Alcohol and Cardiovascular Disease—Modulation of Vascular Cell Function


{kind=link}
Abstract
:1. Introduction/Epidemiological Evidence and Animal Study Evidence
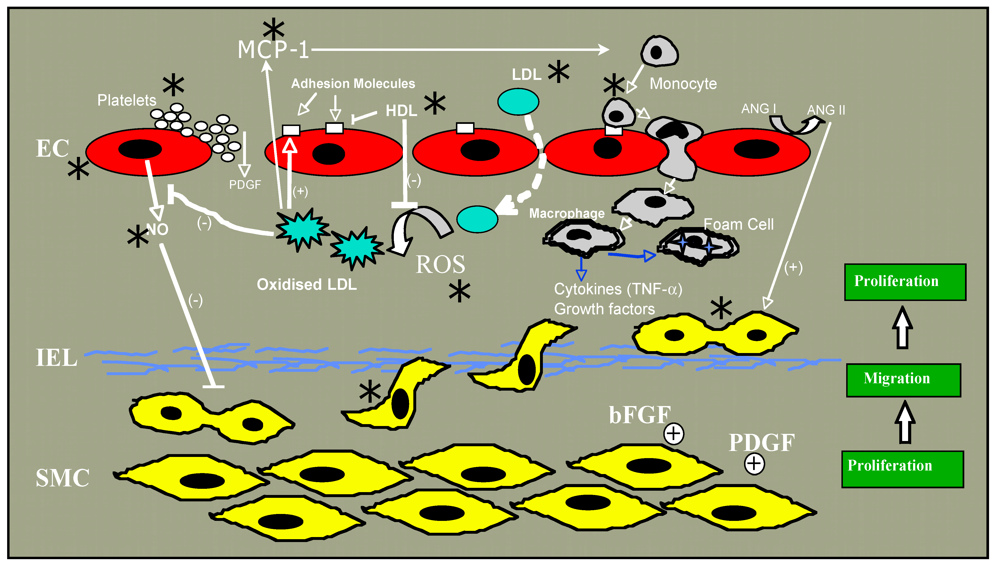
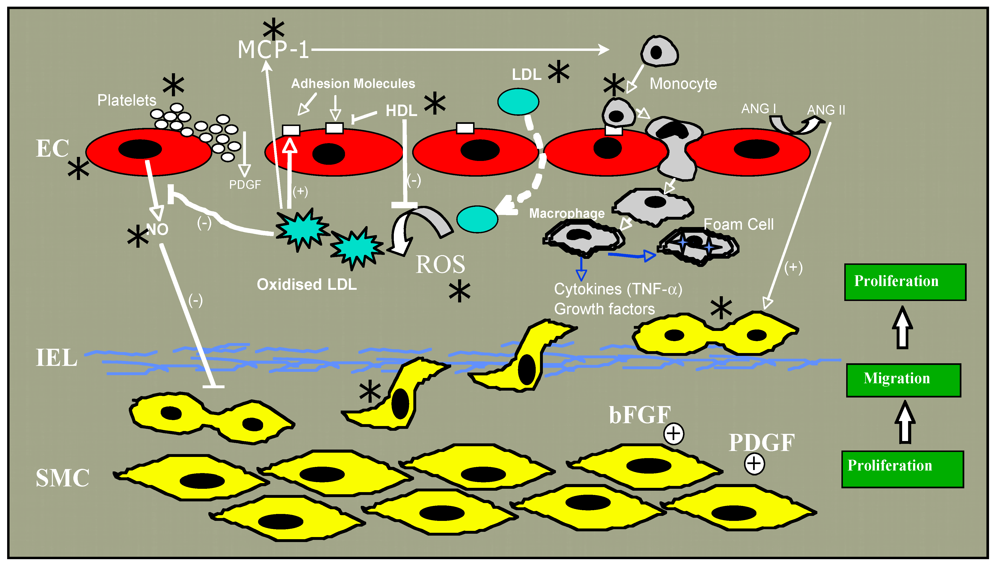
2. Atherosclerosis
3. Alcohol and Nitric Oxide (NO)
4. Endothelial Progenitor Cells
5. Alcohol and Reactive Oxygen Species (ROS). Alcohol: Prooxidant or Antioxidant?
6. Ethanol and Endothelial Proliferation, Migration and Angiogenesis
7. Monocyte Chemotactic Protein-1
8. Smooth Muscle Cell (SMC) Differentiation/Phenotypic Switching and Vascular Disease
Notch, Vascular Injury and SMC Proliferation
9. Molecular Mechanism of Action?
10. Conclusions
Acknowledgments
Conflict of Interest
References
- Corrao, G.; Rubbiati, L.; Bagnardi, V.; Zambon, A.; Poikolainen, K. Alcohol and coronary heart disease: A meta-analysis. Addiction (Abingdon, Engl.) 2000, 95, 1505–1523. [Google Scholar] [CrossRef]
- Ronksley, P.E.; Brien, S.E.; Turner, B.J.; Mukamal, K.J.; Ghali, W.A. Association of alcohol consumption with selected cardiovascular disease outcomes: A systematic review and meta-analysis. BMJ 2011, 342, d671. [Google Scholar]
- Mukamal, K.J.; Conigrave, K.M.; Mittleman, M.A.; Camargo, C.A., Jr.; Stampfer, M.J.; Willett, W.C.; Rimm, E.B. Roles of drinking pattern and type of alcohol consumed in coronary heart disease in men. N. Engl. J. Med. 2003, 348, 109–118. [Google Scholar]
- Mukamal, K.J.; Maclure, M.; Muller, J.E.; Mittleman, M.A. Binge drinking and mortality after acute myocardial infarction. Circulation 2005, 112, 3839–3845. [Google Scholar]
- Tunstall-Pedoe, H.; Kuulasmaa, K.; Mahonen, M.; Tolonen, H.; Ruokokoski, E.; Amouyel, P. Contribution of trends in survival and coronary-event rates to changes in coronary heart disease mortality: 10-year results from 37 WHO MONICA project populations. Monitoring trends and determinants in cardiovascular disease. Lancet 1999, 353, 1547–1557. [Google Scholar]
- Ruidavets, J.B.; Ducimetiere, P.; Evans, A.; Montaye, M.; Haas, B.; Bingham, A.; Yarnell, J.; Amouyel, P.; Arveiler, D.; Kee, F.; et al. Patterns of alcohol consumption and ischaemic heart disease in culturally divergent countries: The Prospective Epidemiological Study of Myocardial Infarction (PRIME). BMJ 2010, 341, c6077. [Google Scholar]
- Gaziano, J.M.; Hennekens, C.H.; Godfried, S.L.; Sesso, H.D.; Glynn, R.J.; Breslow, J.L.; Buring, J.E. Type of alcoholic beverage and risk of myocardial infarction. Am. J. Cardiol. 1999, 83, 52–57. [Google Scholar]
- Tousoulis, D.; Ntarladimas, I.; Antoniades, C.; Vasiliadou, C.; Tentolouris, C.; Papageorgiou, N.; Latsios, G.; Stefanadis, C. Acute effects of different alcoholic beverages on vascular endothelium, inflammatory markers and thrombosis fibrinolysis system. Clin. Nutr. 2008, 27, 594–600. [Google Scholar]
- Chiva-Blanch, G.; Urpi-Sarda, M.; Llorach, R.; Rotches-Ribalta, M.; Guillen, M.; Casas, R.; Arranz, S.; Valderas-Martinez, P.; Portoles, O.; Corella, D.; et al. Differential effects of polyphenols and alcohol of red wine on the expression of adhesion molecules and inflammatory cytokines related to atherosclerosis: A randomized clinical trial. Am. J. Clin. Nutr. 2012, 95, 326–334. [Google Scholar] [CrossRef]
- Cui, J.; Tosaki, A.; Cordis, G.A.; Bertelli, A.A.; Bertelli, A.; Maulik, N.; Das, D.K. Cardioprotective abilities of white wine. Ann. N. Y. Acad. Sci. 2002, 957, 308–316. [Google Scholar]
- Bertelli, A.A.; Migliori, M.; Panichi, V.; Longoni, B.; Origlia, N.; Ferretti, A.; Cuttano, M.G.; Giovannini, L. Oxidative stress and inflammatory reaction modulation by white wine. Ann. N. Y. Acad. Sci. 2002, 957, 295–301. [Google Scholar]
- Dudley, J.I.; Lekli, I.; Mukherjee, S.; Das, M.; Bertelli, A.A.; Das, D.K. Does white wine qualify for French paradox? Comparison of the cardioprotective effects of red and white wines and their constituents: Resveratrol, tyrosol, and hydroxytyrosol. J. Agric. Food Chem. 2008, 56, 9362–9373. [Google Scholar]
- Emeson, E.E.; Manaves, V.; Singer, T.; Tabesh, M. Chronic alcohol feeding inhibits atherogenesis in C57BL/6 hyperlipidemic mice. Am. J. Pathol. 1995, 147, 1749–1758. [Google Scholar]
- Emeson, E.E.; Manaves, V.; Emeson, B.S.; Chen, L.; Jovanovic, I. Alcohol inhibits the progression as well as the initiation of atherosclerotic lesions in C57Bl/6 hyperlipidemic mice. Alcohol. Clin. Exp. Res. 2000, 24, 1456–1466. [Google Scholar]
- Dai, J.; Miller, B.A.; Lin, R.C. Alcohol feeding impedes early atherosclerosis in low-density lipoprotein receptor knockout mice: Factors in addition to high-density lipoprotein-apolipoprotein A1 are involved. Alcohol. Clin. Exp. Res. 1997, 21, 11–18. [Google Scholar]
- Morrow, D.; Cullen, J.P.; Liu, W.; Cahill, P.A.; Redmond, E.M. Alcohol inhibits smooth muscle cell proliferation via regulation of the Notch signaling pathway. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 2597–2603. [Google Scholar]
- Liu, W.; Redmond, E.M.; Morrow, D.; Cullen, J.P. Differential effects of daily-moderate versus weekend binge alcohol consumption on atherosclerotic plaque development in mice. Atherosclerosis 2011, 219, 448–454. [Google Scholar]
- Breslow, R.A.; Smothers, B.A. Drinking patterns and body mass index in never smokers: National Health Interview Survey, 1997–2001. Am. J. Epidemiol. 2005, 161, 368–376. [Google Scholar]
- Bentzon, J.F.; Skovenborg, E.; Hansen, C.; Moller, J.; de Gaulejac, N.S.; Proch, J.; Falk, E. Red wine does not reduce mature atherosclerosis in apolipoprotein E-deficient mice. Circulation 2001, 103, 1681–1687. [Google Scholar]
- Escola-Gil, J.C.; Calpe-Berdiel, L.; Ribas, V.; Blanco-Vaca, F. Moderate beer consumption does not change early or mature atherosclerosis in mice. Nutr. J. 2004, 3, 1. [Google Scholar]
- Paigen, B.; Morrow, A.; Brandon, C.; Mitchell, D.; Holmes, P. Variation in susceptibility to atherosclerosis among inbred strains of mice. Atherosclerosis 1985, 57, 65–73. [Google Scholar]
- Deeg, M.A. Dietary cholate is required for antiatherogenic effects of ethanol in mouse models. Alcohol. Clin. Exp. Res. 2003, 27, 1499–1506. [Google Scholar]
- Ross, R. Atherosclerosis--an inflammatory disease. N. Engl. J. Med. 1999, 340, 115–126. [Google Scholar]
- Mestas, J.; Ley, K. Monocyte-endothelial cell interactions in the development of atherosclerosis. Trends Cardiovasc. Med. 2008, 18, 228–232. [Google Scholar]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and challenges in translating the biology of atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar]
- Blasi, C. The autoimmune origin of atherosclerosis. Atherosclerosis 2008, 201, 17–32. [Google Scholar]
- Morel, D.W.; DiCorleto, P.E.; Chisolm, G.M. Endothelial and smooth muscle cells alter low density lipoprotein in vitro by free radical oxidation. Arteriosclerosis 1984, 4, 357–364. [Google Scholar] [CrossRef]
- Steinbrecher, U.P.; Parthasarathy, S.; Leake, D.S.; Witztum, J.L.; Steinberg, D. Modification of low density lipoprotein by endothelial cells involves lipid peroxidation and degradation of low density lipoprotein phospholipids. Proc. Natl. Acad. Sci. USA 1984, 81, 3883–3887. [Google Scholar]
- Cathcart, M.K.; Morel, D.W.; Chisolm, G.M., III. Monocytes and neutrophils oxidize low density lipoprotein making it cytotoxic. J. Leukoc. Biol. 1985, 38, 341–350. [Google Scholar]
- Parthasarathy, S.; Printz, D.J.; Boyd, D.; Joy, L.; Steinberg, D. Macrophage oxidation of low density lipoprotein generates a modified form recognized by the scavenger receptor. Arteriosclerosis 1986, 6, 505–510. [Google Scholar]
- Mitra, S.; Goyal, T.; Mehta, J.L. Oxidized LDL, LOX-1 and atherosclerosis. Cardiovasc. Drugs Ther. 2011, 25, 419–429. [Google Scholar]
- Brinton, E.A. Effects of ethanol intake on lipoproteins and atherosclerosis. Curr. Opin. Lipidol. 2010, 21, 346–351. [Google Scholar]
- Booyse, F.M.; Pan, W.; Grenett, H.E.; Parks, D.A.; Darley-Usmar, V.M.; Bradley, K.M.; Tabengwa, E.M. Mechanism by which alcohol and wine polyphenols affect coronary heart disease risk. Ann. Epidemiol. 2007, 17, S24–S31. [Google Scholar]
- Pahor, M.; Guralnik, J.M.; Havlik, R.J.; Carbonin, P.; Salive, M.E.; Ferrucci, L.; Corti, M.C.; Hennekens, C.H. Alcohol consumption and risk of deep venous thrombosis and pulmonary embolism in older persons. J. Am. Geriatr. Soc. 1996, 44, 1030–1037. [Google Scholar]
- Pomp, E.R.; Rosendaal, F.R.; Doggen, C.J. Alcohol consumption is associated with a decreased risk of venous thrombosis. Thromb. Haemost. 2008, 99, 59–63. [Google Scholar]
- Hansen-Krone, I.J.; Braekkan, S.K.; Enga, K.F.; Wilsgaard, T.; Hansen, J.B. Alcohol consumption, types of alcoholic beverages and risk of venous thromboembolism—the Tromso Study. Thromb. Haemost. 2011, 106, 272–278. [Google Scholar]
- Furchgott, R.F.; Vanhoutte, P.M. Endothelium-derived relaxing and contracting factors. FASEB J. 1989, 3, 2007–2018. [Google Scholar]
- Moncada, S.; Higgs, E.A. Endogenous nitric oxide: Physiology, pathology and clinical relevance. Eur. J. Clin. Invest. 1991, 21, 361–374. [Google Scholar]
- Gkaliagkousi, E.; Ferro, A. Nitric oxide signalling in the regulation of cardiovascular and platelet function. Front. Biosci. 2011, 16, 1873–1897. [Google Scholar]
- Davda, R.K.; Chandler, L.J.; Crews, F.T.; Guzman, N.J. Ethanol enhances the endothelial nitric oxide synthase response to agonists. Hypertension 1993, 21, 939–943. [Google Scholar]
- Hendrickson, R.J.; Cahill, P.A.; Sitzmann, J.V.; Redmond, E.M. Ethanol enhances basal and flow-stimulated nitric oxide synthase activity in vitro by activating an inhibitory guanine nucleotide binding protein. J. Pharmacol. Exp. Ther. 1999, 289, 1293–1300. [Google Scholar]
- Venkov, C.D.; Myers, P.R.; Tanner, M.A.; Su, M.; Vaughan, D.E. Ethanol increases endothelial nitric oxide production through modulation of nitric oxide synthase expression. Thromb. Haemost. 1999, 81, 638–642. [Google Scholar]
- Abou-Agag, L.H.; Khoo, N.K.; Binsack, R.; White, C.R.; Darley-Usmar, V.; Grenett, H.E.; Booyse, F.M.; Digerness, S.B.; Zhou, F.; Parks, D.A. Evidence of cardiovascular protection by moderate alcohol: Role of nitric oxide. Free Radic. Biol. Med. 2005, 39, 540–548. [Google Scholar]
- Kleinhenz, D.J.; Sutliff, R.L.; Polikandriotis, J.A.; Walp, E.R.; Dikalov, S.I.; Guidot, D.M.; Hart, C.M. Chronic ethanol ingestion increases aortic endothelial nitric oxide synthase expression and nitric oxide production in the rat. Alcohol. Clin. Exp. Res. 2008, 32, 148–154. [Google Scholar]
- Tirapelli, C.R.; Fukada, S.Y.; Yogi, A.; Chignalia, A.Z.; Tostes, R.C.; Bonaventura, D.; Lanchote, V.L.; Cunha, F.Q.; de Oliveira, A.M. Gender-specific vascular effects elicited by chronic ethanol consumption in rats: A role for inducible nitric oxide synthase. Br. J. Pharmacol. 2008, 153, 468–479. [Google Scholar]
- Husain, K.; Vazquez-Ortiz, M.; Lalla, J. Down regulation of aortic nitric oxide and antioxidant systems in chronic alcohol-induced hypertension in rats. Hum. Exp. Toxicol. 2007, 26, 427–434. [Google Scholar]
- Husain, K.; Ferder, L.; Ansari, R.A.; Lalla, J. Chronic ethanol ingestion induces aortic inflammation/oxidative endothelial injury and hypertension in rats. Hum. Exp. Toxicol. 2011, 30, 930–939. [Google Scholar]
- Polikandriotis, J.A.; Rupnow, H.L.; Hart, C.M. Chronic ethanol exposure stimulates endothelial cell nitric oxide production through PI-3 kinase-and hsp90-dependent mechanisms. Alcohol. Clin. Exp. Res. 2005, 29, 1932–1938. [Google Scholar]
- El-Mas, M.M.; Fan, M.; Abdel-Rahman, A.A. Facilitation of myocardial PI3K/Akt/nNOS signaling contributes to ethanol-evoked hypotension in female rats. Alcohol. Clin. Exp. Res. 2009, 33, 1158–1168. [Google Scholar]
- Matsuo, S.; Nakamura, Y.; Takahashi, M.; Ouchi, Y.; Hosoda, K.; Nozawa, M.; Kinoshita, M. Effect of red wine and ethanol on production of nitric oxide in healthy subjects. Am. J. Cardiol. 2001, 87, 1029–1031. [Google Scholar]
- Huang, P.H.; Chen, Y.H.; Tsai, H.Y.; Chen, J.S.; Wu, T.C.; Lin, F.Y.; Sata, M.; Chen, J.W.; Lin, S.J. Intake of red wine increases the number and functional capacity of circulating endothelial progenitor cells by enhancing nitric oxide bioavailability. Arterioscler. Thromb. Vasc. Biol. 2010, 30, 869–877. [Google Scholar]
- Xue, L.; Xu, F.; Meng, L.; Wei, S.; Wang, J.; Hao, P.; Bian, Y.; Zhang, Y.; Chen, Y. Acetylation-dependent regulation of mitochondrial ALDH2 activation by SIRT3 mediates acute ethanol-induced eNOS activation. FEBS Lett. 2012, 586, 137–142. [Google Scholar]
- Kuhlmann, C.R.; Li, F.; Ludders, D.W.; Schaefer, C.A.; Most, A.K.; Backenkohler, U.; Neumann, T.; Tillmanns, H.; Waldecker, B.; Erdogan, A.; et al. Dose-dependent activation of Ca2+-activated K+ channels by ethanol contributes to improved endothelial cell functions. Alcohol. Clin. Exp. Res. 2004, 28, 1005–1011. [Google Scholar]
- Ru, X.C.; Qian, L.B.; Gao, Q.; Li, Y.F.; Bruce, I.C.; Xia, Q. Alcohol induces relaxation of rat thoracic aorta and mesenteric arterial bed. Alcohol Alcohol. 2008, 43, 537–543. [Google Scholar]
- Vlachopoulos, C.; Tsekoura, D.; Tsiamis, E.; Panagiotakos, D.; Stefanadis, C. Effect of alcohol on endothelial function in healthy subjects. Vasc. Med. 2003, 8, 263–265. [Google Scholar]
- Rendig, S.V.; Symons, J.D.; Longhurst, J.C.; Amsterdam, E.A. Effects of red wine, alcohol, and quercetin on coronary resistance and conductance arteries. J. Cardiovasc. Pharmacol. 2001, 38, 219–227. [Google Scholar] [CrossRef]
- Altura, B.M.; Altura, B.T.; Carella, A. Ethanol produces coronary vasospasm: Evidence for a direct action of ethanol on vascular muscle. Br. J. Pharmacol. 1983, 78, 260–262. [Google Scholar]
- Landolfi, R.; Steiner, M. Ethanol raises prostacyclin in vivo and in vitro. Blood 1984, 64, 679–682. [Google Scholar]
- Bau, P.F.; Bau, C.H.; Rosito, G.A.; Manfroi, W.C.; Fuchs, F.D. Alcohol consumption, cardiovascular health, and endothelial function markers. Alcohol 2007, 41, 479–488. [Google Scholar] [CrossRef]
- Yogi, A.; Callera, G.E.; Hipolito, U.V.; Silva, C.R.; Touyz, R.M.; Tirapelli, C.R. Ethanol-induced vasoconstriction is mediated via redox-sensitive cyclo-oxygenase-dependent mechanisms. Clin. Sci. 2010, 118, 657–668. [Google Scholar]
- Vasdev, S.; Ford, C.A.; Longerich, L.; Parai, S.; Gadag, V. Antihypertensive effect of low ethanol intake in spontaneously hypertensive rats. Mol. Cell. Biochem. 1999, 200, 85–92. [Google Scholar]
- Altura, B.M.; Zou, L.Y.; Altura, B.T.; Jelicks, L.; Wittenberg, B.A.; Gupta, R.K. Beneficial vs. detrimental actions of ethanol on heart and coronary vascular muscle: Roles of Mg2+ and Ca2+. Alcohol 1996, 13, 499–513. [Google Scholar] [CrossRef]
- Fujiyama, S.; Amano, K.; Uehira, K.; Yoshida, M.; Nishiwaki, Y.; Nozawa, Y.; Jin, D.; Takai, S.; Miyazaki, M.; Egashira, K.; et al. Bone marrow monocyte lineage cells adhere on injured endothelium in a monocyte chemoattractant protein-1-dependent manner and accelerate reendothelialization as endothelial progenitor cells. Circ. Res. 2003, 93, 980–989. [Google Scholar] [CrossRef]
- Werner, N.; Priller, J.; Laufs, U.; Endres, M.; Bohm, M.; Dirnagl, U.; Nickenig, G. Bone marrow-derived progenitor cells modulate vascular reendothelialization and neointimal formation: Effect of 3-hydroxy-3-methylglutaryl coenzyme a reductase inhibition. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 1567–1572. [Google Scholar]
- Liu, P.; Zhou, B.; Gu, D.; Zhang, L.; Han, Z. Endothelial progenitor cell therapy in atherosclerosis: A double-edged sword? Ageing Res. Rev. 2009, 8, 83–93. [Google Scholar] [CrossRef]
- Lefevre, J.; Michaud, S.E.; Haddad, P.; Dussault, S.; Menard, C.; Groleau, J.; Turgeon, J.; Rivard, A. Moderate consumption of red wine (cabernet sauvignon) improves ischemia-induced neovascularization in ApoE-deficient mice: Effect on endothelial progenitor cells and nitric oxide. FASEB J. 2007, 21, 3845–3852. [Google Scholar]
- Gil-Bernabe, P.; Boveda-Ruiz, D.; D'Alessandro-Gabazza, C.; Toda, M.; Miyake, Y.; Mifuji-Moroka, R.; Iwasa, M.; Morser, J.; Gabazza, E.C.; Takei, Y. Atherosclerosis amelioration by moderate alcohol consumption is associated with increased circulating levels of stromal cell-derived factor-1. Circ. J. 2011, 75, 2269–2279. [Google Scholar]
- Kaneto, H.; Katakami, N.; Matsuhisa, M.; Matsuoka, T.A. Role of reactive oxygen species in the progression of type 2 diabetes and atherosclerosis. Mediators Inflamm. 2010, 2010, 453892. [Google Scholar]
- Victor, V.M.; Rocha, M.; Sola, E.; Banuls, C.; Garcia-Malpartida, K.; Hernandez-Mijares, A. Oxidative stress, endothelial dysfunction and atherosclerosis. Curr. Pharm. Des. 2009, 15, 2988–3002. [Google Scholar]
- Rocha, J.T.; Hipolito, U.V.; Callera, G.E.; Yogi, A.; Filho Mdos, A.; Bendhack, L.M.; Touyz, R.M.; Tirapelli, C.R. Ethanol induces vascular relaxation via redox-sensitive and nitric oxide-dependent pathways. Vasc. pharmacol. 2012, 56, 74–83. [Google Scholar] [Green Version]
- Rakotovao, A.; Berthonneche, C.; Guiraud, A.; de Lorgeril, M.; Salen, P.; de Leiris, J.; Boucher, F. Ethanol, wine, and experimental cardioprotection in ischemia/reperfusion: Role of the prooxidant/antioxidant balance. Antioxid. Redox Signal. 2004, 6, 431–438. [Google Scholar] [CrossRef]
- Krenz, M.; Korthuis, R.J. Moderate ethanol ingestion and cardiovascular protection: From epidemiologic associations to cellular mechanisms. J. Mol. Cell. Cardiol. 2012, 52, 93–104. [Google Scholar]
- Morrow, D.; Cullen, J.P.; Cahill, P.A.; Redmond, E.M. Ethanol stimulates endothelial cell angiogenic activity via a Notch- and angiopoietin-1-dependent pathway. Cardiovasc. Res. 2008, 79, 313–321. [Google Scholar]
- Artavanis-Tsakonas, S.; Rand, M.D.; Lake, R.J. Notch signaling: Cell fate control and signal integration in development. Science 1999, 284, 770–776. [Google Scholar]
- Shawber, C.J.; Kitajewski, J. Notch function in the vasculature: Insights from zebrafish, mouse and man. Bioessays 2004, 26, 225–234. [Google Scholar]
- Liu, Z.J.; Xiao, M.; Balint, K.; Soma, A.; Pinnix, C.C.; Capobianco, A.J.; Velazquez, O.C.; Herlyn, M. Inhibition of endothelial cell proliferation by Notch1 signaling is mediated by repressing MAPK and PI3K/Akt pathways and requires MAML1. FASEB J. 2006, 20, 1009–1011. [Google Scholar]
- Hainaud, P.; Contreres, J.O.; Villemain, A.; Liu, L.X.; Plouet, J.; Tobelem, G.; Dupuy, E. The Role of the vascular endothelial growth factor-delta-like 4 Ligand/Notch4-Ephrin B2 cascade in tumor vessel remodeling and endothelial cell functions. Cancer Res. 2006, 66, 8501–8510. [Google Scholar]
- Morrow, D.; Cullen, J.P.; Cahill, P.A.; Redmond, E.M. Cyclic strain regulates the Notch/CBF-1 signaling pathway in endothelial cells: Role in angiogenic activity. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1289–1296. [Google Scholar]
- Liu, Z.J.; Shirakawa, T.; Li, Y.; Soma, A.; Oka, M.; Dotto, G.P.; Fairman, R.M.; Velazquez, O.C.; Herlyn, M. Regulation of Notch1 and Dll4 by vascular endothelial growth factor in arterial endothelial cells: Implications for modulating arteriogenesis and angiogenesis. Mol. Cell. Biol. 2003, 23, 14–25. [Google Scholar]
- Leong, K.G.; Hu, X.; Li, L.; Noseda, M.; Larrivee, B.; Hull, C.; Hood, L.; Wong, F.; Karsan, A. Activated Notch4 inhibits angiogenesis: Role of beta 1-integrin activation. Mol. Cell. Biol. 2002, 22, 2830–2841. [Google Scholar]
- Taylor, K.L.; Henderson, A.M.; Hughes, C.C. Notch activation during endothelial cell network formation in vitro targets the basic HLH transcription factor HESR-1 and downregulates VEGFR-2/KDR expression. Microvasc. Res. 2002, 64, 372–383. [Google Scholar] [CrossRef]
- Zimrin, A.B.; Pepper, M.S.; McMahon, G.A.; Nguyen, F.; Montesano, R.; Maciag, T. An antisense oligonucleotide to the notch ligand jagged enhances fibroblast growth factor-induced angiogenesis in vitro. J. Biol. Chem. 1996, 271, 32499–32502. [Google Scholar]
- Carmeliet, P.; Jain, R.K. Angiogenesis in cancer and other diseases. Nature 2000, 407, 249–257. [Google Scholar]
- Jeziorska, M.; Woolley, D.E. Local neovascularization and cellular composition within vulnerable regions of atherosclerotic plaques of human carotid arteries. J. Pathol. 1999, 188, 189–196. [Google Scholar]
- Herrmann, J.; Lerman, L.O.; Mukhopadhyay, D.; Napoli, C.; Lerman, A. Angiogenesis in atherogenesis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1948–1957. [Google Scholar]
- Moreno, P.R.; Purushothaman, K.R.; Fuster, V.; Echeverri, D.; Truszczynska, H.; Sharma, S.K.; Badimon, J.J.; O’Connor, W.N. Plaque neovascularization is increased in ruptured atherosclerotic lesions of human aorta: Implications for plaque vulnerability. Circulation 2004, 110, 2032–2038. [Google Scholar]
- Koerselman, J.; van der Graaf, Y.; de Jaegere, P.P.; Grobbee, D.E. Coronary collaterals: An important and underexposed aspect of coronary artery disease. Circulation 2003, 107, 2507–2511. [Google Scholar]
- Hansen, J.F. Coronary collateral circulation: Clinical significance and influence on survival in patients with coronary artery occlusion. Am. Heart J. 1989, 117, 290–295. [Google Scholar]
- Radek, K.A.; Matthies, A.M.; Burns, A.L.; Heinrich, S.A.; Kovacs, E.J.; Dipietro, L.A. Acute ethanol exposure impairs angiogenesis and the proliferative phase of wound healing. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1084–H1090. [Google Scholar]
- Gu, J.W.; Elam, J.; Sartin, A.; Li, W.; Roach, R.; Adair, T.H. Moderate levels of ethanol induce expression of vascular endothelial growth factor and stimulate angiogenesis. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2001, 281, R365–R372. [Google Scholar]
- Jones, M.K.; Sarfeh, I.J.; Tarnawski, A.S. Induction of in vitro angiogenesis in the endothelial-derived cell line, EA hy926, by ethanol is mediated through PKC and MAPK. Biochem. Biophys. Res. Commun. 1998, 249, 118–123. [Google Scholar] [CrossRef]
- Gu, J.W.; Bailey, A.P.; Sartin, A.; Makey, I.; Brady, A.L. Ethanol stimulates tumor progression and expression of vascular endothelial growth factor in chick embryos. Cancer 2005, 103, 422–431. [Google Scholar]
- Gavin, T.P.; Wagner, P.D. Acute ethanol increases angiogenic growth factor gene expression in rat skeletal muscle. J. Appl. Physiol. 2002, 92, 1176–1182. [Google Scholar]
- Bora, P.S.; Kaliappan, S.; Xu, Q.; Kumar, S.; Wang, Y.; Kaplan, H.J.; Bora, N.S. Alcohol linked to enhanced angiogenesis in rat model of choroidal neovascularization. FEBS J. 2006, 273, 1403–1414. [Google Scholar]
- Qian, Y.; Luo, J.; Leonard, S.S.; Harris, G.K.; Millecchia, L.; Flynn, D.C.; Shi, X. Hydrogen peroxide formation and actin filament reorganization by Cdc42 are essential for ethanol-induced in vitro angiogenesis. J. Biol. Chem. 2003, 278, 16189–16197. [Google Scholar]
- Luedemann, C.; Bord, E.; Qin, G.; Zhu, Y.; Goukassian, D.; Losordo, D.W.; Kishore, R. Ethanol modulation of TNF-alpha biosynthesis and signaling in endothelial cells: Synergistic augmentation of TNF-alpha mediated endothelial cell dysfunctions by chronic ethanol. Alcohol. Clin. Exp. Res. 2005, 29, 930–938. [Google Scholar]
- Maulik, N. Reactive oxygen species drives myocardial angiogenesis? Antioxid. Redox Signal. 2006, 8, 2161–2168. [Google Scholar] [CrossRef]
- Koerselman, J.; de Jaegere, P.P.; Verhaar, M.C.; Grobbee, D.E.; van der Graaf, Y. Coronary collateral circulation: The effects of smoking and alcohol. Atherosclerosis 2007, 191, 191–198. [Google Scholar]
- Van Coillie, E.; Van Damme, J.; Opdenakker, G. The MCP/eotaxin subfamily of CC chemokines. Cytokine Growth Factor Rev. 1999, 10, 61–86. [Google Scholar]
- Boring, L.; Gosling, J.; Cleary, M.; Charo, I.F. Decreased lesion formation in CCR2 −/− mice reveals a role for chemokines in the initiation of atherosclerosis. Nature 1998, 394, 894–897. [Google Scholar]
- Han, K.H.; Han, K.O.; Green, S.R.; Quehenberger, O. Expression of the monocyte chemoattractant protein-1 receptor CCR2 is increased in hypercholesterolemia. Differential effects of plasma lipoproteins on monocyte function. J. Lipid Res. 1999, 40, 1053–1063. [Google Scholar]
- Wang, G.; O, K. Homocysteine stimulates the expression of monocyte chemoattractant protein-1 receptor (CCR2) in human monocytes: Possible involvement of oxygen free radicals. Biochem. J. 2001, 357, 233–240. [Google Scholar]
- Cullen, J.P.; Sayeed, S.; Jin, Y.; Theodorakis, N.G.; Sitzmann, J.V.; Cahill, P.A.; Redmond, E.M. Ethanol inhibits monocyte chemotactic protein-1 expression in interleukin-1{beta}-activated human endothelial cells. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H1669–H1675. [Google Scholar]
- Imhof, A.; Blagieva, R.; Marx, N.; Koenig, W. Drinking modulates monocyte migration in healthy subjects: A randomised intervention study of water, ethanol, red wine and beer with or without alcohol. Diabetes Vasc. Dis. Res. 2008, 5, 48–53. [Google Scholar] [CrossRef]
- Blanco-Colio, L.M.; Munoz-Garcia, B.; Martin-Ventura, J.L.; Alvarez-Sala, L.A.; Castilla, M.; Bustamante, A.; Lamuela-Raventos, R.M.; Gomez-Gerique, J.; Fernandez-Cruz, A.; Millan, J.; et al. Ethanol beverages containing polyphenols decrease nuclear factor kappa-B activation in mononuclear cells and circulating MCP-1 concentrations in healthy volunteers during a fat-enriched diet. Atherosclerosis 2007, 192, 335–341. [Google Scholar] [CrossRef]
- Vazquez-Agell, M.; Sacanella, E.; Tobias, E.; Monagas, M.; Antunez, E.; Zamora-Ros, R.; Andres-Lacueva, C.; Lamuela-Raventos, R.M.; Fernandez-Sola, J.; Nicolas, J.M.; et al. Inflammatory markers of atherosclerosis are decreased after moderate consumption of cava (sparkling wine) in men with low cardiovascular risk. J. Nutr. 2007, 137, 2279–2284. [Google Scholar]
- Eriksson, C.J. The role of acetaldehyde in the actions of alcohol (update 2000). Alcohol. Clin. Exp. Res. 2001, 25, 15S–32S. [Google Scholar]
- Redmond, E.M.; Morrow, D.; Kundimi, S.; Miller-Graziano, C.L.; Cullen, J.P. Acetaldehyde stimulates monocyte adhesion in a P-selectin- and TNFalpha-dependent manner. Atherosclerosis 2009, 204, 372–380. [Google Scholar]
- Tsukamoto, S.; Muto, T.; Nagoya, T.; Shimamura, M.; Saito, M.; Tainaka, H. Determinations of ethanol, acetaldehyde and acetate in blood and urine during alcohol oxidation in man. Alcohol Alcohol. 1989, 24, 101–108. [Google Scholar]
- Owens, G.K. Molecular control of vascular smooth muscle cell differentiation and phenotypic plasticity. Novartis Found. Symp. 2007, 283, 174–191, discussion 191–193, 238–241.. [Google Scholar] [CrossRef]
- Schwartz, S.M.; deBlois, D.; O’Brien, E.R. The intima. Soil for atherosclerosis and restenosis. Circ. Res. 1995, 77, 445–465. [Google Scholar]
- Owens, G.K.; Kumar, M.S.; Wamhoff, B.R. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 2004, 84, 767–801. [Google Scholar]
- Liu, M.W.; Lin, S.J.; Chen, Y.L. Local alcohol delivery may reduce phenotype conversion of smooth muscle cells and neointimal formation in rabbit iliac arteries after balloon injury. Atherosclerosis 1996, 127, 221–227. [Google Scholar]
- Merritt, R.; Guruge, B.L.; Miller, D.D.; Chaitman, B.R.; Bora, P.S. Moderate alcohol feeding attenuates postinjury vascular cell proliferation in rabbit angioplasty model. J. Cardiovasc. Pharmacol. 1997, 30, 19–25. [Google Scholar]
- Liu, M.W.; Anderson, P.G.; Luo, J.F.; Roubin, G.S. Local delivery of ethanol inhibits intimal hyperplasia in pig coronary arteries after balloon injury. Circulation 1997, 96, 2295–2301. [Google Scholar]
- Hou, D.; Zhang, P.; Marsh, A.E.; March, K.L. Intrapericardial ethanol delivery inhibits neointimal proliferation after porcine coronary overstretch. J. Chin. Med. Assoc. 2003, 66, 637–642. [Google Scholar]
- Niroomand, F.; Hauer, O.; Tiefenbacher, C.P.; Katus, H.A.; Kuebler, W. Influence of alcohol consumption on restenosis rate after percutaneous transluminal coronary angioplasty and stent implantation. Heart 2004, 90, 1189–1193. [Google Scholar]
- Hendrickson, R.J.; Cahill, P.A.; McKillop, I.H.; Sitzmann, J.V.; Redmond, E.M. Ethanol inhibits mitogen activated protein kinase activity and growth of vascular smooth muscle cells in vitro. Eur. J. Pharmacol. 1998, 362, 251–259. [Google Scholar] [CrossRef]
- Mii, S.; Khalil, R.A.; Morgan, K.G.; Ware, J.A.; Kent, K.C. Mitogen-activated protein kinase and proliferation of human vascular smooth muscle cells. Am. J. Physiol. 1996, 270, H142–H150. [Google Scholar]
- Sayeed, S.; Cullen, J.P.; Coppage, M.; Sitzmann, J.V.; Redmond, E.M. Ethanol differentially modulates the expression and activity of cell cycle regulatory proteins in rat aortic smooth muscle cells. Eur. J. Pharmacol. 2002, 445, 163–170. [Google Scholar]
- Ghiselli, G.; Chen, J.; Kaou, M.; Hallak, H.; Rubin, R. Ethanol inhibits fibroblast growth factor-induced proliferation of aortic smooth muscle cells. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1808–1813. [Google Scholar]
- Locher, R.; Suter, P.M.; Vetter, W. Ethanol suppresses smooth muscle cell proliferation in the postprandial state: A new antiatherosclerotic mechanism of ethanol? Am. J. Clin. Nutr. 1998, 67, 338–341. [Google Scholar]
- Garg, U.C.; Hassid, A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J. Clin. Investig. 1989, 83, 1774–1777. [Google Scholar]
- von der Leyen, H.E.; Gibbons, G.H.; Morishita, R.; Lewis, N.P.; Zhang, L.; Nakajima, M.; Kaneda, Y.; Cooke, J.P.; Dzau, V.J. Gene therapy inhibiting neointimal vascular lesion: In vivo transfer of endothelial cell nitric oxide synthase gene. Proc. Natl. Acad. Sci. USA 1995, 92, 1137–1141. [Google Scholar]
- Sweeney, C.; Morrow, D.; Birney, Y.A.; Coyle, S.; Hennessy, C.; Scheller, A.; Cummins, P.M.; Walls, D.; Redmond, E.M.; Cahill, P.A. Notch 1 and 3 receptor signaling modulates vascular smooth muscle cell growth, apoptosis, and migration via a CBF-1/RBP-Jk dependent pathway. FASEB J. 2004, 18, 1421–1423. [Google Scholar]
- Wang, W.; Campos, A.H.; Prince, C.Z.; Mou, Y.; Pollman, M.J. Coordinate Notch3-hairy-related transcription factor pathway regulation in response to arterial injury. Mediator role of platelet-derived growth factor and ERK. J. Biol. Chem. 2002, 277, 23165–23171. [Google Scholar]
- Wang, W.; Prince, C.Z.; Hu, X.; Pollman, M.J. HRT1 modulates vascular smooth muscle cell proliferation and apoptosis. Biochem. Biophys. Res. Commun. 2003, 308, 596–601. [Google Scholar]
- Morrow, D.; Sweeney, C.; Birney, Y.A.; Cummins, P.M.; Walls, D.; Redmond, E.M.; Cahill, P.A. Cyclic strain inhibits Notch receptor signaling in vascular smooth muscle cells in vitro. Circ. Res. 2005, 96, 567–575. [Google Scholar] [CrossRef]
- Lindner, V.; Booth, C.; Prudovsky, I.; Small, D.; Maciag, T.; Liaw, L. Members of the Jagged/Notch gene families are expressed in injured arteries and regulate cell phenotype via alterations in cell matrix and cell-cell interaction. Am. J. Pathol. 2001, 159, 875–883. [Google Scholar]
- Campos, A.H.; Wang, W.; Pollman, M.J.; Gibbons, G.H. Determinants of Notch-3 receptor expression and signaling in vascular smooth muscle cells: Implications in cell-cycle regulation. Circ. Res. 2002, 91, 999–1006. [Google Scholar]
- Doi, H.; Iso, T.; Yamazaki, M.; Akiyama, H.; Kanai, H.; Sato, H.; Kawai-Kowase, K.; Tanaka, T.; Maeno, T.; Okamoto, E.; et al. HERP1 inhibits myocardin-induced vascular smooth muscle cell differentiation by interfering with SRF binding to CArG box. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2328–2334. [Google Scholar] [CrossRef]
- Sakata, Y.; Xiang, F.; Chen, Z.; Kiriyama, Y.; Kamei, C.N.; Simon, D.I.; Chin, M.T. Transcription factor CHF1/Hey2 regulates neointimal formation in vivo and vascular smooth muscle proliferation and migration in vitro. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2069–2074. [Google Scholar] [CrossRef]
- Havrda, M.C.; Johnson, M.J.; O'Neill, C.F.; Liaw, L. A novel mechanism of transcriptional repression of p27kip1 through Notch/HRT2 signaling in vascular smooth muscle cells. Thromb. Haemost. 2006, 96, 361–370. [Google Scholar]
- Hendrickson, R.J.; Okada, S.S.; Cahill, P.A.; Yankah, E.; Sitzmann, J.V.; Redmond, E.M. Ethanol inhibits basal and flow-induced vascular smooth muscle cell migration in vitro. J. Surg. Res. 1999, 84, 64–70. [Google Scholar] [CrossRef]
- Cullen, J.P.; Sayeed, S.; Kim, Y.; Theodorakis, N.G.; Sitzmann, J.V.; Cahill, P.A.; Redmond, E.M. Ethanol inhibits pulse pressure-induced vascular smooth muscle cell migration by differentially modulating plasminogen activator inhibitor type 1, matrix metalloproteinase-2 and -9. Thromb. Haemost. 2005, 94, 639–645. [Google Scholar]
- Fiotti, N.; Tubaro, F.; Altamura, N.; Grassi, G.; Moretti, M.; Dapas, B.; Farra, R.; Mizzau, M.; Guarnieri, G.; Buiatti, S.; et al. Alcohol reduces MMP-2 in humans and isolated smooth muscle cells. Alcohol 2008, 42, 389–395. [Google Scholar] [CrossRef]
- Harris, R.A.; Trudell, J.R.; Mihic, S.J. Ethanol’s molecular targets. Sci. Signal. 2008, 1, re7. [Google Scholar]
- Insel, P.A.; Patel, H.H. Membrane rafts and caveolae in cardiovascular signaling. Curr. Opin. Nephrol. Hypertens. 2009, 18, 50–56. [Google Scholar]
- Dolganiuc, A.; Bakis, G.; Kodys, K.; Mandrekar, P.; Szabo, G. Acute ethanol treatment modulates Toll-like receptor-4 association with lipid rafts. Alcohol. Clin. Exp. Res. 2006, 30, 76–85. [Google Scholar]
- Fernandez-Lizarbe, S.; Pascual, M.; Gascon, M.S.; Blanco, A.; Guerri, C. Lipid rafts regulate ethanol-induced activation of TLR4 signaling in murine macrophages. Mol. Immunol. 2008, 45, 2007–2016. [Google Scholar]
- Miranda, R.C.; Pietrzykowski, A.Z.; Tang, Y.; Sathyan, P.; Mayfield, D.; Keshavarzian, A.; Sampson, W.; Hereld, D. MicroRNAs: Master regulators of ethanol abuse and toxicity? Alcohol. Clin. Exp. Res. 2010, 34, 575–587. [Google Scholar] [CrossRef]
- Zernecke, A. MicroRNAs in the regulation of immune cell functions—implications for atherosclerotic vascular disease. Thromb. Haemost. 2012, 107, 626–633. [Google Scholar]
- Small, E.M.; Frost, R.J.; Olson, E.N. MicroRNAs add a new dimension to cardiovascular disease. Circulation 2010, 121, 1022–1032. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Cahill, P.A.; Redmond, E.M. Alcohol and Cardiovascular Disease—Modulation of Vascular Cell Function. Nutrients 2012, 4, 297-318. https://doi.org/10.3390/nu4040297
Cahill PA, Redmond EM. Alcohol and Cardiovascular Disease—Modulation of Vascular Cell Function. Nutrients. 2012; 4(4):297-318. https://doi.org/10.3390/nu4040297
Chicago/Turabian StyleCahill, Paul A., and Eileen M. Redmond. 2012. "Alcohol and Cardiovascular Disease—Modulation of Vascular Cell Function" Nutrients 4, no. 4: 297-318. https://doi.org/10.3390/nu4040297
APA StyleCahill, P. A., & Redmond, E. M. (2012). Alcohol and Cardiovascular Disease—Modulation of Vascular Cell Function. Nutrients, 4(4), 297-318. https://doi.org/10.3390/nu4040297