Arginine and Citrulline and the Immune Response in Sepsis

Abstract
:1. Introduction
2. Arginine during Physiological Conditions
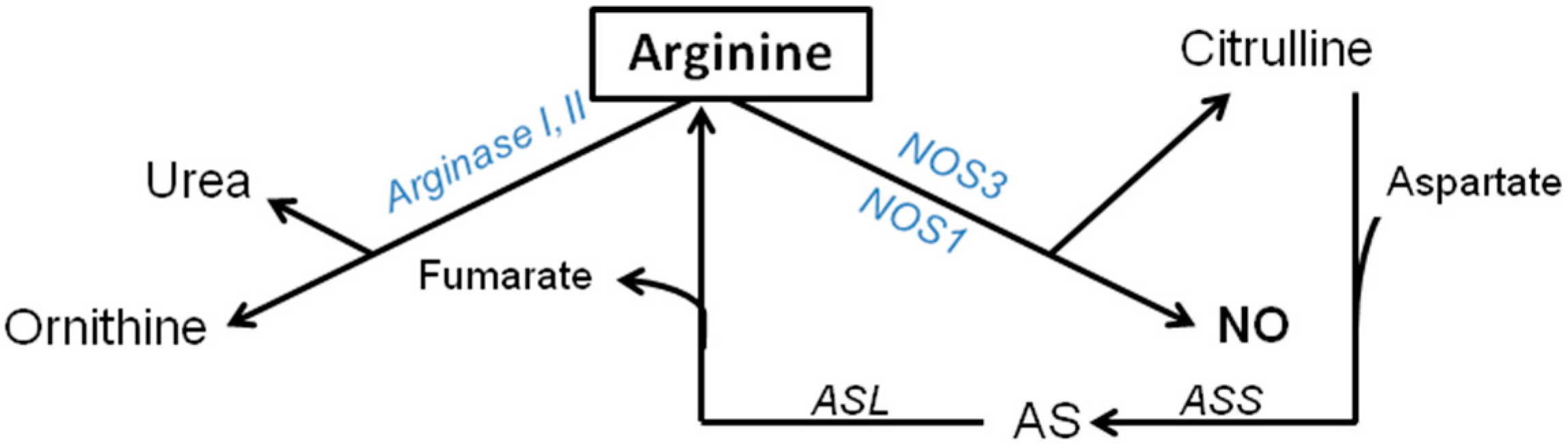
2.1. Arginine de novo Synthesis; Conversion of Citrulline into Arginine by Argininosuccinate Synthetase and Argininosuccinate Lyase
2.2. Arginine and Citrulline Transport Systems
2.3. Catabolic Pathways of Arginine during Basal Conditions
2.4. Arginase Pathway: Synthesis of Urea, Ornithine, Polyamine and Proline
2.5. NOS Pathway (NOS1, NOS2, NOS3); Synthesis of Citrulline and NO
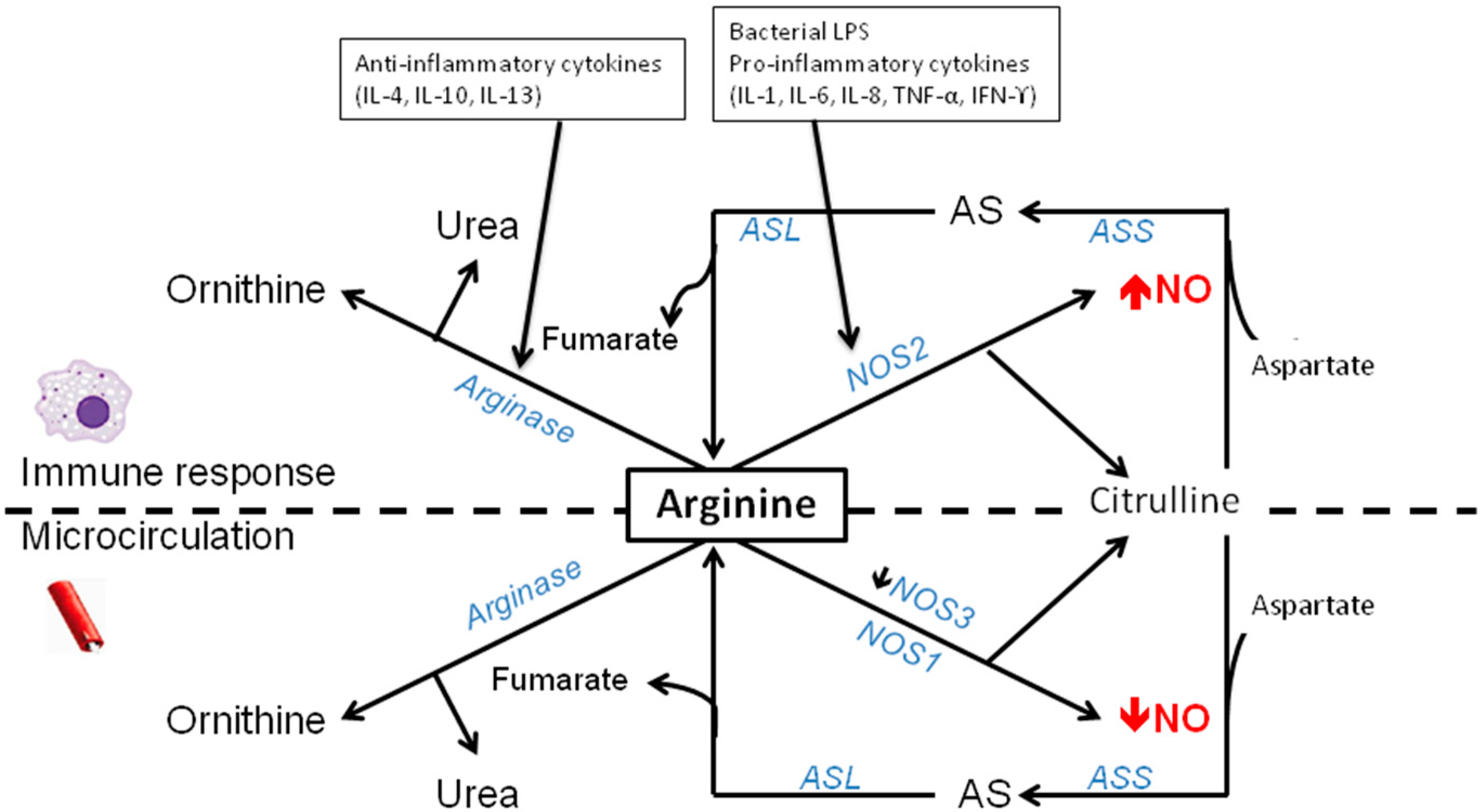
3. Arginine-NO Metabolism during Sepsis and Inflammatory Conditions

3.1. Alterations in the Arginine and Citrulline Transport Systems in Sepsis
3.2. Decreased Citrulline Availability as Cause for Diminished Arginine de novo Synthesis in Sepsis/Infection
3.3. Decreased Arginine Availability by Enhanced Arginase Activity in Sepsis
3.4. Regulation of NOS2 during Sepsis/Inflammation
3.5. Regulation and Expression of Constitutive NOS during Infection/Sepsis
3.6. ASS during Sepsis/Inflammation
4. Modulation of Enzymes
4.1. Modulation of NOS
4.2. Modulation of Arginase during Inflammatory Conditions
4.3. Modulation of ASS in Sepsis
4.4. Modulation of ASL in Sepsis
5. Enhancement of Arginine and Citrulline Availability in Sepsis and Inflammatory Conditions with Supplementation
5.1. Arginine Supplementation during Inflammatory Conditions
5.2. Citrulline Supplementation during Inflammatory Conditions
6. Modulations in the Arginine-NO Metabolism

{kind=link}
{kind=link}
| Outcome | Control | Arg1fl/fl/Tie2-Cretg/− | ||||
|---|---|---|---|---|---|---|
| Basal | LPS | LPS-Cit | Basal | LPS | LPS-Cit | |
| Arginine | 75 | ↓ | ↑/↑ | ↑ | =/↑ | ↑/=/= |
| ADMA | 0.5 | ↑ | =/= | = | ↑/= | =/=/= |
| Arginine/ADMA | 127 | ↓ | ↑/↑ | ↑ | ↓/↑ | =/↑/↑ |
| NO production | 6 | ↓ | ↑/↑ | = | ↑/↑ | =/↓/↓ |
| Microcirculation | 401 | ↓ | =/↑ | = | ↓/= | =/=/= |
7. Conclusions
Acknowledgments
Author contributions
Conflict of Interest
References
- Argaman, Z.; Young, V.R.; Noviski, N.; Castillo-Rosas, L.; Lu, X.M.; Zurakowski, D.; Cooper, M.; Davison, C.; Tharakan, J.F.; Ajami, A.; et al. Arginine and nitric oxide metabolism in critically ill septic pediatric patients. Crit. Care Med. 2003, 31, 591–597. [Google Scholar] [CrossRef]
- Luiking, Y.C.; Poeze, M.; Ramsay, G.; Deutz, N.E. Reduced citrulline production in sepsis is related to diminished de novo arginine and nitric oxide production. Am. J. Clin. Nutr. 2009, 89, 142–152. [Google Scholar] [CrossRef] [PubMed]
- Wijnands, K.A.; Vink, H.; Briede, J.J.; van Faassen, E.E.; Lamers, W.H.; Buurman, W.A.; Poeze, M. Citrulline a more suitable substrate than arginine to restore no production and the microcirculation during endotoxemia. PLoS One 2012, 7, e37439. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.S.; Anstey, N.M. Is plasma arginine concentration decreased in patients with sepsis? A systematic review and meta-analysis. Crit. Care Med. 2011, 39, 380–385. [Google Scholar] [CrossRef] [PubMed]
- Heys, S.D.; Gardner, E. Nutrients and the surgical patient: Current and potential therapeutic applications to clinical practice. J. R. Coll. Surg. Edinb. 1999, 44, 283–293. [Google Scholar] [PubMed]
- Windmueller, H.G.; Spaeth, A.E. Source and fate of circulating citrulline. Am. J. Physiol. 1981, 241, E473–E480. [Google Scholar] [PubMed]
- Yu, Y.M.; Burke, J.F.; Tompkins, R.G.; Martin, R.; Young, V.R. Quantitative aspects of interorgan relationships among arginine and citrulline metabolism. Am. J. Physiol. 1996, 271, E1098–E1109. [Google Scholar] [PubMed]
- Castillo, L.; Chapman, T.E.; Sanchez, M.; Yu, Y.M.; Burke, J.F.; Ajami, A.M.; Vogt, J.; Young, V.R. Plasma arginine and citrulline kinetics in adults given adequate and arginine-free diets. Proc. Natl. Acad. Sci. USA 1993, 90, 7749–7753. [Google Scholar] [CrossRef] [PubMed]
- Dhanakoti, S.N.; Brosnan, J.T.; Herzberg, G.R.; Brosnan, M.E. Renal arginine synthesis: Studies in vitro and in vivo. Am. J. Physiol. 1990, 259, E437–E442. [Google Scholar]
- Featherston, W.R.; Rogers, Q.R.; Freedland, R.A. Relative importance of kidney and liver in synthesis of arginine by the rat. Am. J. Physiol. 1973, 224, 127–129. [Google Scholar] [PubMed]
- Castillo, L.; Beaumier, L.; Ajami, A.M.; Young, V.R. Whole body nitric oxide synthesis in healthy men determined from [15N]arginine-to-[15N]citrulline labeling. Proc. Natl. Acad. Sci. USA 1996, 93, 11460–11465. [Google Scholar] [CrossRef]
- Grimble, G.K. Adverse gastrointestinal effects of arginine and related amino acids. J. Nutr. 2007, 137, 1693S–1701S. [Google Scholar] [PubMed]
- Morris, S.M., Jr. Regulation of enzymes of the urea cycle and arginine metabolism. Annu. Rev. Nutr. 2002, 22, 87–105. [Google Scholar] [CrossRef]
- Wu, G.; Morris, S.M., Jr. Arginine metabolism: Nitric oxide and beyond. Biochem. J. 1998, 336, 1–17. [Google Scholar]
- Thibault, R.; Flet, L.; Vavasseur, F.; Lemerle, M.; Ferchaud-Roucher, V.; Picot, D.; Darmaun, D. Oral citrulline does not affect whole body protein metabolism in healthy human volunteers: Results of a prospective, randomized, double-blind, cross-over study. Clin. Nutr. 2011, 30, 807–811. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.; Bazer, F.W.; Davis, T.A.; Kim, S.W.; Li, P.; Marc Rhoads, J.; Carey Satterfield, M.; Smith, S.B.; Spencer, T.E.; Yin, Y. Arginine metabolism and nutrition in growth, health and disease. Amino Acids 2009, 37, 153–168. [Google Scholar] [CrossRef] [PubMed]
- Cynober, L. Pharmacokinetics of arginine and related amino acids. J. Nutr. 2007, 137, 1646S–1649S. [Google Scholar] [PubMed]
- Morris, S.M., Jr. Arginine metabolism: Boundaries of our knowledge. J. Nutr. 2007, 137, 1602S–1609S. [Google Scholar]
- Morris, S.M., Jr. Enzymes of arginine metabolism. J. Nutr. 2004, 134, 2743S–2747S. [Google Scholar]
- Hammermann, R.; Bliesener, N.; Mossner, J.; Klasen, S.; Wiesinger, H.; Wessler, I.; Racke, K. Inability of rat alveolar macrophages to recycle l-citrulline to l-arginine despite induction of argininosuccinate synthetase mRNA and protein, and inhibition of nitric oxide synthesis by exogenous l-citrulline. Naunyn. Schmiedebergs Arch. Pharmacol. 1998, 358, 601–607. [Google Scholar] [CrossRef]
- Ratner, S.; Petrack, B. Biosynthesis of urea. III. Further studies on arginine synthesis from citrulline. J. Biol. Chem. 1951, 191, 693–705. [Google Scholar] [PubMed]
- Levillain, O.; Hus-Citharel, A.; Morel, F.; Bankir, L. Localization of arginine synthesis along rat nephron. Am. J. Physiol. 1990, 259, F916–F923. [Google Scholar] [PubMed]
- Wakui, H.; Komatsuda, A.; Itoh, H.; Kobayashi, R.; Nakamoto, Y.; Miura, A.B. Renal argininosuccinate synthetase: Purification, immunohistochemical localization, and elastin-binding property. Ren. Physiol. Biochem. 1992, 15, 1–9. [Google Scholar] [PubMed]
- Rochovansky, O.; Ratner, S. Biosynthesis of urea. XII. Further studies on argininosuccinate synthetase: Substrate affinity and mechanism of action. J. Biol. Chem. 1967, 242, 3839–3849. [Google Scholar] [PubMed]
- Van de Poll, M.C.; Ligthart-Melis, G.C.; Boelens, P.G.; Deutz, N.E.; van Leeuwen, P.A.; Dejong, C.H. Intestinal and hepatic metabolism of glutamine and citrulline in humans. J. Physiol. 2007, 581, 819–827. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Yanaga, K. Association between glutamine extraction and release of citrulline and glycine by the human small intestine. Life Sci. 2007, 80, 1846–1850. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.H.; Wierdsma, N.J.; Teerlink, T.; van Leeuwen, P.A.; Mulder, C.J.; van Bodegraven, A.A. The citrulline generation test: Proposal for a new enterocyte function test. Aliment. Pharmacol. Ther. 2008, 27, 1300–1310. [Google Scholar] [CrossRef]
- Wu, G.; Knabe, D.A.; Flynn, N.E. Synthesis of citrulline from glutamine in pig enterocytes. Biochem. J. 1994, 299 (Pt 1), 115–121. [Google Scholar]
- Wu, G. Synthesis of citrulline and arginine from proline in enterocytes of postnatal pigs. Am. J. Physiol. 1997, 272, G1382–G1390. [Google Scholar] [PubMed]
- Van de Poll, M.C.; Siroen, M.P.; van Leeuwen, P.A.; Soeters, P.B.; Melis, G.C.; Boelens, P.G.; Deutz, N.E.; Dejong, C.H. Interorgan amino acid exchange in humans: Consequences for arginine and citrulline metabolism. Am. J. Clin. Nutr. 2007, 85, 167–172. [Google Scholar] [PubMed]
- Ligthart-Melis, G.C.; van de Poll, M.C.; Boelens, P.G.; Dejong, C.H.; Deutz, N.E.; van Leeuwen, P.A. Glutamine is an important precursor for de novo synthesis of arginine in humans. Am. J. Clin. Nutr. 2008, 87, 1282–1289. [Google Scholar] [PubMed]
- Ligthart-Melis, G.C.; van de Poll, M.C.; Dejong, C.H.; Boelens, P.G.; Deutz, N.E.; van Leeuwen, P.A. The route of administration (enteral or parenteral) affects the conversion of isotopically labeled l-[2–15N]glutamine into citrulline and arginine in humans. JPEN J. Parenter Enteral. Nutr. 2007, 31, 343–348. [Google Scholar] [CrossRef]
- Boelens, P.G.; Melis, G.C.; van Leeuwen, P.A.; ten Have, G.A.; Deutz, N.E. Route of administration (enteral or parenteral) affects the contribution of l-glutamine to de novo l-arginine synthesis in mice: A stable-isotope study. Am. J. Physiol. Endocrinol. Metab. 2006, 291, E683–E690. [Google Scholar] [CrossRef] [PubMed]
- Boelens, P.G.; van Leeuwen, P.A.; Dejong, C.H.; Deutz, N.E. Intestinal renal metabolism of l-citrulline and l-arginine following enteral or parenteral infusion of l-alanyl-l-[2,15N]glutamine or l-[2,15N]glutamine in mice. Am. J. Physiol. Gastrointest Liver Physiol. 2005, 289, G679–G685. [Google Scholar]
- Piton, G.; Manzon, C.; Monnet, E.; Cypriani, B.; Barbot, O.; Navellou, J.C.; Carbonnel, F.; Capellier, G. Plasma citrulline kinetics and prognostic value in critically ill patients. Intensive Care Med. 2010, 36, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Jianfeng, G.; Weiming, Z.; Ning, L.; Fangnan, L.; Li, T.; Nan, L.; Jieshou, L. Serum citrulline is a simple quantitative marker for small intestinal enterocytes mass and absorption function in short bowel patients. J. Surg. Res. 2005, 127, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Crenn, P.; Coudray-Lucas, C.; Cynober, L.; Messing, B. Post-absorptive plasma citrulline concentration: A marker of intestinal failure in humans. Transplant Proc. 1998, 30, 2528. [Google Scholar] [CrossRef] [PubMed]
- Crenn, P.; Coudray-Lucas, C.; Thuillier, F.; Cynober, L.; Messing, B. Postabsorptive plasma citrulline concentration is a marker of absorptive enterocyte mass and intestinal failure in humans. Gastroenterology 2000, 119, 1496–1505. [Google Scholar] [CrossRef] [PubMed]
- Lau, T.; Owen, W.; Yu, Y.M.; Noviski, N.; Lyons, J.; Zurakowski, D.; Tsay, R.; Ajami, A.; Young, V.R.; Castillo, L. Arginine, citrulline, and nitric oxide metabolism in end-stage renal disease patients. J. Clin. Investig. 2000, 105, 1217–1225. [Google Scholar] [CrossRef] [PubMed]
- Ceballos, I.; Chauveau, P.; Guerin, V.; Bardet, J.; Parvy, P.; Kamoun, P.; Jungers, P. Early alterations of plasma free amino acids in chronic renal failure. Clin. Chim. Acta 1990, 188, 101–108. [Google Scholar] [CrossRef] [PubMed]
- Levillain, O.; Parvy, P.; Hassler, C. Amino acid handling in uremic rats: Citrulline, a reliable marker of renal insufficiency and proximal tubular dysfunction. Metabolism 1997, 46, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Marini, J.C. Arginine and ornithine are the main precursors for citrulline synthesis in mice. J. Nutr. 2012, 142, 572–580. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, C.; Rafii, M.; Ball, R.O.; Pencharz, P.B. Arginine can be synthesized from enteral proline in healthy adult humans. J. Nutr. 2011, 141, 1432–1436. [Google Scholar] [CrossRef] [PubMed]
- Marini, J.C.; Didelija, I.C.; Castillo, L.; Lee, B. Plasma arginine and ornithine are the main citrulline precursors in mice infused with arginine-free diets. J. Nutr. 2010, 140, 1432–1437. [Google Scholar] [CrossRef] [PubMed]
- Ratner, S.; Anslow, W.P., Jr.; Petrack, B. Biosynthesis of urea. VI. Enzymatic cleavage of argininosuccinic acid to arginine and fumaric acid. J. Biol. Chem. 1953, 204, 115–125. [Google Scholar] [PubMed]
- Goodwin, B.L.; Solomonson, L.P.; Eichler, D.C. Argininosuccinate synthase expression is required to maintain nitric oxide production and cell viability in aortic endothelial cells. J. Biol. Chem. 2004, 279, 18353–18360. [Google Scholar] [CrossRef] [PubMed]
- Nussler, A.K.; Billiar, T.R.; Liu, Z.Z.; Morris, S.M., Jr. Coinduction of nitric oxide synthase and argininosuccinate synthetase in a murine macrophage cell line. Implications for regulation of nitric oxide production. J. Biol. Chem. 1994, 269, 1257–1261. [Google Scholar] [PubMed]
- Erez, A.; Nagamani, S.C.; Shchelochkov, O.A.; Premkumar, M.H.; Campeau, P.M.; Chen, Y.; Garg, H.K.; Li, L.; Mian, A.; Bertin, T.K.; et al. Requirement of argininosuccinate lyase for systemic nitric oxide production. Nat. Med. 2011, 17, 1619–1626. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; George, T.J.; Prima, V.; Nelson, D.; Svetlov, S. Argininosuccinate synthase as a novel biomarker for inflammatory conditions. Biomarkers 2013, 18, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Qualls, J.E.; Subramanian, C.; Rafi, W.; Smith, A.M.; Balouzian, L.; DeFreitas, A.A.; Shirey, K.A.; Reutterer, B.; Kernbauer, E.; Stockinger, S.; et al. Sustained generation of nitric oxide and control of mycobacterial infection requires argininosuccinate synthase 1. Cell Host Microbe. 2012, 12, 313–323. [Google Scholar] [CrossRef] [PubMed]
- Wileman, S.M.; Mann, G.E.; Pearson, J.D.; Baydoun, A.R. Role of l-citrulline transport in nitric oxide synthesis in rat aortic smooth muscle cells activated with LPS and interferon-gamma. Br. J. Pharmacol. 2003, 140, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Schmidlin, A.; Fischer, S.; Wiesinger, H. Transport of l-citrulline in neural cell cultures. Dev. Neurosci. 2000, 22, 393–398. [Google Scholar] [CrossRef] [PubMed]
- Hilderman, R.H.; Casey, T.E.; Pojoga, L.H. P(1),P(4)-Diadenosine 5′-tetraphosphate modulates l-arginine and l-citrulline uptake by bovine aortic endothelial cells. Arch. Biochem. Biophys. 2000, 375, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Flam, B.R.; Eichler, D.C.; Solomonson, L.P. Endothelial nitric oxide production is tightly coupled to the citrulline-no cycle. Nitric Oxide 2007, 17, 115–121. [Google Scholar] [CrossRef] [PubMed]
- Hattori, Y.; Campbell, E.B.; Gross, S.S. Argininosuccinate synthetase mrna and activity are induced by immunostimulants in vascular smooth muscle. Role in the regeneration or arginine for nitric oxide synthesis. J. Biol. Chem. 1994, 269, 9405–9408. [Google Scholar] [PubMed]
- Schwartz, D.; Schwartz, I.F.; Gnessin, E.; Wollman, Y.; Chernichovsky, T.; Blum, M.; Iaina, A. Differential regulation of glomerular arginine transporters (CAT-1 and CAT-2) in lipopolysaccharide-treated rats. Am. J. Physiol. Renal. Physiol. 2003, 284, F788–F795. [Google Scholar] [PubMed]
- Closs, E.I.; Boissel, J.P.; Habermeier, A.; Rotmann, A. Structure and function of cationic amino acid transporters (CATs). J. Membr. Biol. 2006, 213, 67–77. [Google Scholar] [CrossRef] [PubMed]
- Fotiadis, D.; Kanai, Y.; Palacin, M. The SLC3 and SLC7 families of amino acid transporters. Mol. Aspects Med. 2013, 34, 139–158. [Google Scholar] [CrossRef] [PubMed]
- Verrey, F.; Closs, E.I.; Wagner, C.A.; Palacin, M.; Endou, H.; Kanai, Y. CATs and HATs: The SLC7 family of amino acid transporters. Pflugers Arch. 2004, 447, 532–542. [Google Scholar] [CrossRef] [PubMed]
- Cynober, L.A. Plasma amino acid levels with a note on membrane transport: Characteristics, regulation, and metabolic significance. Nutrition 2002, 18, 761–766. [Google Scholar] [CrossRef] [PubMed]
- Shen, L.J.; Beloussow, K.; Shen, W.C. Accessibility of endothelial and inducible nitric oxide synthase to the intracellular citrulline-arginine regeneration pathway. Biochem. Pharmacol. 2005, 69, 97–104. [Google Scholar] [CrossRef] [PubMed]
- McDonald, K.K.; Zharikov, S.; Block, E.R.; Kilberg, M.S. A caveolar complex between the cationic amino acid transporter 1 and endothelial nitric-oxide synthase may explain the “Arginine paradox”. J. Biol. Chem. 1997, 272, 31213–31216. [Google Scholar] [CrossRef] [PubMed]
- Yeramian, A.; Martin, L.; Serrat, N.; Arpa, L.; Soler, C.; Bertran, J.; McLeod, C.; Palacin, M.; Modolell, M.; Lloberas, J.; et al. Arginine transport via cationic amino acid transporter 2 plays a critical regulatory role in classical or alternative activation of macrophages. J. Immunol. 2006, 176, 5918–5924. [Google Scholar] [CrossRef] [PubMed]
- Rotoli, B.M.; Bussolati, O.; Sala, R.; Barilli, A.; Talarico, E.; Gazzola, G.C.; Dall’Asta, V. Infgamma stimulates arginine transport through system y+L in human monocytes. FEBS Lett. 2004, 571, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Yeramian, A.; Martin, L.; Arpa, L.; Bertran, J.; Soler, C.; McLeod, C.; Modolell, M.; Palacin, M.; Lloberas, J.; Celada, A. Macrophages require distinct arginine catabolism and transport systems for proliferation and for activation. Eur. J. Immunol. 2006, 36, 1516–1526. [Google Scholar] [CrossRef] [PubMed]
- Vadgama, J.V.; Evered, D.F. Characteristics of l-citrulline transport across rat small intestine in vitro. Pediatr. Res. 1992, 32, 472–478. [Google Scholar] [CrossRef] [PubMed]
- Bahri, S.; Curis, E.; el Wafi, F.Z.; Aussel, C.; Chaumeil, J.C.; Cynober, L.; Zerrouk, N. Mechanisms and kinetics of citrulline uptake in a model of human intestinal epithelial cells. Clin. Nutr. 2008, 27, 872–880. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Zhu, Y.; Hu, M. Mechanisms and kinetics of uptake and efflux of l-methionine in an intestinal epithelial model (Caco-2). J. Nutr. 1994, 124, 1907–1916. [Google Scholar] [PubMed]
- Pochini, L.; Scalise, M.; Galluccio, M.; Indiveri, C. Membrane transporters for the special amino acid glutamine: Structure/function relationships and relevance to human health. Front. Chem. 2014, 2, 61. [Google Scholar] [CrossRef] [PubMed]
- Nagata, N.; Endo, F.; Matsuda, I. Ornithine carbamoyltransferase (OCT) in the jejunal mucosa, as a reference of the liver oct. Clin. Chim. Acta 1983, 134, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Luiking, Y.C.; ten Have, G.A.; Wolfe, R.R.; Deutz, N.E. Arginine de novo and nitric oxide production in disease states. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E1177–E1189. [Google Scholar] [CrossRef] [PubMed]
- Morris, S.M., Jr. Recent advances in arginine metabolism: Roles and regulation of the arginases. Br. J. Pharmacol. 2009, 157, 922–930. [Google Scholar] [CrossRef] [PubMed]
- Curis, E.; Nicolis, I.; Moinard, C.; Osowska, S.; Zerrouk, N.; Benazeth, S.; Cynober, L. Almost all about citrulline in mammals. Amino Acids 2005, 29, 177–205. [Google Scholar] [CrossRef] [PubMed]
- Yu, H.; Yoo, P.K.; Aguirre, C.C.; Tsoa, R.W.; Kern, R.M.; Grody, W.W.; Cederbaum, S.D.; Iyer, R.K. Widespread expression of arginase I in mouse tissues. Biochemical and physiological implications. J. Histochem. Cytochem. 2003, 51, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Poeze, M.; Bruins, M.J.; Kessels, F.; Luiking, Y.C.; Lamers, W.H.; Deutz, N.E. Effects of l-arginine pretreatment on nitric oxide metabolism and hepatosplanchnic perfusion during porcine endotoxemia. Am. J. Clin. Nutr. 2011, 93, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Wijnands, K.A.; Hoeksema, M.A.; Meesters, D.M.; van den Akker, N.M.; Molin, D.G.; Briede, J.J.; Ghosh, M.; Kohler, S.E.; van Zandvoort, M.A.; de Winther, M.P.; et al. Arginase-1 deficiency regulates arginine concentrations and NOS2-mediated no production during endotoxemia. PLoS One 2014, 9, e86135. [Google Scholar] [CrossRef] [PubMed]
- Louis, C.A.; Mody, V.; Henry, W.L., Jr.; Reichner, J.S.; Albina, J.E. Regulation of arginase isoforms I and II by IL-4 in cultured murine peritoneal macrophages. Am. J. Physiol. 1999, 276, R237–R242. [Google Scholar] [PubMed]
- Berkowitz, D.E.; White, R.; Li, D.; Minhas, K.M.; Cernetich, A.; Kim, S.; Burke, S.; Shoukas, A.A.; Nyhan, D.; Champion, H.C.; et al. Arginase reciprocally regulates nitric oxide synthase activity and contributes to endothelial dysfunction in aging blood vessels. Circulation 2003, 108, 2000–2006. [Google Scholar] [CrossRef] [PubMed]
- Chicoine, L.G.; Paffett, M.L.; Young, T.L.; Nelin, L.D. Arginase inhibition increases nitric oxide production in bovine pulmonary arterial endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L60–L68. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Bugaj, L.J.; Oh, Y.J.; Bivalacqua, T.J.; Ryoo, S.; Soucy, K.G.; Santhanam, L.; Webb, A.; Camara, A.; Sikka, G.; et al. Arginase inhibition restores NOS coupling and reverses endothelial dysfunction and vascular stiffness in old rats. J. Appl. Physiol. 2009, 107, 1249–1257. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Meininger, C.J.; Hawker, J.R., Jr.; Haynes, T.E.; Kepka-Lenhart, D.; Mistry, S.K.; Morris, S.M., Jr.; Wu, G. Regulatory role of arginase I and II in nitric oxide, polyamine, and proline syntheses in endothelial cells. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E75–E82. [Google Scholar] [PubMed]
- Ozaki, M.; Gotoh, T.; Nagasaki, A.; Miyanaka, K.; Takeya, M.; Fujiyama, S.; Tomita, K.; Mori, M. Expression of arginase II and related enzymes in the rat small intestine and kidney. J. Biochem. 1999, 125, 586–593. [Google Scholar] [CrossRef] [PubMed]
- Mori, M. Regulation of nitric oxide synthesis and apoptosis by arginase and arginine recycling. J. Nutr. 2007, 137, 1616S–1620S. [Google Scholar] [PubMed]
- Morris, S.M., Jr.; Bhamidipati, D.; Kepka-Lenhart, D. Human type II arginase: Sequence analysis and tissue-specific expression. Gene 1997, 193, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, T.; Sonoki, T.; Nagasaki, A.; Terada, K.; Takiguchi, M.; Mori, M. Molecular cloning of cdna for nonhepatic mitochondrial arginase (arginase II) and comparison of its induction with nitric oxide synthase in a murine macrophage-like cell line. FEBS Lett. 1996, 395, 119–122. [Google Scholar] [CrossRef] [PubMed]
- Vockley, J.G.; Jenkinson, C.P.; Shukla, H.; Kern, R.M.; Grody, W.W.; Cederbaum, S.D. Cloning and characterization of the human type II arginase gene. Genomics 1996, 38, 118–123. [Google Scholar] [CrossRef] [PubMed]
- Buga, G.M.; Singh, R.; Pervin, S.; Rogers, N.E.; Schmitz, D.A.; Jenkinson, C.P.; Cederbaum, S.D.; Ignarro, L.J. Arginase activity in endothelial cells: Inhibition by NG-hydroxy-l-arginine during high-output NO production. Am. J. Physiol. 1996, 271, H1988–H1998. [Google Scholar] [PubMed]
- De Jonge, W.J.; Hallemeesch, M.M.; Kwikkers, K.L.; Ruijter, J.M.; de Gier-de Vries, C.; van Roon, M.A.; Meijer, A.J.; Marescau, B.; de Deyn, P.P.; Deutz, N.E.; et al. Overexpression of arginase I in enterocytes of transgenic mice elicits a selective arginine deficiency and affects skin, muscle, and lymphoid development. Am. J. Clin. Nutr. 2002, 76, 128–140. [Google Scholar] [PubMed]
- Mayer, B.; John, M.; Bohme, E. Purification of a Ca2+/calmodulin-dependent nitric oxide synthase from porcine cerebellum. Cofactor-role of tetrahydrobiopterin. FEBS Lett. 1990, 277, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.H.; Pollock, J.S.; Nakane, M.; Gorsky, L.D.; Forstermann, U.; Murad, F. Purification of a soluble isoform of guanylyl cyclase-activating-factor synthase. Proc. Natl. Acad. Sci. USA 1991, 88, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Pollock, J.S.; Schmidt, H.H.; Heller, M.; Murad, F. Calmodulin-dependent endothelium-derived relaxing factor/nitric oxide synthase activity is present in the particulate and cytosolic fractions of bovine aortic endothelial cells. Proc. Natl. Acad. Sci. USA 1991, 88, 1788–1792. [Google Scholar] [CrossRef] [PubMed]
- Stuehr, D.J.; Gross, S.S.; Sakuma, I.; Levi, R.; Nathan, C.F. Activated murine macrophages secrete a metabolite of arginine with the bioactivity of endothelium-derived relaxing factor and the chemical reactivity of nitric oxide. J. Exp. Med. 1989, 169, 1011–1020. [Google Scholar] [CrossRef] [PubMed]
- Knowles, R.G.; Moncada, S. Nitric oxide synthases in mammals. Biochem. J. 1994, 298, 249–258. [Google Scholar] [PubMed]
- Bredt, D.S.; Snyder, S.H. Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc. Natl. Acad. Sci. USA 1990, 87, 682–685. [Google Scholar] [CrossRef] [PubMed]
- Moncada, S.; Higgs, A. The l-arginine-nitric oxide pathway. N. Engl. J. Med. 1993, 329, 2002–2012. [Google Scholar] [CrossRef] [PubMed]
- Mulsch, A.; Bassenge, E.; Busse, R. Nitric oxide synthesis in endothelial cytosol: Evidence for a calcium-dependent and a calcium-independent mechanism. Naunyn. Schmiedebergs Arch. Pharmacol. 1989, 340, 767–770. [Google Scholar] [PubMed]
- Hibbs, J.B., Jr.; Taintor, R.R.; Vavrin, Z.; Rachlin, E.M. Nitric oxide: A cytotoxic activated macrophage effector molecule. Biochem. Biophys. Res. Commun. 1988, 157, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Hibbs, J.B., Jr.; Taintor, R.R.; Vavrin, Z. Macrophage cytotoxicity: Role for l-arginine deiminase and imino nitrogen oxidation to nitrite. Science 1987, 235, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Downen, M.; Zhao, M.L.; Lee, P.; Weidenheim, K.M.; Dickson, D.W.; Lee, S.C. Neuronal nitric oxide synthase expression in developing and adult human CNS. J. Neuropathol. Exp. Neurol. 1999, 58, 12–21. [Google Scholar] [CrossRef] [PubMed]
- Dun, N.J.; Dun, S.L.; Wu, S.Y.; Forstermann, U. Nitric oxide synthase immunoreactivity in rat superior cervical ganglia and adrenal glands. Neurosci. Lett. 1993, 158, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.H.; Gagne, G.D.; Nakane, M.; Pollock, J.S.; Miller, M.F.; Murad, F. Mapping of neural nitric oxide synthase in the rat suggests frequent co-localization with nadph diaphorase but not with soluble guanylyl cyclase, and novel paraneural functions for nitrinergic signal transduction. J. Histochem. Cytochem. 1992, 40, 1439–1456. [Google Scholar] [CrossRef] [PubMed]
- Kavdia, M.; Popel, A.S. Contribution of nNOS- and eNOS-derived NO to microvascular smooth muscle no exposure. J. Appl. Physiol. 2004, 97, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Toda, N.; Okamura, T. The pharmacology of nitric oxide in the peripheral nervous system of blood vessels. Pharmacol. Rev. 2003, 55, 271–324. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.H.; Warner, T.D.; Ishii, K.; Sheng, H.; Murad, F. Insulin secretion from pancreatic B cells caused by l-arginine-derived nitrogen oxides. Science 1992, 255, 721–723. [Google Scholar] [CrossRef] [PubMed]
- Han, G.; Ma, H.; Chintala, R.; Miyake, K.; Fulton, D.J.; Barman, S.A.; White, R.E. Nongenomic, endothelium-independent effects of estrogen on human coronary smooth muscle are mediated by type I (neuronal) NOS and PI3-kinase-Akt signaling. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H314–H321. [Google Scholar] [CrossRef] [PubMed]
- Seddon, M.D.; Chowienczyk, P.J.; Brett, S.E.; Casadei, B.; Shah, A.M. Neuronal nitric oxide synthase regulates basal microvascular tone in humans in vivo. Circulation 2008, 117, 1991–1996. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Closs, E.I.; Pollock, J.S.; Nakane, M.; Schwarz, P.; Gath, I.; Kleinert, H. Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension 1994, 23, 1121–1131. [Google Scholar] [CrossRef] [PubMed]
- Förstermann, U.; Sessa, W.C. Nitric oxide synthases: Regulation and function. Eur. Heart J. 2012, 33, 829–837, 837a–837d. [Google Scholar] [CrossRef] [PubMed]
- Bohme, G.A.; Bon, C.; Stutzmann, J.M.; Doble, A.; Blanchard, J.C. Possible involvement of nitric oxide in long-term potentiation. Eur. J. Pharmacol. 1991, 199, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Togashi, H.; Sakuma, I.; Yoshioka, M.; Kobayashi, T.; Yasuda, H.; Kitabatake, A.; Saito, H.; Gross, S.S.; Levi, R. A central nervous system action of nitric oxide in blood pressure regulation. J. Pharmacol. Exp. Ther. 1992, 262, 343–347. [Google Scholar] [PubMed]
- Bult, H.; Boeckxstaens, G.E.; Pelckmans, P.A.; Jordaens, F.H.; van Maercke, Y.M.; Herman, A.G. Nitric oxide as an inhibitory non-adrenergic non-cholinergic neurotransmitter. Nature 1990, 345, 346–347. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.R.; Marletta, M.A. Spectral and kinetic studies on the activation of soluble guanylate cyclase by nitric oxide. Biochemistry 1996, 35, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Rapoport, R.M.; Draznin, M.B.; Murad, F. Endothelium-dependent relaxation in rat aorta may be mediated through cyclic GMP-dependent protein phosphorylation. Nature 1983, 306, 174–176. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Mulsch, A.; Bohme, E.; Busse, R. Stimulation of soluble guanylate cyclase by an acetylcholine-induced endothelium-derived factor from rabbit and canine arteries. Circ. Res. 1986, 58, 531–538. [Google Scholar] [CrossRef] [PubMed]
- Shesely, E.G.; Maeda, N.; Kim, H.S.; Desai, K.M.; Krege, J.H.; Laubach, V.E.; Sherman, P.A.; Sessa, W.C.; Smithies, O. Elevated blood pressures in mice lacking endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. USA 1996, 93, 13176–13181. [Google Scholar] [CrossRef] [PubMed]
- Huang, P.L.; Huang, Z.; Mashimo, H.; Bloch, K.D.; Moskowitz, M.A.; Bevan, J.A.; Fishman, M.C. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 1995, 377, 239–242. [Google Scholar] [CrossRef] [PubMed]
- Radomski, M.W.; Palmer, R.M.; Moncada, S. The anti-aggregating properties of vascular endothelium: Interactions between prostacyclin and nitric oxide. Br. J. Pharmacol. 1987, 92, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Murohara, T.; Asahara, T.; Silver, M.; Bauters, C.; Masuda, H.; Kalka, C.; Kearney, M.; Chen, D.; Symes, J.F.; Fishman, M.C.; et al. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J. Clin. Investig. 1998, 101, 2567–2578. [Google Scholar] [CrossRef] [PubMed]
- Kietadisorn, R.; Juni, R.P.; Moens, A.L. Tackling endothelial dysfunction by modulating NOS uncoupling: New insights into its pathogenesis and therapeutic possibilities. Am. J. Physiol. Endocrinol. Metab. 2012, 302, E481–E495. [Google Scholar] [CrossRef] [PubMed]
- Chennupati, R.; Meens, M.J.; Marion, V.; Janssen, B.J.; Lamers, W.H.; de Mey, J.G.; Kohler, S.E. Endothelial arginine resynthesis contributes to the maintenance of vasomotor function in male diabetic mice. PLoS One 2014, 9, e102264. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, A.; Black, S.M.; Catravas, J.D. Endothelial nitric oxide (NO) and its pathophysiologic regulation. Vascul. Pharmacol. 2008, 49, 134–140. [Google Scholar] [CrossRef] [PubMed]
- Crabtree, M.J.; Channon, K.M. Synthesis and recycling of tetrahydrobiopterin in endothelial function and vascular disease. Nitric Oxide 2011, 25, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Stroes, E.; Hijmering, M.; van Zandvoort, M.; Wever, R.; Rabelink, T.J.; van Faassen, E.E. Origin of superoxide production by endothelial nitric oxide synthase. FEBS Lett. 1998, 438, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Vincent, J.L. Clinical sepsis and septic shock—Definition, diagnosis and management principles. Langenbecks Arch. Surg. 2008, 393, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.M.; Fink, M.P.; Marshall, J.C.; Abraham, E.; Angus, D.; Cook, D.; Cohen, J.; Opal, S.M.; Vincent, J.L.; Ramsay, G.; et al. 2001 SCCM/ESICM/ACCP/ATS/SIS international sepsis definitions conference. Crit. Care Med. 2003, 31, 1250–1256. [Google Scholar] [CrossRef] [PubMed]
- McCuskey, R.S. Hepatic and splanchnic microvascular responses to inflammation and shock. Hepatogastroenterology 1999, 46, 1464–1467. [Google Scholar] [PubMed]
- Ince, C. The microcirculation is the motor of sepsis. Crit. Care 2005, 9, S13–S19. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.; Secchi, A.; Wellmann, R.; Bach, A.; Bohrer, H.; Gebhard, M.M.; Martin, E. Effect of endotoxemia on intestinal villus microcirculation in rats. J. Surg. Res. 1996, 61, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Tadie, J.M.; Cynober, L.; Peigne, V.; Caumont-Prim, A.; Neveux, N.; Gey, A.; Guerot, E.; Diehl, J.L.; Fagon, J.Y.; Tartour, E.; et al. Arginine administration to critically ill patients with a low nitric oxide fraction in the airways: A pilot study. Intensive Care Med. 2013, 39, 1663–1665. [Google Scholar] [CrossRef] [PubMed]
- Freund, H.; Atamian, S.; Holroyde, J.; Fischer, J.E. Plasma amino acids as predictors of the severity and outcome of sepsis. Ann. Surg. 1979, 190, 571–576. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.C.; Bandi, V.; Guntupalli, K.K.; Wu, M.; Castillo, L.; Jahoor, F. Arginine, citrulline and nitric oxide metabolism in sepsis. Clin. Sci. 2009, 117, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Wijnands, K.A.P.; Poeze, M.; Luiking, Y.C.; Breedveld, P.; Dejong, C.H.C.; Ramsay, G.; Deutz, N.E.P. Reduced arginine production and arginase activity are independent of the cause of sepsis. In proceedings of the 21st ESICM Annual Congress, Lisbon, Portugal, 21–24 September 2008.
- Bansal, V.; Ochoa, J.B. Arginine availability, arginase, and the immune response. Curr. Opin. Clin. Nutr. Metab. Care 2003, 6, 223–228. [Google Scholar] [CrossRef] [PubMed]
- Calder, P.C. Immunonutrition in surgical and critically ill patients. Br. J. Nutr. 2007, 98, S133–S139. [Google Scholar] [PubMed]
- Galban, C.; Montejo, J.C.; Mesejo, A.; Marco, P.; Celaya, S.; Sanchez-Segura, J.M.; Farre, M.; Bryg, D.J. An immune-enhancing enteral diet reduces mortality rate and episodes of bacteremia in septic intensive care unit patients. Crit. Care Med. 2000, 28, 643–648. [Google Scholar] [CrossRef] [PubMed]
- Ware, L.B.; Magarik, J.A.; Wickersham, N.; Cunningham, G.; Rice, T.W.; Christman, B.W.; Wheeler, A.P.; Bernard, G.R.; Summar, M.L. Low plasma citrulline levels are associated with acute respiratory distress syndrome in patients with severe sepsis. Crit. Care 2013, 17, R10. [Google Scholar] [CrossRef] [PubMed]
- Kao, C.; Hsu, J.; Bandi, V.; Jahoor, F. Alterations in glutamine metabolism and its conversion to citrulline in sepsis. Am. J. Physiol. Endocrinol. Metab. 2013, 304, E1359–E1364. [Google Scholar] [CrossRef] [PubMed]
- Tizianello, A.; de Ferrari, G.; Garibotto, G.; Gurreri, G.; Robaudo, C. Renal metabolism of amino acids and ammonia in subjects with normal renal function and in patients with chronic renal insufficiency. J. Clin. Investig. 1980, 65, 1162–1173. [Google Scholar] [CrossRef] [PubMed]
- Durante, W.; Johnson, F.K.; Johnson, R.A. Arginase: A critical regulator of nitric oxide synthesis and vascular function. Clin. Exp. Pharmacol. Physiol. 2007, 34, 906–911. [Google Scholar] [CrossRef] [PubMed]
- Ochoa, J.B.; Udekwu, A.O.; Billiar, T.R.; Curran, R.D.; Cerra, F.B.; Simmons, R.L.; Peitzman, A.B. Nitrogen oxide levels in patients after trauma and during sepsis. Ann. Surg. 1991, 214, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Bruins, M.J.; Soeters, P.B.; Deutz, N.E. Endotoxemia affects organ protein metabolism differently during prolonged feeding in pigs. J. Nutr. 2000, 130, 3003–3013. [Google Scholar] [PubMed]
- Salimuddin; Nagasaki, A.; Gotoh, T.; Isobe, H.; Mori, M. Regulation of the genes for arginase isoforms and related enzymes in mouse macrophages by lipopolysaccharide. Am. J. Physiol. 1999, 277, E110–E117. [Google Scholar] [PubMed]
- Darcy, C.J.; Woodberry, T.; Davis, J.S.; Piera, K.A.; McNeil, Y.R.; Chen, Y.; Yeo, T.W.; Weinberg, J.B.; Anstey, N.M. Increased plasma arginase activity in human sepsis: Association with increased circulating neutrophils. Clin. Chem. Lab. Med. 2014, 52, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Luiking, Y.C.; Engelen, M.P.; Deutz, N.E. Regulation of nitric oxide production in health and disease. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 97–104. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Jimenez, J.; Salgado, A.; Mourelle, M.; Martin, M.C.; Segura, R.M.; Peracaula, R.; Moncada, S. l-arginine: Nitric oxide pathway in endotoxemia and human septic shock. Crit. Care Med. 1995, 23, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.; Asensi, V.; Montes, A.H.; Collazos, J.; Alvarez, V.; Perez-Is, L.; Carton, J.A.; Taboada, F.; Valle-Garay, E. Endothelial (NOS3 E298D) and inducible (NOS2 exon 22) nitric oxide synthase polymorphisms, as well as plasma NOX, influence sepsis development. Nitric Oxide 2014, 42, 79–86. [Google Scholar] [CrossRef] [PubMed]
- Groeneveld, A.B.; Hartemink, K.J.; de Groot, M.C.; Visser, J.; Thijs, L.G. Circulating endothelin and nitrate-nitrite relate to hemodynamic and metabolic variables in human septic shock. Shock 1999, 11, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Cauwels, A.; Bultinck, J.; de Zwaef, R.; Vandendriessche, B.; Magez, S.; Brouckaert, P. Nitric oxide production by endotoxin preparations in TLR4-deficient mice. Nitric Oxide 2014, 36, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Bultinck, J.; Sips, P.; Vakaet, L.; Brouckaert, P.; Cauwels, A. Systemic no production during (septic) shock depends on parenchymal and not on hematopoietic cells: In vivo inos expression pattern in (septic) shock. Faseb. J. 2006, 20, 2363–2365. [Google Scholar] [CrossRef] [PubMed]
- Villalpando, S.; Gopal, J.; Balasubramanyam, A.; Bandi, V.P.; Guntupalli, K.; Jahoor, F. In vivo arginine production and intravascular nitric oxide synthesis in hypotensive sepsis. Am. J. Clin. Nutr. 2006, 84, 197–203. [Google Scholar] [PubMed]
- Adrie, C.; Monchi, M.; Dinh-Xuan, A.T.; Dall’Ava-Santucci, J.; Dhainaut, J.F.; Pinsky, M.R. Exhaled and nasal nitric oxide as a marker of pneumonia in ventilated patients. Am. J. Respir. Crit. Care Med. 2001, 163, 1143–1149. [Google Scholar] [CrossRef] [PubMed]
- Luiking, Y.C.; Deutz, N.E. Isotopic investigation of nitric oxide metabolism in disease. Curr. Opin. Clin. Nutr. Metab. Care 2003, 6, 103–108. [Google Scholar] [CrossRef] [PubMed]
- Braulio, V.B.; Ten Have, G.A.; Vissers, Y.L.; Deutz, N.E. Time course of nitric oxide production after endotoxin challenge in mice. Am. J. Physiol. Endocrinol. Metab. 2004, 287, E912–E918. [Google Scholar] [CrossRef] [PubMed]
- Hallemeesch, M.M.; Janssen, B.J.; de Jonge, W.J.; Soeters, P.B.; Lamers, W.H.; Deutz, N.E. NO production by cNOS and iNOS reflects blood pressure changes in LPS-challenged mice. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E871–E875. [Google Scholar] [PubMed]
- Van Eijk, H.M.; Luiking, Y.C.; Deutz, N.E. Methods using stable isotopes to measure nitric oxide (NO) synthesis in the l-arginine/NO pathway in health and disease. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2007, 851, 172–185. [Google Scholar] [CrossRef]
- Tadie, J.M.; Trinquart, L.; Janniere-Nartey, C.; Guerot, E.; Louis, B.; Fagon, J.Y.; Diehl, J.L.; Delclaux, C. Prediction of nosocomial infection acquisition in ventilated patients by nasal nitric oxide: Proof-of-concept study. Shock 2010, 34, 217–221. [Google Scholar] [CrossRef] [PubMed]
- Deja, M.; Busch, T.; Bachmann, S.; Riskowski, K.; Campean, V.; Wiedmann, B.; Schwabe, M.; Hell, B.; Pfeilschifter, J.; Falke, K.J.; et al. Reduced nitric oxide in sinus epithelium of patients with radiologic maxillary sinusitis and sepsis. Am. J. Respir. Crit. Care Med. 2003, 168, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Lundberg, J.O.; Farkas-Szallasi, T.; Weitzberg, E.; Rinder, J.; Lidholm, J.; Anggaard, A.; Hokfelt, T.; Lundberg, J.M.; Alving, K. High nitric oxide production in human paranasal sinuses. Nat. Med. 1995, 1, 370–373. [Google Scholar] [CrossRef] [PubMed]
- Brett, S.J.; Evans, T.W. Measurement of endogenous nitric oxide in the lungs of patients with the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 1998, 157, 993–997. [Google Scholar] [CrossRef] [PubMed]
- McClintock, D.E.; Ware, L.B.; Eisner, M.D.; Wickersham, N.; Thompson, B.T.; Matthay, M.A. Higher urine nitric oxide is associated with improved outcomes in patients with acute lung injury. Am. J. Respir. Crit. Care Med. 2007, 175, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Gotoh, T. Regulation of nitric oxide production by arginine metabolic enzymes. Biochem. Biophys. Res. Commun. 2000, 275, 715–719. [Google Scholar] [CrossRef]
- Siegemund, M.; van Bommel, J.; Schwarte, L.A.; Studer, W.; Girard, T.; Marsch, S.; Radermacher, P.; Ince, C. Inducible nitric oxide synthase inhibition improves intestinal microcirculatory oxygenation and CO2 balance during endotoxemia in pigs. Intensive Care Med. 2005, 31, 985–992. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.L.; Schmiege, L.M., 3rd; Kuo, L.; Liao, J.C. Downregulation of endothelial constitutive nitric oxide synthase expression by lipopolysaccharide. Biochem. Biophys. Res. Commun. 1996, 225, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Soeters, P.B.; Hallemeesch, M.M.; Bruins, M.J.; van Eijk, H.M.; Deutz, N.E. Quantitative in vivo assessment of arginine utilization and nitric oxide production in endotoxemia. Am. J. Surg. 2002, 183, 480–488. [Google Scholar] [CrossRef] [PubMed]
- Xaus, J.; Comalada, M.; Valledor, A.F.; Lloberas, J.; Lopez-Soriano, F.; Argiles, J.M.; Bogdan, C.; Celada, A. LPS induces apoptosis in macrophages mostly through the autocrine production of TNF-alpha. Blood 2000, 95, 3823–3831. [Google Scholar] [PubMed]
- Mills, C.D. M1 and M2 macrophages: Oracles of health and disease. Crit. Rev. Immunol. 2012, 32, 463–488. [Google Scholar] [CrossRef] [PubMed]
- Reade, M.C.; Clark, M.F.; Young, J.D.; Boyd, C.A. Increased cationic amino acid flux through a newly expressed transporter in cells overproducing nitric oxide from patients with septic shock. Clin. Sci. (Lond.) 2002, 102, 645–650. [Google Scholar] [CrossRef]
- Munder, M. Arginase: An emerging key player in the mammalian immune system. Br. J. Pharmacol. 2009, 158, 638–651. [Google Scholar] [CrossRef] [PubMed]
- Munder, M.; Engelhardt, M.; Knies, D.; Medenhoff, S.; Wabnitz, G.; Luckner-Minden, C.; Feldmeyer, N.; Voss, R.H.; Kropf, P.; Muller, I.; et al. Cytotoxicity of tumor antigen specific human T cells is unimpaired by arginine depletion. PLoS One 2013, 8, e63521. [Google Scholar] [CrossRef] [PubMed]
- Rath, M.; Muller, I.; Kropf, P.; Closs, E.I.; Munder, M. Metabolism via arginase or nitric oxide synthase: Two competing arginine pathways in macrophages. Front. Immunol. 2014, 5, 532. [Google Scholar] [CrossRef] [PubMed]
- Baydoun, A.R.; Bogle, R.G.; Pearson, J.D.; Mann, G.E. Discrimination between citrulline and arginine transport in activated murine macrophages: Inefficient synthesis of no from recycling of citrulline to arginine. Br. J. Pharmacol. 1994, 112, 487–492. [Google Scholar] [CrossRef] [PubMed]
- Munder, M.; Choi, B.S.; Rogers, M.; Kropf, P. l-arginine deprivation impairs leishmania major-specific T-cell responses. Eur. J. Immunol. 2009, 39, 2161–2172. [Google Scholar] [CrossRef] [PubMed]
- Feldmeyer, N.; Wabnitz, G.; Leicht, S.; Luckner-Minden, C.; Schiller, M.; Franz, T.; Conradi, R.; Kropf, P.; Muller, I.; Ho, A.D.; et al. Arginine deficiency leads to impaired cofilin dephosphorylation in activated human T lymphocytes. Int. Immunol. 2012, 24, 303–313. [Google Scholar] [CrossRef] [PubMed]
- Modolell, M.; Choi, B.S.; Ryan, R.O.; Hancock, M.; Titus, R.G.; Abebe, T.; Hailu, A.; Muller, I.; Rogers, M.E.; Bangham, C.R.; et al. Local suppression of T cell responses by arginase-induced l-arginine depletion in nonhealing leishmaniasis. PLoS Negl. Trop. Dis. 2009, 3, e480. [Google Scholar] [CrossRef] [PubMed]
- Herbert, D.R.; Orekov, T.; Roloson, A.; Ilies, M.; Perkins, C.; OʼBrien, W.; Cederbaum, S.; Christianson, D.W.; Zimmermann, N.; Rothenberg, M.E.; et al. Arginase I suppresses IL-12/IL-23p40-driven intestinal inflammation during acute schistosomiasis. J. Immunol. 2010, 184, 6438–6446. [Google Scholar] [CrossRef] [PubMed]
- Lewis, N.D.; Asim, M.; Barry, D.P.; de Sablet, T.; Singh, K.; Piazuelo, M.B.; Gobert, A.P.; Chaturvedi, R.; Wilson, K.T. Immune evasion by helicobacter pylori is mediated by induction of macrophage arginase II. J. Immunol. 2011, 186, 3632–3641. [Google Scholar] [CrossRef] [PubMed]
- Kakuda, D.K.; Sweet, M.J.; Mac Leod, C.L.; Hume, D.A.; Markovich, D. CAT2-mediated l-arginine transport and nitric oxide production in activated macrophages. Biochem. J. 1999, 340, 549–553. [Google Scholar] [CrossRef] [PubMed]
- Nicholson, B.; Manner, C.K.; Kleeman, J.; MacLeod, C.L. Sustained nitric oxide production in macrophages requires the arginine transporter CAT2. J. Biol. Chem. 2001, 276, 15881–15885. [Google Scholar] [CrossRef] [PubMed]
- Luiking, Y.C.; Poeze, M.; Dejong, C.H.; Ramsay, G.; Deutz, N.E. Sepsis: An arginine deficiency state? Crit. Care Med. 2004, 32, 2135–2145. [Google Scholar] [CrossRef] [PubMed]
- Luiking, Y.C.; Poeze, M.; Deutz, N.E. Arginine infusion in patients with septic shock increases nitric oxide production without haemodynamic instability. Clin. Sci. (Lond.) 2015, 128, 57–67. [Google Scholar] [CrossRef]
- Crenn, P.; Neveux, N.; Chevret, S.; Jaffray, P.; Cynober, L.; Melchior, J.C.; Annane, D.; COIITSS Study Group. Plasma l-citrulline concentrations and its relationship with inflammation at the onset of septic shock: A pilot study. J. Crit. Care 2014, 29, 315.e1–315.e6. [Google Scholar] [CrossRef] [PubMed]
- Elwafi, F.; Curis, E.; Zerrouk, N.; Neveux, N.; Chaumeil, J.C.; Arnaud, P.; Cynober, L.; Moinard, C. Endotoxemia affects citrulline, arginine and glutamine bioavailability. Eur. J. Clin. Investig. 2012, 42, 282–289. [Google Scholar] [CrossRef]
- Cynober, L. Citrulline: Just a biomarker or a conditionally essential amino acid and a pharmaconutrient in critically ill patients? Crit. Care 2013, 17, 122. [Google Scholar] [CrossRef] [PubMed]
- Piton, G.; Manzon, C.; Cypriani, B.; Carbonnel, F.; Capellier, G. Acute intestinal failure in critically ill patients: Is plasma citrulline the right marker? Intensive Care Med. 2011, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Bruins, M.J.; Luiking, Y.C.; Soeters, P.B.; Akkermans, L.M.; Deutz, N.E. Effect of prolonged hyperdynamic endotoxemia on jejunal motility in fasted and enterally fed pigs. Ann. Surg. 2003, 237, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Cynober, L.; Moinard, C.; de Bandt, J.P. The 2009 ESPEN sir david cuthbertson. Citrulline: A new major signaling molecule or just another player in the pharmaconutrition game? Clin. Nutr. 2010, 29, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Moinard, C.; Cynober, L. Citrulline: A new player in the control of nitrogen homeostasis. J. Nutr. 2007, 137, 1621S–1625S. [Google Scholar] [PubMed]
- Morris, S.M., Jr.; Kepka-Lenhart, D.; Chen, L.C. Differential regulation of arginases and inducible nitric oxide synthase in murine macrophage cells. Am. J. Physiol. 1998, 275, E740–E747. [Google Scholar] [PubMed]
- Tabuchi, S.; Gotoh, T.; Miyanaka, K.; Tomita, K.; Mori, M. Regulation of genes for inducible nitric oxide synthase and urea cycle enzymes in rat liver in endotoxin shock. Biochem. Biophys. Res. Commun. 2000, 268, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Corraliza, I.M.; Soler, G.; Eichmann, K.; Modolell, M. Arginase induction by suppressors of nitric oxide synthesis (IL-4, IL-10 and PGE2) in murine bone-marrow-derived macrophages. Biochem. Biophys. Res. Commun. 1995, 206, 667–673. [Google Scholar] [CrossRef] [PubMed]
- Modolell, M.; Corraliza, I.M.; Link, F.; Soler, G.; Eichmann, K. Reciprocal regulation of the nitric oxide synthase/arginase balance in mouse bone marrow-derived macrophages by Th1 and Th2 cytokines. Eur. J. Immunol. 1995, 25, 1101–1104. [Google Scholar] [CrossRef] [PubMed]
- Hey, C.; Boucher, J.L.; Vadon-Le Goff, S.; Ketterer, G.; Wessler, I.; Racke, K. Inhibition of arginase in rat and rabbit alveolar macrophages by N omega-hydroxy-d,l-indospicine, effects on l-arginine utilization by nitric oxide synthase. Br. J. Pharmacol. 1997, 121, 395–400. [Google Scholar] [CrossRef] [PubMed]
- Hecker, M.; Nematollahi, H.; Hey, C.; Busse, R.; Racke, K. Inhibition of arginase by NG-hydroxy-l-arginine in alveolar macrophages: Implications for the utilization of l-arginine for nitric oxide synthesis. FEBS Lett. 1995, 359, 251–254. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.I.; Liao, J.C.; Kuo, L. Arginase modulates nitric oxide production in activated macrophages. Am. J. Physiol. 1998, 274, H342–H348. [Google Scholar] [PubMed]
- Bivalacqua, T.J.; Hellstrom, W.J.; Kadowitz, P.J.; Champion, H.C. Increased expression of arginase II in human diabetic corpus cavernosum: In diabetic-associated erectile dysfunction. Biochem. Biophys. Res. Commun. 2001, 283, 923–927. [Google Scholar] [CrossRef] [PubMed]
- White, A.R.; Ryoo, S.; Li, D.; Champion, H.C.; Steppan, J.; Wang, D.; Nyhan, D.; Shoukas, A.A.; Hare, J.M.; Berkowitz, D.E. Knockdown of arginase I restores NO signaling in the vasculature of old rats. Hypertension 2006, 47, 245–251. [Google Scholar] [CrossRef] [PubMed]
- Tumer, C.; Bilgin, H.M.; Obay, B.D.; Diken, H.; Atmaca, M.; Kelle, M. Effect of nitric oxide on phagocytic activity of lipopolysaccharide-induced macrophages: Possible role of exogenous l-arginine. Cell Biol. Int. 2007, 31, 565–569. [Google Scholar] [CrossRef] [PubMed]
- Rafiee, P.; Ogawa, H.; Heidemann, J.; Li, M.S.; Lamirand, T.H.; Fisher, P.J.; Graewin, S.J.; Dwinell, M.B.; Johnson, C.P.; Shaker, R.; et al. Isolation and characterization of human esophageal microvascular endothelial cells: Mechanisms of inflammatory activation. Am. J. Physiol. Gastrointest. Liver Physiol. 2003, 285, G1277–G1292. [Google Scholar] [PubMed]
- Hollenberg, S.M.; Broussard, M.; Osman, J.; Parrillo, J.E. Increased microvascular reactivity and improved mortality in septic mice lacking inducible nitric oxide synthase. Circ. Res 2000, 86, 774–778. [Google Scholar] [CrossRef] [PubMed]
- Kubes, P.; McCafferty, D.M. Nitric oxide and intestinal inflammation. Am. J. Med. 2000, 109, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Cobb, J.P.; Hotchkiss, R.S.; Swanson, P.E.; Chang, K.; Qiu, Y.; Laubach, V.E.; Karl, I.E.; Buchman, T.G. Inducible nitric oxide synthase (iNOS) gene deficiency increases the mortality of sepsis in mice. Surgery 1999, 126, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Groeneveld, P.H.; Kwappenberg, K.M.; Langermans, J.A.; Nibbering, P.H.; Curtis, L. Relation between pro- and anti-inflammatory cytokines and the production of nitric oxide (NO) in severe sepsis. Cytokine 1997, 9, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Hesse, M.; Modolell, M.; la Flamme, A.C.; Schito, M.; Fuentes, J.M.; Cheever, A.W.; Pearce, E.J.; Wynn, T.A. Differential regulation of nitric oxide synthase-2 and arginase-1 by type 1/type 2 cytokines in vivo: Granulomatous pathology is shaped by the pattern of l-arginine metabolism. J. Immunol. 2001, 167, 6533–6544. [Google Scholar] [CrossRef] [PubMed]
- Pinsky, M.R.; Vincent, J.L.; Deviere, J.; Alegre, M.; Kahn, R.J.; Dupont, E. Serum cytokine levels in human septic shock. Relation to multiple-system organ failure and mortality. Chest 1993, 103, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Bruins, M.J.; Lamers, W.H.; Meijer, A.J.; Soeters, P.B.; Deutz, N.E. In vivo measurement of nitric oxide production in porcine gut, liver and muscle during hyperdynamic endotoxaemia. Br. J. Pharmacol. 2002, 137, 1225–1236. [Google Scholar] [CrossRef] [PubMed]
- Xie, L.; Gross, S.S. Argininosuccinate synthetase overexpression in vascular smooth muscle cells potentiates immunostimulant-induced no production. J. Biol. Chem. 1997, 272, 16624–16630. [Google Scholar] [CrossRef] [PubMed]
- Enkhbaatar, P.; Murakami, K.; Traber, L.D.; Cox, R.; Parkinson, J.F.; Westphal, M.; Esechie, A.; Morita, N.; Maybauer, M.O.; Maybauer, D.M.; et al. The inhibition of inducible nitric oxide synthase in ovine sepsis model. Shock 2006, 25, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, I.; Abe, M.; Shibata, K.; Shimizu, N.; Sakata, N.; Katsuragi, T.; Tanaka, K. Evaluating the role of inducible nitric oxide synthase using a novel and selective inducible nitric oxide synthase inhibitor in septic lung injury produced by cecal ligation and puncture. Am. J. Respir. Crit. Care Med. 2000, 162, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Enkhbaatar, P.; Lange, M.; Nakano, Y.; Hamahata, A.; Jonkam, C.; Wang, J.; Jaroch, S.; Traber, L.; Herndon, D.; Traber, D. Role of neuronal nitric oxide synthase in ovine sepsis model. Shock 2009, 32, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Gocan, N.C.; Scott, J.A.; Tyml, K. Nitric oxide produced via neuronal nos may impair vasodilatation in septic rat skeletal muscle. Am. J. Physiol. Heart Circ. Physiol. 2000, 278, H1480–H1489. [Google Scholar] [PubMed]
- Ortiz, F.; Garcia, J.A.; Acuna-Castroviejo, D.; Doerrier, C.; Lopez, A.; Venegas, C.; Volt, H.; Luna-Sanchez, M.; Lopez, L.C.; Escames, G. The beneficial effects of melatonin against heart mitochondrial impairment during sepsis: Inhibition of inos and preservation of nnos. J. Pineal. Res. 2014, 56, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Miller, M.J.; Sandoval, M. Nitric oxide. III. A molecular prelude to intestinal inflammation. Am. J. Physiol. 1999, 276, G795–G799. [Google Scholar] [PubMed]
- Beck, P.L.; Xavier, R.; Wong, J.; Ezedi, I.; Mashimo, H.; Mizoguchi, A.; Mizoguchi, E.; Bhan, A.K.; Podolsky, D.K. Paradoxical roles of different nitric oxide synthase isoforms in colonic injury. Am. J. Physiol. Gastrointest Liver Physiol. 2004, 286, G137–G147. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.W.; Thaete, L.G.; Rozenfeld, R.A.; Zhu, Y.; de Plaen, I.G.; Caplan, M.S.; Hsueh, W. Tetrahydrobiopterin prevents platelet-activating factor-induced intestinal hypoperfusion and necrosis: Role of neuronal nitric oxide synthase. Crit. Care Med. 2005, 33, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, S.; Chan, C.K.; Jiang, Y.; Davidge, S.T. Neuronal nitric oxide synthase regulates endothelial inflammation. J. Leukoc. Biol. 2012, 91, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Bachetti, T.; Comini, L.; Curello, S.; Bastianon, D.; Palmieri, M.; Bresciani, G.; Callea, F.; Ferrari, R. Co-expression and modulation of neuronal and endothelial nitric oxide synthase in human endothelial cells. J. Mol. Cell Cardiol. 2004, 37, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Domenico, R. Pharmacology of nitric oxide: Molecular mechanisms and therapeutic strategies. Curr. Pharm. Des. 2004, 10, 1667–1676. [Google Scholar] [CrossRef] [PubMed]
- Forstermann, U.; Li, H. Therapeutic effect of enhancing endothelial nitric oxide synthase (eNOS) expression and preventing eNOS uncoupling. Br. J. Pharmacol. 2011, 164, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Venugopal, S.K.; Devaraj, S.; Yuhanna, I.; Shaul, P.; Jialal, I. Demonstration that C-reactive protein decreases enos expression and bioactivity in human aortic endothelial cells. Circulation 2002, 106, 1439–1441. [Google Scholar] [CrossRef] [PubMed]
- Moens, A.L.; Kietadisorn, R.; Lin, J.Y.; Kass, D. Targeting endothelial and myocardial dysfunction with tetrahydrobiopterin. J. Mol. Cell Cardiol. 2011, 51, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Ince, C.; Sinaasappel, M. Microcirculatory oxygenation and shunting in sepsis and shock. Crit. Care Med. 1999, 27, 1369–1377. [Google Scholar] [CrossRef] [PubMed]
- Derikx, J.P.; Poeze, M.; van Bijnen, A.A.; Buurman, W.A.; Heineman, E. Evidence for intestinal and liver epithelial cell injury in the early phase of sepsis. Shock 2007, 28, 544–548. [Google Scholar] [PubMed]
- Chen, K.; Inoue, M.; Wasa, M.; Fukuzawa, M.; Kamata, S.; Okada, A. Expression of endothelial constitutive nitric oxide synthase mRNA in gastrointestinal mucosa and its downregulation by endotoxin. Life Sci. 1997, 61, 1323–1329. [Google Scholar] [CrossRef] [PubMed]
- Leiper, J.; Vallance, P. Biological significance of endogenous methylarginines that inhibit nitric oxide synthases. Cardiovasc. Res. 1999, 43, 542–548. [Google Scholar] [CrossRef] [PubMed]
- Leiper, J.; Murray-Rust, J.; McDonald, N.; Vallance, P. S-nitrosylation of dimethylarginine dimethylaminohydrolase regulates enzyme activity: Further interactions between nitric oxide synthase and dimethylarginine dimethylaminohydrolase. Proc. Natl. Acad. Sci. USA 2002, 99, 13527–13532. [Google Scholar] [CrossRef] [PubMed]
- Nijveldt, R.J.; Teerlink, T.; van der Hoven, B.; Siroen, M.P.; Kuik, D.J.; Rauwerda, J.A.; van Leeuwen, P.A. Asymmetrical dimethylarginine (ADMA) in critically ill patients: High plasma ADMA concentration is an independent risk factor of ICU mortality. Clin. Nutr. 2003, 22, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Nijveldt, R.J.; Teerlink, T.; van Leeuwen, P.A. The asymmetrical dimethylarginine (ADMA)-multiple organ failure hypothesis. Clin. Nutr. 2003, 22, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Davis, J.S.; Darcy, C.J.; Yeo, T.W.; Jones, C.; McNeil, Y.R.; Stephens, D.P.; Celermajer, D.S.; Anstey, N.M. Asymmetric dimethylarginine, endothelial nitric oxide bioavailability and mortality in sepsis. PLoS One 2011, 6, e17260. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, K.; Gotoh, T.; Oyadomari, S.; Kajizono, M.; Kuniyasu, A.; Ohsawa, K.; Imai, Y.; Kohsaka, S.; Nakayama, H.; Mori, M. Co-induction of argininosuccinate synthetase, cationic amino acid transporter-2, and nitric oxide synthase in activated murine microglial cells. Brain Res. Mol. Brain Res. 2001, 90, 165–173. [Google Scholar] [CrossRef] [PubMed]
- Prima, V.; Wang, A.; Molina, G.; Wang, K.K.; Svetlov, S.I. Inhibition of LPS toxicity by hepatic argininosuccinate synthase (ASS): Novel roles for ASS in innate immune responses to bacterial infection. Int. Immunopharmacol. 2011, 11, 1180–1188. [Google Scholar] [CrossRef] [PubMed]
- Nagasaki, A.; Gotoh, T.; Takeya, M.; Yu, Y.; Takiguchi, M.; Matsuzaki, H.; Takatsuki, K.; Mori, M. Coinduction of nitric oxide synthase, argininosuccinate synthetase, and argininosuccinate lyase in lipopolysaccharide-treated rats. RNA blot, immunoblot, and immunohistochemical analyses. J. Biol. Chem. 1996, 271, 2658–2662. [Google Scholar] [CrossRef] [PubMed]
- Bryk, J.; Ochoa, J.B.; Correia, M.I.; Munera-Seeley, V.; Popovic, P.J. Effect of citrulline and glutamine on nitric oxide production in RAW 264.7 cells in an arginine-depleted environment. JPEN J. Parenter Enteral. Nutr. 2008, 32, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Gotoh, T.; Nagasaki, A.; Takiguchi, M.; Sonoki, T. Regulation of the urea cycle enzyme genes in nitric oxide synthesis. J. Inherit. Metab. Dis. 1998, 21, 59–71. [Google Scholar] [CrossRef] [PubMed]
- Wu, G.Y.; Brosnan, J.T. Macrophages can convert citrulline into arginine. Biochem. J. 1992, 281, 45–48. [Google Scholar] [PubMed]
- Satoh, M.; Iwahori, T.; Sugawara, N.; Yamazaki, M. Liver argininosuccinate synthase binds to bacterial lipopolysaccharides and lipid a and inactivates their biological activities. J. Endotoxin. Res. 2006, 12, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Satoh, M.; Ando, S.; Shinoda, T.; Yamazaki, M. Clearance of bacterial lipopolysaccharides and lipid a by the liver and the role of argininosuccinate synthase. Innate Immun. 2008, 14, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Hibbs, J.B., Jr.; Westenfelder, C.; Taintor, R.; Vavrin, Z.; Kablitz, C.; Baranowski, R.L.; Ward, J.H.; Menlove, R.L.; McMurry, M.P.; Kushner, J.P.; et al. Evidence for cytokine-inducible nitric oxide synthesis from l-arginine in patients receiving interleukin-2 therapy. J. Clin. Investig. 1992, 89, 867–877. [Google Scholar] [CrossRef] [PubMed]
- Nathan, C.; Xie, Q.W. Nitric oxide synthases: Roles, tolls, and controls. Cell 1994, 78, 915–918. [Google Scholar] [CrossRef] [PubMed]
- Kirkeboen, K.A.; Strand, O.A. The role of nitric oxide in sepsis—An overview. Acta Anaesthesiol. Scand. 1999, 43, 275–288. [Google Scholar] [CrossRef] [PubMed]
- Bersten, A.D.; Hersch, M.; Cheung, H.; Rutledge, F.S.; Sibbald, W.J. The effect of various sympathomimetics on the regional circulations in hyperdynamic sepsis. Surgery 1992, 112, 549–561. [Google Scholar] [PubMed]
- Thiemermann, C. Nitric oxide and septic shock. Gen. Pharmacol. 1997, 29, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Garvey, E.P.; Oplinger, J.A.; Furfine, E.S.; Kiff, R.J.; Laszlo, F.; Whittle, B.J.; Knowles, R.G. 1400W is a slow, tight binding, and highly selective inhibitor of inducible nitric-oxide synthase in vitro and in vivo. J. Biol. Chem. 1997, 272, 4959–4963. [Google Scholar] [CrossRef] [PubMed]
- Pittner, A.; Nalos, M.; Asfar, P.; Yang, Y.; Ince, C.; Georgieff, M.; Bruckner, U.B.; Radermacher, P.; Froba, G. Mechanisms of inducible nitric oxide synthase (iNOS) inhibition-related improvement of gut mucosal acidosis during hyperdynamic porcine endotoxemia. Intensive Care Med. 2003, 29, 312–316. [Google Scholar] [PubMed]
- Ploner, F.; Radermacher, P.; Theisen, M.; Tugtekin, I.F.; Matejovic, M.; Stehr, A.; Szabo, C.; Southan, G.J.; Georgieff, M.; Bruckner, U.B.; et al. Effects of combined selective inos inhibition and peroxynitrite blockade during endotoxemia in pigs. Shock 2001, 16, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Saetre, T.; Hovig, T.; Roger, M.; Gundersen, Y.; Aasen, A.O. Hepatocellular damage in porcine endotoxemia: Beneficial effects of selective versus non-selective nitric oxide synthase inhibition? Scand. J. Clin. Lab. Investig. 2001, 61, 503–512. [Google Scholar] [CrossRef]
- Wei, X.Q.; Charles, I.G.; Smith, A.; Ure, J.; Feng, G.J.; Huang, F.P.; Xu, D.; Muller, W.; Moncada, S.; Liew, F.Y. Altered immune responses in mice lacking inducible nitric oxide synthase. Nature 1995, 375, 408–411. [Google Scholar] [CrossRef] [PubMed]
- MacMicking, J.D.; Nathan, C.; Hom, G.; Chartrain, N.; Fletcher, D.S.; Trumbauer, M.; Stevens, K.; Xie, Q.W.; Sokol, K.; Hutchinson, N.; et al. Altered responses to bacterial infection and endotoxic shock in mice lacking inducible nitric oxide synthase. Cell 1995, 81, 641–650. [Google Scholar] [CrossRef] [PubMed]
- Southan, G.J.; Szabo, C.; Thiemermann, C. Isothioureas: Potent inhibitors of nitric oxide synthases with variable isoform selectivity. Br. J. Pharmacol. 1995, 114, 510–516. [Google Scholar] [CrossRef] [PubMed]
- Misko, T.P.; Moore, W.M.; Kasten, T.P.; Nickols, G.A.; Corbett, J.A.; Tilton, R.G.; McDaniel, M.L.; Williamson, J.R.; Currie, M.G. Selective inhibition of the inducible nitric oxide synthase by aminoguanidine. Eur. J. Pharmacol. 1993, 233, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Ruetten, H.; Thiemermann, C. Prevention of the expression of inducible nitric oxide synthase by aminoguanidine or aminoethyl-isothiourea in macrophages and in the rat. Biochem. Biophys. Res. Commun. 1996, 225, 525–530. [Google Scholar] [CrossRef] [PubMed]
- Murakami, K.; McGuire, R.; Jodoin, J.; Katahira, J.; Schmalstieg, F.C.; Traber, L.D.; Traber, D.L. Aminoguanidine did not attenuate septic changes and acute lung injury in sheep. In proceedings of the 7th World Congress for Microcirculation, Sydney, Australia, 19–22 August 2001; pp. 465–469.
- Ishikawa, K.; Calzavacca, P.; Bellomo, R.; Bailey, M.; May, C.N. Effect of selective inhibition of renal inducible nitric oxide synthase on renal blood flow and function in experimental hyperdynamic sepsis. Crit. Care Med. 2012, 40, 2368–2375. [Google Scholar] [CrossRef] [PubMed]
- Thiemermann, C.; Ruetten, H.; Wu, C.C.; Vane, J.R. The multiple organ dysfunction syndrome caused by endotoxin in the rat: Attenuation of liver dysfunction by inhibitors of nitric oxide synthase. Br. J. Pharmacol. 1995, 116, 2845–2851. [Google Scholar] [CrossRef] [PubMed]
- Bone, H.G.; Waurick, R.; van Aken, H.; Booke, M.; Prien, T.; Meyer, J. Comparison of the haemodynamic effects of nitric oxide synthase inhibition and nitric oxide scavenging in endotoxaemic sheep. Intensive Care Med. 1998, 24, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Rogiers, P.; Smail, N.; Cabral, A.; Preiser, J.C.; Peny, M.O.; Vincent, J.L. Effects of nitric oxide on blood flow distribution and O2 extraction capabilities during endotoxic shock. J. Appl. Physiol. 1997, 83, 1164–1173. [Google Scholar] [PubMed]
- Cobb, J.P.; Natanson, C.; Hoffman, W.D.; Lodato, R.F.; Banks, S.; Koev, C.A.; Solomon, M.A.; Elin, R.J.; Hosseini, J.M.; Danner, R.L. N omega-amino-l-arginine, an inhibitor of nitric oxide synthase, raises vascular resistance but increases mortality rates in awake canines challenged with endotoxin. J. Exp. Med. 1992, 176, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Avontuur, J.A.; Tutein Nolthenius, R.P.; van Bodegom, J.W.; Bruining, H.A. Prolonged inhibition of nitric oxide synthesis in severe septic shock: A clinical study. Crit. Care Med. 1998, 26, 660–667. [Google Scholar] [CrossRef] [PubMed]
- Lopez, A.; Lorente, J.A.; Steingrub, J.; Bakker, J.; McLuckie, A.; Willatts, S.; Brockway, M.; Anzueto, A.; Holzapfel, L.; Breen, D.; et al. Multiple-center, randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546c88: Effect on survival in patients with septic shock. Crit. Care Med. 2004, 32, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Scott, J.A.; Mehta, S.; Duggan, M.; Bihari, A.; McCormack, D.G. Functional inhibition of constitutive nitric oxide synthase in a rat model of sepsis. Am. J. Respir. Crit. Care Med. 2002, 165, 1426–1432. [Google Scholar] [CrossRef] [PubMed]
- Kilbourn, R.G.; Szabo, C.; Traber, D.L. Beneficial versus detrimental effects of nitric oxide synthase inhibitors in circulatory shock: Lessons learned from experimental and clinical studies. Shock 1997, 7, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Saetre, T.; Smiseth, O.A.; Scholz, T.; Carlsen, H.; Nordsletten, L.; Fahlstrom, E.; Aasen, A.O. Nitric oxide synthase inhibition reduces venous return in porcine endotoxemia. Am. J. Physiol. 1996, 271, H1325–H1332. [Google Scholar] [PubMed]
- Lange, M.; Hamahata, A.; Traber, D.L.; Nakano, Y.; Traber, L.D.; Enkhbaatar, P. Specific inhibition of nitric oxide synthases at different time points in a murine model of pulmonary sepsis. Biochem. Biophys. Res. Commun. 2011, 404, 877–881. [Google Scholar] [CrossRef] [PubMed]
- Lange, M.; Connelly, R.; Traber, D.L.; Hamahata, A.; Cox, R.A.; Nakano, Y.; Bansal, K.; Esechie, A.; von Borzyskowski, S.; Jonkam, C.; et al. Combined neuronal and inducible nitric oxide synthase inhibition in ovine acute lung injury. Crit. Care Med. 2009, 37, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Vanhoutte, P.M. Arginine and arginase: Endothelial no synthase double crossed? Circ. Res. 2008, 102, 866–868. [Google Scholar] [CrossRef] [PubMed]
- Patejunas, G.; Bradley, A.; Beaudet, A.L.; O’Brien, W.E. Generation of a mouse model for citrullinemia by targeted disruption of the argininosuccinate synthetase gene. Somat Cell Mol. Genet. 1994, 20, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Ye, X.; Whiteman, B.; Jerebtsova, M.; Batshaw, M.L. Correction of argininosuccinate synthetase (AS) deficiency in a murine model of citrullinemia with recombinant adenovirus carrying human AS cDNA. Gene Ther. 2000, 7, 1777–1782. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Leung, T.M.; Ward, S.C.; Nieto, N. Partial deletion of argininosuccinate synthase protects from pyrazole plus lipopolysaccharide-induced liver injury by decreasing nitrosative stress. Am. J. Physiol. Gastrointest Liver Physiol. 2012, 302, G287–G295. [Google Scholar] [CrossRef] [PubMed]
- Premkumar, M.H.; Sule, G.; Nagamani, S.C.; Chakkalakal, S.; Nordin, A.; Jain, M.; Ruan, M.Z.; Bertin, T.; Dawson, B.C.; Zhang, J.; et al. Argininosuccinate lyase in enterocytes protects from development of necrotizing enterocolitis. Am. J. Physiol. Gastrointest Liver Physiol. 2014, 307, G347–G354. [Google Scholar] [CrossRef] [PubMed]
- Barbul, A.; Lazarou, S.A.; Efron, D.T.; Wasserkrug, H.L.; Efron, G. Arginine enhances wound healing and lymphocyte immune responses in humans. Surgery 1990, 108, 331–336. [Google Scholar] [PubMed]
- Boger, R.H. The pharmacodynamics of l-arginine. Altern. Ther. Health Med. 2014, 20, 48–54. [Google Scholar] [PubMed]
- Vissers, Y.L.; Debats, I.B.; Luiking, Y.C.; Jalan, R.; van der Hulst, R.R.; Dejong, C.H.; Deutz, N.E. Pros and cons of l-arginine supplementation in disease. Nutr. Res. Rev. 2004, 17, 193–210. [Google Scholar] [CrossRef] [PubMed]
- Bode-Boger, S.M.; Boger, R.H.; Galland, A.; Tsikas, D.; Frolich, J.C. l-arginine-induced vasodilation in healthy humans: Pharmacokinetic-pharmacodynamic relationship. Br. J. Clin. Pharmacol. 1998, 46, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, G.; Luciani, D.; Biolo, G. Immunonutrition in septic patients: A philosophical view of the current situation. Clin. Nutr. 2007, 26, 25–29. [Google Scholar] [CrossRef] [PubMed]
- Deutz, N.E. The 2007 ESPEN Sir David Cuthbertson lecture: Amino acids between and within organs. The glutamate-glutamine-citrulline-arginine pathway. Clin. Nutr. 2008, 27, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Grover, Z.; Tubman, R.; McGuire, W. Glutamine supplementation for young infants with severe gastrointestinal disease. Cochrane Database Syst. Rev. 2007, CD005947. [Google Scholar] [CrossRef]
- Preiser, J.C.; Berre, P.J.; van Gossum, A.; Cynober, L.; Vray, B.; Carpentier, Y.; Vincent, J.L. Metabolic effects of arginine addition to the enteral feeding of critically ill patients. JPEN J. Parenter Enteral. Nutr. 2001, 25, 182–187. [Google Scholar] [CrossRef] [PubMed]
- Kieft, H.; Roos, A.N.; van Drunen, J.D.; Bindels, A.J.; Bindels, J.G.; Hofman, Z. Clinical outcome of immunonutrition in a heterogeneous intensive care population. Intensive Care Med. 2005, 31, 524–532. [Google Scholar] [CrossRef] [PubMed]
- Tsuei, B.J.; Bernard, A.C.; Barksdale, A.R.; Rockich, A.K.; Meier, C.F.; Kearney, P.A. Supplemental enteral arginine is metabolized to ornithine in injured patients. J. Surg. Res. 2005, 123, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Bertolini, G.; Iapichino, G.; Radrizzani, D.; Facchini, R.; Simini, B.; Bruzzone, P.; Zanforlin, G.; Tognoni, G. Early enteral immunonutrition in patients with severe sepsis: Results of an interim analysis of a randomized multicentre clinical trial. Intensive Care Med. 2003, 29, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.M.; Li, F.; Zhang, M.M.; Wu, X.T. Glutamine dipeptide for parenteral nutrition in abdominal surgery: A meta-analysis of randomized controlled trials. World J. Gastroenterol. 2006, 12, 7537–7541. [Google Scholar] [PubMed]
- Heyland, D.K.; Novak, F.; Drover, J.W.; Jain, M.; Su, X.; Suchner, U. Should immunonutrition become routine in critically ill patients? A systematic review of the evidence. JAMA 2001, 286, 944–953. [Google Scholar] [CrossRef] [PubMed]
- Cynober, L. Can arginine and ornithine support gut functions? Gut 1994, 35, S42–S45. [Google Scholar] [CrossRef] [PubMed]
- Stechmiller, J.K.; Treloar, D.; Allen, N. Gut dysfunction in critically ill patients: A review of the literature. Am. J. Crit. Care 1997, 6, 204–209. [Google Scholar] [PubMed]
- Heyland, D.K.; Dhaliwal, R.; Drover, J.W.; Gramlich, L.; Dodek, P. Canadian clinical practice guidelines for nutrition support in mechanically ventilated, critically ill adult patients. JPEN J. Parenter Enteral. Nutr. 2003, 27, 355–373. [Google Scholar] [CrossRef] [PubMed]
- Dhaliwal, R.; Cahill, N.; Lemieux, M.; Heyland, D.K. The canadian critical care nutrition guidelines in 2013: An update on current recommendations and implementation strategies. Nutr. Clin. Pract. 2014, 29, 29–43. [Google Scholar] [CrossRef] [PubMed]
- Bruins, M.J.; Soeters, P.B.; Lamers, W.H.; Deutz, N.E. l-arginine supplementation in pigs decreases liver protein turnover and increases hindquarter protein turnover both during and after endotoxemia. Am. J. Clin. Nutr. 2002, 75, 1031–1044. [Google Scholar] [PubMed]
- Schwedhelm, E.; Maas, R.; Freese, R.; Jung, D.; Lukacs, Z.; Jambrecina, A.; Spickler, W.; Schulze, F.; Boger, R.H. Pharmacokinetic and pharmacodynamic properties of oral l-citrulline and l-arginine: Impact on nitric oxide metabolism. Br. J. Clin. Pharmacol. 2008, 65, 51–59. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wijnands, K.A.P.; Castermans, T.M.R.; Hommen, M.P.J.; Meesters, D.M.; Poeze, M. Arginine and Citrulline and the Immune Response in Sepsis. Nutrients 2015, 7, 1426-1463. https://doi.org/10.3390/nu7031426
Wijnands KAP, Castermans TMR, Hommen MPJ, Meesters DM, Poeze M. Arginine and Citrulline and the Immune Response in Sepsis. Nutrients. 2015; 7(3):1426-1463. https://doi.org/10.3390/nu7031426
Chicago/Turabian StyleWijnands, Karolina A.P., Tessy M.R. Castermans, Merel P.J. Hommen, Dennis M. Meesters, and Martijn Poeze. 2015. "Arginine and Citrulline and the Immune Response in Sepsis" Nutrients 7, no. 3: 1426-1463. https://doi.org/10.3390/nu7031426
APA StyleWijnands, K. A. P., Castermans, T. M. R., Hommen, M. P. J., Meesters, D. M., & Poeze, M. (2015). Arginine and Citrulline and the Immune Response in Sepsis. Nutrients, 7(3), 1426-1463. https://doi.org/10.3390/nu7031426