Metabogenic and Nutriceutical Approaches to Address Energy Dysregulation and Skeletal Muscle Wasting in Duchenne Muscular Dystrophy

 ,
,  ,
,  and
and
Abstract
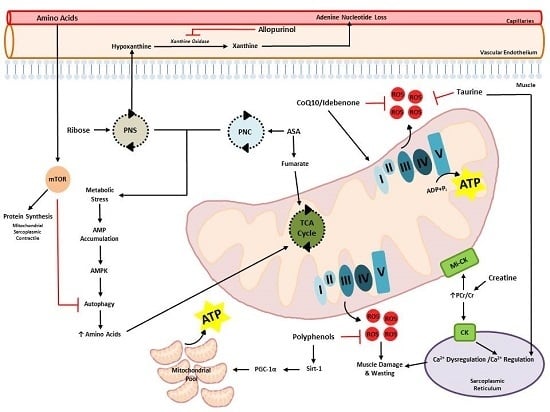
:
1. Introduction
2. Dysregulation of Energy and Protein Metabolism in DMD
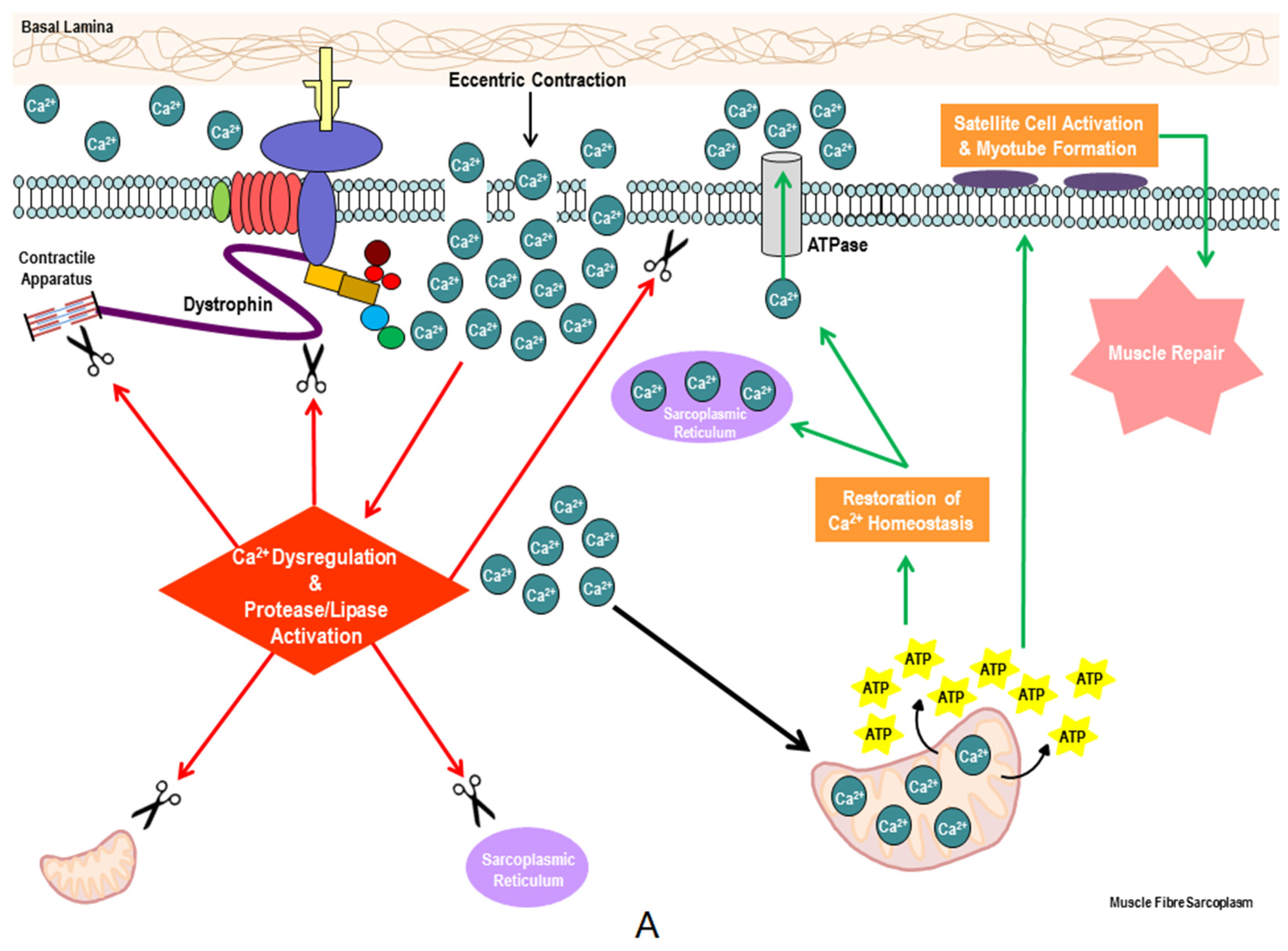
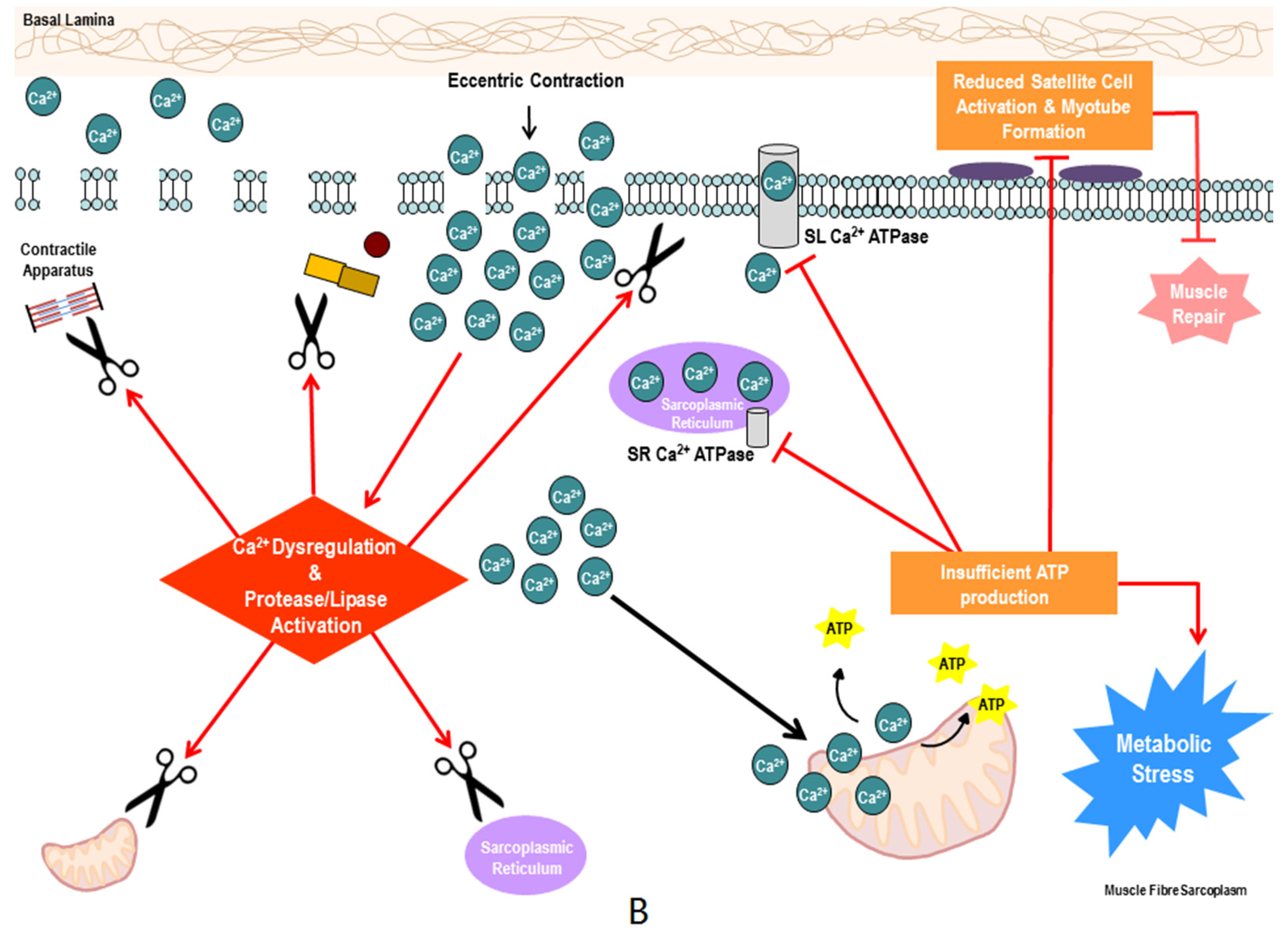
3. Therapeutic Potential of Metabogenic and Nutriceutical Supplements
3.1. Amino Acids and Protein Isolates
3.1.1. Creatine
3.1.2. Glutamine
3.1.3. Taurine
3.1.4. Arginine
3.1.5. Whey Protein Isolates
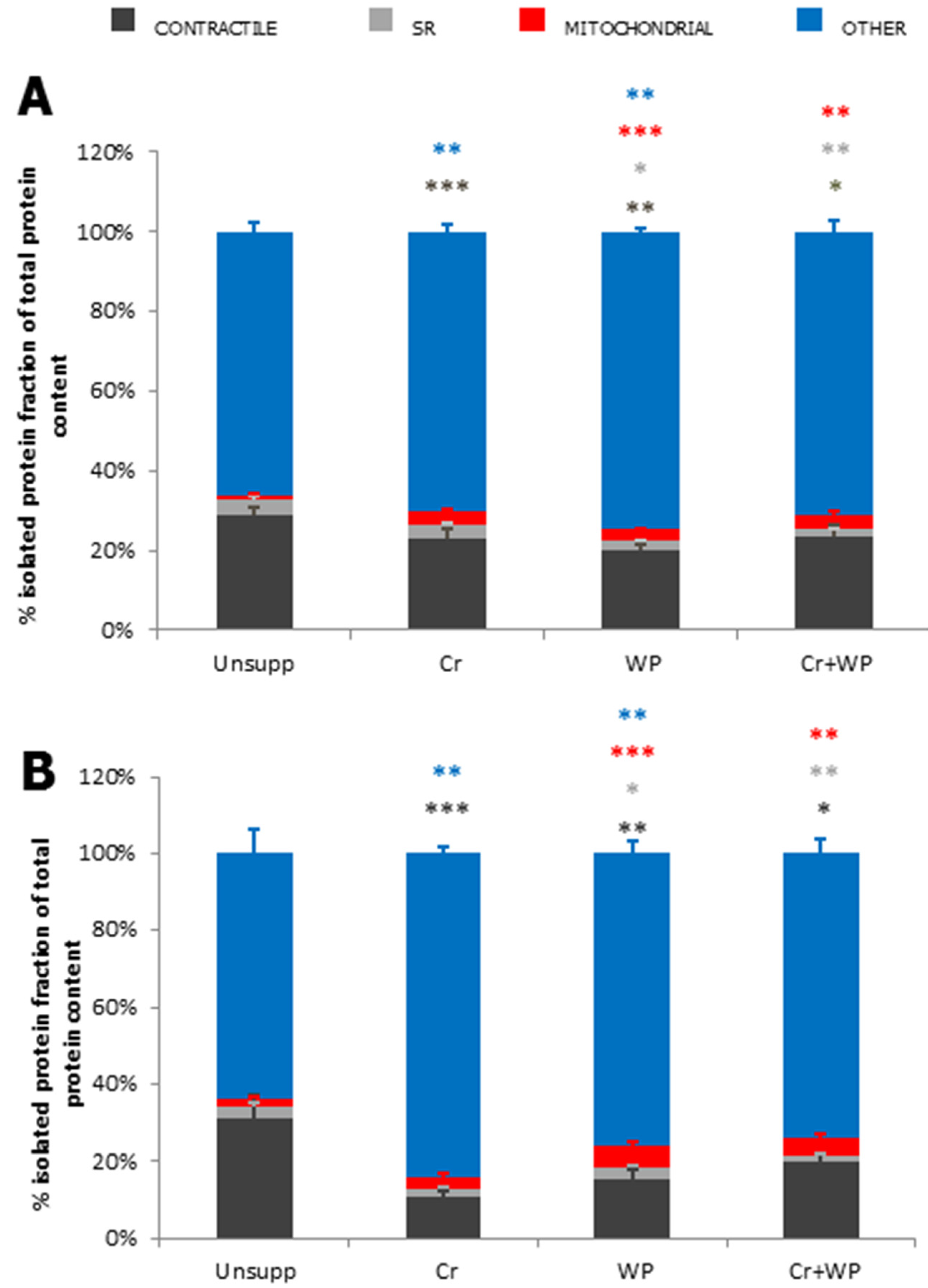
3.2. Mitochondrial Co-Factors and Modulators
3.2.1. Coenzyme Q10 (CoQ10)
3.2.2. Polyphenolic Compounds
3.2.3. Resveratrol
3.2.4. Quercetin
3.2.5. Epigallocatechin Gallate (EGCG)
3.3. Adenine Nucleotide Salvage and de Novo Synthesis Promoters
3.3.1. Adenylosuccinic Acid
3.3.2. Ribose
3.3.3. Allopurinol
4. Summary and Recommendations
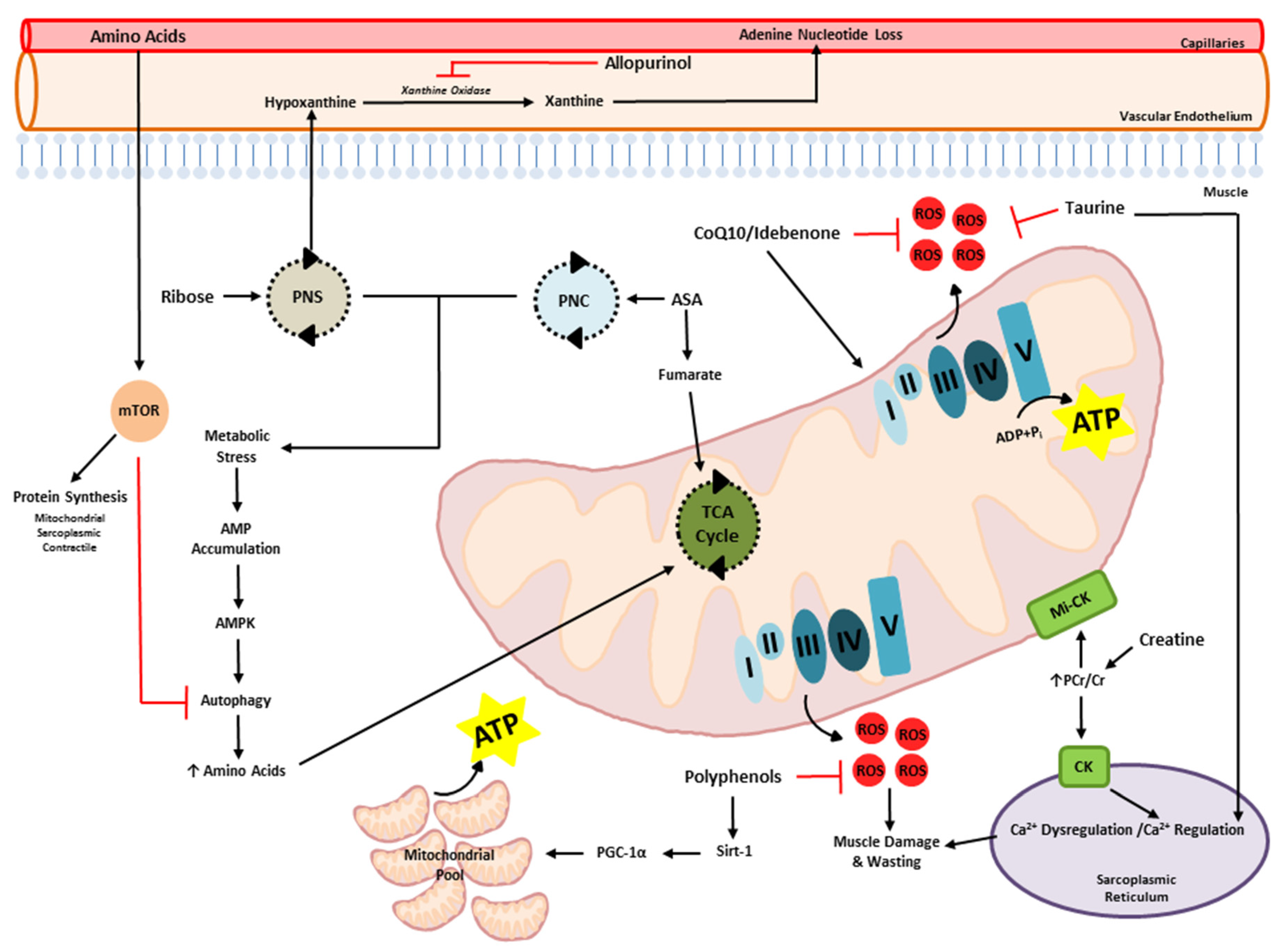

{kind=link}
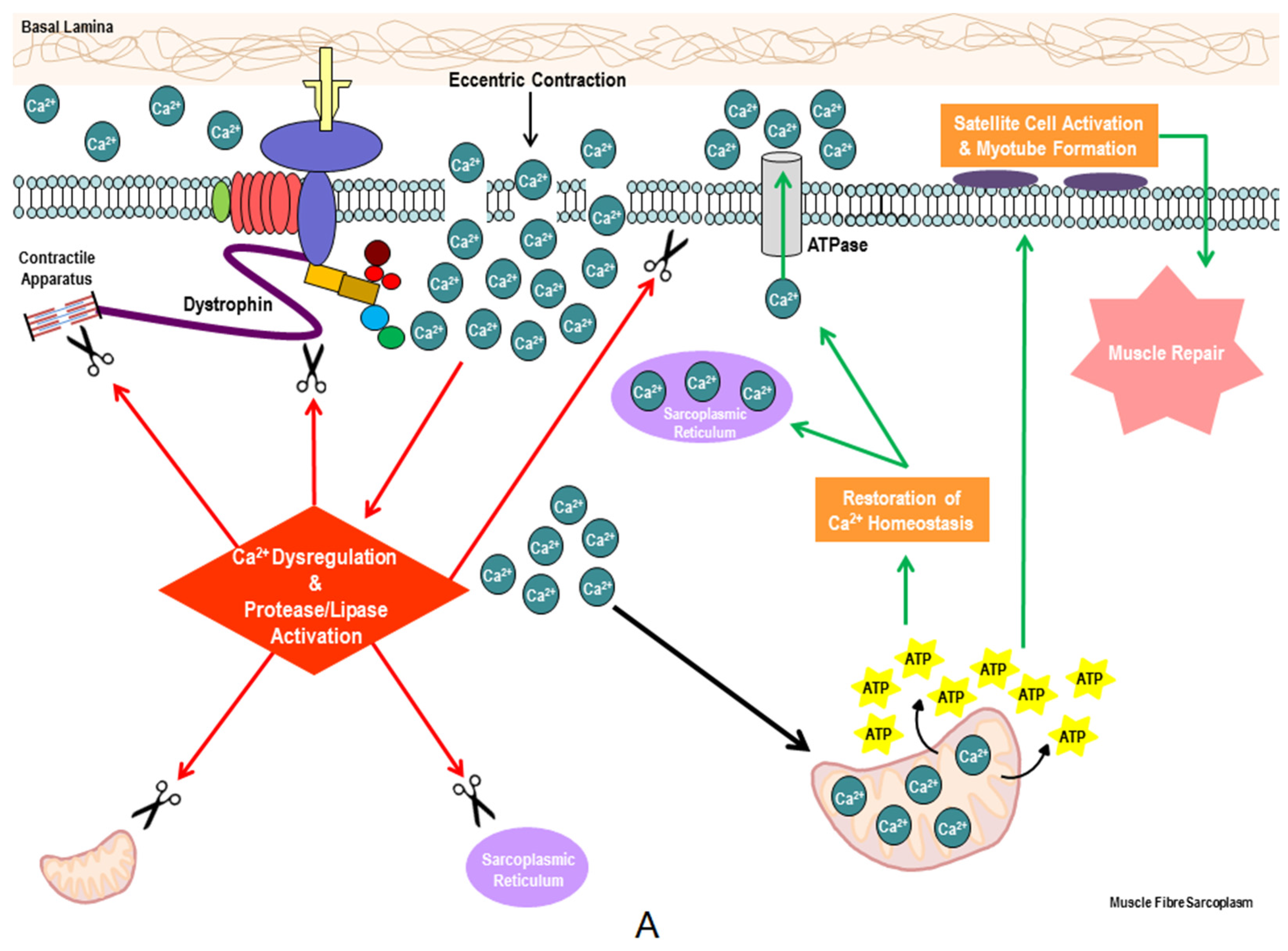{kind=link}
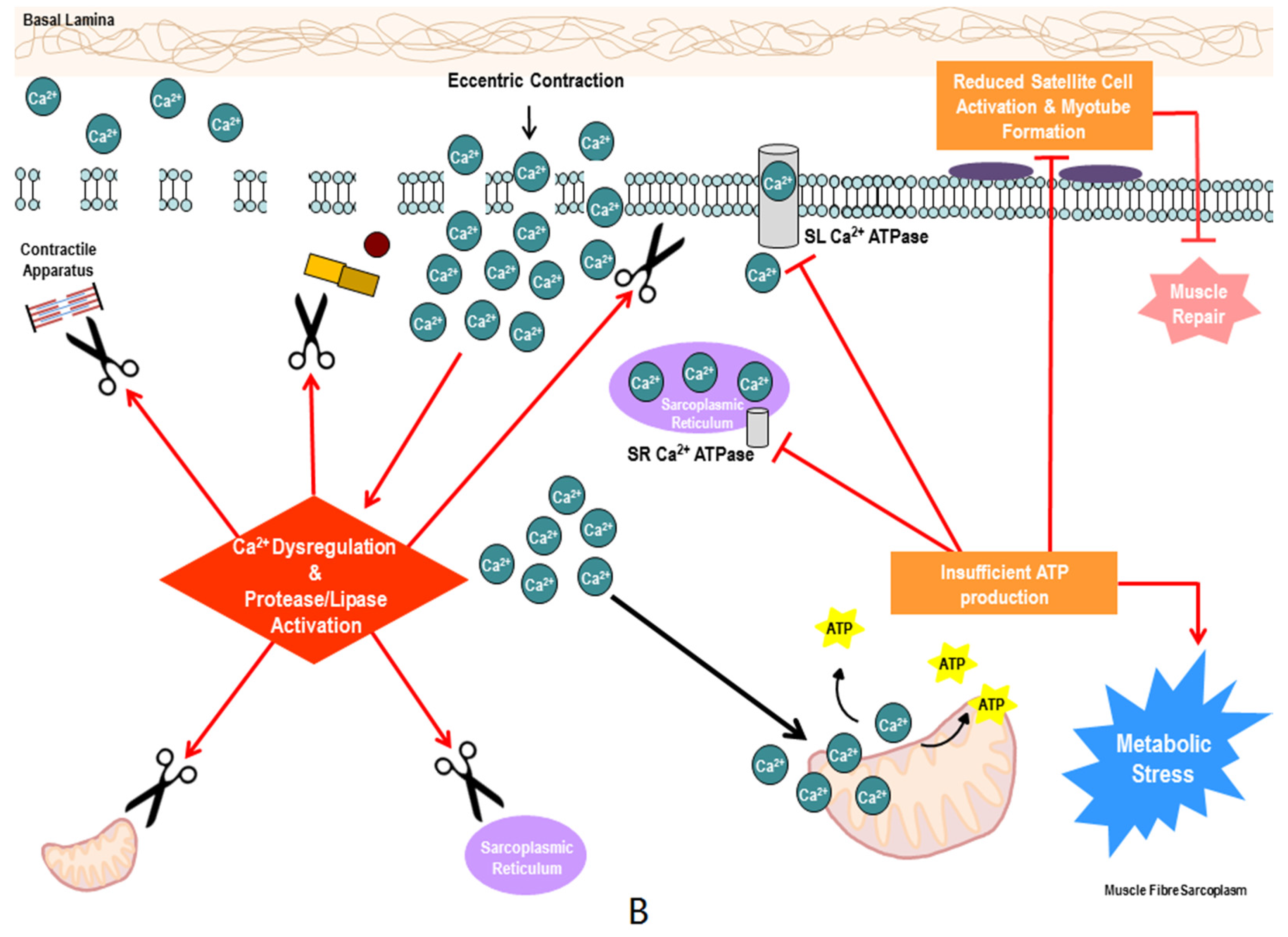{kind=link}
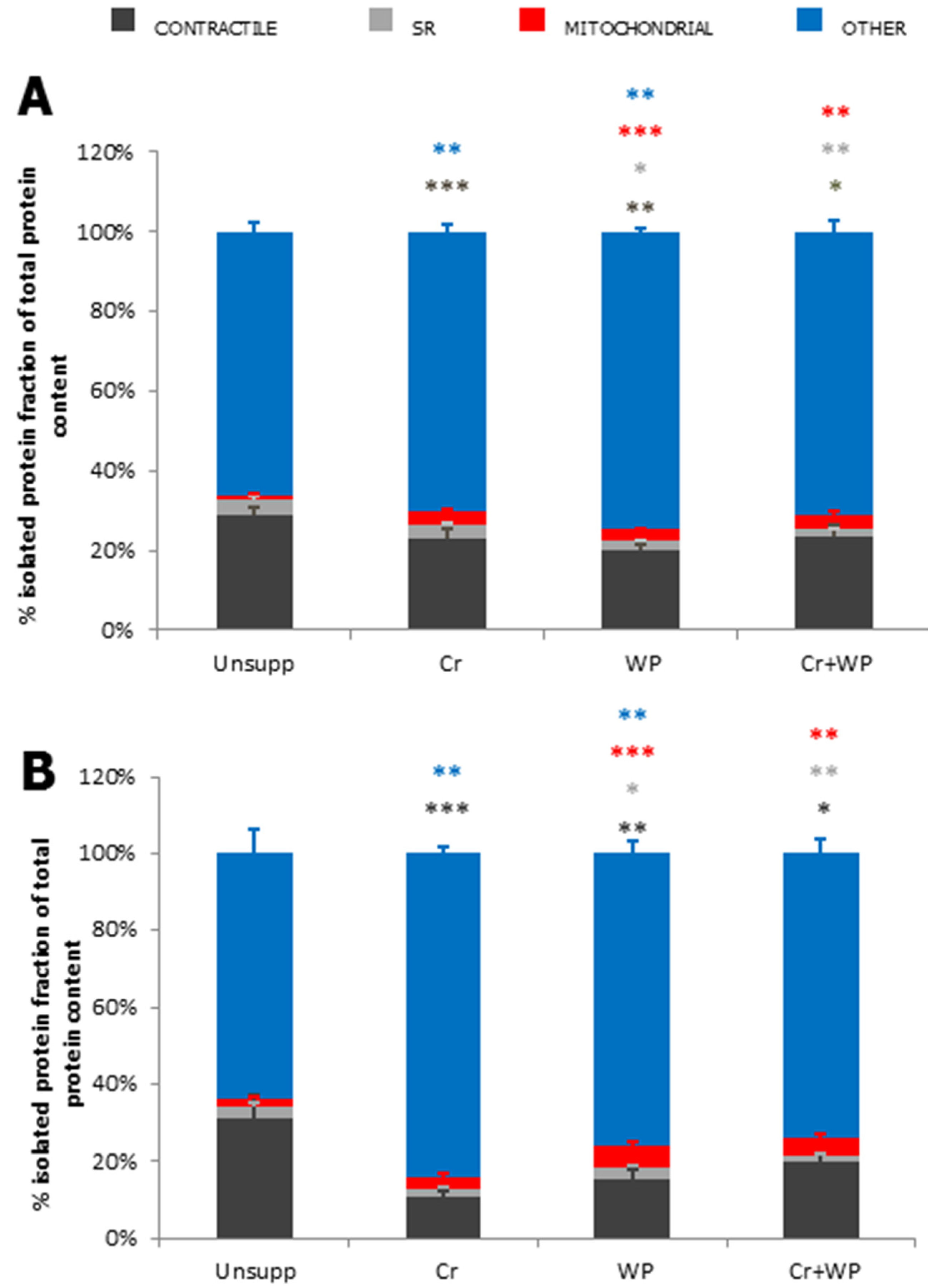{kind=link}
{kind=link}
| Category | Compound | Dosage Range | Summary of Beneficial Effects | Other |
|---|---|---|---|---|
| Apparently effective/safe to consume (in human DMD patients) | Creatine | 3–5 g·day−1 | Improved strength, maintenance of strength, increased lean muscle mass, attenuating exercise-induced fatigue, increased intramuscular energy stores | |
| CoenzymeQ10/Idebenone | >400 mg·day−1 | Decreased plasma CK levels, Improved respiratory functional measures | In conjunction with corticosteroid therapy: improved muscle strength, | |
| Adenylosuccinic acid | 25–600 mg·kg−1·day−1 | Increased energy, endurance and stamina, maintenance of muscle strength and function, reduced serum CK, reduced muscle necrosis and enhanced regeneration | Importantly ASA therapy was administered during preclinical DMD (2.5 years) and was well tolerated for 10 years. | |
| Possibly effective | Glutamine | 0.5–0.8 g·kg−1·day−1 | Increased muscle protein synthesis rates following short-term treatment | No effect on functional assessment tests, body composition/lean muscle mass, muscle protein breakdown following long-term treatment |
| Resveratrol | 100 mg·kg−1·day−1 in drinking water (mice) | Possibly, increased mitochondrial biogenesis, decreased inflammation, small reductions in oxidative stress and muscle fibre damage | Over- or under-dosing limits efficacy | |
| Quercetin | 0.2% of diet (mice) | Possibly, reduced muscle degeneration, inflammation and fibrosis, attenuation of cardiomyopathy | ||
| Epigallocatechin gallate | Current clinical trial is establishing efficacy and safety at 10 mg·kg−1·day−1; Animal data suggests 180 mg·kg−1·day−1 equivalent human dose induces best benefits, albeit safety not established at this concentration | Possibly: Reduced serum CK levels, protection against muscle degeneration and fibrosis in fast-twitch muscle, reduced oxidative stress, reduced inflammation, increased force production and fatigue resistance | ||
| Too early to tell/unclear | Taurine | Not established | Possible benefits include reduced oxidative stress/ROS damage, increased muscle contraction force and strength | |
| l-arginine | Possibly, induction of slow fibre type transitions & utrophin expression (protective against damage) | |||
| Whey protein isolate | Not established | Possibly, induction of mitochondrial biogenesis | Currently on AUS clinical trial registry for efficacy evaluation with and without co-creatine supplementation | |
| Allopurinol | 10 mg·kg−1·day−1 | Improved skeletal muscle energy status, statbilisation or improvement of muscle strength | Several other trials have found no effect, Allopurinol might be most efficacious when combined with other metabogenic compounds | |
| Not effective/not safe | Ribose | 500 mg·day−1 | None observed | In other metabolic diseases low dose therapy (500 mg·day−1) is ineffective, but efficacy is observed at a dosage of 8 g·day−1 |
5. Conclusions
Author Contributions
Conflicts of Interest
References
- Emery, A. Population frequencies of inherited neuromuscular diseases—A world survey. Neuromuscul. Disord. 1991, 1, 19. [Google Scholar] [CrossRef]
- Menke, A.; Jockusch, H. Decreased osmotic stability of dystrophin-less muscle cells from the mdx mouse. Nature 1991, 349, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, C.; Wong, S.; Elson, E.L. Mechanical function of dystrophin in muscle cells. J. Cell Biol. 1995, 128, 355–361. [Google Scholar] [CrossRef] [PubMed]
- Petrof, B.J.; Shrager, J.B.; Stedman, H.H.; Kelly, A.M.; Sweeney, H.L. Dystrophin protects the sarcolemma from stresses developed during muscle contraction. Proc. Natl. Acad. Sci. USA 1993, 90, 3710–3714. [Google Scholar] [CrossRef] [PubMed]
- Imbert, N.; Cognard, C.; Duport, G.; Guillou, C.; Raymond, G. Abnormal calcium homeostasis in duchenne muscular dystrophy myotubes contracting in vitro. Cell Calcium 1995, 18, 177–186. [Google Scholar] [CrossRef]
- Fraysse, B.; Liantonio, A.; Cetrone, M.; Burdi, R.; Pierno, S.; Frigeri, A.; Pisoni, M.; Camerino, C.; de Luca, A. The alteration of calcium homeostasis in adult dystrophic mdx muscle fibers is worsened by a chronic exercise in vivo. Neurobiol. Dis. 2004, 17, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Turner, P.R.; Fong, P.; Denetclaw, W.F.; Steinhardt, R.A. Increased calcium influx in dystrophic muscle. J. Cell Biol. 1991, 115, 1701–1712. [Google Scholar] [CrossRef] [PubMed]
- Cole, M.; Rafael, J.; Taylor, D.; Lodi, R.; Davies, K.; Styles, P. A quantitative study of bioenergetics in skeletal muscle lacking utrophin and dystrophin. Neuromuscul. Disord. 2002, 12, 247–257. [Google Scholar] [CrossRef]
- Passaquin, A.C.; Renard, M.; Kay, L.; Challet, C.; Mokhtarian, A.; Wallimann, T.; Ruegg, U.T. Creatine supplementation reduces skeletal muscle degeneration and enhances mitochondrial function in mdx mice. Neuromuscul. Disord. 2002, 12, 174–182. [Google Scholar] [CrossRef]
- Kuznetsov, A.V.; Winkler, K.; Wiedemann, F.; von Bossanyi, P.; Dietzmann, K.; Kunz, W.S. Impaired mitochondrial oxidative phosphorylation in skeletal muscle of the dystrophin-deficient mdx mouse. Mol. Cell. Biochem. 1998, 183, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Onopiuk, M.; Brutkowski, W.; Wierzbicka, K.; Wojciechowska, S.; Szczepanowska, J.; Fronk, J.; Lochmüller, H.; Górecki, D.C.; Zabłocki, K. Mutation in dystrophin-encoding gene affects energy metabolism in mouse myoblasts. Biochem. Biophys. Res. Commun. 2009, 386, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Rybalka, E.; Timpani, C.A.; Cooke, M.B.; Williams, A.D.; Hayes, A. Defects in mitochondrial ATP synthesis in dystrophin-deficient mdx skeletal muscles may be caused by complex I insufficiency. PLoS ONE 2014, 9, e115763. [Google Scholar] [CrossRef] [PubMed]
- Heslop, L.; Morgan, J.E.; Partridge, T.A. Evidence for a myogenic stem cell that is exhausted in dystrophic muscle. J. Cell Sci. 2000, 113, 2299–2308. [Google Scholar] [PubMed]
- Luz, M.; Marques, M.; Santo Neto, H. Impaired regeneration of dystrophin-deficient muscle fibers is caused by exhaustion of myogenic cells. Braz. J. Med. Biol. Res. 2002, 35, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Kinali, M.; Arechavala-Gomeza, V.; Cirak, S.; Glover, A.; Guglieri, M.; Feng, L.; Hollingsworth, K.; Hunt, D.; Jungbluth, H.; Roper, H. Muscle histology vs MRI in Duchenne muscular dystrophy. Neurology 2011, 76, 346–353. [Google Scholar] [CrossRef] [PubMed]
- Scott, O.; Hyde, S.; Goddard, C.; Dubowitz, V. Quantitation of muscle function in children: A prospective study in duchenne muscular dystrophy. Muscle Nerve 1982, 5, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Bach, J.R.; O’Brien, J.; Krotenberg, R.; Alba, A.S. Management of end stage respiratory failure in Duchenne muscular dystrophy. Muscle Nerve 1987, 10, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Nigro, G.; Comi, L.; Politano, L.; Bain, R. The incidence and evolution of cardiomyopathy in Duchenne muscular dystrophy. Int. J. Cardiol. 1990, 26, 271–277. [Google Scholar] [CrossRef]
- Timpani, C.A.; Hayes, A.; Rybalka, E. Revisiting the dystrophin-ATP connection: How half a century of research still implicates mitochondrial dysfunction in duchenne muscular dystrophy aetiology. Med. Hypotheses 2015, 85, 1021–1033. [Google Scholar] [CrossRef] [PubMed]
- Barbiroli, B.; Funicello, R.; Ferlini, A.; Montagna, P.; Zaniol, P. Muscle energy metabolism in female dmd/bmd carriers: A 31P-MR spectroscopy study. Muscle Nerve 1992, 15, 344–348. [Google Scholar] [CrossRef] [PubMed]
- Dreyfus, J.-C.; Schapira, G.; Schapira, F. Biochemical study of muscle in progressive muscular dystrophy. J. Clin. Investig. 1954, 33, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Di Mauro, S.; Angelini, C.; Catani, C. Enzymes of the glycogen cycle and glycolysis in various human neuromuscular disorders. J. Neurol. Neurosurg. Psychiat. 1967, 30, 411–415. [Google Scholar] [CrossRef] [PubMed]
- Hess, J. Phosphorylase activity and glycogen, glucose-6-phosphate, and lactic acid content of human skeletal muscle in various myopathies. J. Lab. Clin. Med. 1965, 66, 452–463. [Google Scholar] [PubMed]
- Chi, M.M.Y.; Hintz, C.S.; McKee, D.; Felder, S.; Grant, N.; Kaiser, K.K.; Lowry, O.H. Effect of Duchenne muscular dystrophy on enzymes of energy metabolism in individual muscle fibers. Metabolism 1987, 36, 761–767. [Google Scholar] [CrossRef]
- Chinet, A.; Even, P.; Decrouy, A. Dystrophin-dependent efficiency of metabolic pathways in mouse skeletal muscles. Cell. Mol. Life Sci. 1994, 50, 602–605. [Google Scholar] [CrossRef]
- Van Bennekom, C.; Oerlemans, F.T.; Kulakowski, S.; de Bruyn, C.H. Enzymes of purine metabolism in muscle specimens from patients with Duchenne-type muscular dystrophy. Adv. Exp. Med. Biol. 1984, 165, 447–450. [Google Scholar] [PubMed]
- Glesby, M.J.; Rosenmann, E.; Nylen, E.G.; Wrogemann, K. Serum ck, calcium, magnesium, and oxidative phosphorylation in mdx mouse muscular dystrophy. Muscle Nerve 1988, 11, 852–856. [Google Scholar] [CrossRef] [PubMed]
- Austin, L.; de Niese, M.; McGregor, A.; Arthur, H.; Gurusinghe, A.; Gould, M. Potential oxyradical damage and energy status in individual muscle fibres from degenerating muscle diseases. Neuromuscul. Disord. 1992, 2, 27–33. [Google Scholar] [CrossRef]
- Wells, D.J.; Wells, K.E.; Asante, E.A.; Turner, G.; Sunada, Y.; Campbell, K.P.; Walsh, F.S.; Dickson, G. Expression of human full-length and minidystrophin in transgenic mdx mice: Implications for gene therapy of Duchenne muscular dystrophy. Hum. Mol. Genet. 1995, 4, 1245–1250. [Google Scholar] [CrossRef] [PubMed]
- Clemens, P.; Kochanek, S.; Sunada, Y.; Chan, S.; Chen, H.; Campbell, K.; Caskey, C. In vivo muscle gene transfer of full-length dystrophin with an adenoviral vector that lacks all viral genes. Gene Ther. 1996, 3, 965–972. [Google Scholar] [PubMed]
- Sakamoto, M.; Yuasa, K.; Yoshimura, M.; Yokota, T.; Ikemoto, T.; Suzuki, M.; Dickson, G.; Miyagoe-Suzuki, Y.; Takeda, S.I. Micro-dystrophin cDNA ameliorates dystrophic phenotypes when introduced into mdx mice as a transgene. Biochem. Biophys. Res. Commun. 2002, 293, 1265–1272. [Google Scholar] [CrossRef]
- Moxley, R.; Ashwal, S.; Pandya, S.; Connolly, A.; Florence, J.; Mathews, K.; Baumbach, L.; McDonald, C.; Sussman, M.; Wade, C. Practice parameter: Corticosteroid treatment of duchenne dystrophy report of the quality standards subcommittee of the american academy of neurology and the practice committee of the child neurology society. Neurology 2005, 64, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Sicinski, P.; Geng, Y.; Ryder-Cook, A.S.; Barnard, E.A.; Darlison, M.G.; Barnard, P.J. The molecular basis of muscular dystrophy in the mdx mouse: A point mutation. Science 1989, 244, 1578–1580. [Google Scholar] [CrossRef] [PubMed]
- Stedman, H.H.; Sweeney, H.L.; Shrager, J.B.; Maguire, H.C.; Panettieri, R.A.; Petrof, B.; Narusawa, M.; Leferovich, J.M.; Sladky, J.T.; Kelly, A.M. The mdx mouse diaphragm reproduces the degenerative changes of Duchenne muscular dystrophy. Nature 1991, 352, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Brenman, J.E.; Chao, D.S.; Xia, H.; Aldape, K.; Bredt, D.S. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in Duchenne muscular dystrophy. Cell 1995, 82, 743–752. [Google Scholar] [CrossRef]
- Chang, W.J.; Iannaccone, S.T.; Lau, K.S.; Masters, B.S.; McCabe, T.J.; McMillan, K.; Padre, R.C.; Spencer, M.J.; Tidball, J.G.; Stull, J.T. Neuronal nitric oxide synthase and dystrophin-deficient muscular dystrophy. Proc. Natl. Acad. Sci. USA 1996, 93, 9142–9147. [Google Scholar] [CrossRef] [PubMed]
- Balon, T.W.; Nadler, J.L. Evidence that nitric oxide increases glucose transport in skeletal muscle. J. Appl. Physiol. 1997, 82, 359–363. [Google Scholar] [PubMed]
- Bradley, S.J.; Kingwell, B.A.; McConell, G.K. Nitric oxide synthase inhibition reduces leg glucose uptake but not blood flow during dynamic exercise in humans. Diabetes 1999, 48, 1815–1821. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Hudson, A.J.; Strickland, K.P. Fatty acid oxidation by skeletal muscle mitochondria in Duchenne muscular dystrophy. Life Sci. II 1972, 11, 355–362. [Google Scholar] [CrossRef]
- Shumate, J.B.; Carroll, J.E.; Brooke, M.H.; Choksi, R.M. Palmitate oxidation in human muscle: Comparison to CPT and carnitine. Muscle Nerve 1982, 5, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Carroll, J.E.; Norris, B.J.; Brooke, M.H. Defective [U-14 C] palmitic acid oxidation in Duchenne muscular dystrophy. Neurology 1985, 35, 96–97. [Google Scholar] [CrossRef] [PubMed]
- Sharma, U.; Atri, S.; Sharma, M.; Sarkar, C.; Jagannathan, N. Skeletal muscle metabolism in Duchenne muscular dystrophy (DMD): An in vitro proton NMR spectroscopy study. Magn. Reson. Imaging 2003, 21, 145–153. [Google Scholar] [CrossRef]
- Borum, P.R.; Broquist, H.P.; Roelofs, R.I. Muscle carnitine levels in neuromuscular disease. J. Neurol. Sci. 1977, 34, 279–286. [Google Scholar] [CrossRef]
- Carrier, H.N.; Berthillier, G. Carnitine levels in normal children and adults and in patients with diseased muscle. Muscle Nerve 1980, 3, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Le Borgne, F.; Guyot, S.; Logerot, M.; Beney, L.; Gervais, P.; Demarquoy, J. Exploration of lipid metabolism in relation with plasma membrane properties of Duchenne muscular dystrophy cells: Influence of l-carnitine. PLoS ONE 2012, 7, e49346. [Google Scholar] [CrossRef] [PubMed]
- Hardie, D.G.; Ross, F.A.; Hawley, S.A. AMPK: A nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012, 13, 251–262. [Google Scholar] [CrossRef] [PubMed]
- Pauly, M.; Daussin, F.; Burelle, Y.; Li, T.; Godin, R.; Fauconnier, J.; Koechlin-Ramonatxo, C.; Hugon, G.; Lacampagne, A.; Coisy-Quivy, M. AMPK activation stimulates autophagy and ameliorates muscular dystrophy in the mdx mouse diaphragm. Am. J. Pathol. 2012, 181, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Rennie, M.; Edwards, R.; Millward, D.; Wolman, S.; Halliday, D.; Matthews, D. Effects of Duchenne muscular dystrophy on muscle protein synthesis. Nature 1982, 296, 165–167. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Manabe, S.; Sakamoto, S.; Ohnaka, M.; Niiyama, Y. Protein and energy metabolism in patients with progressive muscular dystrophy. J. Nutr. Sci. Vitaminol. 1992, 38, 141–154. [Google Scholar] [CrossRef] [PubMed]
- Bolster, D.R.; Crozier, S.J.; Kimball, S.R.; Jefferson, L.S. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J. Biol. Chem. 2002, 277, 23977–23980. [Google Scholar] [CrossRef] [PubMed]
- Saha, A.K.; Xu, X.J.; Lawson, E.; Deoliveira, R.; Brandon, A.E.; Kraegen, E.W.; Ruderman, N.B. Downregulation of AMPK accompanies leucine-and glucose-induced increases in protein synthesis and insulin resistance in rat skeletal muscle. Diabetes 2010, 59, 2426–2434. [Google Scholar] [CrossRef] [PubMed]
- RE, M. Increase in anterior tibialis muscle protein synthesis in healthy man during mixed amino acid infusion: Studies of incorporation of [1–13C] leucine. Clin. Sci. 1989, 76, 447–454. [Google Scholar]
- Millward, D.J.; Fereday, A.; Gibson, N.R.; Pacy, P.J. Post-prandial protein metabolism. Baillière’s Clin. Endocrinol. Metab. 1996, 10, 533–549. [Google Scholar] [CrossRef]
- Wolfe, R.; Miller, S. Amino acid availability controls muscle protein metabolism. Diabetes Nutr. Metab. 1999, 12, 322–328. [Google Scholar] [PubMed]
- Kimball, S.R.; Farrell, P.A.; Nguyen, H.V.; Jefferson, L.S.; Davis, T.A. Developmental decline in components of signal transduction pathways regulating protein synthesis in pig muscle. Am. J. Physiol.-Endocrinol. Metab. 2002, 282, E585–E592. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bolster, D.R.; Kubica, N.; Crozier, S.J.; Williamson, D.L.; Farrell, P.A.; Kimball, S.R.; Jefferson, L.S. Immediate response of mammalian target of rapamycin (mTOR)-mediated signalling following acute resistance exercise in rat skeletal muscle. J. Physiol. 2003, 553, 213–220. [Google Scholar] [CrossRef] [PubMed]
- Bolster, D.R.; Jefferson, L.S.; Kimball, S.R. Regulation of protein synthesis associated with skeletal muscle hypertrophy by insulin-, amino acid-and exercise-induced signalling. Proc. Nutr. Soc. 2004, 63, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Reeds, P.J.; Fjeld, C.R.; Jahoor, F. Do the differences between the amino acid compositions of acute-phase and muscle proteins have a bearing on nitrogen loss in traumatic states? J. Nutr. 1994, 124, 906–910. [Google Scholar] [PubMed]
- Castillo, C.E.; Katz, A.; Spencer, M.K.; Yan, Z.; Nyomba, B.L. Fasting Inhibits Insulin-Mediated Glycolysis and Anaplerosis in Human Skeletal Muscle. Am. J. Physiol. 1991, 261, E598–E605. [Google Scholar] [PubMed]
- Gibala, M.J.; MacLean, D.A.; Graham, T.E.; Saltin, B. Tricarboxylic acid cycle intermediate pool size and estimated cycle flux in human muscle during exercise. Am. J. Physiol. Endocrinol. Metab. 1998, 275, E235–E242. [Google Scholar]
- Harris, R.C.; Soderlund, K.; Hultman, E. Elevation of creatine in resting and exercised muscle of normal subjects by creatine supplementation. Clin. Sci. 1992, 83, 367–374. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; Montain, S.J.; Matott, R.P.; Zientara, G.P.; Jolesz, F.A.; Fielding, R.A. Effects of creatine supplementation on the energy cost of muscle contraction: A 31P-MRS study. J. Appl. Physiol. 1999, 87, 116–123. [Google Scholar] [PubMed]
- Op‘t Eijnde, B.; Richter, E.A.; Henquin, J.C.; Kiens, B.; Hespel, P. Effect of creatine supplementation on creatine and glycogen content in rat skeletal muscle. Acta Physiol. Scand. 2001, 171, 169–176. [Google Scholar] [CrossRef] [PubMed]
- Bessman, S.P.; Geiger, P.J. Transport of energy in muscle: The phosphorylcreatine shuttle. Science 1981, 211, 448–452. [Google Scholar] [CrossRef] [PubMed]
- Bessman, S.P.; Carpenter, C.L. The creatine-creatine phosphate energy shuttle. Annu. Rev. Biochem. 1985, 54, 831–862. [Google Scholar] [CrossRef] [PubMed]
- Kammermeier, H. Why do cells need phosphocreatine and a phosphocreatine shuttle. J. Mol. Cell. Cardiol. 1987, 19, 115–118. [Google Scholar] [CrossRef]
- Balsom, P.D.; Söderlund, K.; Ekblom, B. Creatine in humans with special reference to creatine supplementation. Sports Med. 1994, 18, 268–280. [Google Scholar] [CrossRef] [PubMed]
- Krzanowski, J.; Matschinsky, F. Regulation of phosphofructokinase by phosphocreatine and phosphorylated glycolytic intermediates. Biochem. Biophys. Res. Commun. 1969, 34, 816–823. [Google Scholar] [CrossRef]
- Ceddia, R.B.; Sweeney, G. Creatine supplementation increases glucose oxidation and AMPK phosphorylation and reduces lactate production in l6 rat skeletal muscle cells. J. Physiol. 2004, 555, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Hultman, E.; Soderlund, K.; Timmons, J.; Cederblad, G.; Greenhaff, P. Muscle creatine loading in men. J. Appl. Physiol. 1996, 81, 232–237. [Google Scholar] [PubMed]
- Greenhaff, P.L. Creatine supplementation: Recent developments. Br. J. Sports Med. 1996, 30, 276. [Google Scholar] [CrossRef] [PubMed]
- Kreider, R.B. Effects of creatine supplementation on performance and training adaptations. Mol. Cell. Biochem. 2003, 244, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Wallimann, T.; Wyss, M.; Brdiczka, D.; Nicolay, K.; Eppenberger, H. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: The ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem. J. 1992, 281, 21. [Google Scholar] [CrossRef] [PubMed]
- Willoughby, D.S.; Rosene, J. Effects of oral creatine and resistance training on myosin heavy chain expression. Med. Sci. Sports Exerc. 2001, 33, 1674–1681. [Google Scholar] [CrossRef] [PubMed]
- Tarnopolsky, M.; Martin, J. Creatine monohydrate increases strength in patients with neuromuscular disease. Neurology 1999, 52, 854–857. [Google Scholar] [CrossRef] [PubMed]
- Tarnopolsky, M.; Parise, G.; Fu, M.-H.; Brose, A.; Parshad, A.; Speer, O.; Wallimann, T. Acute and moderate-term creatine monohydrate supplementation does not affect creatine transporter mRNA or protein content in either young or elderly humans. In Guanidino Compounds in Biology and Medicine; Springer: Heidelberg, Germany, 2003; pp. 159–166. [Google Scholar]
- Tarnopolsky, M.; Mahoney, D.; Vajsar, J.; Rodriguez, C.; Doherty, T.; Roy, B.; Biggar, D. Creatine monohydrate enhances strength and body composition in Duchenne muscular dystrophy. Neurology 2004, 62, 1771–1777. [Google Scholar] [CrossRef] [PubMed]
- Louis, M.; Poortmans, J.R.; Francaux, M.; Berré, J.; Boisseau, N.; Brassine, E.; Cuthbertson, D.J.; Smith, K.; Babraj, J.A.; Waddell, T. No effect of creatine supplementation on human myofibrillar and sarcoplasmic protein synthesis after resistance exercise. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E1089–E1094. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.; Lochmüller, H.; Reilich, P.; Klopstock, T.; Huber, R.; Hartard, M.; Hennig, M.; Pongratz, D.; Müller-Felber, W. Creatine monohydrate in muscular dystrophies: A double-blind, placebo-controlled clinical study. Neurology 2000, 54, 1848–1850. [Google Scholar] [CrossRef] [PubMed]
- Pulido, S.; Passaquin, A.; Leijendekker, W.; Challet, C.; Wallimann, T.; Rüegg, U. Creatine supplementation improves intracellular ca2+ handling and survival in mdx skeletal muscle cells. FEBS Lett. 1998, 439, 357–362. [Google Scholar] [CrossRef]
- Louis, M.; Raymackers, J.M.; Debaix, H.; Lebacq, J.; Francaux, M. Effect of creatine supplementation on skeletal muscle of mdx mice. Muscle Nerve 2004, 29, 687–692. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, B.; Sharma, U.; Balasubramanian, K.; Kalaivani, M.; Kalra, V.; Jagannathan, N.R. Effect of creatine monohydrate in improving cellular energetics and muscle strength in ambulatory Duchenne muscular dystrophy patients: A randomized, placebo-controlled 31P MRS study. Magn. Reson. Imaging 2010, 28, 698–707. [Google Scholar] [CrossRef] [PubMed]
- Passaquin, A.; Metzinger, L.; Leger, J.; Warter, J.M.; Poindron, P. Prednisolone enhances myogenesis and dystrophin-related protein in skeletal muscle cell cultures from mdx mouse. J. Neurosci. Res. 2004, 35, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Rossi, A.; Eppenberger, H.; Volpe, P.; Cotrufo, R.; Wallimann, T. Muscle-type MM creatine kinase is specifically bound to sarcoplasmic reticulum and can support Ca2+ uptake and regulate local ATP/ADP ratios. J. Biol. Chem. 1990, 265, 5258–5266. [Google Scholar] [PubMed]
- Minajeva, A.; Ventura-Clapier, R.; Veksler, V. Ca2+ uptake by cardiac sarcoplasmic reticulum atpase in situ strongly depends on bound creatine kinase. Pflüg. Arch. 1996, 432, 904–912. [Google Scholar] [CrossRef]
- Mok, E.; Hankard, R. Glutamine supplementation in sick children: Is it beneficial? J. Nutr. Metab. 2011, 2011, 617597. [Google Scholar] [CrossRef] [PubMed]
- Hankard, R.G.; Hammond, D.; Haymond, M.W.; Darmaun, D. Oral glutamine slows down whole body protein breakdown in duchenne muscular dystrophy. Pediatr. Res. 1998, 43, 222–226. [Google Scholar] [CrossRef] [PubMed]
- Mok, E.; Eleouet-Da Violante, C.; Daubrosse, C.; Gottrand, F.; Rigal, O.; Fontan, J.E.; Cuisset, J.M.; Guilhot, J.; Hankard, R. Oral glutamine and amino acid supplementation inhibit whole-body protein degradation in children with Duchenne muscular dystrophy. Am. J. Clin. Nutr. 2006, 83, 823–828. [Google Scholar] [PubMed]
- Mok, E.; Constantin, B.; Favreau, F.; Neveux, N.; Magaud, C.; Delwail, A.; Hankard, R. l-glutamine administration reduces oxidized glutathione and map kinase signaling in dystrophic muscle of mdx mice. Pediatr. Res. 2008, 63, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Mok, E.; Letellier, G.; Cuisset, J.M.; Denjean, A.; Gottrand, F.; Alberti, C.; Hankard, R. Lack of functional benefit with glutamine versus placebo in Duchenne muscular dystrophy: A randomized crossover trial. PLoS ONE 2009, 4, e5448. [Google Scholar] [CrossRef] [PubMed]
- Escolar, D.M.; Buyse, G.; Henricson, E.; Leshner, R.; Florence, J.; Mayhew, J.; Tesi-Rocha, C.; Gorni, K.; Pasquali, L.; Patel, K.M.; et al. Cinrg randomized controlled trial of creatine and glutamine in Duchenne muscular dystrophy. Ann. Neurol. 2005, 58, 151–155. [Google Scholar] [CrossRef] [PubMed]
- Granchelli, J.A.; Pollina, C.; Hudecki, M.S. Pre-clinical screening of drugs using the mdx mouse. Neuromuscul. Disord. 2000, 10, 235–239. [Google Scholar] [CrossRef]
- Stipanuk, M.H. Role of the liver in regulation of body cysteine and taurine levels: A brief review. Neurochem. Res. 2004, 29, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Conte Camerino, D.; Tricarico, D.; Pierno, S.; Desaphy, J.F.; Liantonio, A.; Pusch, M.; Burdi, R.; Camerino, C.; Fraysse, B.; de Luca, A. Taurine and skeletal muscle disorders. Neurochem. Res. 2004, 29, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Bakker, A.J.; Berg, H.M. Effect of taurine on sarcoplasmic reticulum function and force in skinned fast-twitch skeletal muscle fibres of the rat. J. Physiol. 2002, 538, 185–194. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, E.J.; Berg, H.M.; Easton, C.J.; Bakker, A.J. The effect of taurine depletion on the contractile properties and fatigue in fast-twitch skeletal muscle of the mouse. Amino Acids 2006, 31, 273–278. [Google Scholar] [CrossRef] [PubMed]
- Warskulat, U.; Flogel, U.; Jacoby, C.; Hartwig, H.G.; Thewissen, M.; Merx, M.W.; Molojavyi, A.; Heller-Stilb, B.; Schrader, J.; Haussinger, D. Taurine transporter knockout depletes muscle taurine levels and results in severe skeletal muscle impairment but leaves cardiac function uncompromised. FASEB J. 2004, 18, 577–579. [Google Scholar] [CrossRef] [PubMed]
- Terrill, J.R.; Boyatzis, A.; Grounds, M.D.; Arthur, P.G. Treatment with the cysteine precursor l-2-oxothiazolidine-4-carboxylate (OTC) implicates taurine deficiency in severity of dystropathology in mdx mice. Int. J. Biochem. Cell Biol. 2013, 45, 2097–2108. [Google Scholar] [CrossRef] [PubMed]
- Cozzoli, A.; Rolland, J.F.; Capogrosso, R.F.; Sblendorio, V.T.; Longo, V.; Simonetti, S.; Nico, B.; de Luca, A. Evaluation of potential synergistic action of a combined treatment with alpha-methyl-prednisolone and taurine on the mdx mouse model of Duchenne muscular dystrophy. Neuropathol. Appl. Neurobiol. 2011, 37, 243–256. [Google Scholar] [CrossRef] [PubMed]
- De Luca, A.; Pierno, S.; Liantonio, A.; Cetrone, M.; Camerino, C.; Fraysse, B.; Mirabella, M.; Servidei, S.; Ruegg, U.T.; Conte Camerino, D. Enhanced dystrophic progression in mdx mice by exercise and beneficial effects of taurine and insulin-like growth factor-1. J. Pharmacol. Exp. Ther. 2003, 304, 453–463. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, L.; Granberg, K.E.; Briere, K.M.; Anderson, J.E. Nuclear magnetic resonance spectroscopy study of muscle growth, mdx dystrophy and glucocorticoid treatments: Correlation with repair. NMR Biomed. 1998, 11, 1–10. [Google Scholar] [CrossRef]
- Horvath, D. The Effect of Taurine on Dystrophic Muscle Tissue Function. Ph.D. Thesis, Victoria University, Melbourne, VIC, Australia, 2011. [Google Scholar]
- Voisin, V.; Sebrie, C.; Matecki, S.; Yu, H.; Gillet, B.; Ramonatxo, M.; Israel, M.; de la Porte, S. l-arginine improves dystrophic phenotype in mdx mice. Neurobiol. Dis. 2005, 20, 123–130. [Google Scholar] [CrossRef] [PubMed]
- Archer, J.D.; Vargas, C.C.; Anderson, J.E. Persistent and improved functional gain in mdx dystrophic mice after treatment with l-arginine and deflazacort. FASEB J. 2006, 20, 738–740. [Google Scholar] [CrossRef] [PubMed]
- Marques, M.J.; Luz, M.A.; Minatel, E.; Neto, H.S. Muscle regeneration in dystrophic mdx mice is enhanced by isosorbide dinitrate. Neurosci. Lett. 2005, 382, 342–345. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.D.; Ye, J.; de Nardi, C.; Monopoli, A.; Ongini, E.; Victor, R.G. Treatment with a nitric oxide-donating NSAID alleviates functional muscle ischemia in the mouse model of Duchenne muscular dystrophy. PLoS ONE 2012, 7, e49350. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, M.G.; Gandossini, S.; Boneschi, F.M.; Sciorati, C.; Bonato, S.; Brighina, E.; Comi, G.P.; Turconi, A.C.; Magri, F.; Stefanoni, G. Nitric oxide donor and non steroidal anti inflammatory drugs as a therapy for muscular dystrophies: Evidence from a safety study with pilot efficacy measures in adult dystrophic patients. Pharmacol. Res. 2012, 65, 472–479. [Google Scholar] [CrossRef] [PubMed]
- Thomas, G.D.; Monopoli, A.; de Nardi, C.; Ongini, E.; Victor, R.G. Treatment with a nitric oxide-donating NSAID counteracts functional muscle ischemia in dystrophin-deficient mdx mice. FASEB J. 2012, 26, 1092–1096. [Google Scholar]
- Sciorati, C.; Miglietta, D.; Buono, R.; Pisa, V.; Cattaneo, D.; Azzoni, E.; Brunelli, S.; Clementi, E. A dual acting compound releasing nitric oxide (no) and ibuprofen, NCX 320, shows significant therapeutic effects in a mouse model of muscular dystrophy. Pharmacol. Res. 2011, 64, 210–217. [Google Scholar] [CrossRef] [PubMed]
- Percival, J.M.; Whitehead, N.P.; Adams, M.E.; Adamo, C.M.; Beavo, J.A.; Froehner, S.C. Sildenafil reduces respiratory muscle weakness and fibrosis in the mdx mouse model of Duchenne muscular dystrophy. J. Pathol. 2012, 228, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.D.; Rader, F.; Tang, X.; Tavyev, J.; Nelson, S.F.; Miceli, M.C.; Elashoff, R.M.; Sweeney, H.L.; Victor, R.G. PDE5 inhibition alleviates functional muscle ischemia in boys with Duchenne muscular dystrophy. Neurology 2014, 82, 2085–2091. [Google Scholar] [CrossRef] [PubMed]
- Kawahara, G.; Gasperini, M.J.; Myers, J.A.; Widrick, J.J.; Eran, A.; Serafini, P.R.; Alexander, M.S.; Pletcher, M.T.; Morris, C.A.; Kunkel, L.M. Dystrophic muscle improvement in zebrafish via increased heme oxygenase signaling. Hum. Mol. Genet. 2015, 24, 4480–4481. [Google Scholar] [CrossRef] [PubMed]
- Witting, N.; Kruuse, C.; Nyhuus, B.; Prahm, K.P.; Citirak, G.; Lundgaard, S.J.; Huth, S.; Vejlstrup, N.; Lindberg, U.; Krag, T.O. Effect of sildenafil on skeletal and cardiac muscle in becker muscular dystrophy. Ann. Neurol. 2014, 76, 550–557. [Google Scholar] [CrossRef] [PubMed]
- Carlström, M.; Larsen, F.J.; Nyström, T.; Hezel, M.; Borniquel, S.; Weitzberg, E.; Lundberg, J.O. Dietary inorganic nitrate reverses features of metabolic syndrome in endothelial nitric oxide synthase-deficient mice. Proc. Natl. Acad. Sci. USA 2010, 107, 17716–17720. [Google Scholar] [CrossRef] [PubMed]
- Larsen, F.J.; Schiffer, T.A.; Borniquel, S.; Sahlin, K.; Ekblom, B.; Lundberg, J.O.; Weitzberg, E. Dietary inorganic nitrate improves mitochondrial efficiency in humans. Cell Metab. 2011, 13, 149–159. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, M.; Winyard, P.G.; Aizawa, K.; Anning, C.; Shore, A.; Benjamin, N. Effect of dietary nitrate on blood pressure, endothelial function, and insulin sensitivity in type 2 diabetes. Free Radic. Biol. Med. 2013, 60, 89–97. [Google Scholar] [CrossRef] [PubMed]
- Gilchrist, M.; Winyard, P.G.; Fulford, J.; Anning, C.; Shore, A.C.; Benjamin, N. Dietary nitrate supplementation improves reaction time in type 2 diabetes: Development and application of a novel nitrate-depleted beetroot juice placebo. Nitric Oxide 2014, 40, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Tipton, K.D.; Elliott, T.A.; Cree, M.G.; Wolf, S.E.; Sanford, A.P.; Wolfe, R.R. Ingestion of casein and whey proteins result in muscle anabolism after resistance exercise. Med. Sci. Sports Exerc. 2004, 36, 2073–2081. [Google Scholar] [CrossRef] [PubMed]
- Tipton, K.D.; Elliott, T.A.; Cree, M.G.; Aarsland, A.A.; Sanford, A.P.; Wolfe, R.R. Stimulation of net muscle protein synthesis by whey protein ingestion before and after exercise. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E71–E76. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.E.; Manolakos, J.J.; Kujbida, G.W.; Lysecki, P.J.; Moore, D.R.; Phillips, S.M. Minimal whey protein with carbohydrate stimulates muscle protein synthesis following resistance exercise in trained young men. Appl. Physiol. Nutr. Metab. 2007, 32, 1132–1138. [Google Scholar] [CrossRef] [PubMed]
- Bucci, L.; Unlu, L. Proteins and amino acid supplements in exercise and sport. Energy Yielding Macronutr. Energy Metab. Sports Nutr. 2000, 191–212. [Google Scholar]
- Walzem, R.; Dillard, C.; German, J. Whey components: Millennia of evolution create functionalities for mammalian nutrition: What we know and what we may be overlooking. Crit. Rev. Food Sci. Nutr. 2002, 42, 353–375. [Google Scholar] [CrossRef] [PubMed]
- Mahe, S.; Roos, N.; Benamouzig, R.; Davin, L.; Luengo, C.; Gagnon, L.; Gausserges, N.; Rautureau, J.; Tomé, D. Gastrojejunal kinetics and the digestion of [15N] beta-lactoglobulin and casein in humans: The influence of the nature and quantity of the protein. Am. J. Clin. Nutr. 1996, 63, 546–552. [Google Scholar] [PubMed]
- Boirie, Y.; Dangin, M.; Gachon, P.; Vasson, M.-P.; Maubois, J.-L.; Beaufrère, B. Slow and fast dietary proteins differently modulate postprandial protein accretion. Proc. Natl. Acad. Sci. USA 1997, 94, 14930–14935. [Google Scholar] [CrossRef] [PubMed]
- Dangin, M.; Boirie, Y.; Garcia-Rodenas, C.; Gachon, P.; Fauquant, J.; Callier, P.; Ballèvre, O.; Beaufrère, B. The digestion rate of protein is an independent regulating factor of postprandial protein retention. Am. J. Physiol. Endocrinol. Metab. 2001, 280, E340–E348. [Google Scholar] [PubMed]
- Dangin, M.; Guillet, C.; Garcia-Rodenas, C.; Gachon, P.; Bouteloup-Demange, C.; Reiffers-Magnani, K.; Fauquant, J.; Ballèvre, O.; Beaufrère, B. The rate of protein digestion affects protein gain differently during aging in humans. J. Physiol. 2003, 549, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Bounous, G.; Gold, P. The biological activity of undenatured dietary whey proteins: Role of glutathione. Clin. Investig. Med. 1991, 14, 296–309. [Google Scholar]
- Carbó, N.; Lopez-Soriano, F.; Fiers, W.; Argiles, J. Tumour growth results in changes in placental amino acid transport in the rat: A tumour necrosis factor alpha-mediated effect. Biochem. J. 1996, 313, 77–82. [Google Scholar] [CrossRef] [PubMed]
- Anthony, J.C.; Anthony, T.G.; Kimball, S.R.; Jefferson, L.S. Signaling pathways involved in translational control of protein synthesis in skeletal muscle by leucine. J. Nutr. 2001, 131, 856S–860S. [Google Scholar] [PubMed]
- Rybalka, E. Impaired Metabolism in X-Linked Muscular Dystrophy: Experimental Evaluation of Potential Therapies to Improve Calcium Regulation, Bioenergetics and Muscle Architecture. Ph.D. Thesis, Victoria University, Melbourne, VIC, Australia, 2008. [Google Scholar]
- Cribb, P.J.; Williams, A.D.; Hayes, A. A creatine-protein-carbohydrate supplement enhances responses to resistance training. Med. Sci. Sport Exerc. 2007, 39, 1960–1968. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Zou, L.Y.; Cao, C.M.; Yang, E.S. Coenzyme Q10 protects SHSY5Y neuronal cells from beta amyloid toxicity and oxygen-glucose deprivation by inhibiting the opening of the mitochondrial permeability transition pore. BioFactors (Oxf. Engl.) 2005, 25, 97–107. [Google Scholar] [CrossRef]
- Cooke, M.; Iosia, M.; Buford, T.; Shelmadine, B.; Hudson, G.; Kerksick, C.; Rasmussen, C.; Greenwood, M.; Leutholtz, B.; Willoughby, D.; et al. Effects of acute and 14-day coenzyme Q10 supplementation on exercise performance in both trained and untrained individuals. J. Int. Soc. Sports Nutr. 2008, 5, 8. [Google Scholar] [CrossRef] [PubMed]
- Emmanuele, V.; Lopez, L.C.; Berardo, A.; Naini, A.; Tadesse, S.; Wen, B.; D’Agostino, E.; Solomon, M.; DiMauro, S.; Quinzii, C.; et al. Heterogeneity of coenzyme Q10 deficiency: Patient study and literature review. Arch. Neurol. 2012, 69, 978–983. [Google Scholar] [CrossRef] [PubMed]
- Papucci, L.; Schiavone, N.; Witort, E.; Donnini, M.; Lapucci, A.; Tempestini, A.; Formigli, L.; Zecchi-Orlandini, S.; Orlandini, G.; Carella, G.; et al. Coenzyme Q10 prevents apoptosis by inhibiting mitochondrial depolarization independently of its free radical scavenging property. J. Biol. Chem. 2003, 278, 28220–28228. [Google Scholar] [CrossRef] [PubMed]
- Sacconi, S.; Trevisson, E.; Salviati, L.; Ayme, S.; Rigal, O.; Redondo, A.G.; Mancuso, M.; Siciliano, G.; Tonin, P.; Angelini, C.; et al. Coenzyme Q10 is frequently reduced in muscle of patients with mitochondrial myopathy. Neuromuscul. Disord. 2010, 20, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Littarru, G.P.; Jones, D.; Scholler, J.; Folkers, K. Deficiency of coenzyme Q9 in mice having hereditary muscular dystrophy. Biochem. Biophys. Res. Commun. 1970, 41, 1306–1313. [Google Scholar] [CrossRef]
- Scholler, J.; Jones, D.; Littarru, G.P.; Folkers, K. Therapy of hereditary mouse muscular dystrophy with coenzyme Q7. Biochem. Biophys. Res. Commun. 1970, 41, 1298–1305. [Google Scholar] [CrossRef]
- Folkers, K.; Nakamura, R.; Littarru, G.P.; Zellweger, H.; Brunkhorst, J.B.; Williams, C.W., Jr.; Langston, J.H. Effect of coenzyme Q on serum levels of creatine phosphokinase in preclinical muscular dystrophy. Proc. Natl. Acad. Sci. USA 1974, 71, 2098–2102. [Google Scholar] [CrossRef] [PubMed]
- Folkers, K.; Wolaniuk, J.; Simonsen, R.; Morishita, M.; Vadhanavikit, S. Biochemical rationale and the cardiac response of patients with muscle disease to therapy with coenzyme Q10. Proc. Natl. Acad. Sci. USA 1985, 82, 4513–4516. [Google Scholar] [CrossRef] [PubMed]
- Folkers, K.; Simonsen, R. Two successful double-blind trials with coenzyme Q10 (vitamin Q10) on muscular dystrophies and neurogenic atrophies. Biochim. Biophys. Acta 1995, 1271, 281–286. [Google Scholar] [CrossRef]
- Spurney, C.F.; Rocha, C.T.; Henricson, E.; Florence, J.; Mayhew, J.; Gorni, K.; Pasquali, L.; Pestronk, A.; Martin, G.R.; Hu, F.; et al. Cinrg pilot trial of coenzyme Q10 in steroid-treated Duchenne muscular dystrophy. Muscle Nerve 2011, 44, 174–178. [Google Scholar] [CrossRef] [PubMed]
- Gillis, J.C.; Benefield, P.; McTavish, D. Idebenone. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in age-related cognitive disorders. Drugs Aging 1994, 5, 133–152. [Google Scholar] [CrossRef] [PubMed]
- Buyse, G.M.; van der Mieren, G.; Erb, M.; D’Hooge, J.; Herijgers, P.; Verbeken, E.; Jara, A.; van den Bergh, A.; Mertens, L.; Courdier-Fruh, I.; et al. Long-term blinded placebo-controlled study of SNT-MC17/idebenone in the dystrophin deficient mdx mouse: Cardiac protection and improved exercise performance. Eur. Heart J. 2009, 30, 116–124. [Google Scholar] [CrossRef] [PubMed]
- Buyse, G.M.; Goemans, N.; van den Hauwe, M.; Thijs, D.; de Groot, I.J.; Schara, U.; Ceulemans, B.; Meier, T.; Mertens, L. Idebenone as a novel, therapeutic approach for Duchenne muscular dystrophy: Results from a 12 month, double-blind, randomized placebo-controlled trial. Neuromuscul. Disord. 2011, 21, 396–405. [Google Scholar] [CrossRef] [PubMed]
- Covas, M.-I.; Nyyssönen, K.; Poulsen, H.E.; Kaikkonen, J.; Zunft, H.-J.F.; Kiesewetter, H.; Gaddi, A.; de la Torre, R.; Mursu, J.; Bäumler, H. The effect of polyphenols in olive oil on heart disease risk factors: A randomized trial. Ann. Intern. Med. 2006, 145, 333–341. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.-Y.; Liao, J.; Kim, K.; Yurkow, E.J.; Yang, C.S. Inhibition of growth and induction of apoptosis in human cancer cell lines by tea polyphenols. Carcinogenesis 1998, 19, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Langcake, P.; Pryce, R.J. The production of resveratrol and the viniferins by grapevines in response to ultraviolet irradiation. Phytochemistry 1977, 16, 1193–1196. [Google Scholar] [CrossRef]
- Wibowo, A.; Ahmat, N.; Hamzah, A.; Ismail, N.; Ahmad, R.; Jaafar, F. Resveratrol oligomers from the stem bark of dryobalanops aromatica. Biochem. Syst. Ecol. 2012, 40, 62–64. [Google Scholar] [CrossRef]
- Huang, K.-S.; Lin, M.; Cheng, G.-F. Anti-inflammatory tetramers of resveratrol from the roots of vitis amurensis and the conformations of the seven-membered ring in some oligostilbenes. Phytochemistry 2001, 58, 357–362. [Google Scholar] [CrossRef]
- Morselli, E.; Maiuri, M.; Markaki, M.; Megalou, E.; Pasparaki, A.; Palikaras, K.; Criollo, A.; Galluzzi, L.; Malik, S.; Vitale, I. Caloric restriction and resveratrol promote longevity through the sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010, 1, e10. [Google Scholar] [CrossRef] [PubMed]
- Shakibaei, M.; Shayan, P.; Busch, F.; Aldinger, C.; Buhrmann, C.; Lueders, C.; Mobasheri, A. Resveratrol mediated modulation of SIRT-1/RUNX2 promotes osteogenic differentiation of mesenchymal stem cells: Potential role of RUNX2 deacetylation. PLoS ONE 2012, 7, e35712. [Google Scholar] [CrossRef] [PubMed]
- Breen, D.M.; Sanli, T.; Giacca, A.; Tsiani, E. Stimulation of muscle cell glucose uptake by resveratrol through sirtuins and AMPK. Biochem. Biophys. Res. Commun. 2008, 374, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Tanno, M.; Sakamoto, J.; Miura, T.; Shimamoto, K.; Horio, Y. Nucleocytoplasmic shuttling of the NAD+- dependent histone deacetylase SIRT1. J. Biol. Chem. 2007, 282, 6823–6832. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.S.; Kuno, A.; Hosoda, R.; Horio, Y. Regulation of foxos and P53 by SIRT1 modulators under oxidative stress. PLoS ONE 2013, 8, e73875. [Google Scholar] [CrossRef] [PubMed]
- Lagauge, M.; Argmann, C.; Gerhart-Hines, Z.; Meziane, H.; Lerin, C.; Daussin, F.; Messadeq, N.; Milne, J.; Lambert, P.; Elliott, P. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC1-A. Cell 2006, 127, 1109–1122. [Google Scholar] [CrossRef] [PubMed]
- Selsby, J.T.; Morine, K.J.; Pendrak, K.; Barton, E.R.; Sweeney, H.L. Rescue of dystrophic skeletal muscle by PGC-1α involves a fast to slow fiber type shift in the mdx mouse. PLoS ONE 2012, 7, e30063. [Google Scholar] [CrossRef] [PubMed]
- Thi Man, N.; Thanh, L.; Blake, D.; Davies, K.; Morris, G. Utrophin, the autosomal homologue of dystrophin, is widely-expressed and membrane-associated in cultured cell lines. FEBS Lett. 1992, 313, 19–22. [Google Scholar] [CrossRef]
- Kuno, A.; Tanno, M.; Horio, Y. The effects of resveratrol and SIRT1 activation on dystrophic cardiomyopathy. Ann. N. Y. Acad. Sci. 2015, 1348, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Ljubicic, V.; Burt, M.; Lunde, J.A.; Jasmin, B.J. Resveratrol induces expression of the slow, oxidative phenotype in mdx mouse muscle together with enhanced activity of the SIRT1-PGC-1α axis. Am. J. Physiol. Cell Physiol. 2014, 307, C66–C82. [Google Scholar] [CrossRef] [PubMed]
- Kuno, A.; Hori, Y.S.; Hosoda, R.; Tanno, M.; Miura, T.; Shimamoto, K.; Horio, Y. Resveratrol improves cardiomyopathy in dystrophin-deficient mice through SIRT1 protein-mediated modulation of P300 protein. J. Biol. Chem. 2013, 288, 5963–5972. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.S.; Kuno, A.; Hosoda, R.; Tanno, M.; Miura, T.; Shimamoto, K.; Horio, Y. Resveratrol ameliorates muscular pathology in the dystrophic mdx mouse, a model for Duchenne muscular dystrophy. J. Pharmacol. Exp. Ther. 2011, 338, 784–794. [Google Scholar] [CrossRef] [PubMed]
- Gordon, B.S.; Weed, P.; Learner, E.; Schoenling, D.; Kostek, M.C. Resveratrol decreases inflammation and oxidative stress in the mdx mouse model of Duchenne muscular dystrophy. FASEB J. 2012, 26, 823.812. [Google Scholar]
- Gordon, B.S.; Delgado-Diaz, D.C.; Carson, J.; Fayad, R.; Wilson, L.B.; Kostek, M.C. Resveratrol improves muscle function but not oxidative capacity in young mdx mice. Can. J. Physiol. Pharmacol. 2014, 92, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.Q.; Shehadeh, L.A.; Mitrani, J.M.; Pessanha, M.; Slepak, T.I.; Webster, K.A.; Bishopric, N.H. Quantitative control of adaptive cardiac hypertrophy by acetyltransferase P300. Circulation 2008, 118, 934–946. [Google Scholar] [CrossRef] [PubMed]
- Hollinger, K.; Shanely, R.A.; Quindry, J.C.; Selsby, J.T. Long-term quercetin dietary enrichment decreases muscle injury in mdx mice. Clin. Nutr. 2015, 34, 515–522. [Google Scholar] [CrossRef] [PubMed]
- Ballmann, C.; Hollinger, K.; Selsby, J.T.; Amin, R.; Quindry, J.C. Histological and biochemical outcomes of cardiac pathology in mdx mice with dietary quercetin enrichment. Exp. Physiol. 2015, 100, 12–22. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, H.; Ahmad, N. Green tea in chemoprevention of cancer. Toxicol. Sci. 1999, 52, 111–117. [Google Scholar] [CrossRef] [PubMed]
- Higdon, J.V.; Frei, B. Tea catechins and polyphenols: Health effects, metabolism, and antioxidant functions. Crit. Rev. Food Sci. Nutr. 2003, 43, 89–143. [Google Scholar] [CrossRef] [PubMed]
- Zhao, B.L.; Li, X.J.; He, R.G.; Cheng, S.J.; Xin, W.J. Scavenging effect of extracts of green tea and natural antioxidants on active oxygen radicals. Cell Biophys. 1989, 14, 175–185. [Google Scholar] [CrossRef] [PubMed]
- Benzie, I.F.; Szeto, Y.T.; Strain, J.J.; Tomlinson, B. Consumption of green tea causes rapid increase in plasma antioxidant power in humans. Nutr. Cancer 1999, 34, 83–87. [Google Scholar] [CrossRef] [PubMed]
- Valcic, S.; Muders, A.; Jacobsen, N.E.; Liebler, D.C.; Timmermann, B.N. Antioxidant chemistry of green tea catechins. Identification of products of the reaction of (-)-epigallocatechin gallate with peroxyl radicals. Chem. Res. Toxicol. 1999, 12, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Valenti, D.; de Rasmo, D.; Signorile, A.; Rossi, L.; de Bari, L.; Scala, I.; Granese, B.; Papa, S.; Vacca, R.A. Epigallocatechin-3-gallate prevents oxidative phosphorylation deficit and promotes mitochondrial biogenesis in human cells from subjects with down’s syndrome. Biochim. Biophys. Acta 2013, 1832, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Buetler, T.M.; Renard, M.; Offord, E.A.; Schneider, H.; Ruegg, U.T. Green tea extract decreases muscle necrosis in mdx mice and protects against reactive oxygen species. Am. J. Clin. Nutr. 2002, 75, 749–753. [Google Scholar] [PubMed]
- Dorchies, O.M.; Wagner, S.; Vuadens, O.; Waldhauser, K.; Buetler, T.M.; Kucera, P.; Ruegg, U.T. Green tea extract and its major polyphenol (−)-epigallocatechin gallate improve muscle function in a mouse model for duchenne muscular dystrophy. Am. J. Physiol. Cell Physiol. 2006, 290, C616–C625. [Google Scholar] [CrossRef] [PubMed]
- Dorchies, O.M.; Wagner, S.; Buetler, T.M.; Ruegg, U.T. Protection of dystrophic muscle cells with polyphenols from green tea correlates with improved glutathione balance and increased expression of 67LR, a receptor for (−)-epigallocatechin gallate. BioFactors 2009, 35, 279–294. [Google Scholar] [CrossRef] [PubMed]
- Nakae, Y.; Dorchies, O.M.; Stoward, P.J.; Zimmermann, B.F.; Ritter, C.; Ruegg, U.T. Quantitative evaluation of the beneficial effects in the mdx mouse of epigallocatechin gallate, an antioxidant polyphenol from green tea. Histochem. Cell Biol. 2012, 137, 811–827. [Google Scholar] [CrossRef] [PubMed]
- Nakae, Y.; Hirasaka, K.; Goto, J.; Nikawa, T.; Shono, M.; Yoshida, M.; Stoward, P.J. Subcutaneous injection, from birth, of epigallocatechin-3-gallate, a component of green tea, limits the onset of muscular dystrophy in mdx mice: A quantitative histological, immunohistochemical and electrophysiological study. Histochem. Cell Biol. 2008, 129, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Call, J.A.; Voelker, K.A.; Wolff, A.V.; McMillan, R.P.; Evans, N.P.; Hulver, M.W.; Talmadge, R.J.; Grange, R.W. Endurance capacity in maturing mdx mice is markedly enhanced by combined voluntary wheel running and green tea extract. J. Appl. Physiol. 2008, 105, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Evans, N.P.; Call, J.A.; Bassaganya-Riera, J.; Robertson, J.L.; Grange, R.W. Green tea extract decreases muscle pathology and NF-kappaB immunostaining in regenerating muscle fibers of mdx mice. Clin. Nutr. (Edinb. Scotl.) 2010, 29, 391–398. [Google Scholar] [CrossRef] [PubMed]
- Lesch, M.; Nyhan, W.L. A familial disorder of uric acid metabolism and central nervous system function. Am. J. Med. 1964, 36, 561–570. [Google Scholar] [CrossRef]
- Stathis, C.; Febbraio, M.; Carey, M.; Snow, R. Influence of sprint training on human skeletal muscle purine nucleotide metabolism. J. Appl. Physiol. 1994, 76, 1802–1809. [Google Scholar] [PubMed]
- Bangsbo, J.; Sjödin, B.; Helleten-Westing, Y. Exchange of hypoxanthine in muscle during intense exercise in man. Acta Physiol. Scand. 1992, 146, 549–550. [Google Scholar] [CrossRef] [PubMed]
- Tullson, P.C.; John-Alder, H.B.; Hood, D.A.; Terjung, R.L. De novo synthesis of adenine nucleotides in different skeletal muscle fiber types. Am. J. Physiol. Cell Physiol. 1988, 255, C271–C277. [Google Scholar]
- Hellsten, Y.; Skadhauge, L.; Bangsbo, J. Effect of ribose supplementation on resynthesis of adenine nucleotides after intense intermittent training in humans. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004, 286, R182–R188. [Google Scholar] [CrossRef] [PubMed]
- Karatzaferi, C.; de Haan, A.; Ferguson, R.; van Mechelen, W.; Sargeant, A. Phosphocreatine and ATP content in human single muscle fibres before and after maximum dynamic exercise. Pflügers Archiv 2001, 442, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Tullson, P.C.; Arabadjis, P.G.; Rundell, K.W.; Terjung, R.L. IMP reamination to AMP in rat skeletal muscle fiber types. Am. J. Physiol.-Cell Physiol. 1996, 270, C1067–C1074. [Google Scholar]
- Webster, C.; Silberstein, L.; Hays, A.P.; Blau, H.M. Fast muscle fibers are preferentially affected in Duchenne muscular dystrophy. Cell 1988, 52, 503–513. [Google Scholar] [CrossRef]
- Thomson, W.; Guest, K.E. A trial of therapy by nucleosides and nucleotides in muscular dystrophy. J. Neurol. Neurosurg. Psychiatry 1963, 26, 111. [Google Scholar] [CrossRef] [PubMed]
- Bonsett, C.; Rudman, A. The dystrophin connection—ATP? Med. Hypotheses 1992, 38, 139–154. [Google Scholar] [CrossRef]
- Pearce, G. Electron microscopy in the study of muscular dystrophy. Ann. N. Y. Acad. Sci. 1966, 138, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Harriman, D.; Reed, R. The incidence of lipid droplets in human skeletal muscle in neuromuscular disorders: A histochemical, electron-microscopic and freeze-etch study. J. Pathol. 1972, 106, 1–24. [Google Scholar] [CrossRef] [PubMed]
- Bonsett, C.; Rudman, A.; Elliott, AY. Intracellular lipid in pseudohypertrophic muscular dystrophy tissue culture. J. Indiana State Med. Assoc. 1979, 72, 184–187. [Google Scholar] [PubMed]
- Bonsett, C.; Rudman, A. Duchenne’s muscular dystrophy: A tissue culture perspective. Indiana Med. 1984, 77, 446. [Google Scholar] [PubMed]
- Dodd, S.L.; Johnson, C.A.; Fernholz, K.; st Cyr, J.A. The role of ribose in human skeletal muscle metabolism. Med. Hypotheses 2004, 62, 819–824. [Google Scholar] [CrossRef] [PubMed]
- Pauly, D.; Johnson, C.; Cyr, J.S. The benefits of ribose in cardiovascular disease. Med. Hypotheses 2003, 60, 149–151. [Google Scholar] [CrossRef]
- Griffiths, R.D.; Cady, E.B.; Edwards, R.H.T.; Wilkie, D.R. Muscle energy metabolism in Duchenne dystrophy studied by 31P-NMR: Controlled trials show no effect of allopurinol or ribose. Muscle Nerve 1985, 8, 760–767. [Google Scholar] [CrossRef] [PubMed]
- Gross, M.; Reiter, S.; Zöllner, N. Metabolism of d-ribose administered continuously to healthy persons and to patients with myoadenylate deaminase deficiency. Klin. Wochenschr. 1989, 67, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Zöllner, N.; Reiter, S.; Gross, M.; Pongratz, D.; Reimers, C.; Gerbitz, K.; Paetzke, I.; Deufel, T.; Hübner, G. Myoadenylate deaminase deficiency: Successful symptomatic therapy by high dose oral administration of ribose. Klin. Wochenschr. 1986, 64, 1281–1290. [Google Scholar] [CrossRef] [PubMed]
- Hellsten-Westing, Y. Immunohistochemical localization of xanthine oxidase in human cardiac and skeletal muscle. Histochemistry 1993, 100, 215–222. [Google Scholar] [CrossRef] [PubMed]
- Thomson, W.H.S.; Smith, I. X-linked recessive (Duchenne) muscular dystrophy (DMD) and purine metabolism: Effects of oral allopurinol and adenylate. Metabolism 1978, 27, 151–163. [Google Scholar] [CrossRef]
- Castro-Gago, M.; Lojo, S.; Novo, I.; del Río, R.; Peña, J.; Rodriguez-Segade, S. Effects of chronic allopurinol therapy on purine metabolism in Duchenne muscular dystrophy. Biochem. Biophys. Res. Commun. 1987, 147, 152–157. [Google Scholar] [CrossRef]
- Camiña, F.; Novo-Rodriguez, M.I.; Rodriguez-Segade, S.; Castro-Gago, M. Purine and carnitine metabolism in muscle of patients with Duchenne muscular dystrophy. Clin. Chim. Acta 1995, 243, 151–164. [Google Scholar] [CrossRef]
- Thomson, W.; Smith, I. Allopurinol in Duchenne’S muscular dystrophy. N. Engl. J. Med. 1978, 299, 101. [Google Scholar] [PubMed]
- De Bruyn, C.; Kulakowski, S.; van Bennekom, C.; Renoirte, P.; Müller, M. Purine metabolism in Duchenne muscular dystrophy. Adv. Exp. Med. Biol. 1980, 122, 177. [Google Scholar]
- Kulakowski, S.; Renoirte, P.; de Bruyn, C. Dynamometric and biochemical observations in Duchenne patients receiving allopurinol. Neuropediatrics 1981, 12, 92. [Google Scholar] [PubMed]
- Tamari, H.; Ohtani, Y.; Higashi, A.; Miyoshino, S.; Matsuda, I. Xanthine oxidase inhibitor in Duchenne muscular dystrophy. Brain Dev. 1982, 4, 137–143. [Google Scholar] [CrossRef]
- Bakouche, P.; Chaouat, D.; Nick, J. Allopurinol not effective in muscular dystrophy. N. Engl. J. Med. 1979, 301, 785. [Google Scholar] [PubMed]
- Mendell, J.R.; Wiechers, D.O. Lack of benefit of allopurinol in Duchenne dystrophy. Muscle Nerve 1979, 2, 53–56. [Google Scholar] [CrossRef] [PubMed]
- Doriguzzi, C.; Bertolotto, A.; Ganzit, G.P.; Mongini, T.; Palmucci, L. Ineffectiveness of allopurinol in Duchenne muscular dystrophy. Muscle Nerve 1981, 4, 176–178. [Google Scholar] [PubMed]
- Hunter, J.R.; Galloway, J.R.; Brooke, M.M.; Kutner, M.H.; Rudman, D.; Vogel, R.L.; Wardlaw, C.F.; Gerron, G.G. Effects of allopurinol in Duchenne’S muscular dystrophy. Arch. Neurol. 1983, 40, 294. [Google Scholar] [CrossRef] [PubMed]
- Stern, L.M.; Fewings, J.D.; Bretag, A.H.; Ballard, F.J.; Tomas, F.M.; Cooper, D.M.; Goldblatt, E. The progression of Duchenne muscular dystrophy: Clinical trial of allopurinol therapy. Neurology 1981, 31, 422. [Google Scholar] [CrossRef] [PubMed]
- Bertorini, T.E.; Palmieri, G.M.A.; Griffin, J.; Chesney, C.; Pifer, D.; Verling, L.; Airozo, D.; Fox, I.H. Chronic allopurinol and adenine therapy in Duchenne muscular dystrophy: Effects on muscle function, nucleotide degradation, and muscle ATP and ADP content. Neurology 1985, 35, 61. [Google Scholar] [CrossRef] [PubMed]
- Ziter, F.A.; Allsop, K.G.; Tyler, F.H. Assessment of muscle strength in Duchenne muscular dystrophy. Neurology 1977, 27, 981–984. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rybalka, E.; Timpani, C.A.; Stathis, C.G.; Hayes, A.; Cooke, M.B. Metabogenic and Nutriceutical Approaches to Address Energy Dysregulation and Skeletal Muscle Wasting in Duchenne Muscular Dystrophy. Nutrients 2015, 7, 9734-9767. https://doi.org/10.3390/nu7125498
Rybalka E, Timpani CA, Stathis CG, Hayes A, Cooke MB. Metabogenic and Nutriceutical Approaches to Address Energy Dysregulation and Skeletal Muscle Wasting in Duchenne Muscular Dystrophy. Nutrients. 2015; 7(12):9734-9767. https://doi.org/10.3390/nu7125498
Chicago/Turabian StyleRybalka, Emma, Cara A. Timpani, Christos G. Stathis, Alan Hayes, and Matthew B. Cooke. 2015. "Metabogenic and Nutriceutical Approaches to Address Energy Dysregulation and Skeletal Muscle Wasting in Duchenne Muscular Dystrophy" Nutrients 7, no. 12: 9734-9767. https://doi.org/10.3390/nu7125498
APA StyleRybalka, E., Timpani, C. A., Stathis, C. G., Hayes, A., & Cooke, M. B. (2015). Metabogenic and Nutriceutical Approaches to Address Energy Dysregulation and Skeletal Muscle Wasting in Duchenne Muscular Dystrophy. Nutrients, 7(12), 9734-9767. https://doi.org/10.3390/nu7125498