Metazoan Remaining Genes for Essential Amino Acid Biosynthesis: Sequence Conservation and Evolutionary Analyses


Abstract
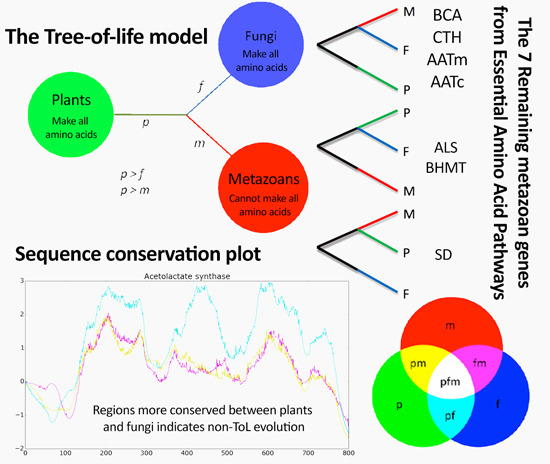
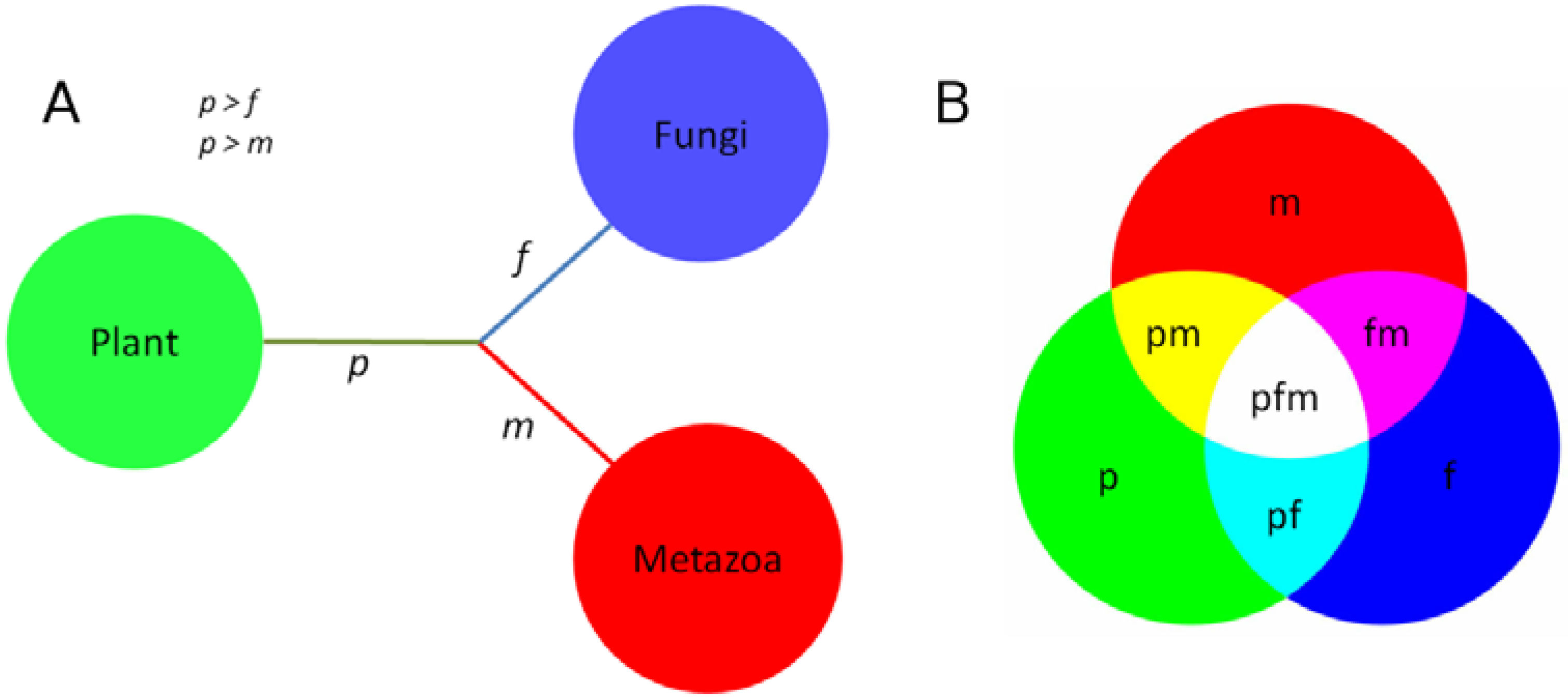
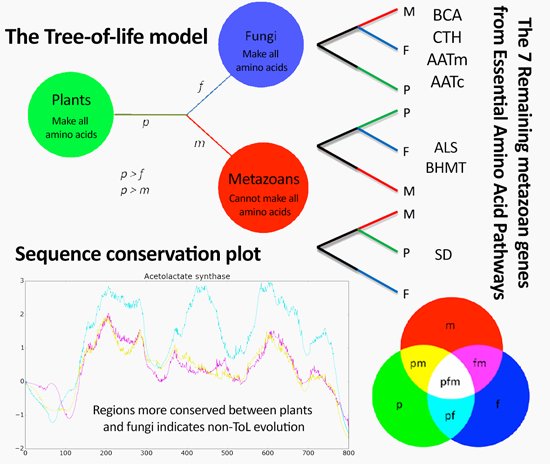
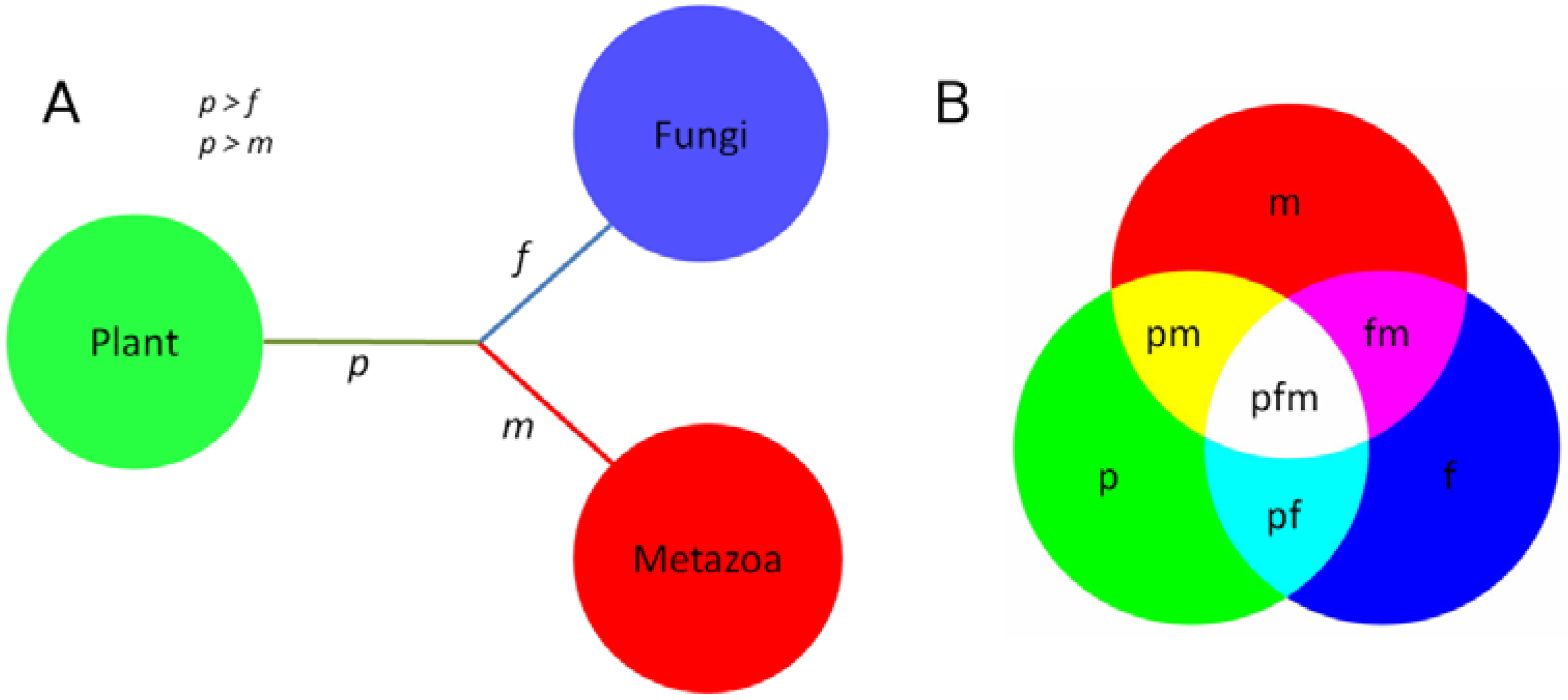
:
1. Introduction
2. Experimental Section
2.1. Database Searches for Genes Involved in Essential Amino Acid Biosynthesis
2.2. Curation of Metazoan Homologs of Yeast EAA Biosynthetic Enzymes
2.3. Phylogenetic Analysis of ReGens
2.4. Conservation Pattern
2.5. Back Translation
2.6. Ka/Ks Estimation Using PAML
3. Results
3.1. Finding and Describing Human ReGens

{kind=link}
{kind=link}
{kind=link}
| Enzyme Name | Acronym | Yeast ID | Human ID | EC Number | EAA | Pathway Functions |
|---|---|---|---|---|---|---|
| Acetolactate synthase | ALS | P07342.1 | NP_006835.2 | 2.2.1.6 | Val, Leu, Ile | Second reaction of the branched-chain amino acid biosynthesis pathway |
| Betaine-homocysteine S-methyltransferase | BHMT | Q12525.1 | NP_001704.2 | Met | Last reaction of the Met biosynthesis pathway | |
| 2.1.1.10 | ||||||
| Branched-chain-amino acid aminotransferase cytosolic | BCA | P47176.1 | NP_005495.2 | 2.6.1.42 | Val, Leu, Ile | Last reaction of the branched-chain amino acid biosynthesis pathway |
| Saccharopine dehydrogenase | SD | P38999.1 | NP_005754.2 | 1.5.1.7 | Lys | Last reaction of the Lys biosynthesis pathway |
| Cystathionine gamma-lyase | CTH | P31373.2 | NP_001893.2 | 4.4.1.1 | Met | Biosynthesis of Cys and Met |
| Aspartate aminotransferase, mitochondrial | AATm | Q01802.2 | NP_002071.2 | 2.6.1.1 | Phe | Ala, Asp, Glu, Cys, Met, Arg, Pro, Tyr, and Phe metabolism |
| Aspartate aminotransferase, cytoplasmic | AATc | P23542.3 | NP_002070.1 | 2.6.1.1 | Phe | Ala, Asp, Glu, Cys, Met, Arg, Pro, Tyr, and Phe metabolism |
| Aromatic/aminoadipate aminotransferase 1 | AadAT * | P53090 | NP_057312.1 | 2.6.1.39 | Lys | Fifth reaction of the Lys biosynthesis pathway |
3.2. Known Functions of the SD, BHMT and ALS Proteins
3.3. Conservation of Protein Sequences between Clades
| Organism Name | Clade |
|---|---|
| Neurospora crassa OR74A | Fungi |
| Pyrenophora tritici-repentis | Fungi |
| Sclerotinia sclerotiorum 1980 UF-70 | Fungi |
| Schizosaccharomyces pombe | Fungi |
| Gibberella zeae PH-1 | Fungi |
| Aspergillus niger | Fungi |
| Fusarium oxysporum f. sp. lycopersici | Fungi |
| Puccinia graminis f. sp. Tritici | Fungi |
| Saccharomyces cerevisiae S288c | Fungi |
| Ustilago maydis 521 | Fungi |
| Homo sapiens | Metazoan |
| Pan troglodytes | Metazoan |
| Mus musculus | Metazoan |
| Monodelphis domestica | Metazoan |
| Taeniopygia guttata | Metazoan |
| Anolis carolinensis | Metazoan |
| Xenopus tropicalis | Metazoan |
| Danio rerio | Metazoan |
| Drosophila melanogaster | Metazoan |
| Ciona intestinalis | Metazoan |
| Caenorhabditis elegans | Metazoan |
| Solanum tuberosum | Plant |
| Vitis vinifera | Plant |
| Populus trichocarpa | Plant |
| Arabidopsis thaliana | Plant |
| Physcomitrella patens | Plant |
| Chlamydomonas reinhardtii | Plant |
| Selaginella moellendorffii | Plant |
| Oryza sativa | Plant |
| Sorghum bicolor | Plant |
| Zea mays | Plant |
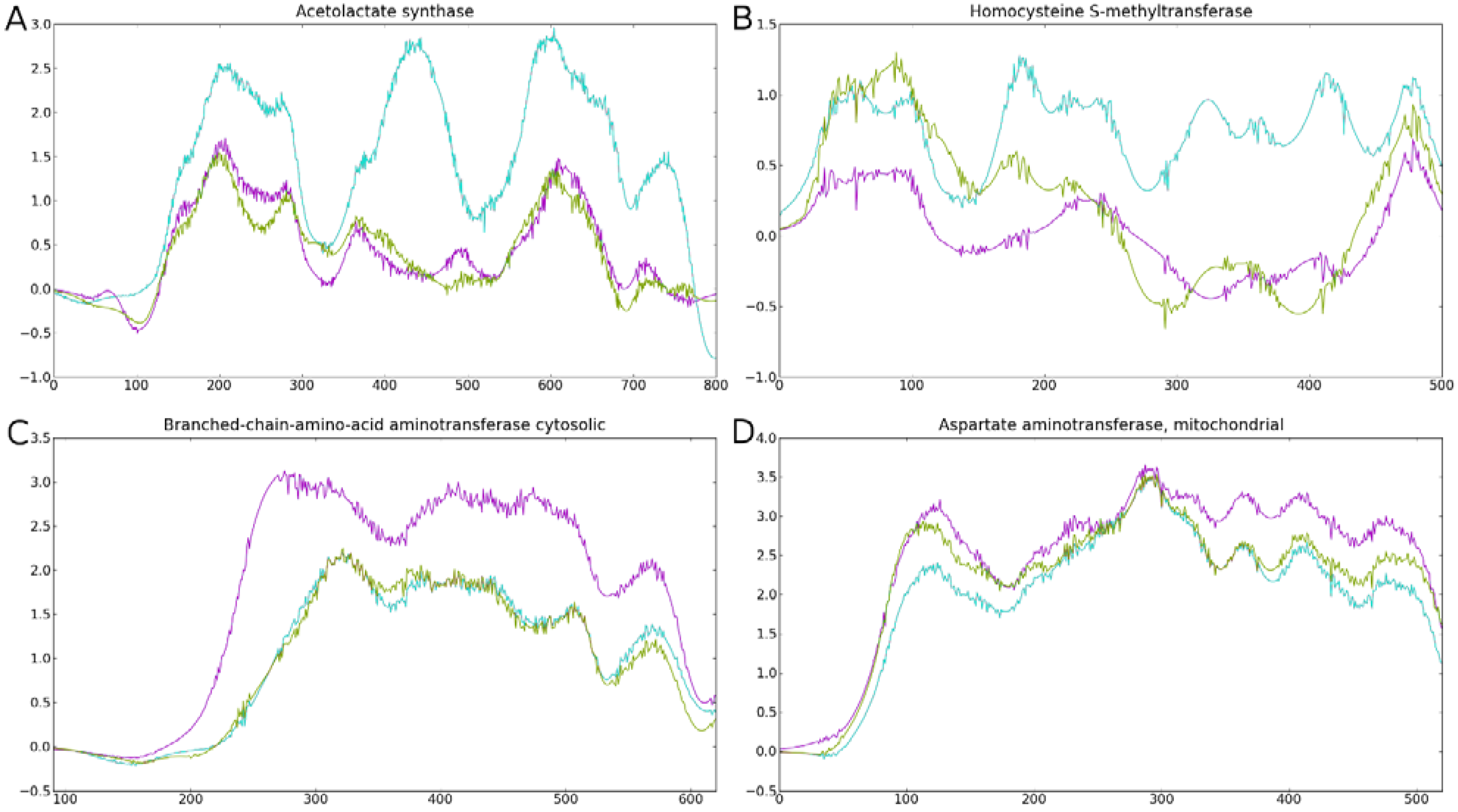
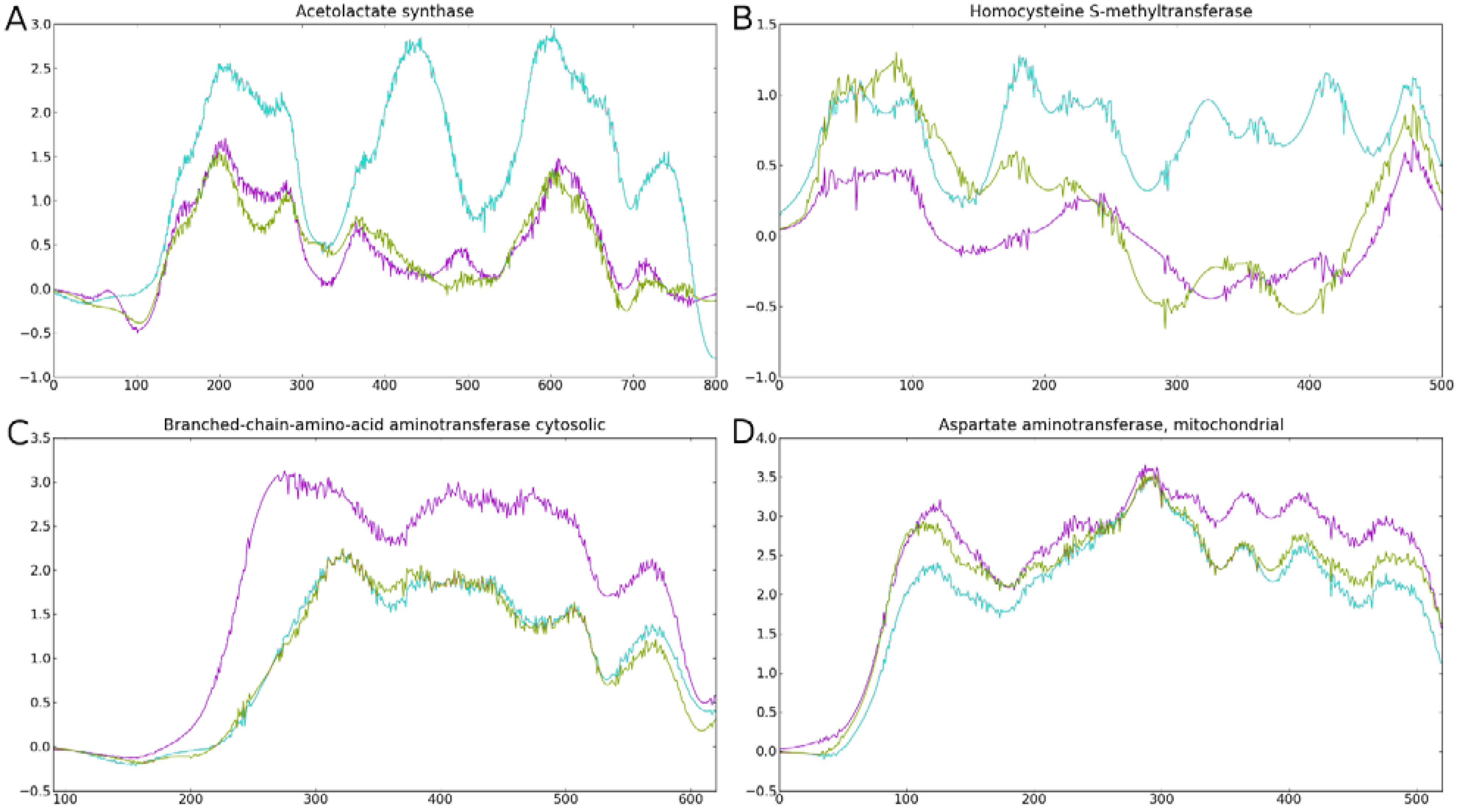
3.4. Phylogenies of ReGens Using Maximum Likelihood
| Topology Name | Topology Schema | ReGen | mf/fp Branch Length Ratio |
|---|---|---|---|
| ToL-like topology |  | BCA | 0.17 |
| CTH | 0.31 | ||
| AATm | 0.34 | ||
| AATc | 1.30 | ||
| Autotrophic paraphyly |  | ALS | 3.80 |
| BHMT | 3.59 | ||
| Fungi as outgroup |  | SD | 0.73 |
3.5. Synonymous and Non-Synonymous Mutation Rates
| ReGen | Clades Compared | Color Code | Ka | Ks |
|---|---|---|---|---|
| ALS | fp |  | 0.58 | 57.29 |
| mf |  | 1.15 | 37.93 | |
| mp |  | 1.26 | 28.65 | |
| BHMT | fp | | 0.89 | 54.54 |
| mf | | 1.74 | 31.84 | |
| mp | | 1.45 | 36.84 | |
| BCA | fp | | 0.70 | 48.79 |
| mf | | 0.45 | 57.63 | |
| mp | | 0.70 | 61.02 | |
| SD | fp | | 0.64 | 35.62 |
| mf | | 0.66 | 44.35 | |
| mp | | 0.67 | 17.31 | |
| CTH | fp | | 0.74 | 48.33 |
| mf | | 0.53 | 53.39 | |
| mp | | 0.92 | 34.77 | |
| AATm | fp | | 0.46 | 64.12 |
| mf | | 0.34 | 61.14 | |
| mp | | 0.40 | 62.58 | |
| AATc | fp | | 0.47 | 65.63 |
| mf | | 0.43 | 56.60 | |
| mp | | 0.46 | 56.57 |
4. Discussion
| ReGen | Tree Topology * | Conservation Diagram | Ka/Ks Branch Test (p < 0.05) | Ka/Ks Clade Average > 1 for Metazoans |
|---|---|---|---|---|
| ALS | Non-ToL | Non-ToL | + | Yes |
| BHMT | Non-ToL | Non-ToL | + | Yes |
| BCA | ToL | ToL | − | No |
| SD | FO | ToL | − | No |
| CTH | ToL | ToL | − | No |
| AATm | ToL | ToL | + | No |
| AATc | ToL | ToL | + | No |
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgment
Author Contributions
Conflicts of Interest
References
- Rose, W.C. The amino acids in nutrition. Yale J. Biol. Med. 1932, 4, 519–536. [Google Scholar] [PubMed]
- Guedes, R.L.; Prosdocimi, F.; Fernandes, G.R.; Moura, L.K.; Ribeiro, H.A.; Ortega, J.M. Amino acids biosynthesis and nitrogen assimilation pathways: A great genomic deletion during eukaryotes evolution. BMC Genomics 2011, 12, S2. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Montes, G.; Diaz-Mejia, J.J.; Perez-Rueda, E.; Segovia, L. The hidden universal distribution of amino acid biosynthetic networks: A genomic perspective on their origins and evolution. Genome Biol. 2008, 9, R95. [Google Scholar] [CrossRef] [PubMed]
- Starcevic, A.; Akthar, S.; Dunlap, W.C.; Shick, J.M.; Hranueli, D.; Cullum, J.; Long, P.F. Enzymes of the shikimic acid pathway encoded in the genome of a basal metazoan, nematostella vectensis, have microbial origins. Proc. Natl. Acad. Sci. USA 2008, 105, 2533–2537. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J. Evolution by gene duplication: An update. Trends Ecol. Evol. 2003, 18, 292–298. [Google Scholar] [CrossRef]
- Lynch, M.; Conery, J.S. The evolutionary fate and consequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Kondrashov, F.A.; Rogozin, I.B.; Wolf, Y.I.; Koonin, E.V. Selection in the evolution of gene duplications. Genome Biol. 2002, 3, RESEARCH0008. [Google Scholar] [CrossRef] [PubMed]
- Prosdocimi, F.; Mudado, M.A.; Ortega, J.M. A set of amino acids found to occur more frequently in human and fly than in plant and yeast proteomes consists of non-essential amino acids. Comput. Biol. Med. 2007, 37, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Santana-Santos, L.; Prosdocimi, F.; Ortega, J.M. Essential amino acid usage and evolutionary nutrigenomics of eukaryotes—Insights into the differential usage of amino acids in protein domains and extra-domains. Genet. Mol. Res. 2008, 7, 839–852. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- The UniProt Consortium. Reorganizing the protein space at the universal protein resource (UniProt). Nucleic Acids Res. 2012, 40, D71–D75. [Google Scholar]
- Scheer, M.; Grote, A.; Chang, A.; Schomburg, I.; Munaretto, C.; Rother, M.; Sohngen, C.; Stelzer, M.; Thiele, J.; Schomburg, D. BRENDA, the enzyme information system in 2011. Nucleic Acids Res. 2011, 39, D670–D676. [Google Scholar] [CrossRef] [PubMed]
- Kanehisa, M. The KEGG database. Novartis Found. Symp. 2002, 247, 91–101. [Google Scholar] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. Muscle: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.T.; Taylor, W.R.; Thornton, J.M. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992, 8, 275–282. [Google Scholar] [PubMed]
- Hunter, J.D. Matplotlib: A 2D graphics environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Brown, G.R.; Maglott, D.R. NCBI reference sequences (RefSeq): Current status, new features and genome annotation policy. Nucleic Acids Res. 2012, 40, D130–D135. [Google Scholar] [CrossRef] [PubMed]
- Apweiler, R.; Bairoch, A.; Wu, C.H. Protein sequence databases. Curr. Opin. Chem. Biol. 2004, 8, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Gene-Ontology-Consortium. The gene ontology: Enhancements for 2011. Nucleic Acids Res. 2012, 40, D559–D564. [Google Scholar]
- Marchler-Bauer, A.; Lu, S.; Anderson, J.B.; Chitsaz, F.; Derbyshire, M.K.; DeWeese-Scott, C.; Fong, J.H.; Geer, L.Y.; Geer, R.C.; Gonzales, N.R.; et al. CDD: A conserved domain database for the functional annotation of proteins. Nucleic Acids Res. 2011, 39, D225–D229. [Google Scholar] [CrossRef] [PubMed]
- Sacksteder, K.A.; Biery, B.J.; Morrell, J.C.; Goodman, B.K.; Geisbrecht, B.V.; Cox, R.P.; Gould, S.J.; Geraghty, M.T. Identification of the alpha-aminoadipic semialdehyde synthase gene, which is defective in familial hyperlysinemia. Am. J. Hum. Genet. 2000, 66, 1736–1743. [Google Scholar] [CrossRef] [PubMed]
- Markovitz, P.J.; Chuang, D.T. The bifunctional aminoadipic semialdehyde synthase in lysine degradation. Separation of reductase and dehydrogenase domains by limited proteolysis and column chromatography. J. Biol. Chem. 1987, 262, 9353–9358. [Google Scholar] [PubMed]
- Serrano, G.C.; Rezende e Silva Figueira, T.; Kiyota, E.; Zanata, N.; Arruda, P. Lysine degradation through the saccharopine pathway in bacteria: LKR and SDH in bacteria and its relationship to the plant and animal enzymes. FEBS Lett. 2012, 586, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, J.D.; Martin, J.J. Homocysteine. Int. J. Biochem. Cell Biol. 2000, 32, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, D.W. Homocysteine and vitamins in cardiovascular disease. Clin. Chem. 1998, 44, 1833–1843. [Google Scholar] [PubMed]
- McCully, K.S. Vascular pathology of homocysteinemia: Implications for the pathogenesis of arteriosclerosis. Am. J. Pathol. 1969, 56, 111–128. [Google Scholar] [PubMed]
- Blom, H.J.; Smulders, Y. Overview of homocysteine and folate metabolism. With special references to cardiovascular disease and neural tube defects. J. Inherit. Metab. Dis. 2011, 34, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, I.S.; Park, E.; Ballman, K.V.; Berger, P.; Nunn, M.; Suh, D.S.; Breksa, A.P., III; Garrow, T.A.; Rozen, R. Investigations of a common genetic variant in betaine-homocysteine methyltransferase (BHMT) in coronary artery disease. Atherosclerosis 2003, 167, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Heil, S.G.; Lievers, K.J.; Boers, G.H.; Verhoef, P.; den Heijer, M.; Trijbels, F.J.; Blom, H.J. Betaine-homocysteine methyltransferase (BHMT): Genomic sequencing and relevance to hyperhomocysteinemia and vascular disease in humans. Mol. Genet. Metab. 2000, 71, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Morin, I.; Platt, R.; Weisberg, I.; Sabbaghian, N.; Wu, Q.; Garrow, T.A.; Rozen, R. Common variant in betaine-homocysteine methyltransferase (BHMT) and risk for spina bifida. Am. J. Med. Genet. A 2003, 119A, 172–176. [Google Scholar] [CrossRef]
- Zhu, H.; Curry, S.; Wen, S.; Wicker, N.J.; Shaw, G.M.; Lammer, E.J.; Yang, W.; Jafarov, T.; Finnell, R.H. Are the betaine-homocysteine methyltransferase (BHMT and BHMT2) genes risk factors for spina bifida and orofacial clefts? Am. J. Med. Genet. A 2005, 135, 274–277. [Google Scholar] [CrossRef] [PubMed]
- Smach, M.A.; Jacob, N.; Golmard, J.L.; Charfeddine, B.; Lammouchi, T.; Ben Othman, L.; Dridi, H.; Bennamou, S.; Limem, K. Folate and homocysteine in the cerebrospinal fluid of patients with Alzheimer’s disease or dementia: A case control study. Eur. Neurol. 2011, 65, 270–278. [Google Scholar] [CrossRef] [PubMed]
- Morris, M.S. Homocysteine and Azheimer’s disease. Lancet Neurol. 2003, 2, 425–428. [Google Scholar] [CrossRef] [PubMed]
- Teng, Y.W.; Mehedint, M.G.; Garrow, T.A.; Zeisel, S.H. Deletion of betaine-homocysteine S-methyltransferase in mice perturbs choline and 1-carbon metabolism, resulting in fatty liver and hepatocellular carcinomas. J. Biol. Chem. 2011, 286, 36258–36267. [Google Scholar] [CrossRef] [PubMed]
- Collinsova, M.; Strakova, J.; Jiracek, J.; Garrow, T.A. Inhibition of betaine-homocysteine S-methyltransferase causes hyperhomocysteinemia in mice. J. Nutr. 2006, 136, 1493–1497. [Google Scholar] [PubMed]
- Pajares, M.A.; Perez-Sala, D. Betaine homocysteine S-methyltransferase: Just a regulator of homocysteine metabolism? Cell. Mol. Life Sci. 2006, 63, 2792–2803. [Google Scholar] [CrossRef] [PubMed]
- Thomas, D.; Becker, A.; Surdin-Kerjan, Y. Reverse methionine biosynthesis from S-adenosylmethionine in eukaryotic cells. J. Biol. Chem. 2000, 275, 40718–40724. [Google Scholar] [CrossRef] [PubMed]
- Ranocha, P.; Bourgis, F.; Ziemak, M.J.; Rhodes, D.; Gage, D.A.; Hanson, A.D. Characterization and functional expression of cDNAs encoding methionine-sensitive and -insensitive homocysteine S-methyltransferases from arabidopsis. J. Biol. Chem. 2000, 275, 15962–15968. [Google Scholar] [CrossRef] [PubMed]
- Szegedi, S.S.; Castro, C.C.; Koutmos, M.; Garrow, T.A. Betaine-homocysteine S-methyltransferase-2 is an S-methylmethionine-homocysteine methyltransferase. J. Biol. Chem. 2008, 283, 8939–8945. [Google Scholar] [CrossRef] [PubMed]
- Chaleff, R.S.; Mauvais, C.J. Acetolactate synthase is the site of action of two sulfonylurea herbicides in higher plants. Science 1984, 224, 1443–1445. [Google Scholar] [CrossRef] [PubMed]
- Saxena, P.K.; King, J. Herbicide resistance in Datura innoxia: Cross-resistance of sulfonylurea-resistant cell lines to imidazolinones. Plant Physiol. 1988, 86, 863–867. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, M.V.; Hung, H.Y.; Dias, J.M.; Miner, V.W.; Butler, J.H.; Jachetta, J.J. Properties of mutant acetolactate synthases resistant to triazolopyrimidine sulfonanilide. Plant Physiol. 1990, 94, 239–244. [Google Scholar] [CrossRef] [PubMed]
- Pang, R.G.D.A.S.S. Acetohydroxyacid synthase. J. Biochem. Mol. Biol. 2000, 33, 1–36. [Google Scholar]
- Joutel, A.; Ducros, A.; Alamowitch, S.; Cruaud, C.; Domenga, V.; Marechal, E.; Vahedi, K.; Chabriat, H.; Bousser, M.G.; Tournier-Lasserve, E. A human homolog of bacterial acetolactate synthase genes maps within the cadasil critical region. Genomics 1996, 38, 192–198. [Google Scholar] [CrossRef] [PubMed]
- Duggleby, R.G.; Kartikasari, A.E.R.; Wunsch, R.M.; Lee, Y.T.; Kil, M.W.; Shin, J.Y.; Chang, S.I. Expression in Escherichia coli of a putative human acetohydroxyacid synthase. J. Biochem. Mol. Biol. 2000, 33, 195–201. [Google Scholar]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. Weblogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Hurst, L.D. The Ka/Ks ratio: Diagnosing the form of sequence evolution. Trends Genet. 2002, 18, 486–487. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Liu, F.; Wang, L.; Huang, S.; Yu, J. Nonsynonymous substitution rate (Ka) is a relatively consistent parameter for defining fast-evolving and slow-evolving protein-coding genes. Biol. Direct. 2011, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Arumugam, M.; Raes, J.; Pelletier, E.; le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Hansen, A.K.; Moran, N.A. Aphid genome expression reveals host-symbiont cooperation in the production of amino acids. Proc. Natl. Acad Sci. USA 2011, 108, 2849–2854. [Google Scholar] [CrossRef] [PubMed]
- McCourt, J.A.; Pang, S.S.; King-Scott, J.; Guddat, L.W.; Duggleby, R.G. Herbicide-binding sites revealed in the structure of plant acetohydroxyacid synthase. Proc. Natl. Acad. Sci. USA 2006, 103, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Furnham, N.; Holliday, G.L.; de Beer, T.A.; Jacobsen, J.O.; Pearson, W.R.; Thornton, J.M. The catalytic site atlas 2.0: Cataloging catalytic sites and residues identified in enzymes. Nucleic Acids Res. 2014, 42, D485–D489. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Costa, I.R.; Thompson, J.D.; Ortega, J.M.; Prosdocimi, F. Metazoan Remaining Genes for Essential Amino Acid Biosynthesis: Sequence Conservation and Evolutionary Analyses. Nutrients 2015, 7, 1-16. https://doi.org/10.3390/nu7010001
Costa IR, Thompson JD, Ortega JM, Prosdocimi F. Metazoan Remaining Genes for Essential Amino Acid Biosynthesis: Sequence Conservation and Evolutionary Analyses. Nutrients. 2015; 7(1):1-16. https://doi.org/10.3390/nu7010001
Chicago/Turabian StyleCosta, Igor R., Julie D. Thompson, José Miguel Ortega, and Francisco Prosdocimi. 2015. "Metazoan Remaining Genes for Essential Amino Acid Biosynthesis: Sequence Conservation and Evolutionary Analyses" Nutrients 7, no. 1: 1-16. https://doi.org/10.3390/nu7010001
APA StyleCosta, I. R., Thompson, J. D., Ortega, J. M., & Prosdocimi, F. (2015). Metazoan Remaining Genes for Essential Amino Acid Biosynthesis: Sequence Conservation and Evolutionary Analyses. Nutrients, 7(1), 1-16. https://doi.org/10.3390/nu7010001