Vitamin E Supplementation in Chemical Colorectal Carcinogenesis: A Two-Edged Knife

Abstract
:1. Introduction
2. Experimental Section
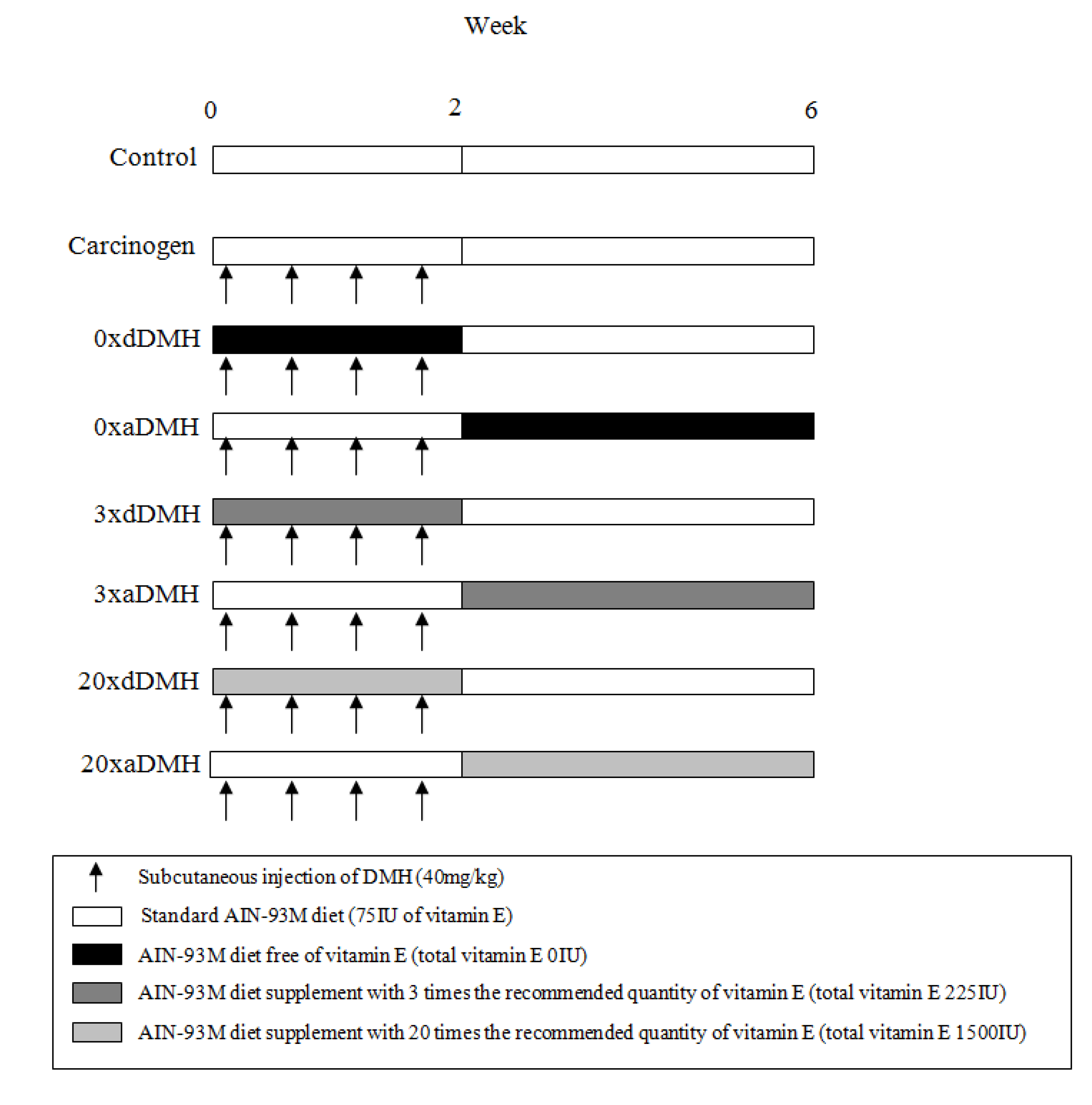
2.1. Protocol for Experimental Tumor Induction
2.2. Diets

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredients | Quantity (%) |
|---|---|
| Cornstarch | 62.25 |
| Casein | 14 |
| Soy Oil | 4 |
| Sucrose | 10 |
| Cellulose | 5 |
| Vitamin Mixture | 1 |
| Mineral Mixture | 3.5 |
| Choline Chloride | 0.25 |
| Composition | ATA Free | Standard | 3× ATA | 20× ATA |
|---|---|---|---|---|
| Folic Acid | 200 mg | 200 mg | 200 mg | 200 mg |
| Nicotinic acid | 3000 mg | 3000 mg | 3000 mg | 3000 mg |
| Biotin | 20 mg | 20 mg | 20 mg | 20 mg |
| Calcium pantothenate | 1600 mg | 1600 mg | 1600 mg | 1600 mg |
| Pyridoxine.HCl | 700 mg | 700 mg | 700 mg | 700 mg |
| Riboflavin | 600 mg | 600 mg | 600 mg | 600 mg |
| Thiamine.HCl | 600 mg | 600 mg | 600 mg | 600 mg |
| Vitamin A | 400,000 IU | 400,000 IU | 400,000 IU | 400,000 IU |
| Vitamin B12 | 2500 mcg | 2500 mcg | 2500 mcg | 2500 mcg |
| Vitamin D3 | 100,000 IU | 100,000 IU | 100,000 UI | 100,000 IU |
| Vitamin E (as dl-alpha tocopheryl acetate) | 0 IU | 7500 IU | 22,500 IU | 150,000 IU |
| Vitamin K1 | 75 mg | 75 mg | 75 mg | 75 mg |
| Boron | 14.26 mg | 14.26 mg | 14.26 mg | 14.26 mg |
| Calcium | 142.94 g | 142.94 g | 142.94 g | 142.94 g |
| Chloride | 44.9 g | 44.9 g | 44.9 g | 44.9 g |
| Copper | 72.41 mg | 72.41 mg | 72.41 mg | 72.41 mg |
| Chromium | 28.65 mg | 28.65 mg | 28.65 mg | 28.65 mg |
| Sulfur | 8.6 g | 8.6 g | 8.6 g | 8.6 g |
| Iron | 1000 mg | 1000 mg | 1000 mg | 1000 mg |
| Fluor | 28.72 mg | 28.72 mg | 28.72 mg | 28.72 mg |
| Phosphorus | 56.9 g | 56.9 g | 56.9 g | 56.9 g |
| Iodine | 5.93 mg | 5.93 mg | 5.93 mg | 5.93 mg |
| Lithium | 2.85 mg | 2.85 mg | 2.85 mg | 2.85 mg |
| Magnesium | 14.48 g | 14.48 g | 14.48 g | 14.48 g |
| Manganese | 300 mg | 300 mg | 300 mg | 300 mg |
| Molybdenum | 4.32 mg | 4.32 mg | 4.32 mg | 4.32 mg |
| Nickel | 14.31 mg | 14.31 mg | 14.31 mg | 14.31 mg |
| Potassium | 102.86 g | 102.86 g | 102.86 g | 102.86 g |
| Selenium | 4.28 mg | 4.28 mg | 4.28 mg | 4.28 mg |
| Silicon | 143.26 mg | 143.26 mg | 143.26 mg | 143.26 mg |
| Sodium | 29.38 mg | 29.38 mg | 29.38 mg | 29.38 mg |
| Vanadium | 2.87 mg | 2.87 mg | 2.87 mg | 2.87 mg |
| Zinc | 860 mg | 860 mg | 860 mg | 860 mg |
2.3. Analyses
2.3.1. Oxidative Stress Parameters
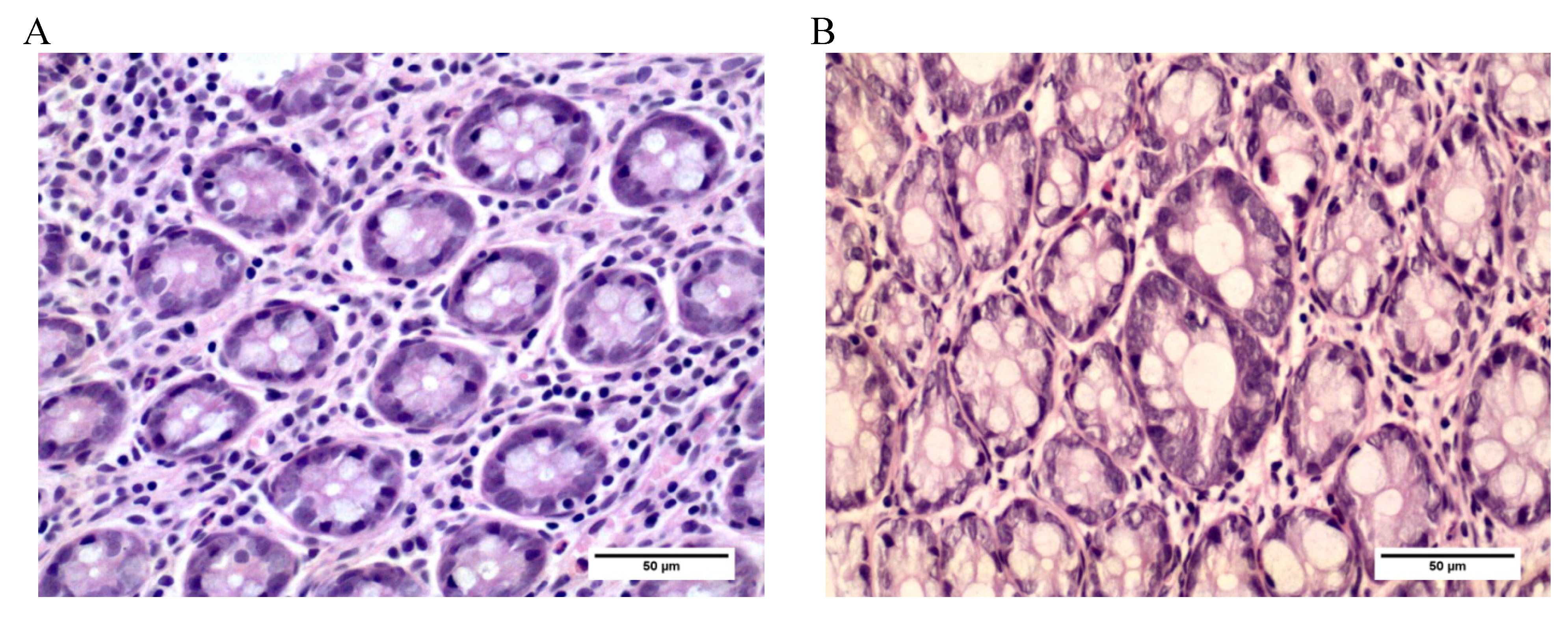
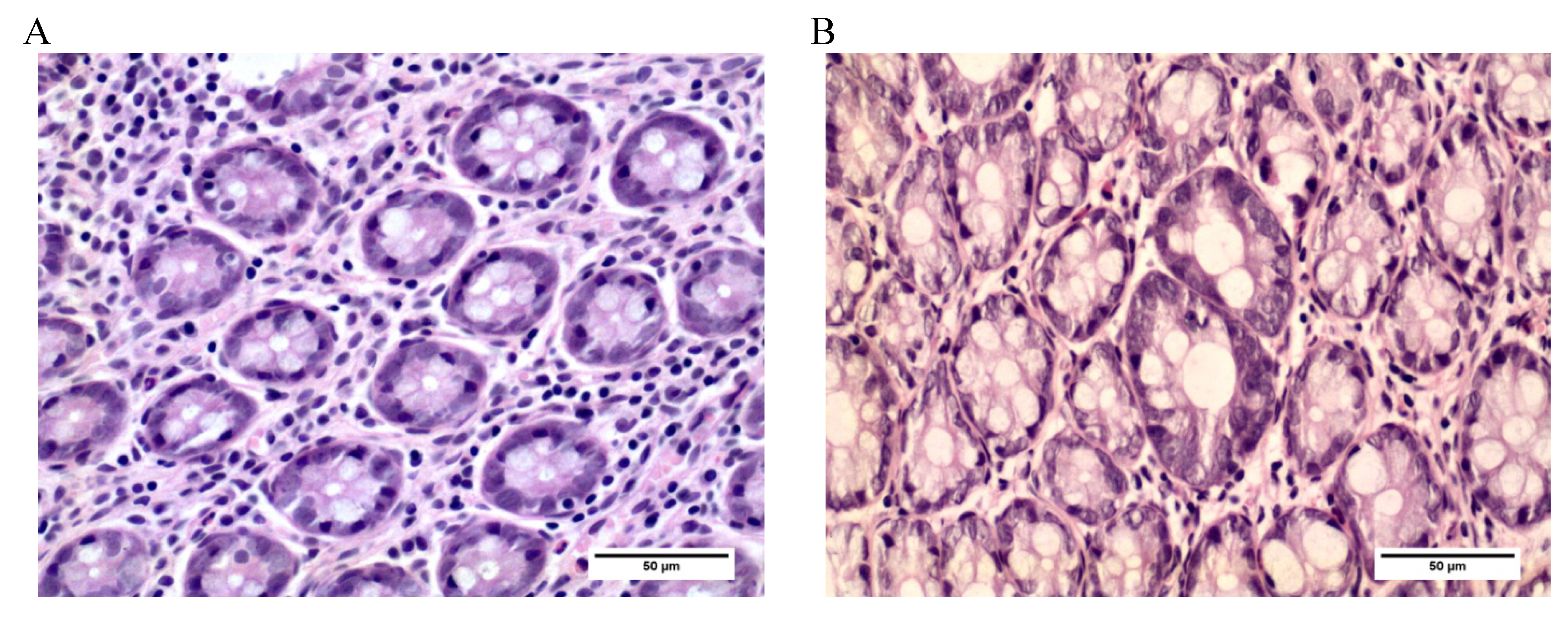
2.3.2. Aberrant Crypt Foci (ACF)
2.3.3. Immunohistochemistry
2.4. Statistical Analysis
3. Results
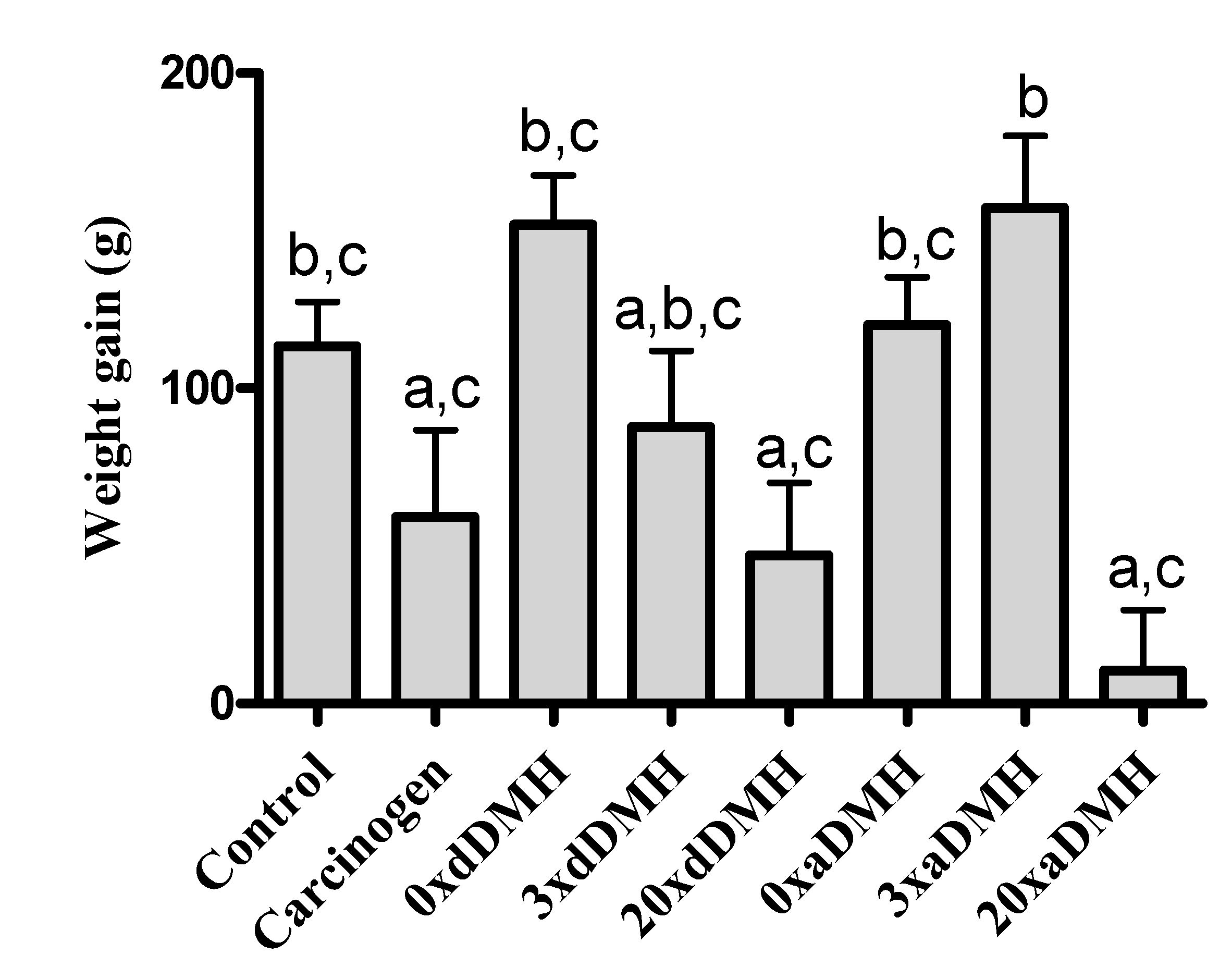
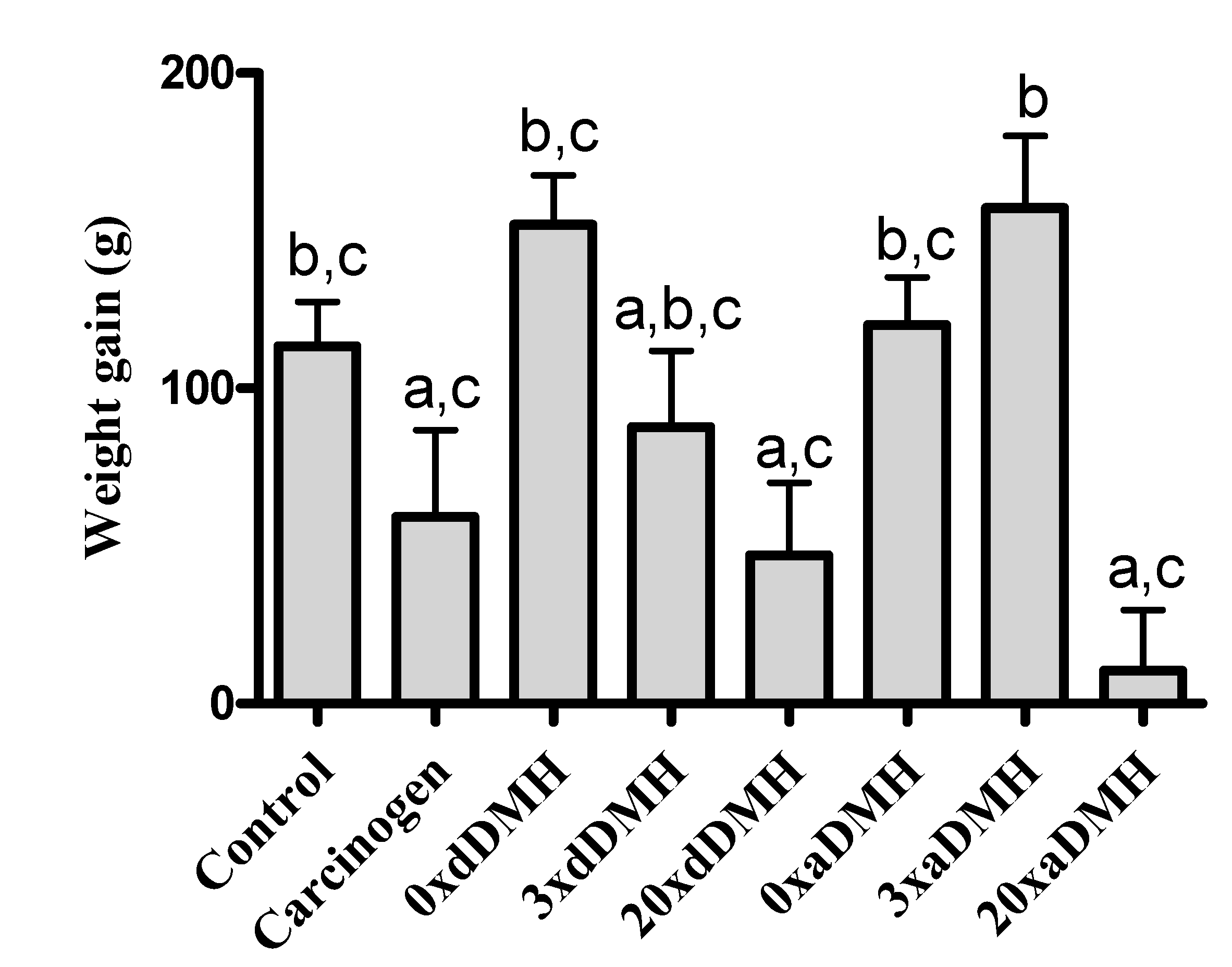
3.1. Daily Ingestion and Weight Gain
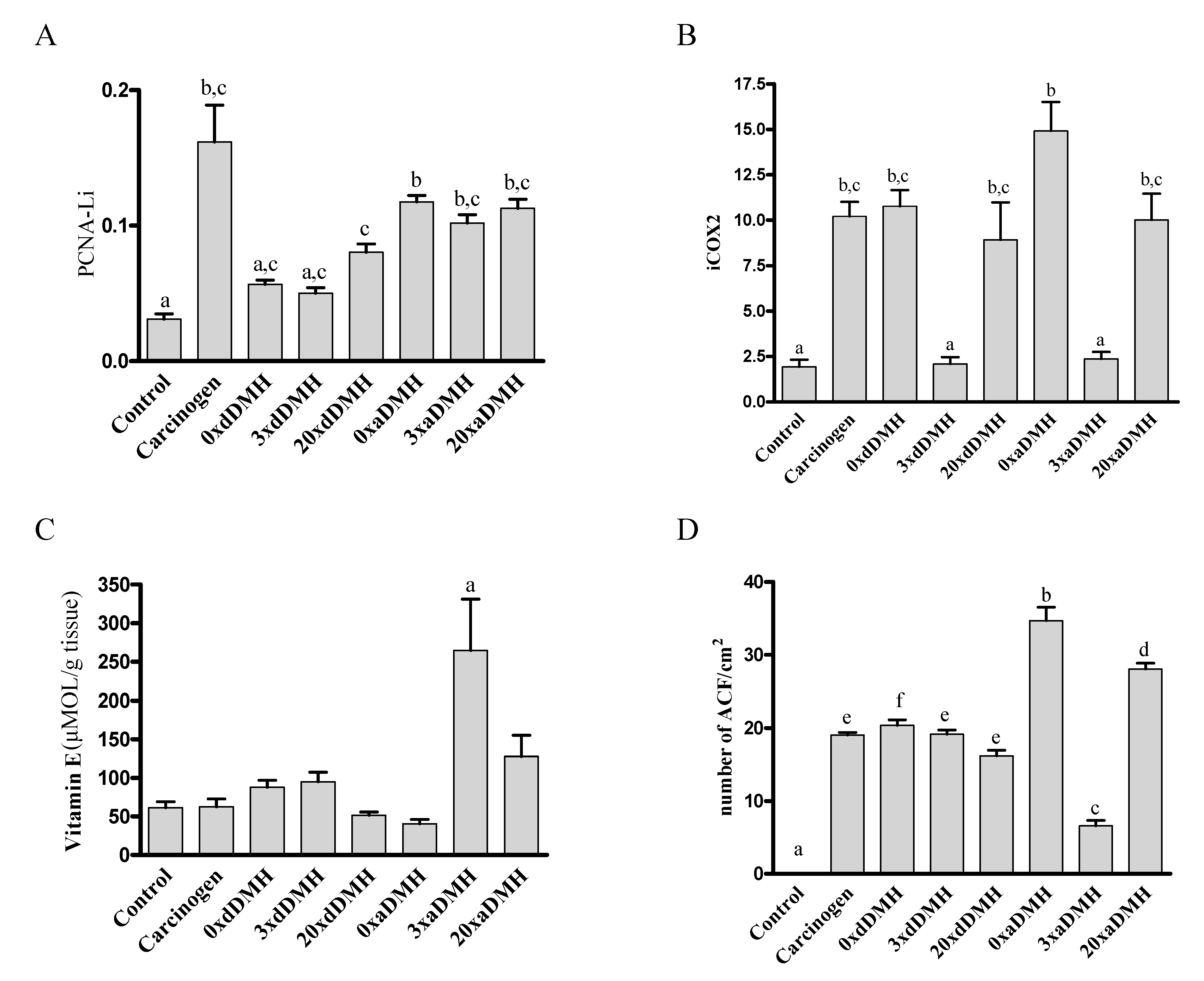
3.2. Oxidative Stress Parameters
3.3. Aberrant Crypt Foci
3.4. Immunohistochemistry Findings
3.5. Multivariate Analysis
| Analysis | Control | Carcinogen | During (dDMH) | After (aDMH) | ||||
|---|---|---|---|---|---|---|---|---|
| 0× | 3× | 20× | 0× | 3× | 20× | |||
| Serum VE (umol/L) | 18.37 ± 0.83 | 19.35 ± 1.75 | 11.83 ± 0.42 a,b,c | 9.50 ± 1.14 a,b,c | 16.85 ± 1.47 d,e | 7.03 ± 0.46 a,b,c | 11.85 ± 1.06 b | 19.80 ± 3.22 |
| GSH (umol/gptn) | 36.34 ± 1.97 | 45.95 ± 4.29 | 25.89 ± 1.99 b | 30.90 ± 2.53 b | 19.20 ± 1.45 a,b | 29.29 ± 3.66 | 26.37 ± 2.21 b | 28.72 ± 3.02 b |
| MDA (nmol/mgptn) | 0.13 ± 0.00 | 0.13 ± 0.01 | 0.17 ± 0.01 a,e | 0.12 ± 0.01 c | 0.15 ± 0.00 d | 0.19 ± 0.00 a,b,e | 0.15 ± 0.01 d | 0.160 ± 0.01 a,b |
| HCy (umol/L) | 6.00 ± 0.32 | 4.90 ± 0.93 | 6.65 ± 0.70 c | 4.20 ± 0.58 | 5.05 ± 0.36 | 5.55 ± 0.70 | 5.00 ± 0.40 | 3.90 ± 0.32 |
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69. [Google Scholar] [CrossRef]
- Kune, G.A.; Bannerman, S.; Watson, L.F. Attributable risk for diet, alcohol and family history in the Melbourne Colorectal Cancer Study. Nutr. Cancer 1992, 18, 231–235. [Google Scholar] [CrossRef]
- Slattery, M.L. Diet, lifestyle, and colon cancer. Semin. Gastrointest. Dis. 2000, 11, 142–146. [Google Scholar]
- Khlat, M. Cancer in Mediterranean migrants—Based on studies in France and Australia. Cancer Causes Control 1995, 6, 525–531. [Google Scholar] [CrossRef]
- Ingles, S.A.; Bird, C.L.; Shikany, J.M. Plasma tocopherol and prevalence of colorectal adenomas in a multiethnic population. Cancer Res. 1998, 58, 661–666. [Google Scholar]
- Bird, R.P. Role of aberrant crypt foci in understanding the pathogenesis of colon cancer. Cancer Lett. 1995, 93, 55–71. [Google Scholar] [CrossRef]
- Park, H.S.; Goodlad, R.A.; Wright, N.A. The incidence of aberrant crypt foci and colonic carcinoma in dimethylhydrazinetreated rats varies in a site-specific manner and depends on tumor histology. Cancer Res. 1997, 57, 4507–4510. [Google Scholar]
- Sequeira, J.L.; Kobayasi, S.K.; Rodrigues, M.A.M. Early and late effects of wound healing on development of colon tumors in a model of colon carcinogenesis by 1,2-dimethylhydrazine in the rat. Pathology 2000, 32, 250–252. [Google Scholar]
- Hall, P.A.; Coates, P.J. Assessment of cell proliferation in pathology—What next? Histopathology 1995, 26, 105–112. [Google Scholar] [CrossRef]
- Preston-Martin, S.; Pike, M.C.; Ross, R.K.; Jones, P.A.; Henderson, B.E. Increased cell division as a cause of human cancer. Cancer Res. 1990, 50, 7415–7421. [Google Scholar]
- Dixon, D.A.; Tolley, N.D.; King, P.H.; Nabors, L.B.; McIntyre, T.M.; Zimmerman, G.A.; Prescott, S.M. Altered expression of the mRNA stability factor HuR promotes cyclooxygenase-2 expression in colon cancer cells. J. Clin. Investig. 2001, 108, 1657–1665. [Google Scholar] [CrossRef]
- Yamauchi, T.; Watanabe, M.; Kubota, T.; Hasegawa, H.; Ishii, Y.; Endo, T.; Kabeshima, Y.; Yorozuya, K.; Yamamoto, K.; Mukai, M.; et al. Cyclooxygenase-2 expression as a new marker for patients with colorectal cancer. Dis. Colon Rectum 2002, 45, 98–103. [Google Scholar]
- Soumaoro, L.T.; Uetake, H.; Higuchi, T.; Takagi, Y.; Enomoto, M.; Sugihara, K. Cyclooxygenase-2 expression: A significant prognostic indicator for patients with colorectal cancer. Clin. Cancer Res. 2004, 10, 8465–8471. [Google Scholar]
- Sies, H. Strategies of antioxidant defense. Eur. J. Biochem. 1993, 215, 213–219. [Google Scholar] [CrossRef]
- Campbell, S.E.; Stone, W.L.; Whaley, S.G.; Qui, M.; Krishnan, K. Gamma (gamma) tocopherol upregulates peroxisome proliferator activated receptor (PPAR) gamma (gamma) expression in SW 480 human colon cancer cell lines. BMC Cancer 2003, 3, 25. [Google Scholar] [CrossRef]
- Dutta, A.; Dutta, S.K. Vitamin E and its role in the prevention of atherosclerosis and carcinogenesis: A review. J. Am. Coll. Nutr. 2003, 22, 258–268. [Google Scholar] [CrossRef]
- Sun, Y.; Ma, A.; Li, Y.; Han, X.; Wang, Q.; Liang, H. Vitamin E supplementation protects erythrocyte membranes from oxidative stress in healthy chinese middle-aged and elderly people. Nutr. Res. 2012, 32, 328–334. [Google Scholar] [CrossRef]
- Cardoso, J.F.R.; Cohen, C.; Jordão, A.A., Jr.; Vannucchi, H.; Garcia, S.B.; Zucoloto, S. Light and Moderate Doses of Ethanol in Chemical Carcinogenesis of the Colon in Rats. Nutr. Cancer 2011, 63, 1029–1035. [Google Scholar] [CrossRef]
- Reeves, P.G.; Nielsen, F.H.; Fahey, G.C., Jr. AIN-93 purified diets for laboratory rodents: Final report of the American Institute of Nutrition ad hoc writing committee on the reformulation of the AIN-76A rodent diet. J. Nutr. 1993, 123, 1939–1951. [Google Scholar]
- Sweetman, S.; Martindale, C. The Complete Drug Reference; Pharmaceutical Press: London, UK, 2009. [Google Scholar]
- Monsen, E.R. Dietary Reference Intakes for antioxidant nutrients: Vitamin C, Vitamin E, Selenium and Carotenoids. J. Am. Diet. Assoc. 2000, 100, 637–640. [Google Scholar] [CrossRef]
- Slover, H.T. Tocopherols in foods and fats. Lipids 1971, 6, 291–296. [Google Scholar] [CrossRef]
- Arnaud, J.; Fortis, I.; Blachier, S.; Kia, D.; Favier, A. Simultaneous determination of retinol, alpha-tocopherol and beta-carotene in serum by isocratic high-performance liquid chromatography. J. Chromatogr. 1991, 572, 103–116. [Google Scholar] [CrossRef]
- Sedlack, J.; Lindsay, R.H. Estimation of total, protein-bound, and non protein sulfhydryl groups in tissue with Ellman’s reagent. Anal. Biochem. 1968, 25, 192–205. [Google Scholar]
- Buege, J.A.; Aust, S.D. Microsomal lipid peroxidation. Methods Enzymol. 1978, 52, 302–310. [Google Scholar] [CrossRef]
- Azzi, A. Molecular mechanism of α-tocopherol action. Free Radic. Biol. Med. 2007, 43, 16–21. [Google Scholar] [CrossRef]
- Komninou, D.; Ayonote, A.; Richie, J.P., Jr.; Rigas, B. Insulin resistance and its contribution to colon carcinogenesis. Exp. Biol. Med. 2003, 228, 396–405. [Google Scholar]
- Meister, A.; Anderson, M.E. Glutathione. Annu. Rev. Biochem. 1983, 52, 711–760. [Google Scholar] [CrossRef]
- Davies, K.J.A. Oxidative stress, antioxidant defenses, and damage removal, repair, and replacement systems. IUBMB Life 2000, 50, 279–289. [Google Scholar] [CrossRef]
- Stocker, R.; Keaney, J.F., Jr. New insights on oxidative stress in the artery wall. J. Thromb. Haemost. 2005, 3, 1825–1834. [Google Scholar] [CrossRef]
- Meagher, E.A.; Barry, O.P.; Lawson, J.A.; Rokach, J.; FitzGerald, G.A. Effects of vitamin E on lipid peroxidation in healthy persons. J. Am. Med. Assoc. 2001, 285, 1178–1182. [Google Scholar]
- Bowry, V.W.; Ingold, K.; Stocker, R. Vitamin E in human low-density lipoprotein. When and how this antioxidant becomes a pro-oxidant. Biochem. J. 1992, 288, 341–344. [Google Scholar]
- Bowry, V.W.; Ingold, K.U. The unexpected role of vitamin E (a-tocopherol) in the peroxidation of human low-density lipoprotein. Acc. Chem. Res. 1999, 32, 27–34. [Google Scholar] [CrossRef]
- Cook, M.G.; McNamara, P. Effect of dietary vitamin E on dimethylhydrazine induced colonic tumors in mice. Cancer Res. 1980, 40, 1329–1331. [Google Scholar]
- Toth, B.; Patii, K. Enhancing effect of vitamin E on murine intestinal tumorigenesis by 1,2-dimethylhydrazine dihydrochloride. J. Natl. Cancer Inst. 1983, 70, 1107–1111. [Google Scholar]
- Sumiyoshi, H. Effects of vitamin E deficiency on 1,2-dimethylhydrazine-induced intestinal carcinogenesis in rats. Hiroshima J. Med. Sci. 1985, 34, 363–369. [Google Scholar]
- Kune, G.A. Causes and Control of Colorectal Cancer: A Model for Cancer Prevention; Kluwer: Boston, MA, USA, 1996. [Google Scholar]
- Lipkin, M. Biomarkers of increased susceptibility to gastrointestinal cancer: New application to studies of cancer prevention in human subjects. Cancer Res. 1988, 48, 235–245. [Google Scholar]
- Paganelli, G.M.; Biasco, G.; Brandi, G.; Santucci, R.; Gizzi, G.; Villani, V.; Miglioli, M.; Barbara, L. Effect of vitamin A, C, and E supplementation on rectal cell proliferation in patients with colorectal adenomas. J. Natl. Cancer Inst. 1992, 84, 47–51. [Google Scholar] [CrossRef]
- Masferrer, J.L.; Leahy, K.M.; Koki, A.T.; Zweifel, B.S.; Settle, S.L.; Woerner, B.M.; Edwards, D.A.; Flickinger, A.G.; Moore, R.J.; Siebert, K. Antiangiogenic and antitumor activities of cyclooxygenase-2 inhibitors. Cancer Res. 2000, 60, 1306–1311. [Google Scholar]
- Gysin, R.; Azzi, A.; Visarius, T. Gamma-tocopherol inhibits human cancer cell cycle progression and cell proliferation by down-regulation of cyclins. FASEB J. 2002, 16, 1952–1954. [Google Scholar]
- O’Leary, K.A.; de Pascual-Teresa, S.; Needs, P.W.; Bao, Y.P.; O’Brien, N.M.; Williamson, G. Effect of flavonoids and vitamin E on cyclooxygenase-2 (COX-2) transcription. Mutat. Res. 2004, 551, 245–254. [Google Scholar] [CrossRef]
- Starkebaum, G.; Harlan, J.M. Endothelial cell injury due to copper-catalysed hydrogen peroxide geration from homocysteine. J. Clin. Investig. 1986, 77, 1370–1376. [Google Scholar] [CrossRef]
- Crott, J.W.; Fenech, M. Preliminary study of the genotoxic potential of homocysteine in human lymphocytes in vitro. Mutagenesis 2001, 16, 213–217. [Google Scholar] [CrossRef]
- Sharma, R.A.; Gescher, A.; Plastaras, J.P.; Leuratti, C.; Singh, R.; Gallacher-Horley, B.; Offord, E.; Marnett, L.J.; Steward, W.P.; Plummer, S.M. Cyclooxygenase-2, malondialdehyde and pyrimidopurinone adducts of deoxyguanosine in human colon cells. Carcinogenesis 2001, 22, 1557–1560. [Google Scholar] [CrossRef]
- Chaudhary, A.K.; Nokubo, M.; Reddy, G.R.; Yeola, S.N.; Morrow, J.D.; Blair, I.A.; Marnett, L.J. Detection of endogenous malondialdehydedeoxyguanosine adducts in human liver. Science 1994, 265, 1580–1582. [Google Scholar]
- Brigelius-Flohe, R.; Traber, M.G. Vitamin E: Function and metabolism. FASEB J. 1999, 13, 1145–1155. [Google Scholar]
- Boscoboinik, D.; Szewczyk, A.; Hensey, C.; Azzi, A. Inhibition of cell proliferation by alpha-tocopherol. Role of protein kinase C. J. Biol. Chem. 1991, 266, 6188–6194. [Google Scholar]
- Tasinato, A.; Boscoboinik, D.; Bartoli, G.M.; Maroni, P.; Azzi, A. d-alpha tocopherol inhibition of vascular smooth muscle cell proliferation occurs at physiological concentrations, correlates with protein kinase C inhibition, and is independent of its antioxidant properties. Proc. Natl. Acad. Sci. USA 1995, 92, 12190–12194. [Google Scholar] [CrossRef]
- Ricciarelli, R.; Zingg, J.M.; Azzi, A. Vitamin E reduces the uptake of oxidized LDL by inhibiting CD36 scavenger receptor expression in cultured aortic smooth muscle cells. Circulation 2000, 102, 82–87. [Google Scholar] [CrossRef]
- Pédeboscq, S.; Rey, C.; Petit, M.; Harpey, C.; de Giorgi, F.; Ichas, F.; Lartigue, L. Non-antioxidant properties of a-tocopherol reduce the anticancer activity of several protein kinase inhibitors in vitro. PLoS ONE 2012, 7, e36811. [Google Scholar]
- Gopalakrishna, R.; Jaken, S. Protein kinase C signaling and oxidative stress. Free Radic. Biol. Med. 2000, 28, 1349–1361. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cohen, C.; Cardoso, J.F.R.; Garcia, S.B.; Vannucchi, H. Vitamin E Supplementation in Chemical Colorectal Carcinogenesis: A Two-Edged Knife. Nutrients 2014, 6, 3214-3229. https://doi.org/10.3390/nu6083214
Cohen C, Cardoso JFR, Garcia SB, Vannucchi H. Vitamin E Supplementation in Chemical Colorectal Carcinogenesis: A Two-Edged Knife. Nutrients. 2014; 6(8):3214-3229. https://doi.org/10.3390/nu6083214
Chicago/Turabian StyleCohen, Celia, João Felipe Rito Cardoso, Sergio Britto Garcia, and Helio Vannucchi. 2014. "Vitamin E Supplementation in Chemical Colorectal Carcinogenesis: A Two-Edged Knife" Nutrients 6, no. 8: 3214-3229. https://doi.org/10.3390/nu6083214
APA StyleCohen, C., Cardoso, J. F. R., Garcia, S. B., & Vannucchi, H. (2014). Vitamin E Supplementation in Chemical Colorectal Carcinogenesis: A Two-Edged Knife. Nutrients, 6(8), 3214-3229. https://doi.org/10.3390/nu6083214