Homocysteine, Iron and Cardiovascular Disease: A Hypothesis

Abstract
:1. Introduction
2. “Iron-CVD” Hypothesis
3. Iron-Hcy Interaction
3.1. Plasma tHcy and Anticoagulants Used for Blood Collection

{kind=link}
{kind=link}
| Researchers | Anticoagulant | Incubation (h)/temperature | Increase in tHcy from Baseline (%) |
|---|---|---|---|
| Stabler et al. (1987) [18] | None | 1/RT b | 0 |
| None | 4; 24/RT | 35; 75 | |
| Ubbink et al. (1992) [19] | NaF c | 24/25 °C; 4 °C | 20; 12 |
| EDTA | 24/25 °C; 4 °C | 188; 15 | |
| Andersson et al. (1992) [20] | EDTA | 24/22 °C | 176 |
| EDTA | 4/37 °C | 134 | |
| Willems et al. (1998) [21] | Citrate | 4/RT | 3 |
| NaF | 4/RT | 14 | |
| EDTA | 4/RT | 26 | |
| Hughes et al. (1998) [22] | EDTA | 4/25 °C | 24 |
| EDTA + NaF | 4/25 °C | 13 | |
| EDTA + LiF d | 4/25 °C | 10 | |
| Calişkan et al. (2001) [25] | Citrate | 3/0 °C; RT | 3; 14 |
| NaF | 3/0 °C; RT | −5; 12 | |
| EDTA | 3/0 °C; RT | 3; 18 | |
| Duarte et al. (2002) [26] | NaF | 1; 4/RT | 8; 24 |
| EDTA | 1; 4/RT | 9; 27 | |
| Scheidhauser et al. (2005) [27] | NaF + heparin | 144/RT | 0 |
| EDTA | 168/RT | 280 | |
| Hübner et al. (2007) [28] | Citrate | 24/RT | 0; 21 |
| EDTA | 24/4 °C; RT | 0; 85 |
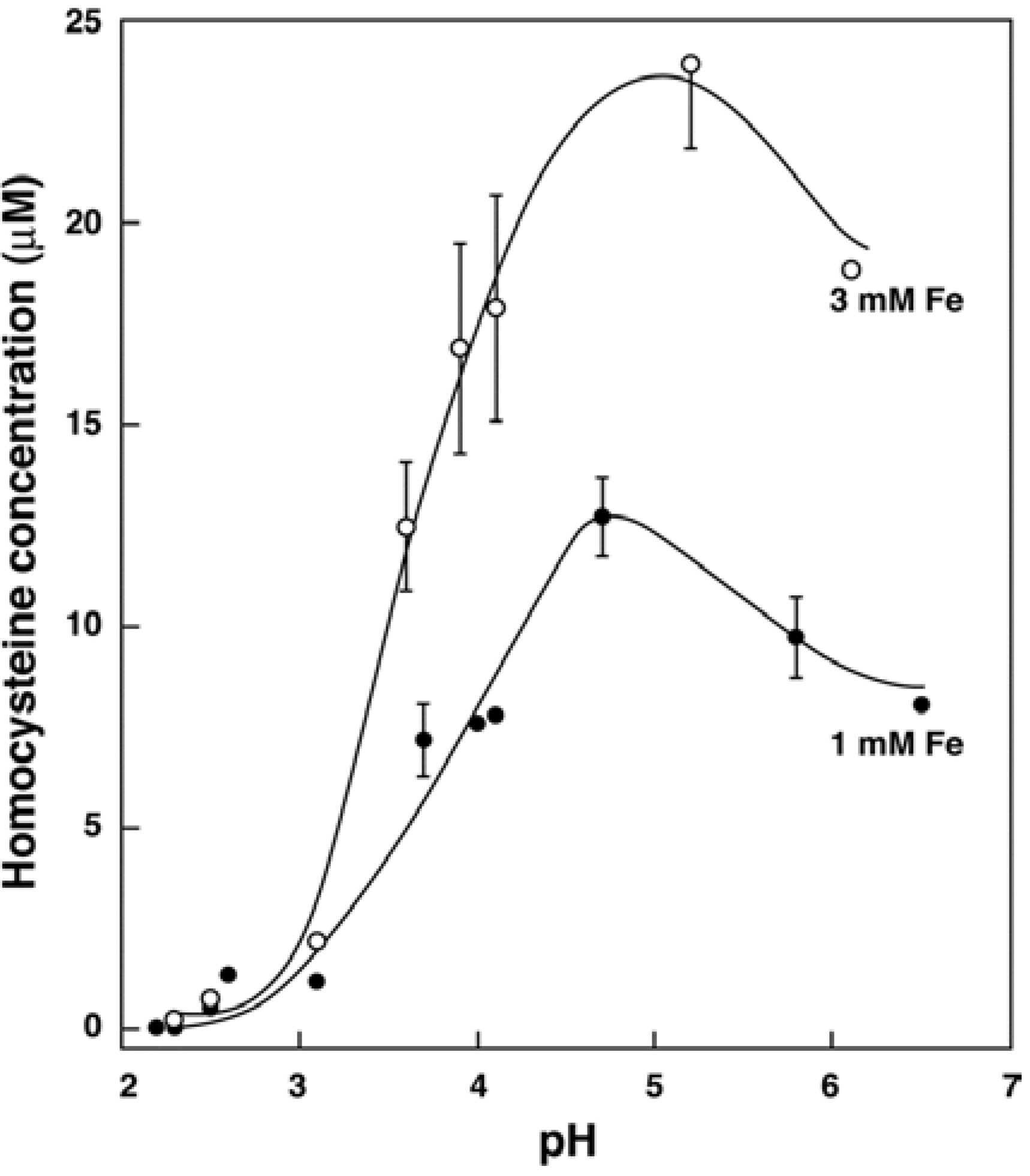
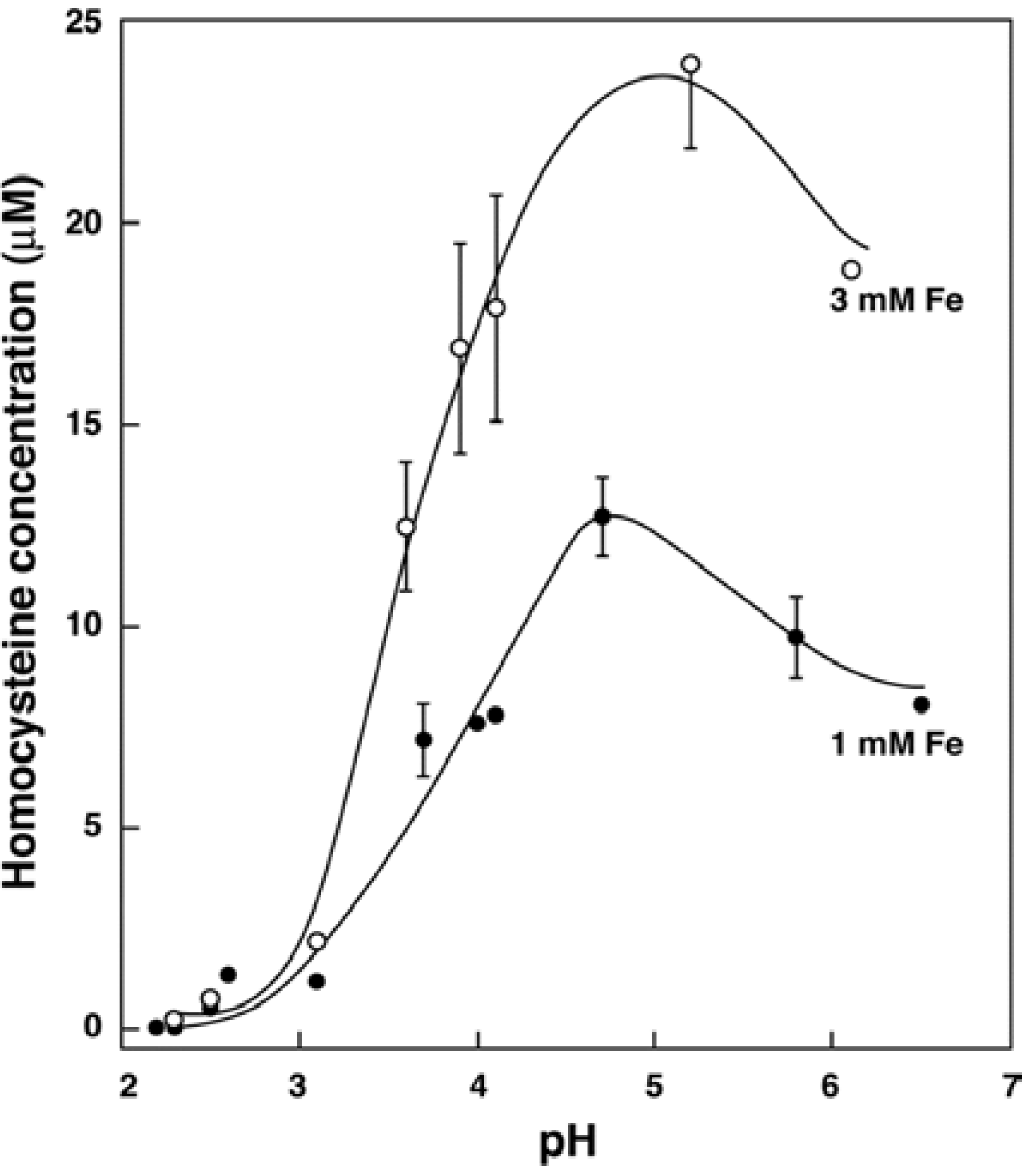
3.2. In Vitro Formation of Hcy from Met, S-adenosylhomocysteine and Cystathionine
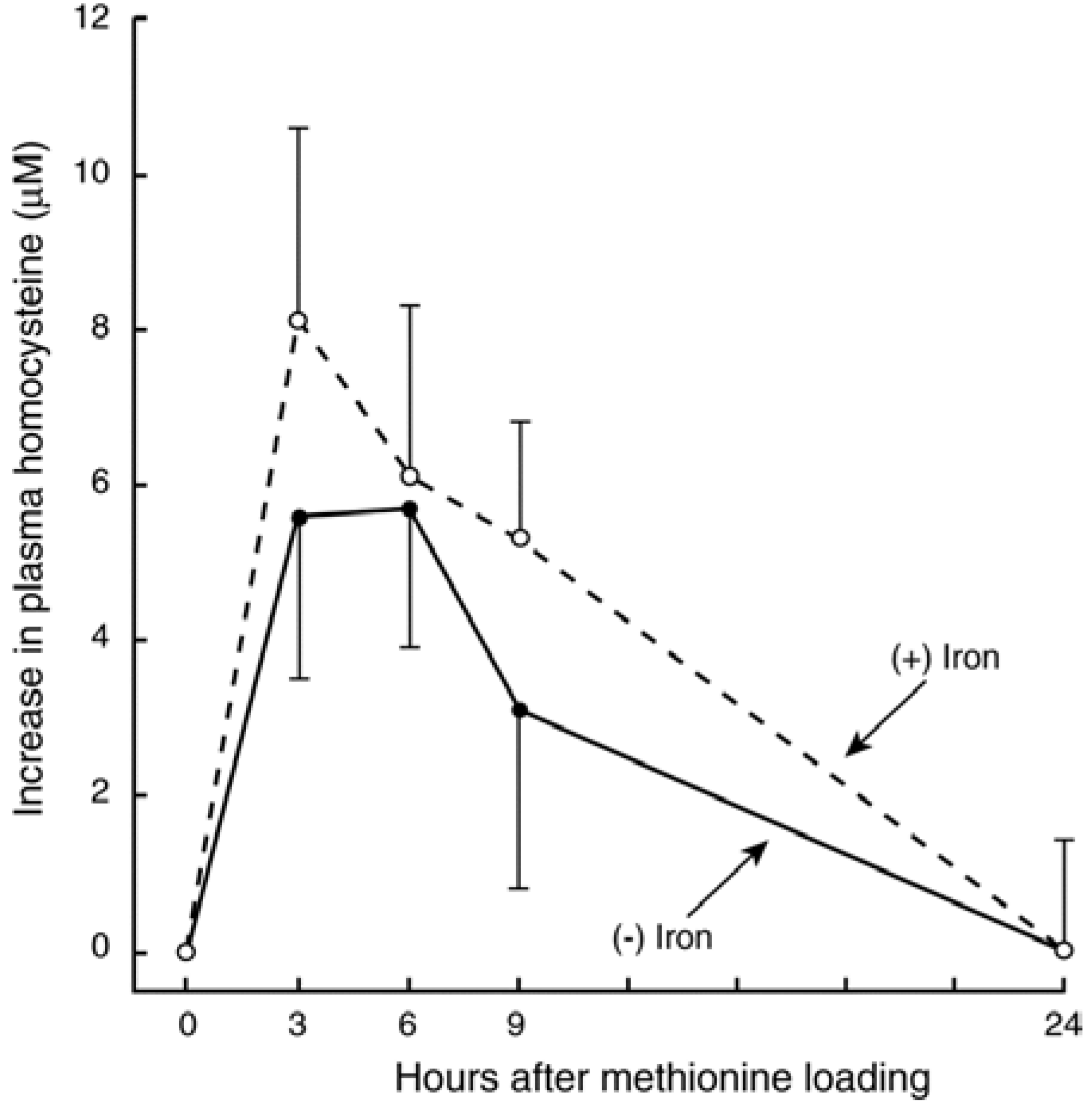
3.3. In Vivo Changes in tHcy after a Met Load with/without Iron
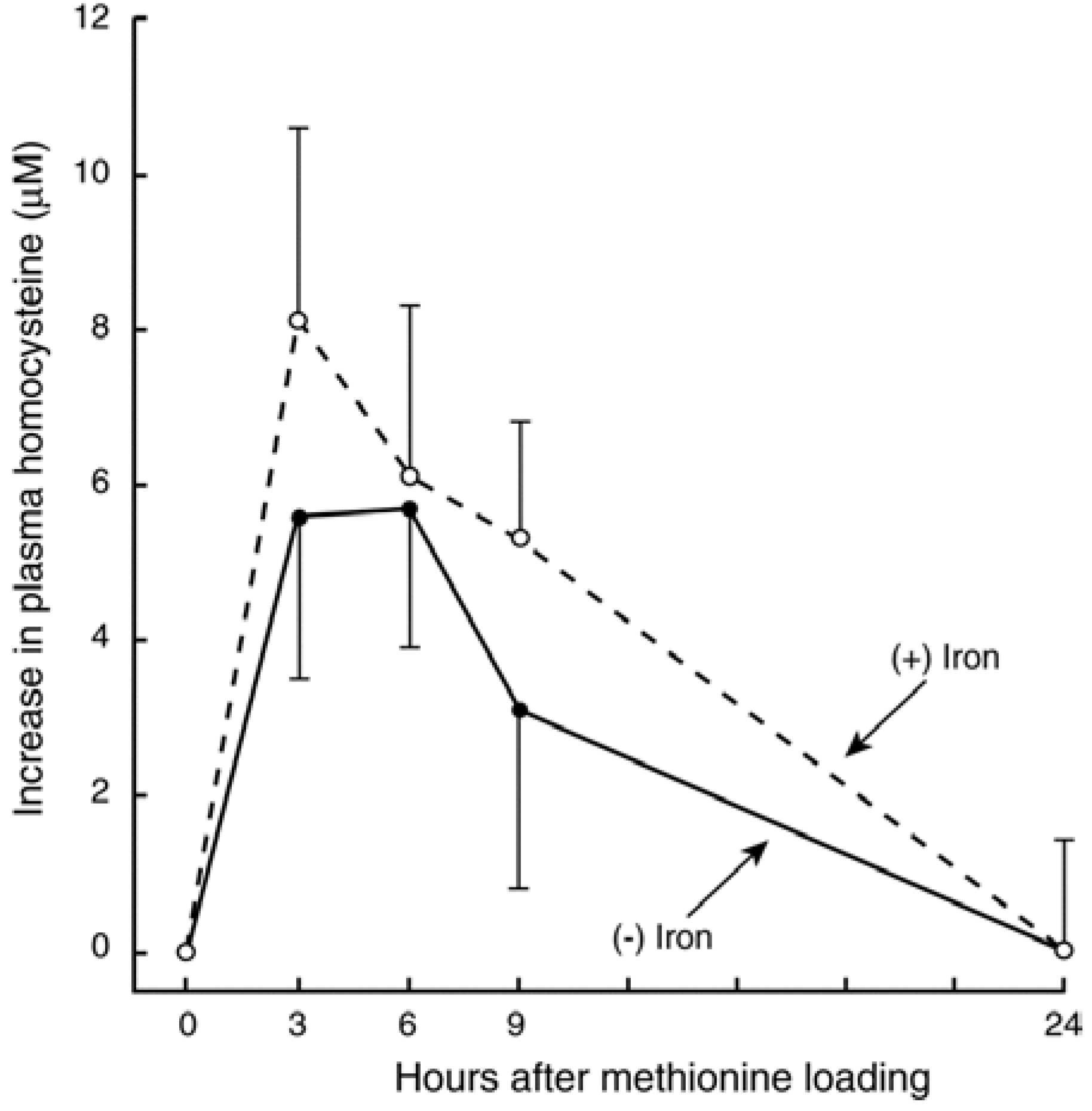
3.4. Recent Studies on Iron and Hcy in Humans
4. Conclusions
Author Contributions
Conflicts of Interest
References
- Finkelstein, J.D. Methionine metabolism in mammals. J. Nutr. Biochem. 1990, 1, 228–237. [Google Scholar] [CrossRef] [PubMed]
- McCully, K.S. Homocystinuria, arteriosclerosis, methylmalonic aciduria, and methyltransferase deficiency: A key case revisited. Nutr. Rev. 1992, 50, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Clarke, R.; Daly, L.; Robinson, K.; Naughten, E.; Cahalane, S.; Fowler, B.; Graham, I. Hyperhomocysteinemia: An independent risk factor for vascular disease. N. Engl. J. Med. 1991, 324, 1149–1155. [Google Scholar] [CrossRef] [PubMed]
- Refsum, H.; Ueland, P.M.; Nygård, O.; Vollset, S.E. Homocysteine and cardiovascular disease. Ann. Rev. Med. 1998, 49, 31–62. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.-T.; Lee, M.; Hong, K.-S.; Ovbiagele, B.; Saver, J.L. Efficacy of folic acid supplementation in cardiovascular disease prevention: An updated meta-analysis of randomized controlled trials. Eur. J. Int. Med. 2012, 23, 745–754. [Google Scholar] [CrossRef]
- Holvoet, P.; Collen, D. Oxidized lipoproteins in atheroscerosis and thrombosis. FASEB J. 1994, 8, 1279–1284. [Google Scholar] [PubMed]
- Aisen, P.; Cohen, G.; Kang, J.O. Iron toxicosis. Int. Rev. Exp. Pathol. 1990, 31, 1–46. [Google Scholar] [PubMed]
- Sullivan, J.L. Iron and the sex difference in heart disease risk. Lancet 1981, 1, 1293–1294. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Bravo, C.; Gutiérrez-Bedmar, M.; Gómez-Aracena, J.; García-Rodoríguez, A.; Navajas, J.F.-C. Iron: Protector or risk factor for cardiovascular disease? Still controversial. Nutrients 2013, 5, 2384–2404. [Google Scholar] [CrossRef] [PubMed]
- Lapice, E.; Masulli, M.; Vaccaro, O. Iron deficiency and cardiovascular disease: An updated review of the evidence. Curr. Atheroscler. Rep. 2013, 15, 358. [Google Scholar] [CrossRef] [PubMed]
- Hsu, H.-S.; Li, C.-I.; Liu, C.-S.; Lin, C.-C.; Haung, K.-C.; Li, T.-C.; Haung, H.-Y.; Lin, W.-Y. Iron deficiency is associated with increased risk for cardiovascular disease and all-cause mortality in the elderly living in long-term care facilities. Nutrition 2013, 29, 737–743. [Google Scholar] [CrossRef] [PubMed]
- Bacon, B.R.; Tavill, A.S. Role of the liver in normal iron metabolism. Sem. Liver Dis. 1984, 4, 181–192. [Google Scholar] [CrossRef]
- Lamb, D.J.; Leake, D.S. Iron released from transferrin at acidic pH can catalyse the oxidation of low density lipoprotein. FEBS Lett. 1994, 352, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Bothwell, T.H.; Charlton, R.W.; Motulsky, A.G. Hemochromatosis. In The Metabolic and Molecular Basis of Inherited Disease, 7th ed.; Scriver, C.R., Beaudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 1995; pp. 2237–2269. [Google Scholar]
- Daher, R.; van Lente, F. Relationship of increased homocysteine with copper, iron, and zinc concentrations in serum. Irish J. Med. Sci. 1995, 164 (Suppl. 15), 21. [Google Scholar]
- Zheng, H.; Huang, X.; Zhang, Q.; Katz, S.D. Iron sucrose augments homocysteine-induced endothelial dysfunction in normal subjects. Kidney Int. 2006, 69, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Facchini, F.S.; Saylor, K.L. Effect of iron depletion on cardiovascular risk factors. Studies in carbohydrate-intolerant patients. Ann. N. Y. Acad. Sci. 2002, 967, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Stabler, S.P.; Marcell, P.D.; Podell, E.R.; Allen, R.H. Quantitation of total homocysteine, total cysteine, and methionine in normal serum and urine using capillary gas chromatography-mass spectrometry. Anal. Biochem. 1987, 162, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Ubbink, J.B.; Vermaak, W.J.H.; van der Merwe, A.; Becker, P.J. The effect of blood sample aging and food consumption on plasma total homocysteine levels. Clin. Chim. Acta 1992, 207, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Andersson, A.; Isaksson, A.; Hultberg, B. Homocysteine export from erythrocytes and its implication for plasma sampling. Clin. Chem. 1992, 38, 1311–1315. [Google Scholar] [PubMed]
- Willems, H.P.J.; Bos, G.M.J.; Gerrits, W.B.J.; den Heijer, M.; Vloet, S.; Blom, H.J. Acidic citrate stabilizes blood samples for assay of total homocysteine. Clin. Chem. 1998, 44, 342–345. [Google Scholar] [PubMed]
- Hughes, M.P.; Carlson, T.H.; McLaughlin, M.K.; Bankson, D.D. Addition of sodium fluoride to whole blood does not stabilize plasma homocysteine but produces dilution effects on plasma constituents and hematocrit. Clin. Chem. 1998, 44, 2204–2206. [Google Scholar] [PubMed]
- Salazar, J.-F.; Herbeth, B.; Siest, G.; Leroy, P. Stability of blood homocysteine and other thiols: EDTA or acidic citrate? Clin. Chem. 1999, 45, 2016–2019. [Google Scholar] [PubMed]
- Palmer-Toy, D.E.; Szczepiorkowski, Z.M.; Shih, V.; van Cott, E.M. Compatibility of the Abbott IMx homocysteine assay with citrate-anticoagulated plasma and stability of homocysteine in citrate whole blood. Clin. Chem. 2001, 47, 1704–1707. [Google Scholar] [PubMed]
- Calişkan, S.; Kuralay, F.; Önvural, B. Effect of anticoagulants on plasma homocysteine determination. Clin. Chim. Acta 2001, 309, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Duarte, N.L.; Wang, X.L.; Wilken, D.E.L. Effects of anticoagulant and time of plasma separation on measurement of homocysteine. Clin. Chem. 2002, 48, 665–668. [Google Scholar] [PubMed]
- Scheidhauer, R.; Guessregen, B.; Hohl, A.; Arndt, T. Effects of prolonged ambient storage of sodium fluoride/heparin specimens on plasma homocysteine. Clin. Chem. 2005, 51, 1564–1565. [Google Scholar] [CrossRef] [PubMed]
- Hübner, U.; Schorr, H.; Eckert, R.; Geisel, J.; Herrmann, W. Stability of plasma homocysteine, S-adenosylmethionine, and S-adenosylhomocysteine in EDTA, acidic citrate, and PrimavetteTM collection tubes. Clin. Chem. 2007, 53, 2217–2218. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Baggott, J.E. In vitro formation of homocysteine in whole blood in the presence of anticoagulants. Clin. Chem. 2008, 54, 1402–1403. [Google Scholar] [CrossRef] [PubMed]
- Martín, J.; Gilbert, M.J.; Vila, M.; Pintos, C.; Obrador, A.; Malo, O. Stabilization of blood homocysteine in an epidemiological setting. Eur. J. Cancer Prev. 2001, 10, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Hill, D.M.; Johnson, L.J.; Burns, P.J.; Neale, A.M.; Harmening, D.M.; Kenney, A.C. Effects of temperature on stability of blood homocysteine in collection tubes containing 3-deazaadenosine. Clin. Chem. 2002, 48, 2017–2022. [Google Scholar] [PubMed]
- Baggott, J.E.; Tamura, T. Iron-dependent formation of homocysteine from methionine and other thioethers. Eur. J. Clin. Nutr. 2007, 61, 1359–1363. [Google Scholar] [CrossRef] [PubMed]
- Nalini, S.; Ramakrishna, B.S.; Mohanty, A.; Balasubramanian, K.A. Hydroxyl radical formation in human gastric juice. J. Gastroenterol. Hepatol. 1992, 7, 497–501. [Google Scholar] [CrossRef] [PubMed]
- Nakivell, B.J.; Tay, Y.C.; Boadle, R.A.; Harris, D.C.H. Lysosomal iron accumulation in diabetic nephropathy. Ren. Fail. 1994, 16, 367–381. [Google Scholar] [CrossRef] [PubMed]
- Mattioli, A.V.; Benetti, L.; Zennaro, M.; Bertoncelli, P.; Mattioli, G. Acute myocardial infarction in young patients: Nutritional status and biochemical factors. Int. J. Cardiol. 2005, 101, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Schiepers, O.J.G.; van Boxtel, M.P.J.; de Groot, R.H.M.; Jolles, J.; de Kort, W.L.A.M.; Swinkels, D.W.; Kok, F.J.; Verhoef, P.; Durga, J. Serum iron parameters, HFEC282Y genotype, and cognitive performance in older adults: Results from the FACIT study. J. Gerontol. A Biol. Sci. Med. Sci. 2010, 65, 1312–1321. [Google Scholar] [CrossRef] [PubMed]
- Baggott, J.E.; Tamura, T. Serum iron parameters and plasma total homocysteine concentrations. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66A, 656. [Google Scholar] [CrossRef] [PubMed]
- Schiepers, O.J.G.; Durga, J. Response to Baggott and Tamura: “Serum iron parameters and plasma total homocysteine concentrations”. J. Gerontol. A Biol. Sci. Med. Sci. 2011, 66A, 657–658. [Google Scholar] [CrossRef]
- Lioupis, C.; Barbatis, C.; Drougou, A.; Koliaraki, V.; Mamalaki, A.; Klonaris, C.; Georgopoulos, S.; Andrikopoulos, V.; Bastounis, E. Association of heptoglobin genotype and common cardiovascular risk factors with the amount of iron in atherosclerotic carotid plaques. Atherosclerosis 2011, 216, 131–138. [Google Scholar] [CrossRef] [PubMed]
- Peña-Duque, M.A.; Baños-González, M.A.; Valente-Acosta, B.; Rogoríguez-Lobato, L.G.; Martínes-Ríos, M.A.; Cardoso-Saldaña, G.; Barragán-García, R.; Herrera-Alarcón, V.; Linares-López, C.; Delgado-Granados, H.; et al. Homocysteine is related to aortic mineralization in patients with ischemic heart disease. J. Atheroscl. Thrombo. 2012, 19, 292–297. [Google Scholar] [CrossRef]
- Pinková, K.; Chrastinová, L.; Suttnar, J.; Štikanová, J.; Kotlín, R.; Čermák, J.; Dyr, J.E. Plasma levels of aminothiols, nitrite, nitrate, and malondialdehyde in myelodysplastic syndromes in the context of clinical outcomes and as a consequence of iron overload. Oxid. Med. Cell. Long. 2014, 2014. [Google Scholar] [CrossRef]
- Greenough, M.A.; Camakaris, J.; Bush, A.I. Metal dyshomeostasis and oxidative stress in Alzheimer’s disease. Neurochem. Int. 2013, 62, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Brattstrӧm, L.; Wilcken, D.E.L. Homocysteine and cardiovascular disease: Cause or effect? Am. J. Clin. Nutr. 2000, 72, 315–323. [Google Scholar] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baggott, J.E.; Tamura, T. Homocysteine, Iron and Cardiovascular Disease: A Hypothesis. Nutrients 2015, 7, 1108-1118. https://doi.org/10.3390/nu7021108
Baggott JE, Tamura T. Homocysteine, Iron and Cardiovascular Disease: A Hypothesis. Nutrients. 2015; 7(2):1108-1118. https://doi.org/10.3390/nu7021108
Chicago/Turabian StyleBaggott, Joseph E., and Tsunenobu Tamura. 2015. "Homocysteine, Iron and Cardiovascular Disease: A Hypothesis" Nutrients 7, no. 2: 1108-1118. https://doi.org/10.3390/nu7021108
APA StyleBaggott, J. E., & Tamura, T. (2015). Homocysteine, Iron and Cardiovascular Disease: A Hypothesis. Nutrients, 7(2), 1108-1118. https://doi.org/10.3390/nu7021108