Dietary Omega-3 Fatty Acid Deficiency and High Fructose Intake in the Development of Metabolic Syndrome, Brain Metabolic Abnormalities, and Non-Alcoholic Fatty Liver Disease

Abstract
:1. Introduction
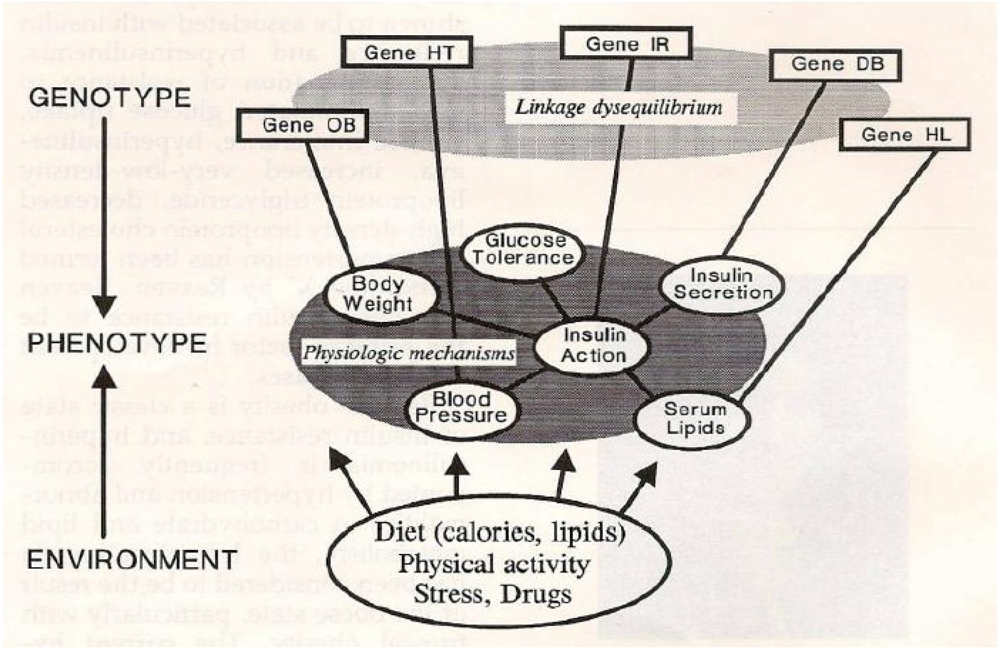
2. The Metabolic Syndrome
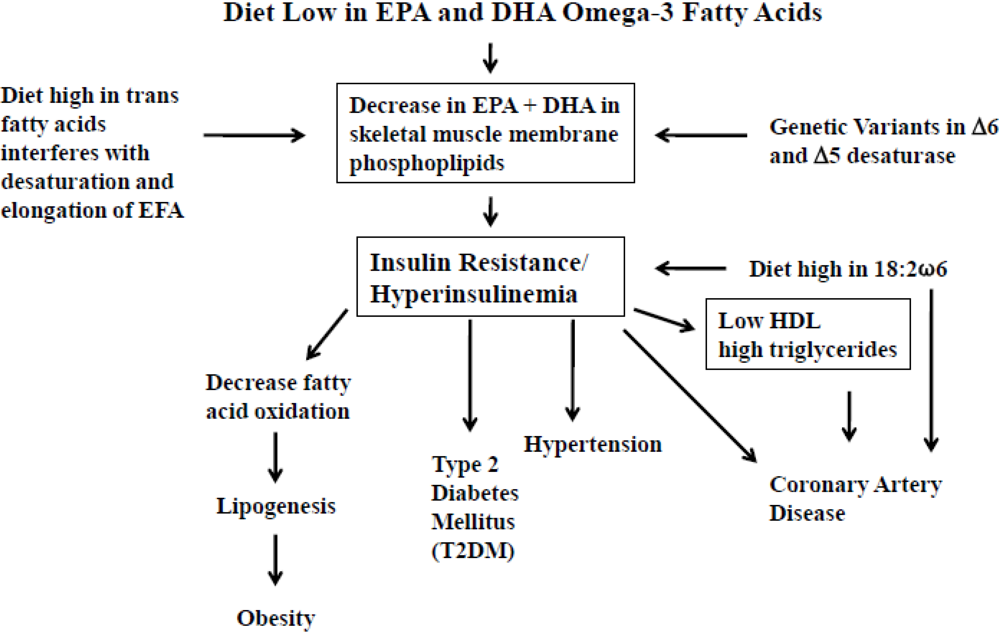
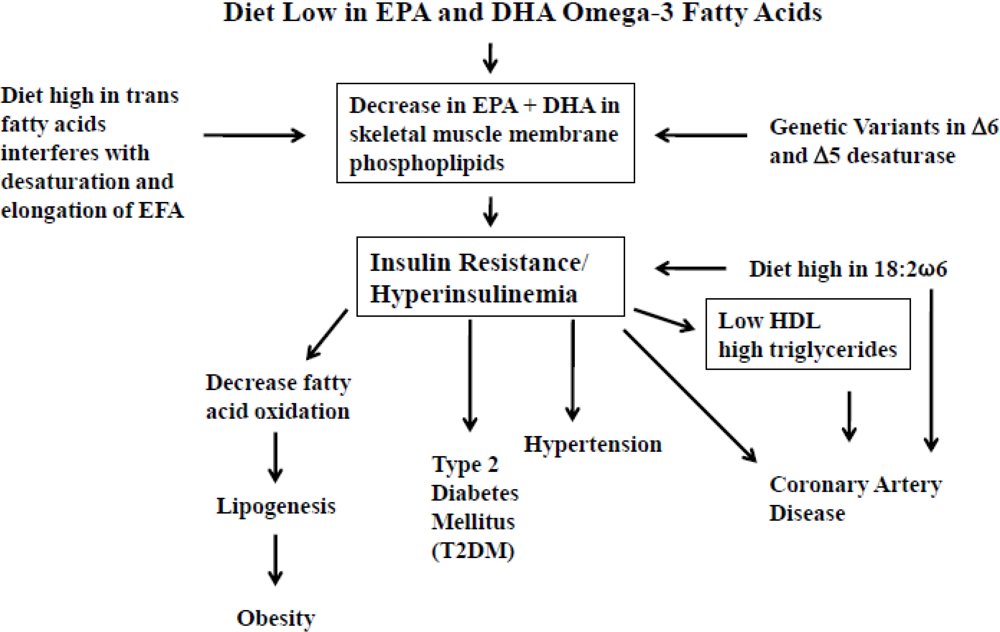
3. Dietary Omega-3 Fatty Acid Deficiency
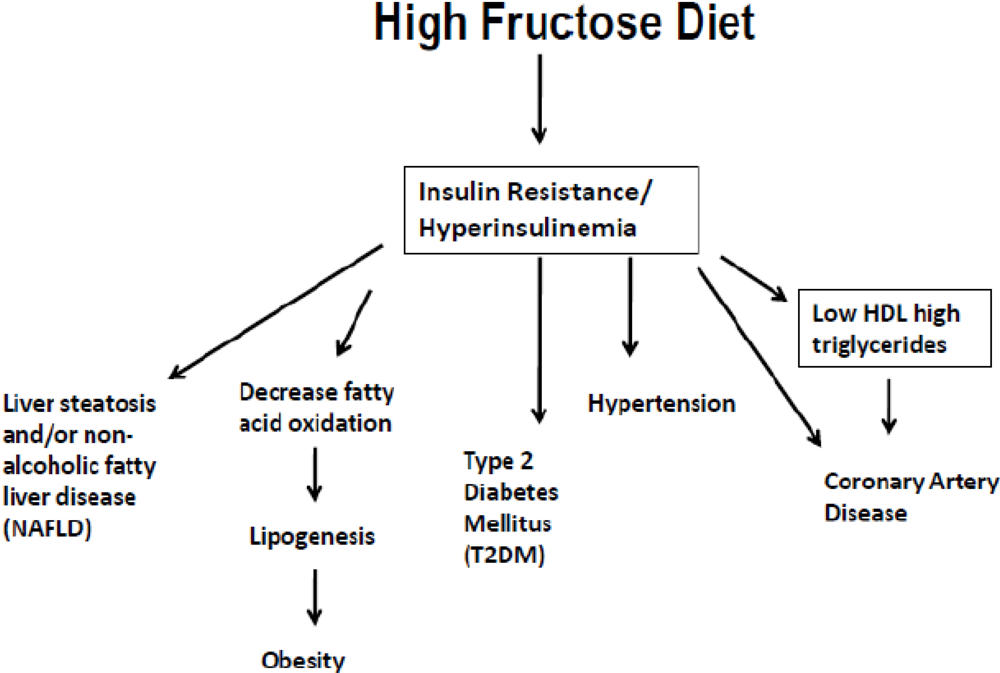
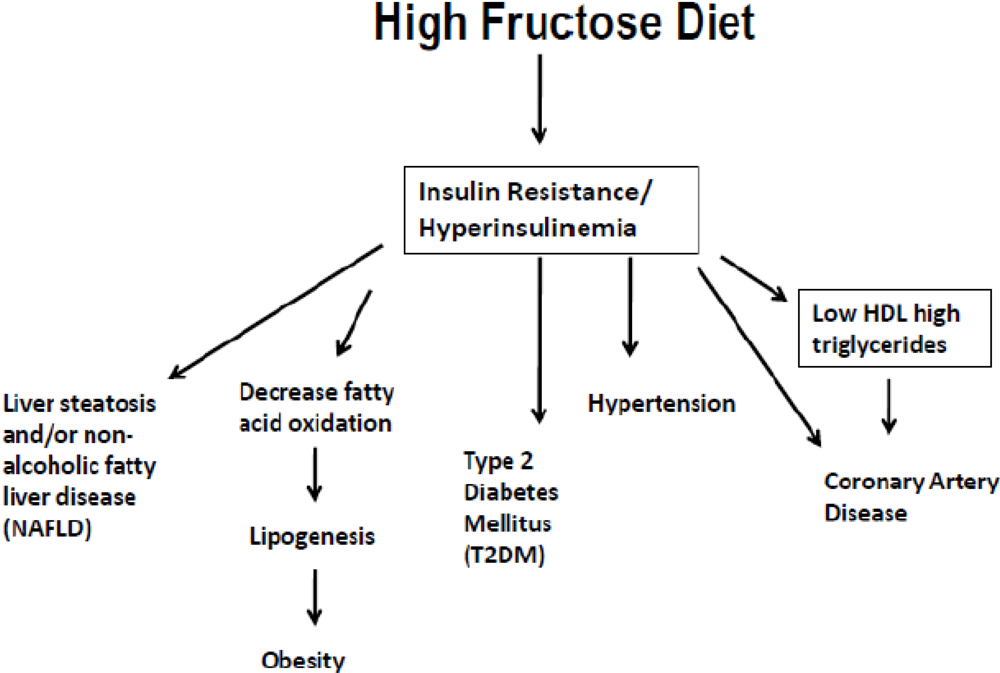
4. High Fructose Intake

{kind=link}
{kind=link}
{kind=link}
|
5. Dietary Omega-3 Deficiency, High Fructose Intake, Insulin Resistance, and the Brain
| Factor | Omega-3 Deficient Diet | Fructose | Omega-3 Diet |
|---|---|---|---|
| Increase in latency time in the Barnes Maze Test indicating memory impairment | ↑ | Potentiates this effect | Ameliorates |
| Triglycerides | ↑ | Potentiates this effect | Ameliorates |
| Insulin Resistance | ↑ | Potentiates this effect | Corrects |
| pTyrIR levels in Hippocampus | ↓ | Decreases this effect | Reversed |
| Akt phosphorylation | ↓ | Exacerbates this effect | Alleviates |
| Phosphorylation in KLβ1 | ↓ | ____ | Increases |
| Phosphorylation of CREB | ↓ | Exacerbates this effect | Counterregulated the fructose induced alteration in synaptic plasticity via CREB Synapsin 1 Synaptophysin |
| Hippocampus and Frontal lobe volume | ↓ | ____ | ____ |
| Sir2 | ↓ | ↓ | Normalizes |
| Liver Steatosis | ↑ | ↑ | Reverses |
6. Dietary Omega-3 Deficiency and High Fructose Intake in the Development of Non-Alcoholic Fatty Liver Disease (NAFLD)
7. Conclusions, Health Implications and Recommendations
- Increase the intake of omega-3 fatty acids ALA, EPA, and DHA, and decrease LA and AA [122]. This can be accomplished by changing the oils in the Western diet and substituting the high omega-6 oils (corn oil, sunflower, safflower, soybean) with low omega-6 fatty acids and oils high in omega-3’s in order to balance the omega-6/omega-3 ratio such as (1) Olive oil which is low in omega-6 fatty acids (LA 6%–12%), (2) Canola oil which has a ratio of omega-6/omega-3 of 2:1. (3) Chia and Perilla oil which contain 55%–60% omega-3 fatty acids. Industry has recognized the need to change the oils by decreasing the high omega-6 content through genetic manipulation, i.e., by developing high monounsaturated sunflower oil. Furthermore through genetic engineering of soybeans, by increasing Stearidonic acid, industry has increased the omega-3 content of soybean oil which has been shown in animal and human studies to be more effective than its precursor, α-linolenic acid, to be metabolized to EPA, thus enriching membrane phospholipids with EPA. Hence, stearidonic acid can serve as a “pro-eicosapentaenoic acid” [123,124]. It is necessary that food labels state the levels of omega-6 and omega-3 fatty acids separately instead of simply reporting them as PUFA. Furthermore studies and all journal publications should distinguish the concentration of omega-6 and omega-3 fatty acids instead of simply PUFA. The omega-6 and omega-3 fatty acids are physiologically and metabolically distinct and have opposing properties. Therefore, their balance is important for health.
- Decrease the amount of added sugar to less than 10% of energy intake, and remove high fructose corn syrup from sweetened beverages, snacks, cookies and other forms of processed foods. Again, industry has recognized the detrimental effects to health, resulting from high fructose intake and is searching for natural ingredients low in sugars (and fructose) to substitute for HFCS. Furthermore, the levels of glucose and fructose should be stated in all food labels [125,126,127,128]. At present, U.S. food labels contain information on total sugars per serving but do not distinguish between sugars that are naturally present in foods and added sugars. Thus, it is impossible for consumers to determine the amount of added sugars in foods or beverages, or some drugs and cough syrups.
References
- Simopoulos, A.P. The importance of the omega-6/omega-3 Fatty Acid ratio in cardiovascular disease and other chronic diseases. Exp. Biol. Med. (Maywood) 2008, 233, 674–688. [Google Scholar] [CrossRef]
- Johnson, R.K.; Appel, L.J.; Brands, M.; Howard, B.V.; Lefevre, M.; Lustig, R.H.; Sacks, F.; Steffen, L.M.; Wylie-Rosett, J. American Heart Association Nutrition Committee of the Council on Nutrition, Physical Activity, and Metabolism and the Council on Epidemiology and Prevention. Dietary sugars intake and cardiovascular health: A scientific statement from the American Heart Association. Circulation. 2009, 120, 1011–1020. [Google Scholar]
- Simopoulos, A.P. Omega-3 fatty acids in health and disease and in growth and development. Am. J. Clin. Nutr. 1991, 54, 438–463. [Google Scholar]
- Stanhope, K.L.; Schwarz, J.M.; Keim, N.L.; Griffen, S.C.; Bremer, A.A.; Graham, J.L.; Hatcher, B.; Cox, C.L.; Dyachenko, A.; Zhang, W.; et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J. Clin. Investig. 2009, 119, 1322–1334. [Google Scholar] [CrossRef]
- Fung, T.T.; Malik, V.; Rexrode, K.M.; Manson, J.E.; Willett, W.C.; Hu, F.B. Sweetened beverage consumption and risk of coronary heart disease in women. Am. J. Clin. Nutr. 2009, 89, 1037–1042. [Google Scholar] [CrossRef]
- Montonen, J.; Järvinen, R.; Knekt, P.; Heliövaara, M.; Reunanen, A. Consumption of sweetened beverages and intakes of fructose and glucose predict type 2 diabetes occurrence. J. Nutr. 2007, 137, 1447–1454. [Google Scholar]
- Blasbalg, T.L.; Hibbeln, J.R.; Ramsden, C.E.; Majchrzak, S.F.; Rawlings, R.R. Changes in consumption of omega-3 and omega-6 fatty acids in the United States during the 20th century. Am. J. Clin. Nutr. 2011, 93, 950–962. [Google Scholar] [CrossRef]
- Storlien, L.H.; Jenkins, A.B.; Chisholm, D.J.; Pascoe, W.S.; Khouri, S.; Kraegen, E.W. Influence of dietary fat composition on development of insulin resistance in rats. Relationship to muscle triglyceride and omega-3 fatty acids in muscle phospholipid. Diabetes 1991, 40, 280–289. [Google Scholar]
- Borkman, M.; Storlien, L.H.; Pan, D.A.; Jenkins, A.B.; Chisholm, D.J.; Campbell, L.V. The relation between insulin sensitivity and the fatty-acid composition of skeletal-muscle phospholipids. N. Engl. J. Med. 1993, 328, 238–244. [Google Scholar] [CrossRef]
- Farooqui, A.A.; Farooqui, T.; Panza, F.; Frisardi, V. Metabolic syndrome as a risk factor for neurological disorders. Cell Mol. Life Sci. 2012, 69, 741–762. [Google Scholar] [CrossRef]
- Yates, K.F.; Sweat, V.; Yau, P.L.; Turchiano, M.M.; Convit, A. Impact of metabolic syndrome on cognition and brain: A selected review of the literature. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2060–2067. [Google Scholar] [CrossRef]
- Williamson, R.; McNeilly, A.; Sutherland, C. Insulin resistance in the brain: An old-age or new-age problem? Biochem. Pharmacol. 2012, 84, 737–745. [Google Scholar] [CrossRef]
- De La Monte, S.M. Metabolic derangements mediate cognitive impairment and Alzheimer’s disease: Role of peripheral insulin-resistance diseases. Panminerva Med. 2012, 54, 171–178. [Google Scholar]
- González-Périz, A.; Horrillo, R.; Ferré, N.; Gronert, K.; Dong, B.; Morán-Salvador, E.; Titos, E.; Martínez-Clemente, M.; López-Parra, M.; Arroyo, V.; et al. Obesity-induced insulin resistance and hepatic steatosis are alleviated by omega-3 fatty acids: A role for resolvins and protectins. FASEB J. 2009, 23, 1946–1957. [Google Scholar] [CrossRef]
- Simopoulos, A.P. Evolutionary Aspects of Diet and Essential Fatty Acids. In Fatty Acids and Lipids—New Findings; Hamazaki, T., Okuyama, H., Eds.; Kagar: Basel, Switzerland, 2001; pp. 18–27. [Google Scholar]
- Palmer, J.R.; Boggs, D.A.; Krishnan, S.; Hu, F.B.; Singer, M.; Rosenberg, L. Sugar sweetened beverages and incidence of type 2 diabetes mellitus in African American women. Arch. Intern. Med. 2008, 168, 1487–1492. [Google Scholar] [CrossRef]
- Schulze, M.B.; Manson, J.E.; Ludwig, D.S.; Colditz, G.A.; Stampfer, M.J.; Willett, W.C.; Hu, F.B. Sugar-sweetened beverages, weight gain, and incidence of type 2 diabetes in young and middle-aged women. JAMA 2004, 292, 927–934. [Google Scholar] [CrossRef]
- Bremer, A.A.; Auinger, P.; Byrd, R.S. Sugar-sweetened beverage intake trends in U.S. adolescents and their association with insulin resistance-related parameters. J. Nutr. Metab. 2009. [Google Scholar] [CrossRef]
- Yoshida, M.; McKeown, N.M.; Rogers, G.; Meigs, J.B.; Saltzman, E.; D’Agostino, R.; Jacques, P.F. Surrogate markers of insulin resistance are associated with consumption of sugar-sweetened drinks and fruit juice in middle and older-aged adults. J. Nutr. 2007, 137, 2121–2127. [Google Scholar]
- Aeberli, I.; Hochuli, M.; Gerber, P.A.; Sze, L.; Murer, S.B.; Tappy, L.; Spinas, G.A.; Berneis, K. Moderate amounts of fructose consumption impair insulin sensitivity in healthy young men: A randomized controlled trial. Diabetes Care 2013, 36, 150–156. [Google Scholar] [CrossRef]
- Tappy, L.; Lê, K.A. Metabolic effects of fructose and the worldwide increase in obesity. Physiol. Rev. 2010, 90, 23–46. [Google Scholar] [CrossRef]
- Tappy, L.; Mittendorfer, B. Fructose toxicity: Is the science ready for public health actions? Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 357–361. [Google Scholar] [CrossRef]
- Lecoultre, V.; Egli, L.; Carrel, G.; Theytaz, F.; Kreis, R.; Schneiter, P.; Boss, A.; Zwygart, K.; Lê, K.A.; Bortolotti, M.; et al. Effects of fructose and glucose overfeeding on hepatic insulin sensitivity and intrahepatic lipids in healthy humans. Obesity 2013, 21, 782–785. [Google Scholar] [CrossRef]
- Reaven, G.M. Banting lecture 1988: Role of insulin resistance in human disease. Diabetes 1988, 37, 1595–1607. [Google Scholar]
- Foster, D.W. Insulin resistance—A secret killer? N. Engl. J. Med. 1989, 320, 733–734. [Google Scholar] [CrossRef]
- Angulo, P. Nonalcoholic fatty liver disease. N. Engl. J. Med. 2002, 346, 1221–1231. [Google Scholar] [CrossRef]
- Sanyal, A.J. American Gastroenterological Association. AGA technical review on nonalcoholic fatty liver disease. Gastroenterology 2002, 123, 1705–1725. [Google Scholar] [CrossRef]
- Ford, E.S.; Li, C.; Zhao, G. Prevalence and correlates of metabolic syndrome based on a harmonious definition among adults in the US. J. Diabetes 2010, 2, 180–193. [Google Scholar] [CrossRef]
- Ferrannini, E. Metabolic abnormalities of hypertension. A lesson in complexity. Hypertension 1991, 18, 636–639. [Google Scholar] [CrossRef]
- Simopoulos, A.P. Fatty acid composition of skeletal-muscle membrane phospholipids, insulin resistance and obesity. Nutr. Today 1994, 29, 12–16. [Google Scholar] [CrossRef]
- Simopoulos, A.P. Is insulin resistance influenced by dietary linoleic acid and trans fatty acids? Free Radic. Biol. Med. 1994, 17, 367–372. [Google Scholar] [CrossRef]
- Lauritzen, L.; Hansen, H.S.; Jorgensen, M.H.; Michaelsen, K.F. The essentiality of long chain n-3 fatty acids in relation to development and function of the brain and retina. Prog. Lipid Res. 2001, 40, 1–94. [Google Scholar] [CrossRef]
- Parker, G.; Gibson, N.A.; Brotchie, H.; Heruc, G.; Rees, A.M.; Hadzi-Pavlovic, D. Omega-3 fatty acids and mood disorders. Am. J. Psychiatry 2006, 163, 969–978. [Google Scholar] [CrossRef]
- Di Marzo, V.; Goparaju, S.K.; Wang, L.; Liu, J.; Bátkai, S.; Járai, Z.; Fezza, F.; Miura, G.I.; Palmiter, R.D.; Sugiura, T.; et al. Leptin-regulated endocannabinoids are involved in maintaining food intake. Nature 2001, 410, 822–825. [Google Scholar] [CrossRef]
- Osei-Hyiaman, D.; DePetrillo, M.; Pacher, P.; Liu, J.; Radaeva, S.; Bátkai, S.; Harvey-White, J.; Mackie, K.; Offertáler, L.; Wang, L.; et al. Endocannabinoid activation at hepatic CB1 receptors stimulates fatty acid synthesis and contributes to diet-induced obesity. J. Clin. Investig. 2005, 115, 1298–1305. [Google Scholar]
- Pagotto, U.; Marsicano, G.; Cota, D.; Lutz, B.; Pasquali, R. The emerging role of the endocannabinoid system in endocrine regulation and energy balance. Endocr. Rev. 2006, 27, 73–100. [Google Scholar]
- Christensen, R.; Kristensen, P.K.; Bartels, E.M.; Bliddal, H.; Astrup, A. Efficacy and safety of the weight-loss drug rimonabant: A meta-analysis of randomised trials. Lancet 2007, 370, 1706–1713. [Google Scholar] [CrossRef]
- Samat, A.; Tomlinson, B.; Taheri, S.; Thomas, G.N. Rimonabant for the treatment of obesity. Recent Pat. Cardiovasc. Drug Discov. 2008, 3, 187–193. [Google Scholar]
- Alvheim, A.R.; Malde, M.K.; Osei-Hyiaman, D.; Lin, Y.H.; Pawlosky, R.J.; Madsen, L.; Kristiansen, K.; Frøyland, L.; Hibbeln, J.R. Dietary linoleic acid elevates endogenous 2-AG and anandamide and induces obesity. Obesity 2012, 20, 1984–1994. [Google Scholar] [CrossRef]
- Evans, D.L.; Charney, D.S.; Lewis, L.; Golden, R.N.; Gorman, J.M.; Krishnan, K.R.; Nemeroff, C.B.; Bremner, J.D.; Carney, R.M.; Coyne, J.C.; et al. Mood disorders in the medically ill: Scientific review and recommendations. Biol. Psychiatry 2005, 58, 175–189. [Google Scholar] [CrossRef]
- Lafourcade, M.; Larrieu, T.; Mato, S.; Duffaud, A.; Sepers, M.; Matias, I.; de Smedt-Peyrusse, V.; Labrousse, V.F.; Bretillon, L.; Matute, C.; et al. Nutritional omega-3 deficiency abolishes endocannabinoid-mediated neuronal functions. Nat. Neurosci. 2011, 14, 345–350. [Google Scholar] [CrossRef]
- World Health Organization, Diet, Nutrition and the Prevention of Chronic Diseases. Report of the Joint WHO/FAO Expert Consultation; WHO Technical Report Series No. 916; WHO: Geneva, Switzerland, 2003.
- Parks, E.J.; Hellerstein, M.K. Carbohydrate-induced hypertriacylglycerolemia: Historical perspective and review of biological mechanisms. Am. J. Clin. Nutr. 2000, 71, 412–433. [Google Scholar]
- Schwarz, J.M.; Neese, R.A.; Turner, S.; Dare, D.; Hellerstein, M.K. Short-term alterations in carbohydrate energy intake in humans. Striking effects on hepatic glucose production, de novo lipogenesis, lipolysis, and wholebody fuel selection. J. Clin. Investig. 1995, 96, 2735–2743. [Google Scholar]
- Sevastianova, K.; Santos, A.; Kotronen, A.; Hakkarainen, A.; Makkonen, J.; Silander, K.; Peltonen, M.; Romeo, S.; Lundbom, J.; Lundbom, N.; et al. Effect of short-term carbohydrate overfeeding and long-term weight loss on liver fat in overweight humans. Am. J. Clin. Nutr. 2012, 96, 727–734. [Google Scholar] [CrossRef]
- Stanhope, K.L.; Bremer, A.A.; Medici, V.; Nakajima, K.; Ito, Y.; Nakano, T.; Chen, G.; Fong, T.H.; Lee, V.; Menorca, R.I.; et al. Consumption of fructose and high fructose corn syrup increase postprandial triglycerides, LDL-cholesterol, and apolipoprotein-B in young men and women. J. Clin. Endocrinol. Metab. 2011, 96, E1596–E1605. [Google Scholar] [CrossRef]
- Ruxton, C.H.; Gardner, E.J.; McNulty, H.M. Is sugar consumption detrimental to health? A review of the evidence 1995–2006. Crit. Rev. Food Sci. Nutr. 2010, 50, 1–19. [Google Scholar] [CrossRef]
- Dolan, L.C.; Potter, S.M.; Burdock, G.A. Evidence-based review on the effect of normal dietary consumption of fructose on development of hyperlipidemia and obesity in healthy, normal weight individuals. Crit. Rev. Food Sci. Nutr. 2010, 50, 53–84. [Google Scholar]
- Dolan, L.C.; Potter, S.M.; Burdock, G.A. Evidence-based review on the effect of normal dietary consumption of fructose on blood lipids and body weight of overweight and obese individuals. Crit. Rev. Food Sci. Nutr. 2010, 50, 889–918. [Google Scholar] [CrossRef]
- Stanhope, K.L.; Griffen, S.C.; Bremer, A.A.; Vink, R.G.; Schaefer, E.J.; Nakajima, K.; Schwarz, J.M.; Beysen, C.; Berglund, L.; Keim, N.L.; et al. Metabolic responses to prolonged consumption of glucose- and fructose-sweetened beverages are not associated with postprandial or 24-h glucose and insulin excursions. Am. J. Clin. Nutr. 2011, 94, 112–119. [Google Scholar] [CrossRef]
- Ding, E.L.; Malik, V.S. Convergence of obesity and high glycemic diet on compounding diabetes and cardiovascular risks in modernizing China: An emerging public health dilemma. Glob. Health 2008, 4, 4. [Google Scholar] [CrossRef]
- Hu, F.B.; Malik, V.S. Sugar-sweetened beverages and risk of obesity and type 2 diabetes: Epidemiologic evidence. Physiol. Behav. 2010, 100, 47–54. [Google Scholar] [CrossRef]
- Schernhammer, E.S.; Hu, F.B.; Giovannucci, E.; Michaud, D.S.; Colditz, G.A.; Stampfer, M.J.; Fuchs, C.S. Sugar-sweetened soft drink consumption and risk of pancreatic cancer in two prospective cohorts. Cancer Epidemiol. Biomarkers Prev. 2005, 14, 2098–2105. [Google Scholar] [CrossRef]
- Lê, K.A.; Faeh, D.; Stettler, R.; Ith, M.; Kreis, R.; Vermathen, P.; Boesch, C.; Ravussin, E.; Tappy, L. A 4-wk high-fructose diet alters lipid metabolism without affecting insulin sensitivity or ectopic lipids in healthy humans. Am. J. Clin. Nutr. 2006, 84, 1374–1379. [Google Scholar]
- Silbernagel, G.; Machann, J.; Unmuth, S.; Schick, F.; Stefan, N.; Häring, H.U.; Fritsche, A. Effects of 4-week very-high-fructose/glucose diets on insulin sensitivity, visceral fat and intrahepatic lipids: An exploratory trial. Br. J. Nutr. 2011, 106, 79–86. [Google Scholar] [CrossRef]
- USDA database for the added sugars content of selected foods. February 2006. Available online: http://www.nal.usda.gov/fnic/foodcamp/Data/add_sug/addsug01.pdf (accessed on 15 February 2013).
- Smart Choices Program: Guiding Food Choices. Smart Choices Program Web site. Available online: http://smartchoicesprogram.com (accessed on 15 February 2013).
- Guiding Stars Program. Hannaford Brothers Co Web site. Available online: http://www.hannaford.com/Contents/Healthy_Living/Guiding_Stars/index.shtml (accessed on 15 February 2013).
- NuVal Nutritional Scoring System. Available online: http://www.nuval.com (accessed on 15 February 2013).
- Drewnowski, A. Concept of a nutritious food: Toward a nutrient density score. Am. J. Clin. Nutr. 2005, 82, 721–732. [Google Scholar]
- Nguyen, S.; Choi, H.K.; Lustig, R.H.; Hsu, C.Y. Sugar-sweetened beverages, serum uric acid, and blood pressure in adolescents. J. Pediatr. 2009, 154, 807–813. [Google Scholar] [CrossRef]
- Fried, S.K.; Rao, S.P. Sugars, hypertriglyceridemia, and cardiovascular disease. Am. J. Clin. Nutr. 2003, 78, 873S–880S. [Google Scholar]
- Parks, E.J.; Skokan, L.E.; Timlin, M.T.; Dingfelder, C.S. Dietary sugars stimulate fatty acid synthesis in adults. J. Nutr. 2008, 138, 1039–1046. [Google Scholar]
- Chong, M.F.; Fielding, B.A.; Frayn, K.N. Mechanisms for the acute effect of fructose on postprandial lipemia. Am. J. Clin. Nutr. 2007, 85, 1511–1520. [Google Scholar]
- Ceriello, A.; Bortolotti, N.; Crescentini, A.; Motz, E.; Lizzio, S.; Russo, A.; Ezsol, Z.; Tonutti, L.; Taboga, C. Antioxidant defences are reduced during the oral glucose tolerance test in normal and non-insulin-dependent diabetic subjects. Eur. J. Clin. Investig. 1998, 28, 329–333. [Google Scholar] [CrossRef]
- Ma, S.W.; Tomlinson, B.; Benzie, I.F. A study of the effect of oral glucose loading on plasma oxidant:antioxidant balance in normal subjects. Eur. J. Nutr. 2005, 44, 250–254. [Google Scholar] [CrossRef]
- Jürgens, H.; Haass, W.; Castañeda, T.R.; Schürmann, A.; Koebnick, C.; Dombrowski, F.; Otto, B.; Nawrocki, A.R.; Scherer, P.E.; Spranger, J.; et al. Consuming fructose-sweetened beverages increases body adiposity in mice. Obes. Res. 2005, 13, 1146–1156. [Google Scholar] [CrossRef]
- Stavric, B.; Johnson, W.J.; Clayman, S.; Gadd, R.E.; Chartrand, A. Effect of fructose administration on serum urate levels in the uricase inhibited rat. Experientia 1976, 32, 373–374. [Google Scholar] [CrossRef]
- Vartanian, L.R.; Schwartz, M.B.; Brownell, K.D. Effects of soft drink consumption on nutrition and health: A systematic review and meta-analysis. Am. J. Public Health 2007, 97, 667–675. [Google Scholar] [CrossRef]
- Chen, L.; Appel, L.J.; Loria, C.; Lin, P.H.; Champagne, C.M.; Elmer, P.J.; Ard, J.D.; Mitchell, D.; Batch, B.C.; Svetkey, L.P.; et al. Reduction in consumption of sugar-sweetened beverages is associated with weight loss: The PREMIER trial. Am. J. Clin. Nutr. 2009, 89, 1299–1306. [Google Scholar] [CrossRef]
- Simopoulos, A.P.; Bazan, N.G. Omega-3 Fatty Acids, the Brain and Retina. World Review of Nutrition and Dietetics; Karger: Basel, Switzerland, 2009; Volume 99. [Google Scholar]
- Spangler, R.; Wittkowski, K.M.; Goddard, N.L.; Avena, N.M.; Hoebel, B.G.; Leibowitz, S.F. Opiate-like effects of sugar on gene expression in reward areas of the rat brain. Brain Res. Mol. Brain Res. 2004, 124, 134–142. [Google Scholar] [CrossRef]
- Kelley, A.E.; Bakshi, V.P.; Haber, S.N.; Steininger, T.L.; Will, M.J.; Zhang, M. Opioid modulation of taste hedonics within the ventral striatum. Physiol. Behav. 2002, 76, 365–377. [Google Scholar] [CrossRef]
- Pelchat, M.L.; Johnson, A.; Chan, R.; Valdez, J.; Ragland, J.D. Images of desire: Food-craving activation during fMRI. Neuroimage 2004, 23, 1486–1493. [Google Scholar] [CrossRef]
- Anderzhanova, E.; Covasa, M.; Hajnal, A. Altered basal and stimulated accumbens dopamine release in obese OLETF rats as a function of age and diabetic status. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2007, 293, R603–R611. [Google Scholar] [CrossRef]
- Teff, K.L.; Elliott, S.S.; Tschöp, M.; Kieffer, T.J.; Rader, D.; Heiman, M.; Townsend, R.R.; Keim, N.L.; D’Alessio, D.; Havel, P.J. Dietary fructose reduces circulating insulin and leptin, attenuates postprandial suppression of ghrelin, and increases triglycerides in women. J. Clin. Endocrinol. Metab. 2004, 89, 2963–2972. [Google Scholar] [CrossRef]
- Page, K.A. Effects of fructose vs glucose on regional cerebral blood flow in brain regions involved with appetite and reward pathways. JAMA 2013, 309, 63–70. [Google Scholar] [CrossRef]
- Tabas, I.; Ron, D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat. Cell Biol. 2011, 13, 184–190. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic IKKbeta/NF-kappaB and ER stress link overnutrition to energy imbalance and obesity. Cell 2008, 135, 61–73. [Google Scholar] [CrossRef]
- Purkayastha, S.; Zhang, H.; Zhang, G.; Ahmed, Z.; Wang, Y.; Cai, D. Neural dysregulation of peripheral insulin action and blood pressure by brain endoplasmic reticulum stress. Proc. Natl. Acad. Sci. USA 2011, 108, 2939–2944. [Google Scholar]
- Won, J.C.; Jang, P.G.; Namkoong, C.; Koh, E.H.; Kim, S.K.; Park, J.Y.; Lee, K.U.; Kim, M.S. Central administration of an endoplasmic reticulum stress inducer inhibits the anorexigenic effects of leptin and insulin. Obesity 2009, 17, 1861–1865. [Google Scholar] [CrossRef]
- Denis, R.G.; Arruda, A.P.; Romanatto, T.; Milanski, M.; Coope, A.; Solon, C.; Razolli, D.S.; Velloso, L.A. TNF-alpha transiently induces endoplasmic reticulum stress and an incomplete unfolded protein response in the hypothalamus. Neuroscience 2010, 170, 1035–1044. [Google Scholar] [CrossRef]
- Ozcan, L.; Ergin, A.S.; Lu, A.; Chung, J.; Sarkar, S.; Nie, D.; Myers, M.G., Jr.; Ozcan, U. Endoplasmic reticulum stress plays a central role in development of leptin resistance. Cell Metab. 2009, 9, 35–51. [Google Scholar] [CrossRef]
- Myers, M.G.; Cowley, M.A.; Munzberg, H. Mechanisms of leptin action and leptin resistance. Annu. Rev. Physiol. 2008, 70, 537–556. [Google Scholar] [CrossRef]
- Murphy, K.G.; Bloom, S.R. Gut hormones and the regulation of energy homeostasis. Nature 2006, 444, 854–859. [Google Scholar] [CrossRef]
- Sandoval, D.; Cota, D.; Seeley, R.J. The integrative role of CNS fuel-sensing mechanisms in energy balance and glucose regulation. Annu. Rev. Physiol. 2008, 70, 513–535. [Google Scholar] [CrossRef]
- Coll, A.P.; Farooqi, I.S.; O’Rahilly, S. The hormonal control of food intake. Cell 2007, 129, 251–262. [Google Scholar] [CrossRef]
- Flier, J.S. Neuroscience. Regulating energy balance: The substrate strikes back. Science 2006, 312, 861–864. [Google Scholar] [CrossRef]
- Friedman, J.M. Modern science versus the stigma of obesity. Nat. Med. 2004, 10, 563–569. [Google Scholar] [CrossRef]
- Morton, G.J.; Cummings, D.E.; Baskin, D.G.; Barsh, G.S.; Schwartz, M.W. Central nervous system control of food intake and body weight. Nature 2006, 443, 289–295. [Google Scholar] [CrossRef]
- Cone, R.D. Anatomy and regulation of the central melanocortin system. Nat. Neurosci. 2005, 8, 571–578. [Google Scholar] [CrossRef]
- Elmquist, J.K.; Coppari, R.; Balthasar, N.; Ichinose, M.; Lowell, B.B. Identifying hypothalamic pathways controlling food intake, body weight, and glucose homeostasis. J. Comp. Neurol. 2005, 493, 63–71. [Google Scholar] [CrossRef]
- Kahn, B.B.; Alquier, T.; Carling, D.; Hardie, D.G. AMP-activated protein kinase: Ancient energy gauge provides clues to modern understanding of metabolism. Cell Metab. 2005, 1, 15–25. [Google Scholar] [CrossRef]
- Pissios, P.; Bradley, R.L.; Maratos-Flier, E. Expanding the scales: The multiple roles of MCH in regulating energy balance and other biological functions. Endocr. Rev. 2006, 27, 606–620. [Google Scholar] [CrossRef]
- Belgardt, B.F.; Bruning, J.C. CNS leptin and insulin action in the control of energy homeostasis. Ann. N. Y. Acad. Sci. 2012, 1212, 97–113. [Google Scholar] [CrossRef]
- Virtue, S.; Vidal-Puig, A. Nothing Iffy about HIF in the Hypothalamus. PLoS Biol. 2011, 9, e1001116. [Google Scholar] [CrossRef]
- Meister, B. Neurotransmitters in key neurons of the hypothalamus that regulate feeding behavior and body weight. Physiol. Behav. 2007, 92, 263–271. [Google Scholar] [CrossRef]
- Yi, C.X.; Habegger, K.M.; Chowen, J.A.; Stern, J.; Tschop, M.H. A role for astrocytes in the central control of metabolism. Neuroendocrinology 2011, 93, 143–149. [Google Scholar] [CrossRef]
- Lam, T.K.; Schwartz, G.J.; Rossetti, L. Hypothalamic sensing of fatty acids. Nat. Neurosci. 2005, 8, 579–584. [Google Scholar] [CrossRef]
- Cole, A.R.; Astell, A.; Green, C.; Sutherland, C. Molecular connexions between dementia and diabetes. Neurosci. Biobehav. Rev. 2007, 31, 1046–1063. [Google Scholar] [CrossRef]
- Shulman, G.I. Cellular mechanism of insulin resistance. J. Clin. Investig. 2000, 106, 171–176. [Google Scholar] [CrossRef]
- Agrawal, R.; Gomez-Pinilla, F. “Metabolic syndrome” in the brain: Deficiency in omega-3 fatty acid exacerbates dysfunctions in insulin receptor signalling and cognition. J. Physiol. 2012, 590, 2485–2499. [Google Scholar] [CrossRef]
- Gerrits, P.M.; Tsalikian, E. Diabetes and fructose metabolism. Am. J. Clin. Nutr. 1993, 58, 796S–799S. [Google Scholar]
- Kelley, G.L.; Allan, G.; Azhar, S. High dietary fructose induces a hepatic stress response resulting in cholesterol and lipid dysregulation. Endocrinology. 2004, 145, 548–555. [Google Scholar] [CrossRef]
- Pawar, A.; Jump, D.B. Unsaturated fatty acid regulation of peroxisome proliferator-activated receptor alpha activity in rat primary hepatocytes. J. Biol. Chem. 2003, 278, 35931–35939. [Google Scholar] [CrossRef]
- Schmitz, G.; Ecker, J. The opposing effects of n-3 and n-6 fatty acids. Prog. Lipid Res. 2008, 47, 147–155. [Google Scholar] [CrossRef]
- Sekiya, M.; Yahagi, N.; Matsuzaka, T.; Najima, Y.; Nakakuki, M.; Nagai, R.; Ishibashi, S.; Osuga, J.; Yamada, N.; Shimano, H. Polyunsaturated fatty acids ameliorate hepatic steatosis in obese mice by SREBP-1 suppression. Hepatology 2003, 38, 1529–1539. [Google Scholar]
- Xu, J.; Cho, H.; O’Malley, S.; Park, J.H.; Clarke, S.D. Dietary polyunsaturated fats regulate rat liver sterol regulatory element binding proteins-1 and -2 in three distinct stages and by different mechanisms. J. Nutr. 2002, 132, 3333–3339. [Google Scholar]
- Davidson, M.H. Mechanisms for the hypotriglyceridemic effect of marine omega-3 fatty acids. Am. J. Cardiol. 2006, 98, 27i–33i. [Google Scholar] [CrossRef]
- Appel, L.J.; Miller, E.R., III; Seidler, A.J.; Whelton, P.K. Does supplementation of diet with “fish oil” reduce blood pressure? A meta-analysis of controlled clinical trials. Arch. Intern. Med. 1993, 153, 1429–1438. [Google Scholar]
- Simopoulos, A.P. Omega-3 fatty acids in inflammation an autoimmune diseases. J. Am. Coll. Nutr. 2002, 21, 495–505. [Google Scholar] [CrossRef]
- Zelber-Sagi, S.; Nitzan-Kaluski, D.; Goldsmith, R.; Webb, M.; Blendis, L.; Halpern, Z.; Oren, R. Long term nutritional intake and the risk for non-alcoholic fatty liver disease (NAFLD): A population based study. J. Hepatol. 2007, 47, 711–717. [Google Scholar] [CrossRef]
- Shapiro, H.; Tehilla, M.; Attal-Singer, J.; Bruck, R.; Luzzatti, R.; Singer, P. The therapeutic potential of long-chain omega-3 fatty acids in nonalcoholic fatty liver disease. Clin. Nutr. 2011, 30, 6–19. [Google Scholar] [CrossRef]
- Louchami, K.; Zhang, Y.; Oguzhan, B.; Delporte, C.; Portois, L.; Carpentier, Y.A.; Genten, F.; Danguy, A.; Malaisse, W.J.; Sener, A. Rapid changes in liver lipid composition and pancreatic islet K+ handling and secretory behaviour provoked by the intravenous administration of a medium-chain triglyceride: Fish oil emulsion to long-chain polyunsaturated omega3 fatty acid depleted rats. Int. J. Mol. Med. 2006, 18, 1047–1055. [Google Scholar]
- Pachikian, B.D.; Neyrinck, A.M.; Cani, P.D.; Portois, L.; Deldicque, L.; de Backer, F.C.; Bindels, L.B.; Sohet, F.M.; Malaisse, W.J.; Francaux, M.; et al. Hepatic steatosis in n-3 fatty acid depleted mice: Focus on metabolic alterations related to tissue fatty acid composition. BMC Physiol. 2008, 8, 21. [Google Scholar] [CrossRef]
- Pachikian, B.D.; Essaghir, A.; Demoulin, J.B.; Neyrinck, A.M.; Catry, E.; de Backer, F.C.; Dejeans, N.; Dewulf, E.M.; Sohet, F.M.; Portois, L.; et al. Hepatic n-3 polyunsaturated fatty acid depletion promotes steatosis and insulin resistance in mice: Genomic analysis of cellular targets. PLoS One 2011, 6, e23365. [Google Scholar] [CrossRef]
- Ferre, P.; Foufelle, F. SREBP-1c transcription factor and lipid homeostasis: Clinical perspective. Horm. Res. 2007, 68, 72–82. [Google Scholar] [CrossRef]
- Jump, D.B. N-3 polyunsaturated fatty acid regulation of hepatic gene transcription. Curr. Opin. Lipidol. 2008, 19, 242–247. [Google Scholar] [CrossRef]
- Pettinelli, P.; del Pozo, T.; Araya, J.; Rodrigo, R.; Araya, A.V.; Smok, G.; Csendes, A.; Gutierrez, L.; Rojas, J.; Korn, O.; et al. Enhancement in liver SREBP-1c/PPAR-alpha ratio and steatosis in obese patients: Correlations with insulin resistance and n-3 long-chain polyunsaturated fatty acid depletion. Biochim. Biophys. Acta 2009, 1792, 1080–1086. [Google Scholar]
- Yaqoob, P.; Calder, P.C. Fatty acids and immune function: New insights into mechanisms. Br. J. Nutr. 2007, 98 (Suppl 1.), 41–45. [Google Scholar]
- Cai, D.; Liu, T. Inflammatory cause of metabolic syndrome via brain stress and NF-κB. Aging 2012, 4, 98–115. [Google Scholar]
- Simopoulos, A.P. Nutrigenetics/nutrigenomics. Ann. Rev. Public Health 2010, 31, 53–68. [Google Scholar] [CrossRef]
- Harris, W.S. Stearidonic acid as a “pro-eicosapentaenoic acid”. Curr. Opin. Lipidol. 2012, 23, 30–34. [Google Scholar] [CrossRef]
- Harris, W.S. Stearidonic acid-enhanced soybean oil: A plant-based source of (n-3) fatty acids for foods. J. Nutr. 2012, 142, 600S–604S. [Google Scholar] [CrossRef]
- Simopoulos, A.P. Healthy Agriculture, Healthy Nutrition, Healthy People. World Review of Nutrition and Dietetics; Karger: Basel, Switzerland, 2011; Volume 102. [Google Scholar]
- Simopoulos, A.P.; Faergeman, O.; Bourne, P.G. Action plan for a healthy agriculture, healthy nutrition, healthy people. J. Nutrigenet. Nutrigenomics 2011, 4, 65–68. [Google Scholar] [CrossRef]
- Simopoulos, A.P.; Bourne, P.G.; Faergeman, O. Bellagio report on healthy agriculture, healthy nutrition, healthy people. Nutrients 2013, 5, 411–423. [Google Scholar] [CrossRef]
- Yon, M.A.; Mauger, S.L.; Pickavance, L.C. Relationships between dietary macronutrients and adult neurogenesis in the regulation of energy metabolism. Br. J. Nutr. 2013, 109, 1573–1589. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Simopoulos, A.P. Dietary Omega-3 Fatty Acid Deficiency and High Fructose Intake in the Development of Metabolic Syndrome, Brain Metabolic Abnormalities, and Non-Alcoholic Fatty Liver Disease. Nutrients 2013, 5, 2901-2923. https://doi.org/10.3390/nu5082901
Simopoulos AP. Dietary Omega-3 Fatty Acid Deficiency and High Fructose Intake in the Development of Metabolic Syndrome, Brain Metabolic Abnormalities, and Non-Alcoholic Fatty Liver Disease. Nutrients. 2013; 5(8):2901-2923. https://doi.org/10.3390/nu5082901
Chicago/Turabian StyleSimopoulos, Artemis P. 2013. "Dietary Omega-3 Fatty Acid Deficiency and High Fructose Intake in the Development of Metabolic Syndrome, Brain Metabolic Abnormalities, and Non-Alcoholic Fatty Liver Disease" Nutrients 5, no. 8: 2901-2923. https://doi.org/10.3390/nu5082901
APA StyleSimopoulos, A. P. (2013). Dietary Omega-3 Fatty Acid Deficiency and High Fructose Intake in the Development of Metabolic Syndrome, Brain Metabolic Abnormalities, and Non-Alcoholic Fatty Liver Disease. Nutrients, 5(8), 2901-2923. https://doi.org/10.3390/nu5082901