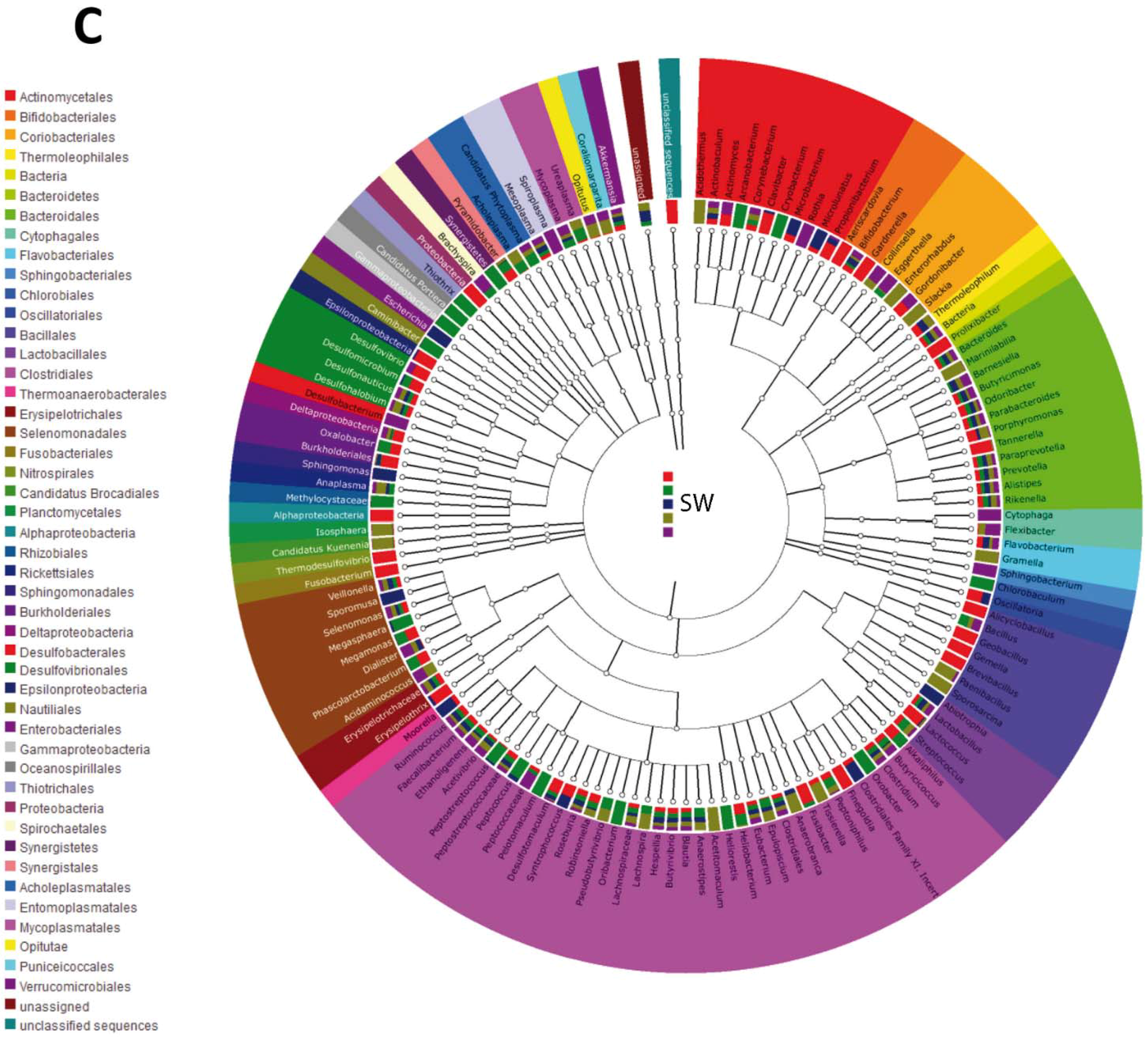{kind=link}
| Subject code | Capsule | Age | Gender | Number of sequences | ||
|---|---|---|---|---|---|---|
| t1 | t2 | t3 | ||||
| A | Placebo | 20 | Female | 3772 | 4924 | 3170 |
| B | Placebo | 27 | Female | 4581 | 5864 | 4674 |
| C | Placebo | 30 | Male | 6108 | 4819 | 5564 |
| D | Placebo | 29 | Male | 4005 | 5455 | 5214 |
| E | Placebo | 27 | Male | 7231 | 6276 | 4801 |
| F | L. rhamnosus CNCM I-4036 | 28 | Female | 1725 | 7814 | 6949 |
| G | L. rhamnosus CNCM I-4036 | 20 | Female | 4971 | 4506 | 4877 |
| H | L. rhamnosus CNCM I-4036 | 21 | Male | 4052 | 3795 | 4146 |
| I | L. rhamnosus CNCM I-4036 | 29 | Female | 3538 | 3595 | 4564 |
| J | L. rhamnosus CNCM I-4036 | 20 | Male | 3523 | 5645 | 5221 |
| K | B. breve CNCM I-4035 | 21 | Male | 10,569 | 4525 | 4782 |
| L | B. breve CNCM I-4035 | 20 | Male | 4924 | 4976 | 4381 |
| M | B. breve CNCM I-4035 | 26 | Male | 3888 | 1940 | 4772 |
| N | B. breve CNCM I-4035 | 35 | Male | 5708 | 5627 | 5370 |
| O | B. breve CNCM I-4035 and L. rhamnosus CNCM I-4036 | 20 | Female | 7098 | 8277 | 6383 |
| P | B. breve CNCM I-4035 and L. rhamnosus CNCM I-4036 | 30 | Male | 7860 | 3646 | 5009 |
| Q | B. breve CNCM I-4035 and L. rhamnosus CNCM I-4036 | 25 | Male | 3437 | 4506 | 4486 |
| R | B. breve CNCM I-4035 and L. rhamnosus CNCM I-4036 | 27 | Female | 5707 | 3011 | 3397 |
| S | L. paracasei CNCM I-4034 | 28 | Male | 6095 | 4015 | 4651 |
| T | L. paracasei CNCM I-4034 | 26 | Male | 9256 | 5426 | 2931 |
| U | L. paracasei CNCM I-4034 | 25 | Female | 3926 | 5271 | 6547 |
| V | L. paracasei CNCM I-4034 | 26 | Male | 3726 | 4208 | 3523 |
| W | L. paracasei CNCM I-4034 | 31 | Female | 2753 | 8642 | 2435 |

| Capsule | Genus | ||||
|---|---|---|---|---|---|
| First washout | Intervention | Second washout | |||
| Placebo | Bacteroides | 18.3 (7.5–37.9) | 15.9 (10.8–27.1) | 12.5 (6.4–29.9) | |
| Faecalibacterium | 10.5 (7.3–15.5) | 11.0 (6.4–19.8) | 15.1 (7.9–19.8) | ||
| Alistipes | 7.0 (4.6–11.2) | 5.2 (3.4–13.8) | 5.7 (3.1–14.3) | ||
| Prevotella | 7.1 (0.0–13.2) | 16.6 (0.0–25.8) | 2.4 (0.0–13.3) | ||
| Clostridium | 6.9 (1.9–8.7) | 4.9 (2.6–10.6) | 6.1 (3.6–9.3) | ||
| Eubacterium | 4.0 (1.6–7.1) | 5.1 (1.9–7.3) | 3.5 (1.6–5.1) | ||
| Ruminococcus | 1.8 (0.8–8.3) | 1.8 (1.2–4.1) | 4.6 (2.1–12.1) | ||
| Phascolarctobacterium | 1.9 (0.0–7.3) | 1.8 (0.0–3.6) | 1.8 (0.0–4.7) | ||
| Parabacteroides | 1.9 (1.2–4.5) | 1.6 (1.0–5.8) | 2.8 (2.4–3.6) | ||
| Veillonella | 0.8 (0.2–5.2) | 0.4 (0.0–4.8) | 1.4 (0.4–2.2) | ||
| Butyrivibrio | 1.1 (0.9–2.6) | 1.3 (0.7–2.0) | 1.6 (0.2–3.1) | ||
| Akkermansia | 0.6 (0.0–2.9) | 0.0 (0.0–0.1) | 0.0 (0.0–1.8) | ||
| Bifidobacterium | 0.4 (0.1–0.6) | 0.2 (0.0–0.8) | 0.0 (0.0–0.8) | ||
| Lactobacillus | 0.0 (0.0–0.5) | 0.0 (0.0–0.4) | 0.0 (0.0–0.8) | ||
| Bacillus | 0.0 (0.0–0.2) | 0.2 (0.0–0.4) | 0.1 (0.0–0.6) | ||
| Lactobacillus rhamnosus CNCM I-4036 | Bacteroides | 18.9 (8.8–32.0) | 13.0 (5.4–29.1) | 14.8 (5.4–23.6) | |
| Faecalibacterium | 9.9 (3.6–22.4) | 6.3 (2.8–20.8) | 8.3 (4.5–16.8) | ||
| Alistipes | 5.5 (0.1–7.0) | 4.0 (2.9–15.1) | 4.7 (2.1–9.7) | ||
| Prevotella | 7.1 (0.0–38.1) | 4.2 (0.1–26.5) | 5.1 (0.0–29.2) | ||
| Clostridium | 5.7 (2.5–7.2) | 6.2 (3.9–8.3) | 8.3 (3.1–11.8) | ||
| Eubacterium | 5.0 (1.4–7.1) | 3.7 (3.2–9.6) | 4.4 (3.7–8.9) | ||
| Ruminococcus | 0.7A (0.5–6.0) | 4.3AB (2.3–5.0) | 6.2B (0.9–7.7) | ||
| Phascolarctobacterium | 0.5 (0.0–1.6) | 0.0 (0.0–0.7) | 1.3 (0.0–2.8) | ||
| Parabacteroides | 2.0 (0.5–3.4) | 1.6 (0.4–4.5) | 1.8 (1.1–3.8) | ||
| Veillonella | 0.5 (0.0–2.5) | 0.8 (0.0–3.2) | 0.5 (0.0–1.3) | ||
| Butyrivibrio | 1.4 (0.2–2.4) | 0.7 (0.3–1.6) | 0.9 (0.3–1.7) | ||
| Akkermansia | 4.1 (0.0–7.7) | 1.3 (0.2–13.4) | 3.1 (1.0–7.7) | ||
| Bifidobacterium | 0.2 (0.0–4.0) | 0.8 (0.2–2.4) | 0.4 (0.0–1.2) | ||
| Lactobacillus | 0.0A (0.0–0.3) | 1.7B (0.5–7.8) | 0.8BC (0.5–4.0) | ||
| Bacillus | 0.5 (0.0–1.8) | 0.1 (0.0–0.2) | 0.1 (0.0–0.5) | ||
| Bifidobacterium breve CNCM I-4035 | Bacteroides | 22.7 (6.9–29.8) | 25.2 (14.2–42.2) | 19.2 (7.1–25.2) | |
| Faecalibacterium | 6.9 (5.2–10.3) | 6.6 (4.4–11.3) | 7.1 (4.1–8.1) | ||
| Alistipes | 5.5 (4.7–9.8) | 5.5 (3.7–9.2) | 4.9 (1.6–10.3) | ||
| Prevotella | 7.7 (0.0–19.8) | 3.0 (0.1–18.4) | 12.1 (6.4–41.2) | ||
| Clostridium | 10.5 (5.5–11.3) | 7.2 (2.2–11.6) | 6.3 (5.4–8.4) | ||
| Eubacterium | 4.8 (3.8–7.7) | 3.7 (2.1–5.4) | 4.2 (2.0–11.8) | ||
| Ruminococcus | 4.0 (0.6–8.7) | 2.0 (0.2–5.6) | 2.4 (1.5–3.4) | ||
| Phascolarctobacterium | 0.0 (0.0–0.0) | 0.0 (0.0–0.0) | 0.0 (0.0–0.0) | ||
| Parabacteroides | 2.1 (1.2–3.6) | 2.3 (0.8–3.2) | 2.3 (0.9–3.6) | ||
| Veillonella | 1.5 (0.3–2.2) | 1.6 (0.0–1.6) | 2.1 (0.8–2.6) | ||
| Butyrivibrio | 1.5 (0.6–3.7) | 1.3 (0.6–3.2) | 2.1 (1.0–2.6) | ||
| Akkermansia | 0.0 (0.0–0.2) | 0.0 (0.0–0.0) | 2.9 (0.0–8.1) | ||
| Bifidobacterium | 0.1 (0.0–0.5) | 1.1 (0.0–2.8) | 0.0 (0.0–0.0) | ||
| Lactobacillus | 0.0 (0.0–0.7) | 0.0 (0.0–0.0) | 0.0 (0.0–0.2) | ||
| Bacillus | 1.5 (0.5–3.9) | 2.4 (0.0–5.4) | 0.3 (0.1–0.6) | ||
| Bifidobacterium breve CNCM I-4035 and Lactobacillus rhamnosus CNCM I-4036 | Bacteroides | 16.5 (12.0–38.7) | 23.2 (12.8–50.0) | 18.4 (8.8–49.6) | |
| Faecalibacterium | 7.3 (2.2–9.8) | 10.3 (7.9–13.4) | 6.5 (2.4–14.3) | ||
| Alistipes | 4.2 (3.8–7.9) | 5.5 (2.8–7.3) | 4.8 (1.0–11.7) | ||
| Prevotella | 7.6 (5.3–23.1) | 7.8 (0.1–17.6) | 10.6 (1.0–12.8) | ||
| Clostridium | 8.4 (2.3–11.1) | 4.4 (2.3–8.2) | 7.1 (4.8–8.4) | ||
| Eubacterium | 4.4 (3.0–10.1) | 3.9 (2.1–6.2) | 6.4 (3.8–10.7) | ||
| Ruminococcus | 4.9 (1.7–10.2) | 2.7 (0.3–3.6) | 4.1 (0.8–7.3) | ||
| Phascolarctobacterium | 0.0 (0.0–0.0) | 0.0 (0.0–1.4) | 0.1 (0.0–0.7) | ||
| Parabacteroides | 3.7 (1.3–4.0) | 3.1 (1.9–5.9) | 4.1 (2.6–8.9) | ||
| Veillonella | 0.4 (0.2–2.1) | 0.9 (0.1–1.6) | 0.5 (0.3–1.1) | ||
| Butyrivibrio | 1.3 (0.9–3.0) | 1.3 (0.4–1.8) | 1.3 (0.7–2.1) | ||
| Akkermansia | 0.8 (0.0–7.1) | 0.1 (0.0–3.3) | 0.0 (0.0–0.9) | ||
| Bifidobacterium | 0.4 (0.2–0.6) | 0.3 (0.0–2.0) | 0.4 (0.2–0.6) | ||
| Lactobacillus | 0.3 (0.0–0.5) | 0.2 (0.0–0.7) | 0.0 (0.0–1.6) | ||
| Bacillus | 0.5 (0.0–3.3) | 0.5 (0.0–3.3) | 0.0 (0.0–0.5) | ||
| Lactobacillus paracasei CNCM I-4034 | Bacteroides | 23.6 (19.7–46.0) | 21.3 (6.0–26.6) | 21.8 (10.1–50.4) | |
| Faecalibacterium | 9.6 (8.3–18.3) | 9.8 (3.8–11.5) | 10.3 (8.0–13.5) | ||
| Alistipes | 5.6 (3.5–12.8) | 8.5 (2.2–11.5) | 5.9 (3.8–8.9) | ||
| Prevotella | 0.1 (0.0–14.2) | 2.4 (0.0–22.0) | 0.0 (0.0–19.1) | ||
| Clostridium | 5.4 (3.1–7.4) | 6.4 (5.7–8.3) | 6.1 (4.0–7.5) | ||
| Eubacterium | 8.7 (3.8–13.8) | 5.5 (3.6–8.3) | 7.1 (5.6–7.8) | ||
| Ruminococcus | 2.4 (1.3–5.2) | 2.7 (2.0–12.5) | 3.4 (1.8–4.7) | ||
| Phascolarctobacterium | 1.0 (0.0–4.7) | 0.6 (0.0–2.7) | 0.1 (0.0–2.7) | ||
| Parabacteroides | 2.2AC (0.0–3.4) | 2.3A (1.1–4.3) | 1.3BC (0.0–1.7) | ||
| Veillonella | 0.3 (0.0–3.1) | 0.7 (0.0–1.5) | 0.7 (0.0–1.4) | ||
| Butyrivibrio | 1.4 (0.7–3.3) | 0.6 (0.6–2.7) | 1.6 (1.2–1.9) | ||
| Akkermansia | 0.0 (0.0–7.0) | 0.1 (0.0–0.1) | 0.0 (0.0–0.1) | ||
| Bifidobacterium | 0.0 (0.0–1.7) | 0.2 (0.0–1.9) | 0.3 (0.0–1.1) | ||
| Lactobacillus | 0.0 (0.0–0.6) | 0.1 (0.0–1.2) | 0.2 (0.0–2.6) | ||
| Bacillus | 0.2 (0.0–0.8) | 0.2 (0.0–2.1) | 0.7 (0.0–3.1) | ||
3.2. Impact of Lactobacillus rhamnosus CNCM I-4036 on the Colon Microbiota Compositions of Healthy Volunteers
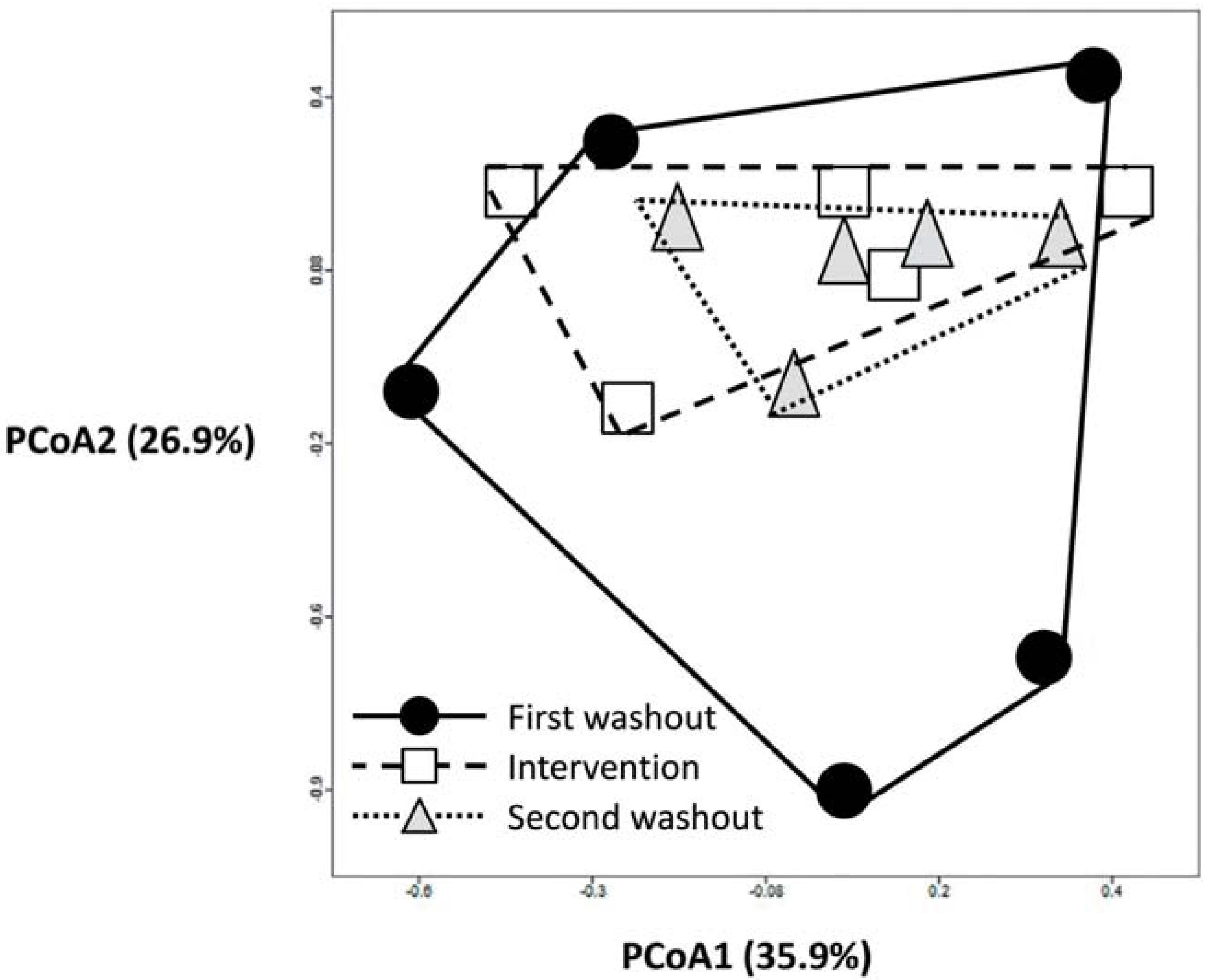
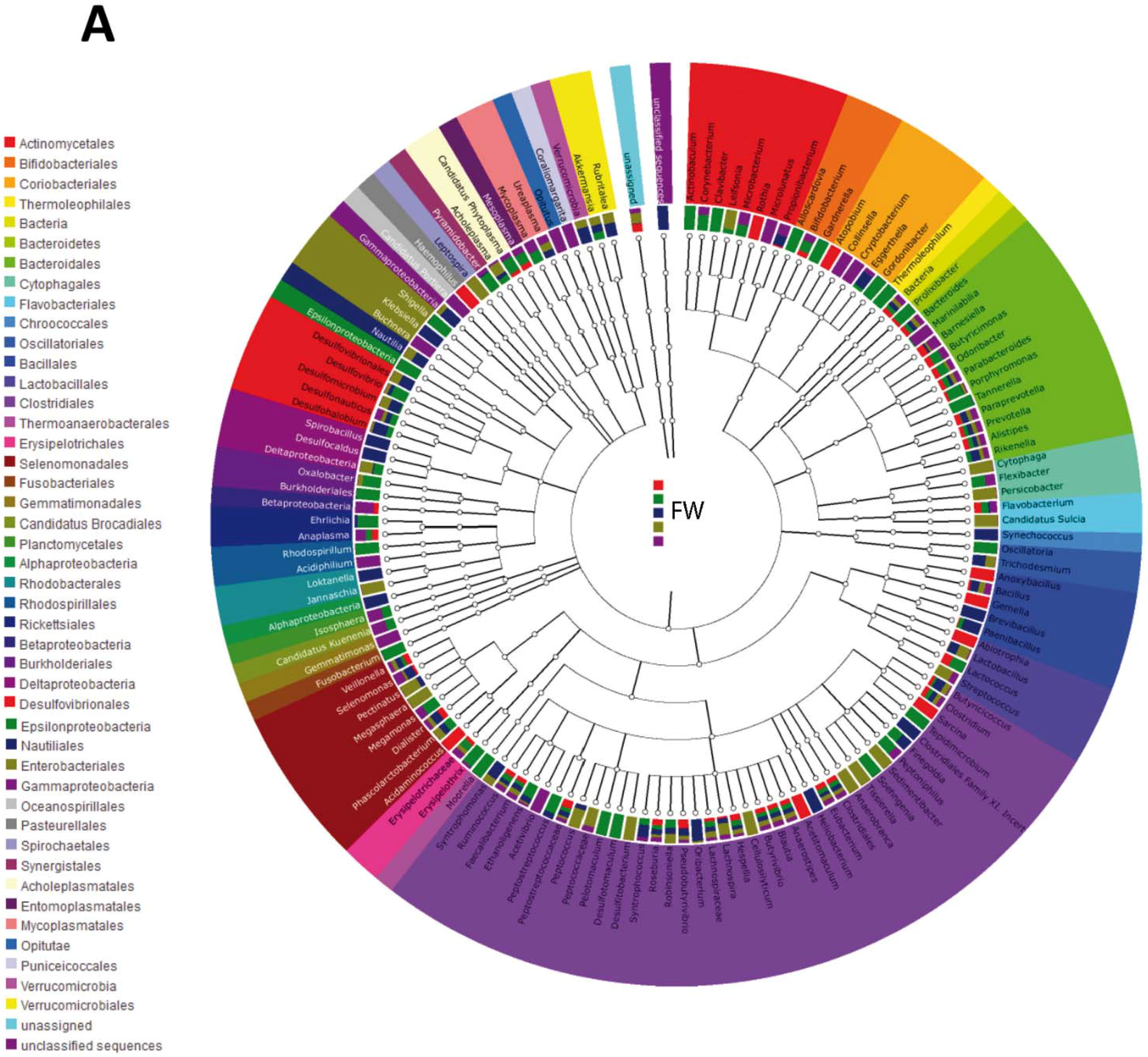
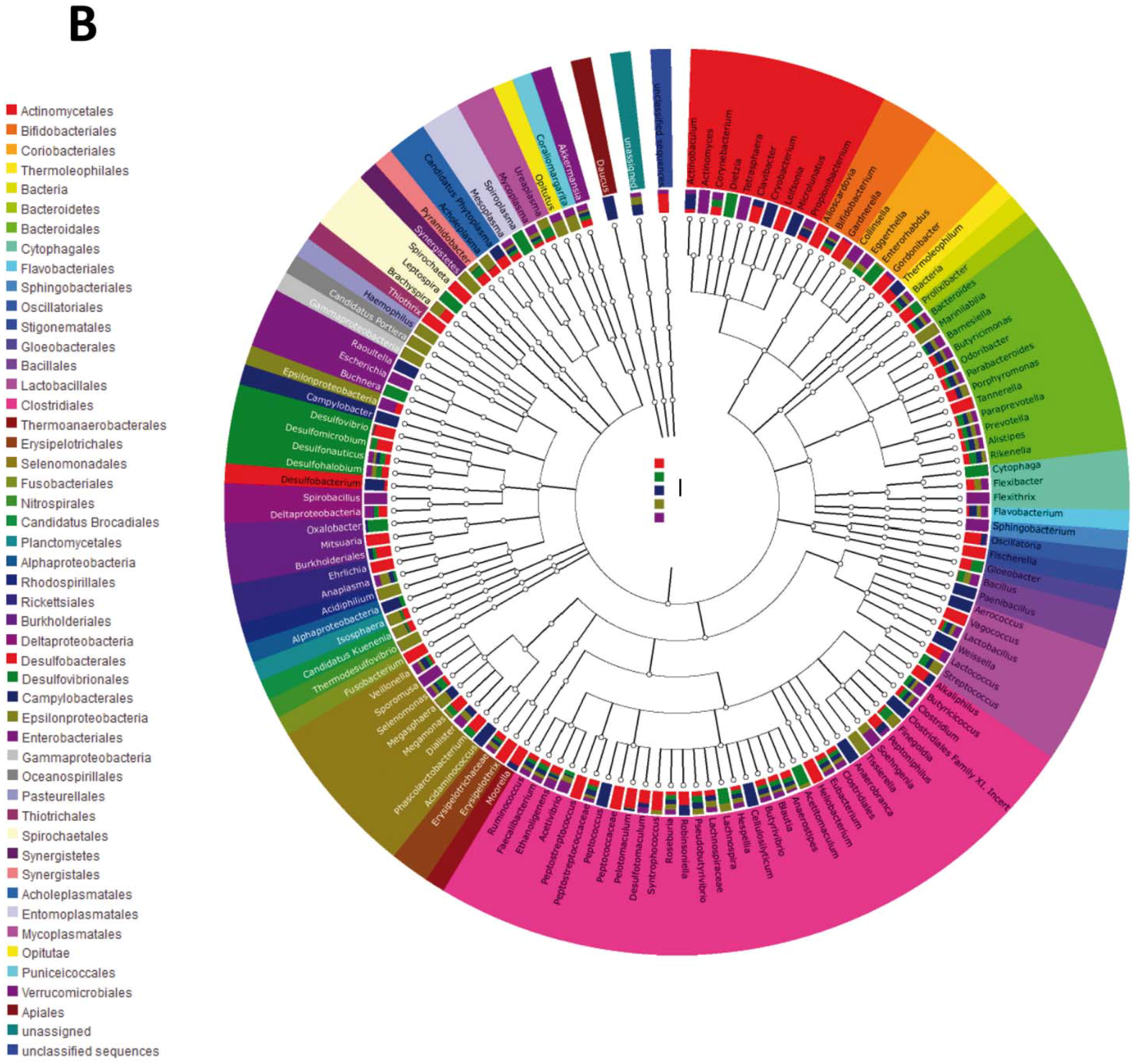
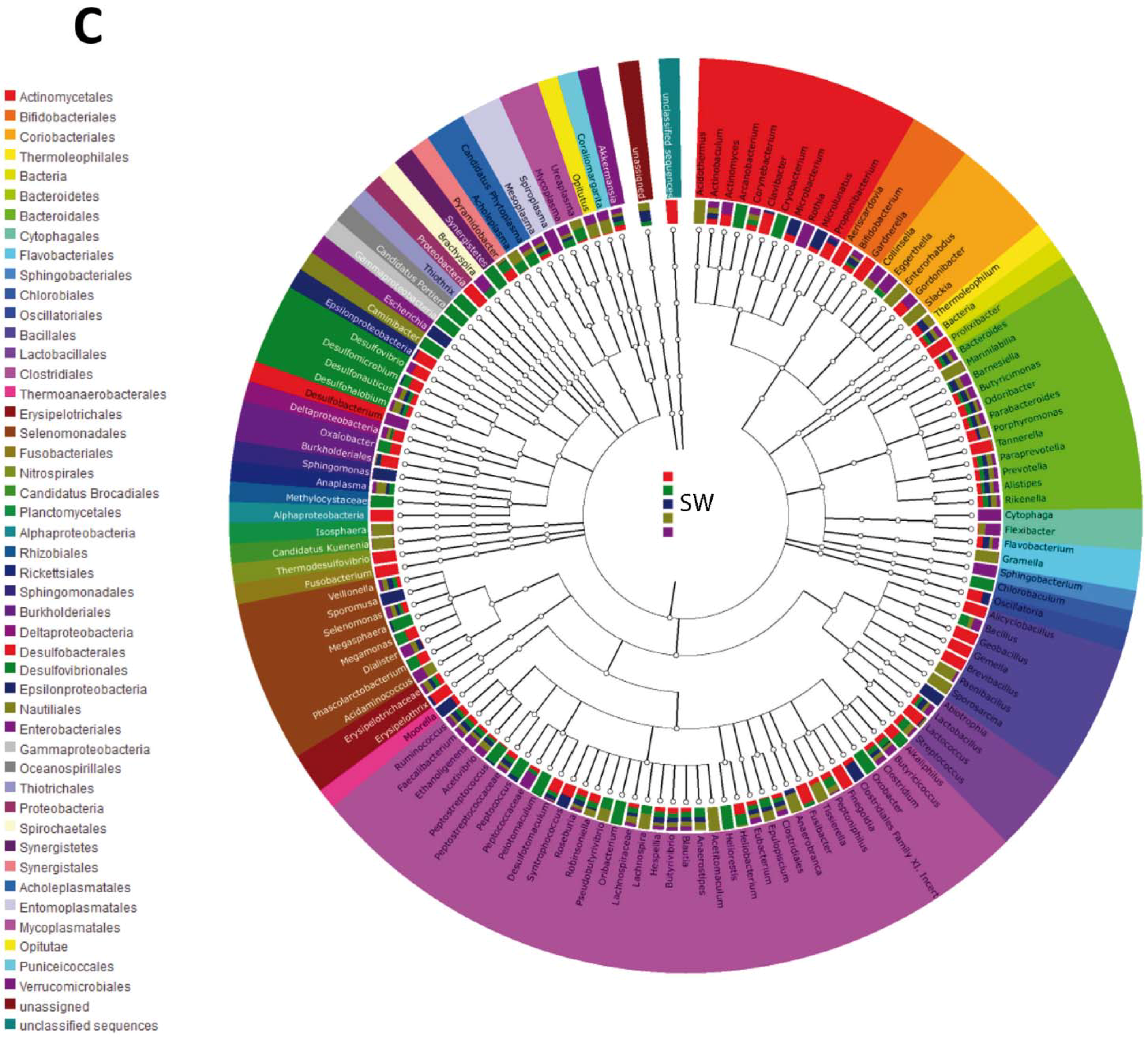
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Savage, D.C. Microbial ecology of the gastrointestinal tract. Annu. Rev. Microbiol. 1977, 31, 107–133. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Peterson, D.A.; Gordon, J.I. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell 2006, 124, 837–848. [Google Scholar] [CrossRef] [PubMed]
- Steinhoff, U. Who controls the crowd? New findings and old questions about the intestinal microflora. Immunol. Lett. 2005, 99, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Backhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar] [CrossRef] [PubMed]
- Hooper, L.V.; Midtvedt, T.; Gordon, J.I. How host-microbial interactions shape the nutrient environment of the mammalian intestine. Annu. Rev. Nutr. 2002, 22, 283–307. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, N.; Moudgal, V. Probiotics: A Review. J. Clin. Outcomes Manage 2012, 19, 76–84. [Google Scholar]
- Kim, S.W.; Suda, W.; Kim, S.; Oshima, K.; Fukuda, S.; Ohno, H.; Morita, H.; Hattori, M. Robustness of gut microbiota of healthy adults in response to probiotic intervention revealed by high-throughput pyrosequencing. DNA Res. 2013, 20, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.; Taverniti, V.; Milani, C.; Fiore, W.; Laureati, M.; De Noni, I.; Stuknyte, M.; Chouaia, B.; Riso, P.; Guglielmetti, S. Modulation of Fecal Clostridiales Bacteria and Butyrate by Probiotic Intervention with Lactobacillus paracasei DG Varies among Healthy Adults. J. Nutr. 2014, 144, 1787–1796. [Google Scholar] [CrossRef] [PubMed]
- Hamady, M.; Knight, R. Microbial community profiling for human microbiome projects: Tools, techniques, and challenges. Genome Res. 2009, 19, 1141–1152. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Backhed, F.; Fulton, L.; Gordon, J.I. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 2008, 3, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Larsen, N.; Vogensen, F.K.; van den Berg, F.W.; Nielsen, D.S.; Andreasen, A.S.; Pedersen, B.K.; Al-Soud, W.A.; Sorensen, S.J.; Hansen, L.H.; Jakobsen, M. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE 2010, 5, e9085. [Google Scholar] [CrossRef] [PubMed]
- Friswell, M.; Campbell, B.; Rhodes, J. The role of bacteria in the pathogenesis of inflammatory bowel disease. Gut Liver 2010, 4, 295–306. [Google Scholar] [CrossRef] [PubMed]
- Ley, R.E.; Backhed, F.; Turnbaugh, P.; Lozupone, C.A.; Knight, R.D.; Gordon, J.I. Obesity alters gut microbial ecology. Proc. Natl. Acad. Sci. USA 2005, 102, 11070–11075. [Google Scholar] [CrossRef] [PubMed]
- Ravussin, Y.; Koren, O.; Spor, A.; Leduc, C.; Gutman, R.; Stombaugh, J.; Knight, R.; Ley, R.E.; Leibel, R.L. Responses of gut microbiota to diet composition and weight loss in lean and obese mice. Obesity 2012, 20, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Plaza-Diaz, J.; Gomez-Llorente, C.; Campaña-Martin, L.; Matencio, E.; Ortuño, I.; Martínez-Silla, R.; Gomez-Gallego, C.; Periago, M.J.; Ros, G.; Chenoll, E.; et al. Safety and immunomodulatory effects of three probiotic strains isolated from the feces of breast-fed infants in healthy adults: SETOPROB study. PLoS ONE 2013, 8, e78111. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Quezada, S.; Chenoll, E.; Vieites, J.M.; Genovés, S.; Maldonado, J.; Bermúdez-Brito, M.; Gomez-Llorente, C.; Matencio, E.; Bernal, M.J.; Romero, F.; et al. Isolation, identification and characterisation of three novel probiotic strains (Lactobacillus paracasei CNCM I-4034, Bifidobacterium breve CNCM I-4035 and Lactobacillus rhamnosus CNCM I-4036) from the faeces of exclusively breast-fed infants. Br. J. Nutr. 2013, 109, S51–S62. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Quezada, S.; Bermudez-Brito, M.; Chenoll, E.; Genovés, S.; Gomez-Llorente, C.; Plaza-Diaz, J.; Matencio, E.; Bernal, M.J.; Romero, F.; Ramón, D.; Gil, A. Competitive inhibition of three novel bacteria isolated from faeces of breast milk-fed infants against selected enteropathogens. Br. J. Nutr. 2013, 109, S63–S69. [Google Scholar] [CrossRef] [PubMed]
- Vieites-Fernández, J.M.; Muñoz-Quezada, S.; Llamas-Company, I.; Maldonado-Lozano, J.; Romero-Braquehais, F.; Suárez-García, A.; Gil-Hernández, A.; Gómez-Llorente, C.; Bermúdez-Brito, M. Isolation, Identification and Characterisation of Strains with Probiotic Activity, from Faeces of Infants Fed Exclusively with Breast Milk. European Patent Application EP 2 407 532 A2; Bulletin 2012/03 18 January 2012; International publication number: WO 2010/103140 (16.09.2010 Gazette 2010/37),
- Meyer, F.; Paarmann, D.; D’Souza, M.; Olson, R.; Glass, E.M.; Kubal, M.; Paczian, T.; Rodriguez, A.; Stevens, R.; Wilke, A.; Wilkening, J.; Edwards, R.A. The metagenomics RAST server—A public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 2008, 9, 386. [Google Scholar] [CrossRef] [PubMed]
- Aziz, Q.; Doré, J.; Emmanuel, A.; Guarner, F.; Quigley, E.M. Gut microbiota and gastrointestinal health: current concepts and future directions. Neurogastroenterol. Motil. 2013, 25, 4–15. [Google Scholar] [CrossRef] [PubMed]
- Manichanh, C.; Borruel, N.; Casellas, F.; Guarner, F. The gut microbiota in IBD. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Koren, O.; Knights, D.; Gonzalez, A.; Waldron, L.; Segata, N.; Knight, R.; Huttenhower, C.; Ley, R.E. A guide to enterotypes across the human body: Meta-analysis of microbial community structures in human microbiome datasets. PLoS Comput. Biol. 2013, 9, e1002863. [Google Scholar] [CrossRef] [PubMed]
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214. [Google Scholar]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Wang, L.; Guo, Z.; Sun, Z.; Gesudu, Q.; Kwok, L.; Menghebilige; Zhang, H. 454 pyrosequencing reveals changes in the faecal microbiota of adults consuming Lactobacillus casei Zhang. FEMS Microbiol. Ecol. 2014, 88, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Azad, M.B.; Konya, T.; Maughan, H.; Guttman, D.S.; Field, C.J.; Chari, R.S.; Sears, M.R.; Becker, A.B.; Scott, J.A.; Kozyrskyj, A.L.; CHILD Study Investigators. Gut microbiota of healthy Canadian infants: profiles by mode of delivery and infant diet at 4 months. CMAJ 2013, 185, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Bermudez-Brito, M.; Muñoz-Quezada, S.; Gomez-Llorente, C.; Matencio, E.; Bernal, M.J.; Romero, F.; Gil, A. Human intestinal dendritic cells decrease cytokine release against Salmonella infection in the presence of Lactobacillus paracasei upon TLR activation. PLoS ONE 2012, 7, e43197. [Google Scholar] [CrossRef] [PubMed]
- Bermudez-Brito, M.; Muñoz-Quezada, S.; Gomez-Llorente, C.; Matencio, E.; Bernal, M.J.; Romero, F.; Gil, A. Cell-free culture supernatant of Bifidobacterium breve CNCM I-4035 decreases pro-inflammatory cytokines in human dendritic cells challenged with Salmonella typhi through TLR activation. PLoS ONE 2013, 8, e59370. [Google Scholar] [CrossRef] [PubMed]
- Azad, M.B.; Konya, T.; Maughan, H.; Guttman, D.S.; Field, C.J.; Sears, M.R.; Becker, A.B.; Scott, J.A.; Kozyrskyj, A.L. Infant gut microbiota and the hygiene hypothesis of allergic disease: Impact of household pets and siblings on microbiota composition and diversity. Allergy Asthma Clin. Immunol. 2013, 9, 15. [Google Scholar] [CrossRef] [PubMed]
- Bailey, M.T.; Dowd, S.E.; Galley, J.D.; Hufnagle, A.R.; Allen, R.G.; Lyte, M. Exposure to a social stressor alters the structure of the intestinal microbiota: Implications for stressor-induced immunomodulation. Brain Behav. Immun. 2011, 25, 397–407. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plaza-Díaz, J.; Fernández-Caballero, J.Á.; Chueca, N.; García, F.; Gómez-Llorente, C.; Sáez-Lara, M.J.; Fontana, L.; Gil, Á. Pyrosequencing Analysis Reveals Changes in Intestinal Microbiota of Healthy Adults Who Received a Daily Dose of Immunomodulatory Probiotic Strains. Nutrients 2015, 7, 3999-4015. https://doi.org/10.3390/nu7063999
Plaza-Díaz J, Fernández-Caballero JÁ, Chueca N, García F, Gómez-Llorente C, Sáez-Lara MJ, Fontana L, Gil Á. Pyrosequencing Analysis Reveals Changes in Intestinal Microbiota of Healthy Adults Who Received a Daily Dose of Immunomodulatory Probiotic Strains. Nutrients. 2015; 7(6):3999-4015. https://doi.org/10.3390/nu7063999
Chicago/Turabian StylePlaza-Díaz, Julio, Jose Ángel Fernández-Caballero, Natalia Chueca, Federico García, Carolina Gómez-Llorente, María José Sáez-Lara, Luis Fontana, and Ángel Gil. 2015. "Pyrosequencing Analysis Reveals Changes in Intestinal Microbiota of Healthy Adults Who Received a Daily Dose of Immunomodulatory Probiotic Strains" Nutrients 7, no. 6: 3999-4015. https://doi.org/10.3390/nu7063999
APA StylePlaza-Díaz, J., Fernández-Caballero, J. Á., Chueca, N., García, F., Gómez-Llorente, C., Sáez-Lara, M. J., Fontana, L., & Gil, Á. (2015). Pyrosequencing Analysis Reveals Changes in Intestinal Microbiota of Healthy Adults Who Received a Daily Dose of Immunomodulatory Probiotic Strains. Nutrients, 7(6), 3999-4015. https://doi.org/10.3390/nu7063999