Molecular Nutrition Research—The Modern Way Of Performing Nutritional Science

Abstract
1. Introduction
1.1. Population Growth and Life Expectancy
1.2. Modern Nutrition Research
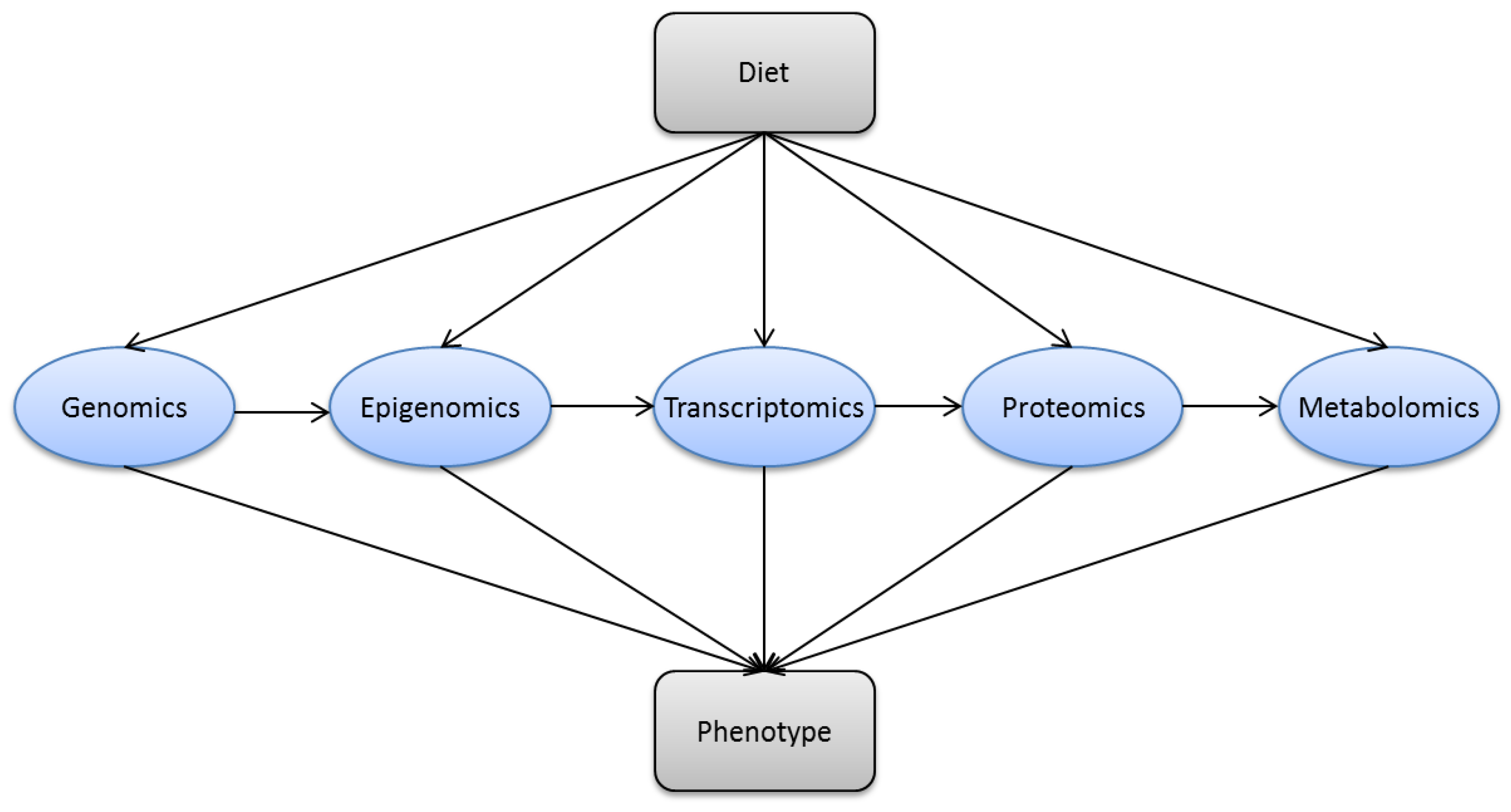
1.3. Nutrigenomics and Molecular Nutrition
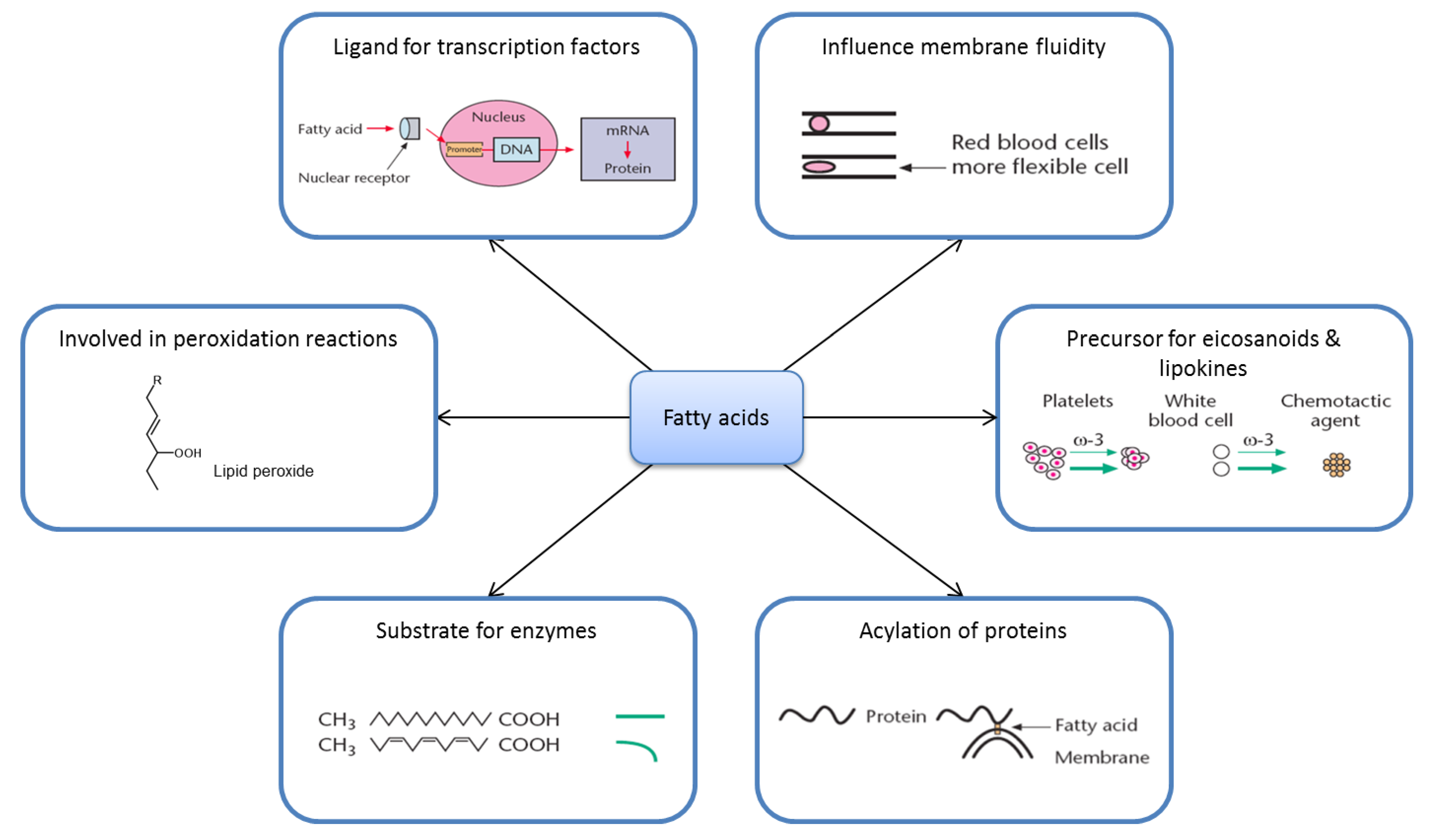
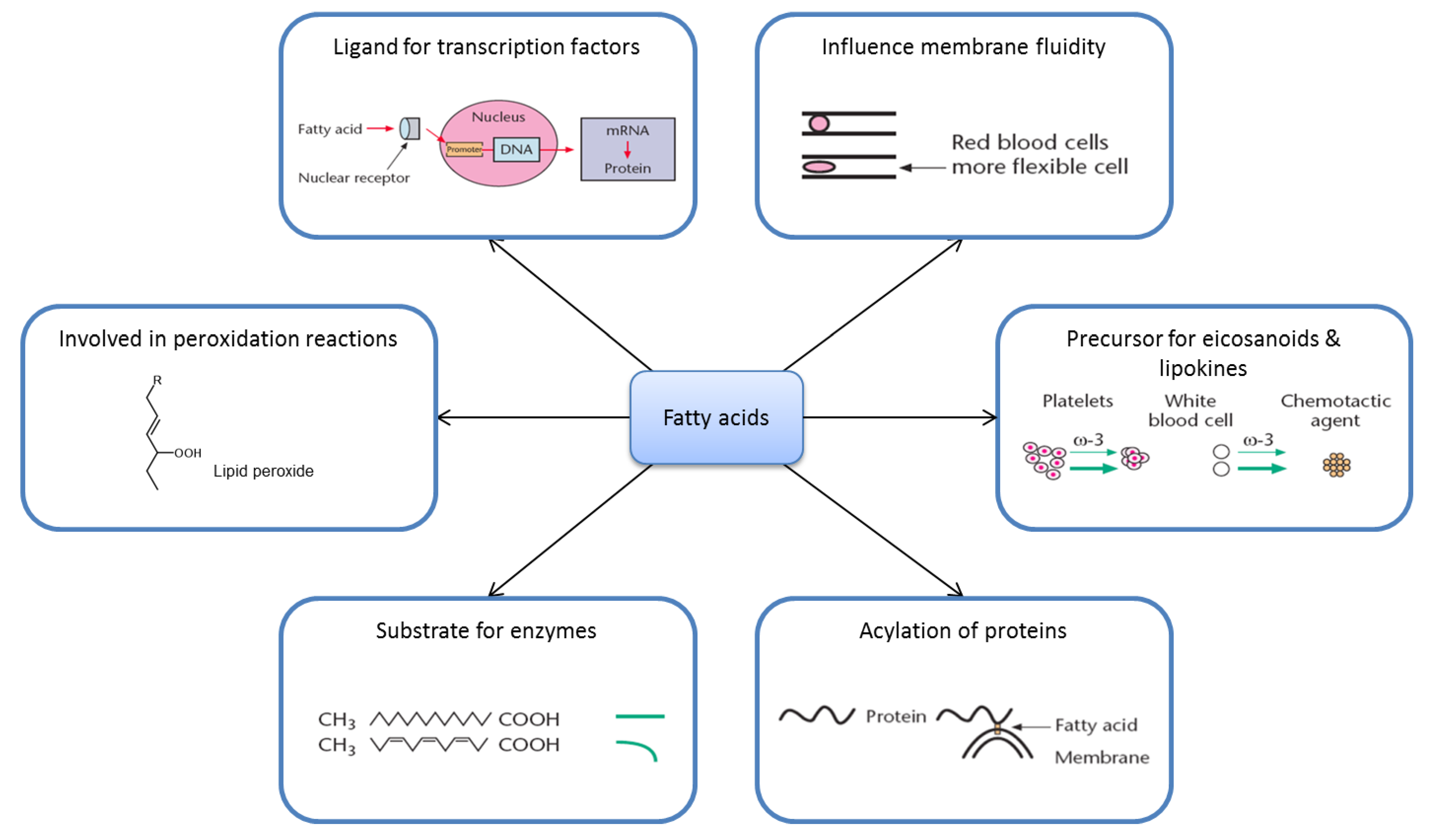
2. Nutrition and Metabolism Are Complicated
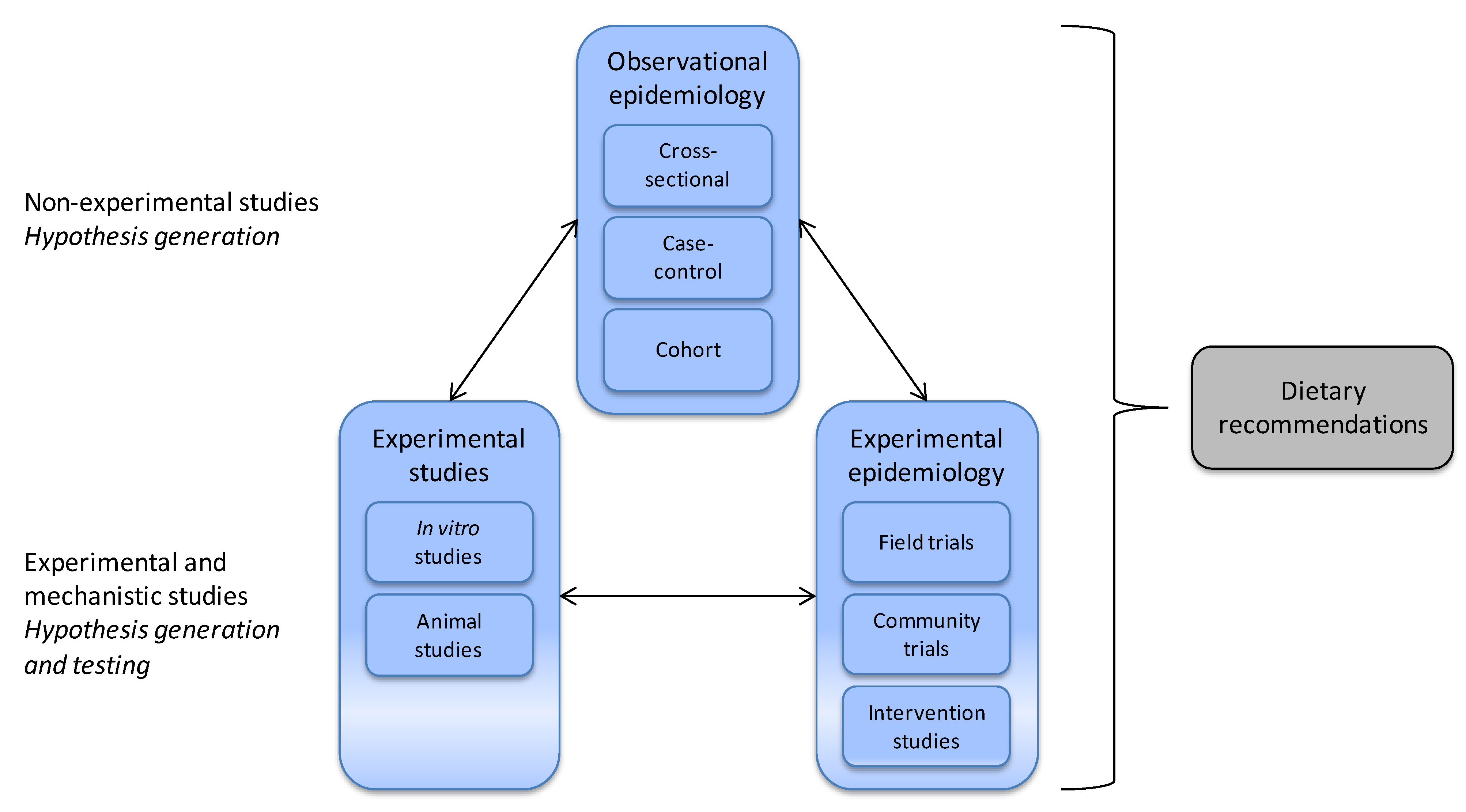
2.1. Nutrition Is Demanding

{kind=link}
{kind=link}
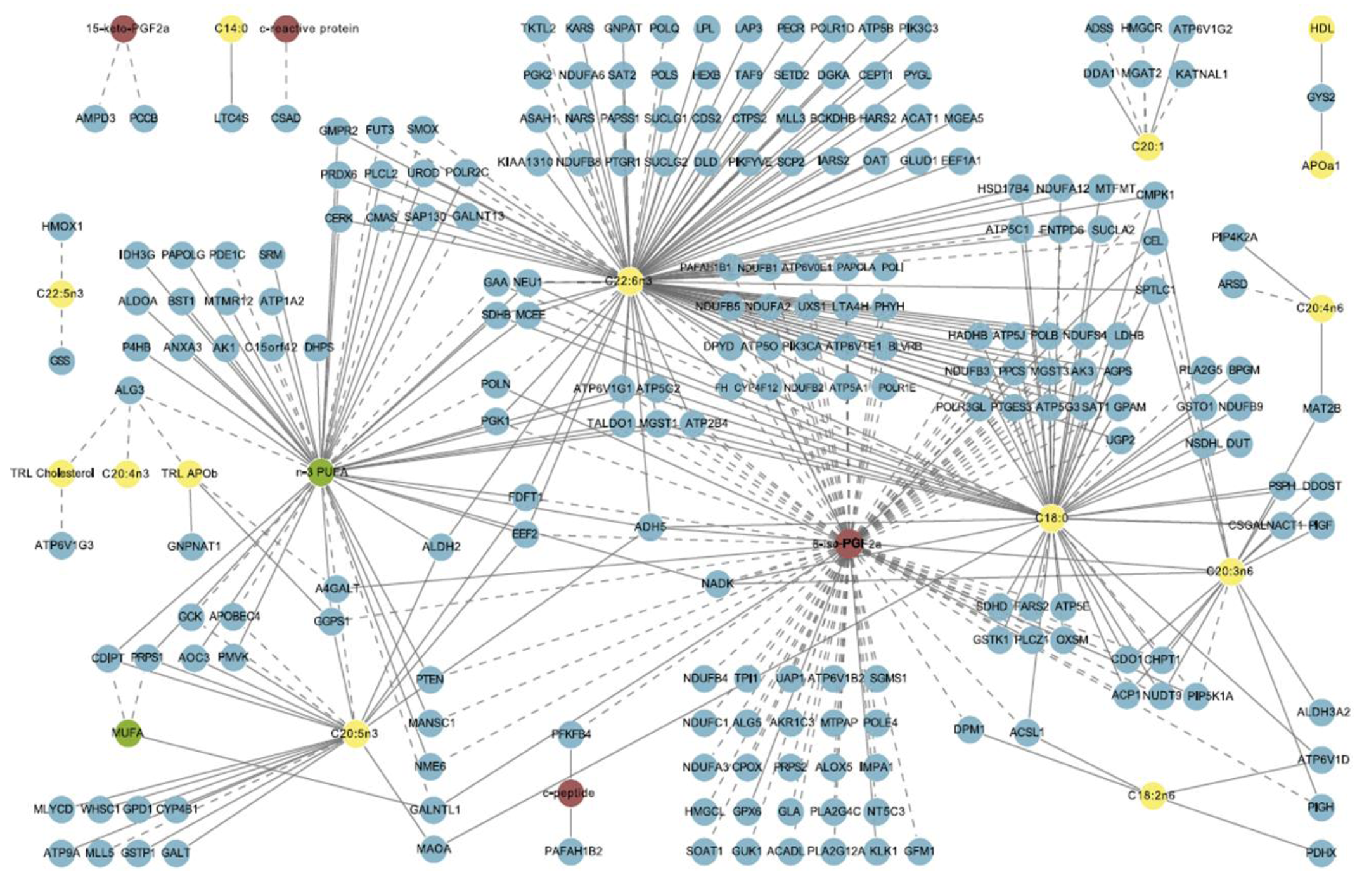{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
|
2.2. Fatty Acid Metabolism Is Complex
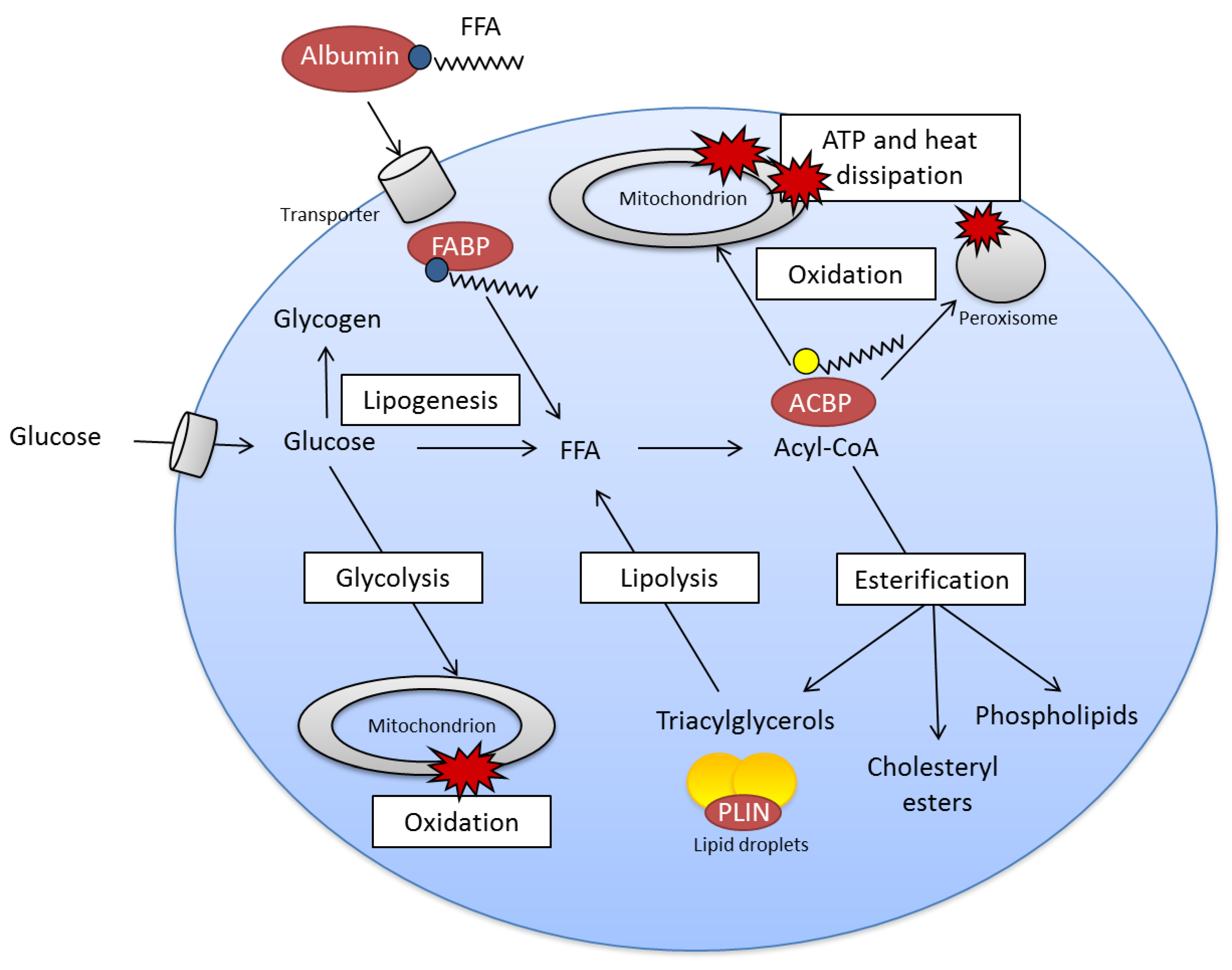
2.3. Ligands for Transcription Factors/Altered Gene Expression
2.4. Eicosanoids
2.5. Substrate Specificity
2.6. Membrane Fluidity
2.7. Lipid Peroxidation
2.8. Acylation of Proteins
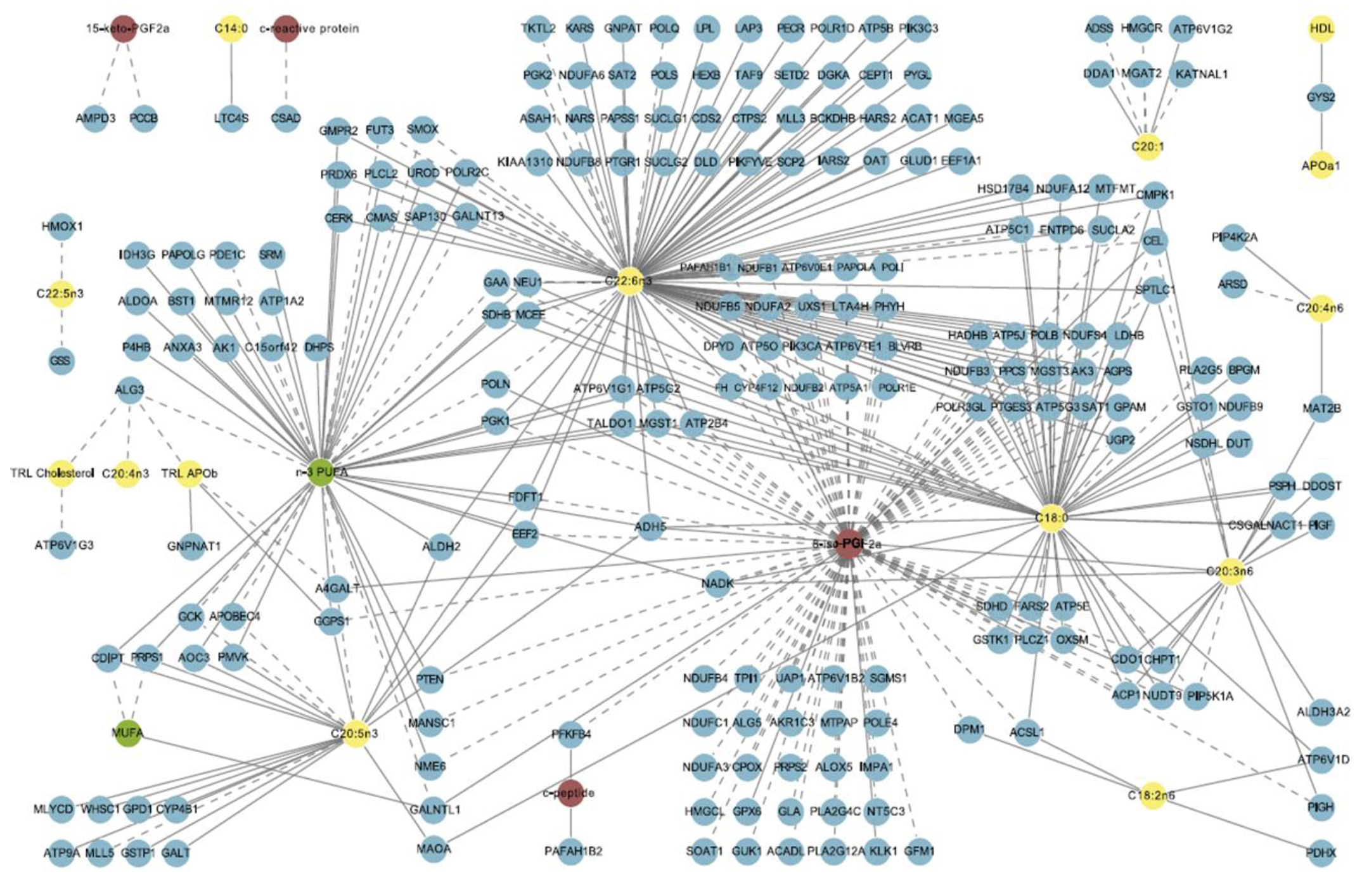
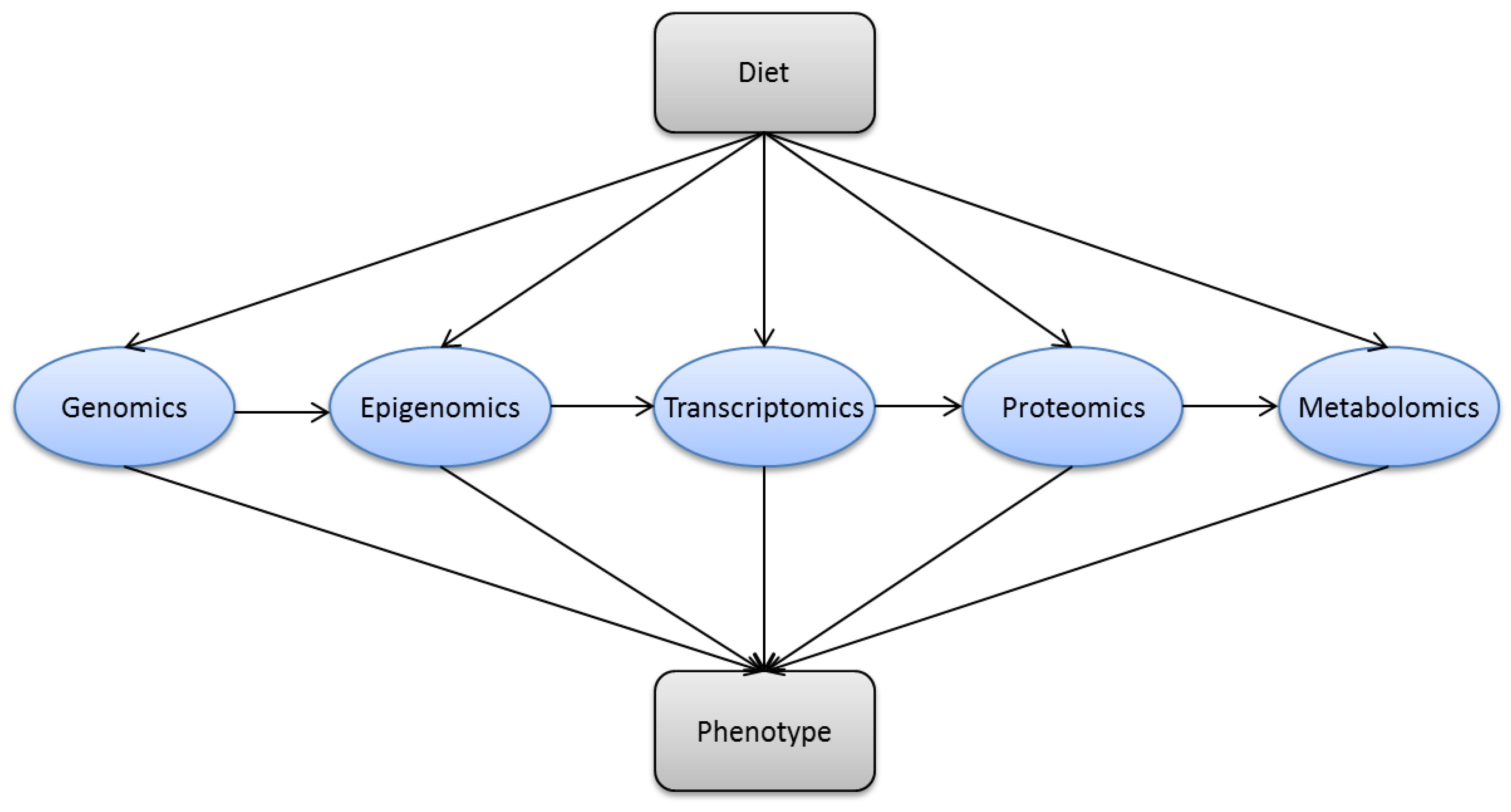
3. Methods in Modern Nutrition Research
| Research Area | Technologies | Assessed Parameters |
|---|---|---|
| Epidemiology | Observational | Association between diet and health outcomes and effects of controlled dietary changes |
| Experimental | ||
| Genomics | Microarray | Association between genetic variation (e.g., SNPs, alleles) and phenotypic traits |
| Next generation sequencing | ||
| Epigenomics | Bisulfite sequencing | DNA methylation and histone modification |
| ChiP-sequencing | ||
| Transcriptomics | Microarray | mRNA levels and splice variants |
| RNA sequencing | ||
| Proteomics | Chromatography | Protein composition and posttranslational modifications |
| Electrophoresis | ||
| Mass spectrometry | ||
| Protein microarrays | ||
| Metabolomics | Gas liquid chromatography | Metabolites |
| Liquid chromatography | ||
| Mass spectrometry | ||
| Nuclear magnetic resonance | ||
| Microbiota | Sequencing the 16S rRNA gene | Microbe species composition; genome, transcriptome, proteome and metabolome of the microbiotic community |
| Metaomics (includes all omics described above) | ||
| Imaging | CT | Whole body dynamic non-invasive detection of body composition (fat and lean mass), gene regulation and molecular tracers and probes |
| MRI | ||
| PET | ||
| SPECT | ||
| Optical imaging | ||
| Calorimetry | Indirect calorimetry | Energy intake and expenditure |
| Direct calorimetry | ||
| Cognition | Cognitive tests (K-ABC, Fagan, ERP, Kendrick object learning, Trail making, Digit symbol, Block design, Mini-mental state examination, Oral word association), EEG | IQ (sequential & simultaneous processing, nonverbal abilities, recognition memory) |
| Systems biology | Mathematical modeling | Integrate large data sets to understand complex physiological systems |
| Statistical methods |
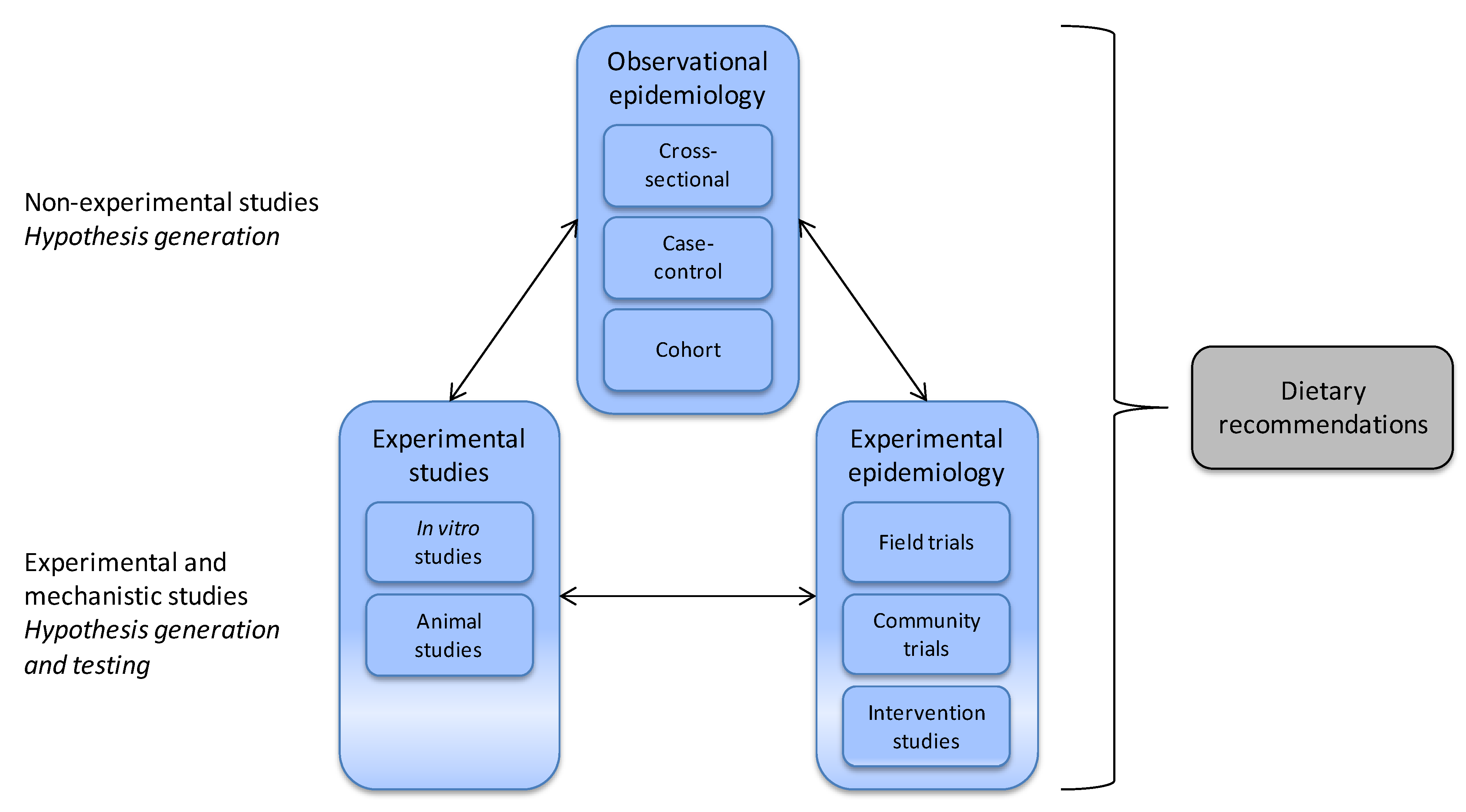
3.1. Nutritional Epidemiology
3.2. Genomics
3.3. Epigenomics
3.4. Transcriptomics
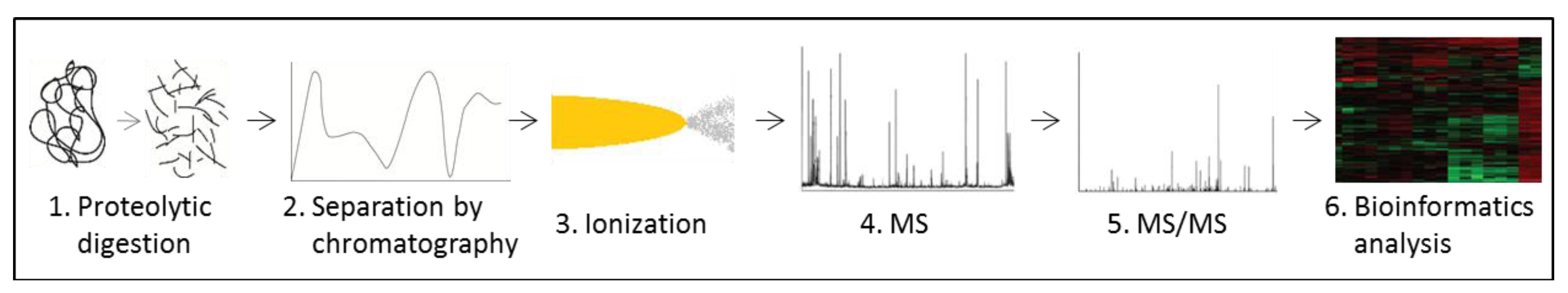
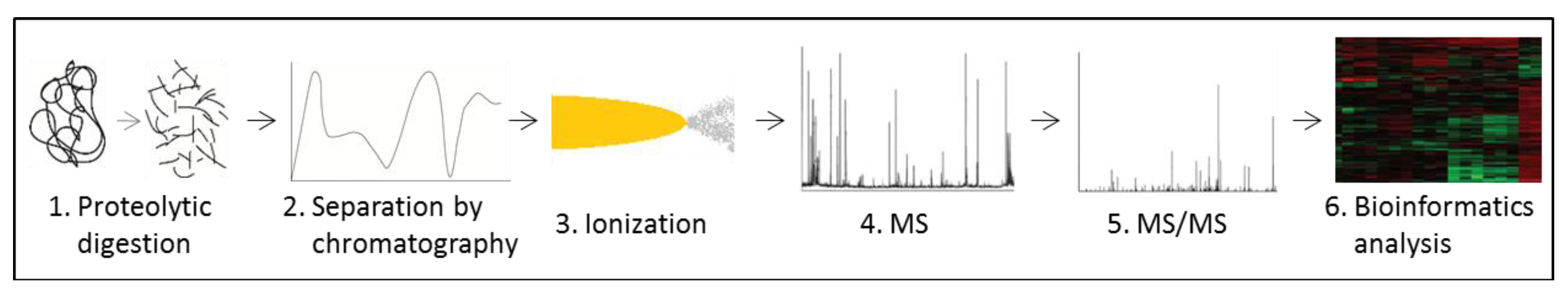
3.5. Proteomics
3.6. Metabolomics
3.7. Microbiota
3.8. Imaging
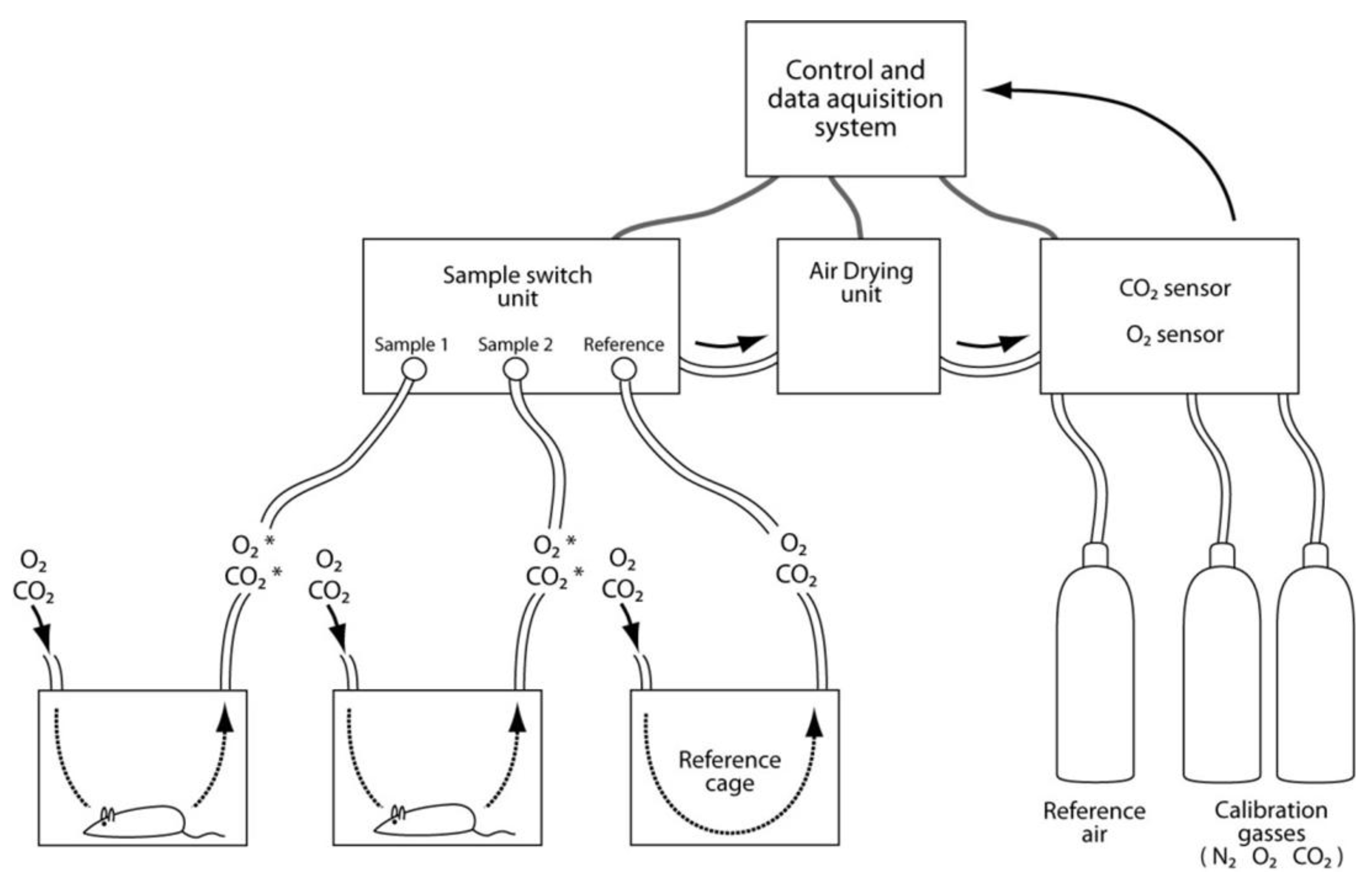
3.9. Assessments of Energy Expenditure Using Calorimetry
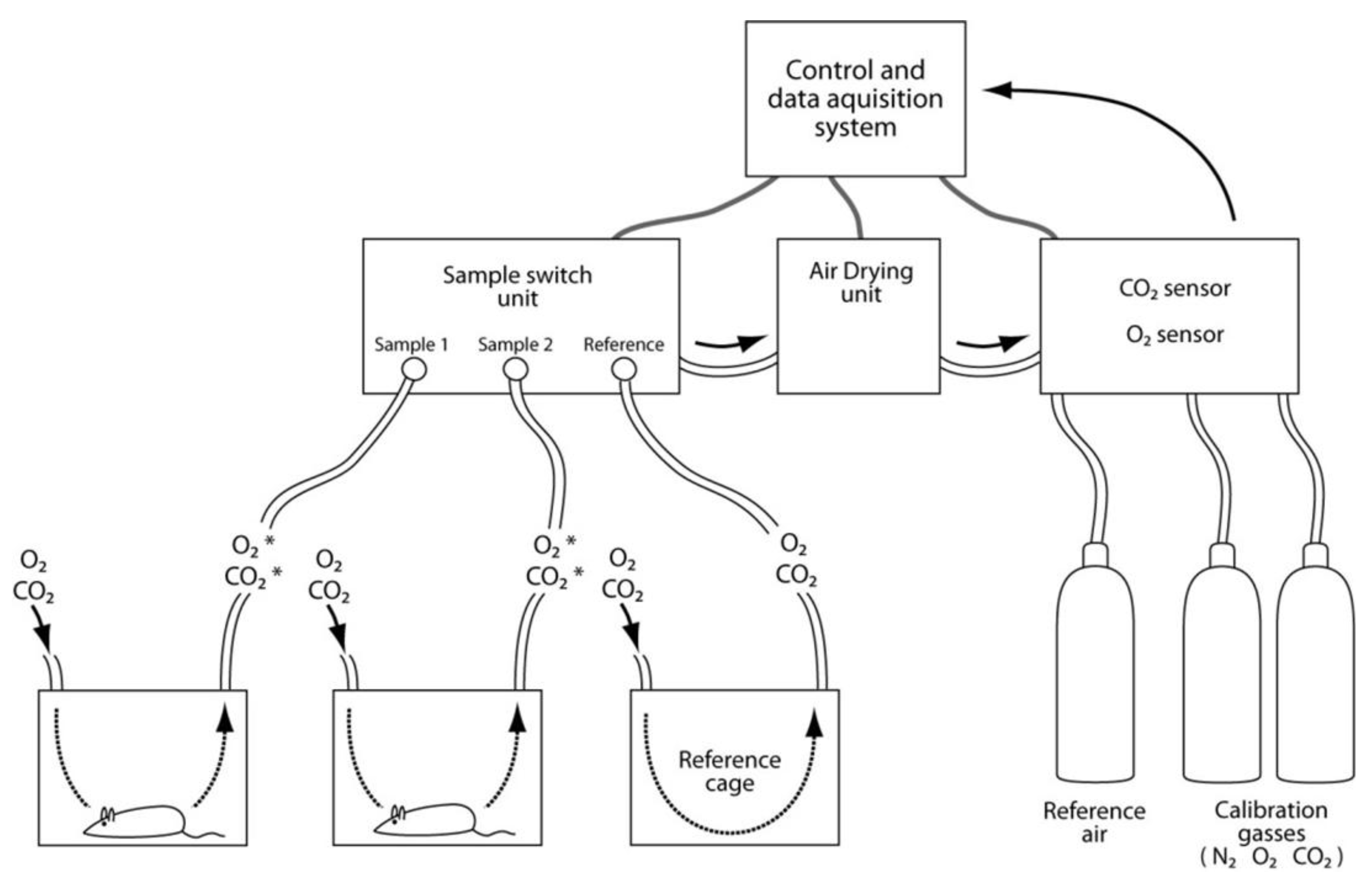
3.10. Systems Biology
4. Lessons Learnt from Molecular Methods Applied in Different Tissues
4.1. Adipose Tissue
4.2. Skeletal Muscle
4.3. Liver
5. Molecular Nutrition Research Applied on the Whole Organism
5.1. Different Diets
5.2. Challenges (Glucose Tolerance Test, Physical Exercise, Meals, Fasting)
5.3. Time Courses—What Is the Effect of Time as Registered by Molecular Nutrition?
6. Conclusion
Conflict of Interest
References
- Karkanas, P.; Shahack-Gross, R.; Ayalon, A.; Bar-Matthews, M.; Barkai, R.; Frumkin, A.; Gopher, A.; Stiner, M.C. Evidence for habitual use of fire at the end of the Lower Paleolithic: Site-formation processes at Qesem Cave, Israel. J. Hum. Evol. 2007, 53, 197–212. [Google Scholar]
- Weiner, S.; Xu, Q.; Goldberg, P.; Liu, J.; Bar-Yosef, O. Evidence for the use of fire at Zhoukoudian, China. Science 1998, 281, 251–253. [Google Scholar] [CrossRef]
- Gupta, A.K. Origin of agriculture and domestication of plants and animals linked to early Holocene climate amelioration. Curr. Sci. 2004, 87, 54–59. [Google Scholar]
- Bloom, D.E. 7 billion and counting. Science 2011, 333, 562–569. [Google Scholar] [CrossRef]
- Bocquet-Appel, J.P. When the world’s population took off: The springboard of the Neolithic demographic transition. Science 2011, 333, 560–561. [Google Scholar]
- World Population Prospects, the 2010 Revision. Available online: http://esa.un.org/unpd/wpp/index.htm (accessed on 15 November 2012).
- Statistics Norway. Available online: http://www.ssb.no (accessed on 15 November 2012).
- Van Ommen, B.; Bouwman, J.; Dragsted, L.O.; Drevon, C.A.; Elliott, R.; de Groot, P.; Kaput, J.; Mathers, J.C.; Muller, M.; Pepping, F.; et al. Challenges of molecular nutrition research 6: The nutritional phenotype database to store, share and evaluate nutritional systems biology studies. Genes Nutr. 2010, 5, 189–203. [Google Scholar] [CrossRef]
- Frequently Asked Questions about Genetic and Genomic Science. Available online: http://www.genome.gov/19016904 (accessed on 15 November 2012).
- Muller, M.; Kersten, S. Nutrigenomics: Goals and strategies. Nat. Rev. Genet. 2003, 4, 315–322. [Google Scholar] [CrossRef]
- Afman, L.; Muller, M. Nutrigenomics: From molecular nutrition to prevention of disease. J. Am. Diet. Assoc. 2006, 106, 569–576. [Google Scholar]
- Nutrigenomics Organisation Homepage. Available online: http://www.nugo.org/everyone (accessed on 15 November 2012).
- Rustan, A.C.; Drevon, C.A. Fatty Acids: Structures and Properties; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2001. [Google Scholar]
- Hihi, A.K.; Michalik, L.; Wahli, W. PPARs: Transcriptional effectors of fatty acids and their derivatives. Cell. Mol. Life Sci. 2002, 59, 790–798. [Google Scholar]
- Schoonjans, K.; Staels, B.; Auwerx, J. Role of the peroxisome proliferator-activated receptor (PPAR) in mediating the effects of fibrates and fatty acids on gene expression. J. Lipid Res. 1996, 37, 907–925. [Google Scholar]
- De Roos, B.; Mavrommatis, Y.; Brouwer, I.A. Long-chain n-3 polyunsaturated fatty acids: New insights into mechanisms relating to inflammation and coronary heart disease. Br. J. Pharmacol. 2009, 158, 413–428. [Google Scholar] [CrossRef]
- Rustan, A.C.; Nossen, J.O.; Osmundsen, H.; Drevon, C.A. Eicosapentaenoic acid inhibits cholesterol esterification in cultured parenchymal cells and isolated microsomes from rat liver. J. Biol. Chem. 1988, 263, 8126–8132. [Google Scholar]
- Rustan, A.C.; Nossen, J.O.; Christiansen, E.N.; Drevon, C.A. Eicosapentaenoic acid reduces hepatic synthesis and secretion of triacylglycerol by decreasing the activity of acyl-coenzyme A:1,2-diacylglycerol acyltransferase. J. Lipid Res. 1988, 29, 1417–1426. [Google Scholar]
- Leikin, A.I.; Brenner, R.R. Microsomal delta 5 desaturation of eicosa-8,11,14-trienoic acid is activated by a cytosolic fraction. Lipids 1989, 24, 101–104. [Google Scholar]
- Nenseter, M.S.; Rustan, A.C.; Lund-Katz, S.; Soyland, E.; Maelandsmo, G.; Phillips, M.C.; Drevon, C.A. Effect of dietary supplementation with n-3 polyunsaturated fatty acids on physical properties and metabolism of low density lipoprotein in humans. Arterioscler. Thromb. 1992, 12, 369–379. [Google Scholar] [CrossRef]
- Brude, I.R.; Drevon, C.A.; Hjermann, I.; Seljeflot, I.; Lund-Katz, S.; Saarem, K.; Sandstad, B.; Solvoll, K.; Halvorsen, B.; Arnesen, H.; Nenseter, M.S. Peroxidation of LDL from combined-hyperlipidemic male smokers supplied with omega-3 fatty acids and antioxidants. Arterioscler. Thromb. Vasc. Biol. 1997, 17, 2576–2588. [Google Scholar]
- Triola, G.; Waldmann, H.; Hedberg, C. Chemical biology of lipidated proteins. ACS Chem. Biol. 2012, 7, 87–99. [Google Scholar]
- Muszbek, L.; Laposata, M. Covalent modification of proteins by arachidonate and eicosapentaenoate in platelets. J. Biol. Chem. 1993, 268, 18243–18248. [Google Scholar]
- Morine, M.J.; Tierney, A.C.; van Ommen, B.; Daniel, H.; Toomey, S.; Gjelstad, I.M.; Gormley, I.C.; Perez-Martinez, P.; Drevon, C.A.; Lopez-Miranda, J.; Roche, H.M. Transcriptomic coordination in the human metabolic network reveals links between n-3 fat intake, adipose tissue gene expression and metabolic health. PLoS Comput. Biol. 2011, 7, e1002223. [Google Scholar] [CrossRef]
- Willett, W. Nutritional Epidemiology; Oxford University Press: New York, NY, USA, 1998. [Google Scholar]
- Sempos, C.T.; Liu, K.; Ernst, N.D. Food and nutrient exposures: What to consider when evaluating epidemiologic evidence. Am. J. Clin. Nutr. 1999, 69, 1330S–1338S. [Google Scholar]
- Margetts, B.; Nelson, M. Design Concepts in Nutritional Epidemiology; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Jeffery, E.H.; Keck, A.S. Translating knowledge generated by epidemiological and in vitro studies into dietary cancer prevention. Mol. Nutr. Food Res. 2008, 52, S7–S17. [Google Scholar]
- Fowke, J.H.; Schlundt, D.; Gong, Y.; Jin, F.; Shu, X.O.; Wen, W.; Liu, D.K.; Gao, Y.T.; Zheng, W. Impact of season of food frequency questionnaire administration on dietary reporting. Ann. Epidemiol. 2004, 14, 778–785. [Google Scholar] [CrossRef]
- Palaniappan, U.; Cue, R.I.; Payette, H.; Gray-Donald, K. Implications of day-to-day variability on measurements of usual food and nutrient intakes. J. Nutr. 2003, 133, 232–235. [Google Scholar]
- Murtaugh, M.A.; Herrick, J.S.; Sweeney, C.; Baumgartner, K.B.; Guiliano, A.R.; Byers, T.; Slattery, M.L. Diet composition and risk of overweight and obesity in women living in the southwestern United States. J. Am. Diet. Assoc. 2007, 107, 1311–1321. [Google Scholar] [CrossRef]
- Rosell, M.; Appleby, P.; Spencer, E.; Key, T. Weight gain over 5 years in 21,966 meat-eating, fish-eating, vegetarian, and vegan men and women in EPIC-Oxford. Int. J. Obes. (Lond.) 2006, 30, 1389–1396. [Google Scholar] [CrossRef]
- Spencer, E.A.; Appleby, P.N.; Davey, G.K.; Key, T.J. Diet and body mass index in 38,000 EPIC-Oxford meat-eaters, fish-eaters, vegetarians and vegans. Int. J. Obes. Relat. Metab. Disord. 2003, 27, 728–734. [Google Scholar] [CrossRef]
- El-Khairy, L.; Ueland, P.M.; Nygard, O.; Refsum, H.; Vollset, S.E. Lifestyle and cardiovascular disease risk factors as determinants of total cysteine in plasma: The Hordaland Homocysteine Study. Am. J. Clin. Nutr. 1999, 70, 1016–1024. [Google Scholar]
- Elshorbagy, A.K.; Nurk, E.; Gjesdal, C.G.; Tell, G.S.; Ueland, P.M.; Nygard, O.; Tverdal, A.; Vollset, S.E.; Refsum, H. Homocysteine, cysteine, and body composition in the Hordaland Homocysteine Study: Does cysteine link amino acid and lipid metabolism? Am. J. Clin. Nutr. 2008, 88, 738–746. [Google Scholar]
- Elshorbagy, A.K.; Valdivia-Garcia, M.; Refsum, H.; Butte, N. The association of cysteine with obesity, inflammatory cytokines and insulin resistance in Hispanic children and adolescents. PloS One 2012, 7, e44166. [Google Scholar]
- Pereira da Silva, N.; Suano de Souza, F.I.; Ifanger Pendezza, A.; Luiz Affonso Fonseca, F.; Hix, S.; Oliveira, A.C.; Oselka Saccardo Sarni, R.; D’Almeida, V. Homocysteine and cysteine levels in prepubertal children: Association with waist circumference and lipid profile. Nutrition 2012. [Google Scholar]
- Olefsky, J.M. Comparison of the effects of insulin and insulin-like agents on different aspects of adipocyte metabolism. Horm. Metab. Res. 1979, 11, 209–213. [Google Scholar] [CrossRef]
- Czech, M.P.; Fain, J.N. Antagonism of insulin action on glucose metabolism in white fat cells by dexamethasone. Endocrinology 1972, 91, 518–522. [Google Scholar]
- Elshorbagy, A.K.; Church, C.; Valdivia-Garcia, M.; Smith, A.D.; Refsum, H.; Cox, R. Dietary cystine level affects metabolic rate and glycaemic control in adult mice. J. Nutr. Biochem. 2012, 23, 332–340. [Google Scholar]
- Elshorbagy, A.K.; Valdivia-Garcia, M.; Mattocks, D.A.; Plummer, J.D.; Smith, A.D.; Drevon, C.A.; Refsum, H.; Perrone, C.E. Cysteine supplementation reverses methionine restriction effects on rat adiposity: significance of stearoyl-coenzyme A desaturase. J. Lipid Res. 2011, 52, 104–112. [Google Scholar] [CrossRef]
- Vang, A.; Singh, P.N.; Lee, J.W.; Haddad, E.H.; Brinegar, C.H. Meats, processed meats, obesity, weight gain and occurrence of diabetes among adults: Findings from Adventist Health Studies. Ann. Nutr. Metab. 2008, 52, 96–104. [Google Scholar] [CrossRef]
- Mudd, S.H.; Levy, H.L.; Skovby, F. Disorders of Transsulfuration; McGraw-Hill, Inc.: New York, NY, USA, 1995. [Google Scholar]
- Pogribna, M.; Melnyk, S.; Pogribny, I.; Chango, A.; Yi, P.; James, S.J. Homocysteine metabolism in children with Down syndrome: In vitro modulation. Am. J. Hum. Genet. 2001, 69, 88–95. [Google Scholar] [CrossRef]
- Furukawa, S.; Fujita, T.; Shimabukuro, M.; Iwaki, M.; Yamada, Y.; Nakajima, Y.; Nakayama, O.; Makishima, M.; Matsuda, M.; Shimomura, I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J. Clin. Invest. 2004, 114, 1752–1761. [Google Scholar]
- Elshorbagy, A.K.; Kozich, V.; Smith, A.D.; Refsum, H. Cysteine and obesity: Consistency of the evidence across epidemiologic, animal and cellular studies. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 49–57. [Google Scholar] [CrossRef]
- Carlson, C.S.; Eberle, M.A.; Kruglyak, L.; Nickerson, D.A. Mapping complex disease loci in whole-genome association studies. Nature 2004, 429, 446–452. [Google Scholar]
- Hirschhorn, J.N.; Daly, M.J. Genome-wide association studies for common diseases and complex traits. Nat. Rev. Genet. 2005, 6, 95–108. [Google Scholar] [CrossRef]
- Manning, A.K.; Hivert, M.F.; Scott, R.A.; Grimsby, J.L.; Bouatia-Naji, N.; Chen, H.; Rybin, D.; Liu, C.T.; Bielak, L.F.; Prokopenko, I.; et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat. Genet. 2012, 44, 659–669. [Google Scholar] [CrossRef]
- Speliotes, E.K.; Willer, C.J.; Berndt, S.I.; Monda, K.L.; Thorleifsson, G.; Jackson, A.U.; Allen, H.L.; Lindgren, C.M.; Luan, J.; Magi, R.; et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 2010, 42, 937–948. [Google Scholar] [CrossRef]
- MacLellan, W.R.; Wang, Y.; Lusis, A.J. Systems-based approaches to cardiovascular disease. Nat. Rev. Cardiol. 2012, 9, 172–184. [Google Scholar]
- Bennett, B.J.; Farber, C.R.; Orozco, L.; Kang, H.M.; Ghazalpour, A.; Siemers, N.; Neubauer, M.; Neuhaus, I.; Yordanova, R.; Guan, B.; et al. A high-resolution association mapping panel for the dissection of complex traits in mice. Genome Res. 2010, 20, 281–290. [Google Scholar] [CrossRef]
- Ghazalpour, A.; Rau, C.D.; Farber, C.R.; Bennett, B.J.; Orozco, L.D.; van Nas, A.; Pan, C.; Allayee, H.; Beaven, S.W.; Civelek, M.; et al. Hybrid mouse diversity panel: A panel of inbred mouse strains suitable for analysis of complex genetic traits. Mamm. Genome 2012, 23, 680–692. [Google Scholar] [CrossRef]
- Giovannini, M.; Verduci, E.; Salvatici, E.; Paci, S.; Riva, E. Phenylketonuria: Nutritional advances and challenges. Nutr. Metab. 2012, 9. [Google Scholar]
- Frayling, T.M.; Timpson, N.J.; Weedon, M.N.; Zeggini, E.; Freathy, R.M.; Lindgren, C.M.; Perry, J.R.; Elliott, K.S.; Lango, H.; Rayner, N.W.; et al. A common variant in the FTO gene is associated with body mass index and predisposes to childhood and adult obesity. Science 2007, 316, 889–894. [Google Scholar]
- Scuteri, A.; Sanna, S.; Chen, W.M.; Uda, M.; Albai, G.; Strait, J.; Najjar, S.; Nagaraja, R.; Orru, M.; Usala, G.; et al. Genome-wide association scan shows genetic variants in the FTO gene are associated with obesity-related traits. PLoS Genet. 2007, 3, e115. [Google Scholar] [CrossRef]
- Tung, Y.C.; Yeo, G.S. From GWAS to biology: Lessons from FTO. Ann. N. Y. Acad. Sci. 2011, 1220, 162–171. [Google Scholar]
- Moleres, A.; Ochoa, M.C.; Rendo-Urteaga, T.; Martinez-Gonzalez, M.A.; Azcona San Julian, M.C.; Martinez, J.A.; Marti, A. Dietary fatty acid distribution modifies obesity risk linked to the rs9939609 polymorphism of the fat mass and obesity-associated gene in a Spanish case-control study of children. Br. J. Nutr. 2012, 107, 533–538. [Google Scholar] [CrossRef]
- Phillips, C.M.; Kesse-Guyot, E.; McManus, R.; Hercberg, S.; Lairon, D.; Planells, R.; Roche, H.M. High dietary saturated fat intake accentuates obesity risk associated with the fat mass and obesity-associated gene in adults. J. Nutr. 2012, 142, 824–831. [Google Scholar]
- Kilpelainen, T.O.; Qi, L.; Brage, S.; Sharp, S.J.; Sonestedt, E.; Demerath, E.; Ahmad, T.; Mora, S.; Kaakinen, M.; Sandholt, C.H.; et al. Physical activity attenuates the influence of FTO variants on obesity risk: A meta-analysis of 218,166 adults and 19,268 children. PLoS Med. 2011, 8, e1001116. [Google Scholar] [CrossRef]
- Barker, D.J.; Osmond, C. Infant mortality, childhood nutrition, and ischaemic heart disease in England and Wales. Lancet 1986, 1, 1077–1081. [Google Scholar] [CrossRef]
- Painter, R.C.; de Rooij, S.R.; Bossuyt, P.M.; Simmers, T.A.; Osmond, C.; Barker, D.J.; Bleker, O.P.; Roseboom, T.J. Early onset of coronary artery disease after prenatal exposure to the Dutch famine. Am. J. Clin. Nutr. 2006, 84, 322–327. [Google Scholar]
- Ruemmele, F.M.; Garnier-Lengline, H. Why are genetics important for nutrition? Lessons from epigenetic research. Ann. Nutr. Metab. 2012, 60, 38–43. [Google Scholar]
- Handel, A.E.; Ebers, G.C.; Ramagopalan, S.V. Epigenetics: Molecular mechanisms and implications for disease. Trends Mol. Med. 2010, 16, 7–16. [Google Scholar] [CrossRef]
- Cedar, H.; Bergman, Y. Linking DNA methylation and histone modification: Patterns and paradigms. Nat. Rev. Genet. 2009, 10, 295–304. [Google Scholar]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suner, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar]
- Milagro, F.I.; Campion, J.; Cordero, P.; Goyenechea, E.; Gomez-Uriz, A.M.; Abete, I.; Zulet, M.A.; Martinez, J.A. A dual epigenomic approach for the search of obesity biomarkers: DNA methylation in relation to diet-induced weight loss. FASEB J. 2011, 25, 1378–1389. [Google Scholar]
- Ling, C.; Poulsen, P.; Simonsson, S.; Ronn, T.; Holmkvist, J.; Almgren, P.; Hagert, P.; Nilsson, E.; Mabey, A.G.; Nilsson, P.; et al. Genetic and epigenetic factors are associated with expression of respiratory chain component NDUFB6 in human skeletal muscle. J. Clin. Invest. 2007, 117, 3427–3435. [Google Scholar] [CrossRef]
- Mootha, V.K.; Lindgren, C.M.; Eriksson, K.F.; Subramanian, A.; Sihag, S.; Lehar, J.; Puigserver, P.; Carlsson, E.; Ridderstrale, M.; Laurila, E.; et al. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 2003, 34, 267–273. [Google Scholar] [CrossRef]
- Patti, M.E.; Butte, A.J.; Crunkhorn, S.; Cusi, K.; Berria, R.; Kashyap, S.; Miyazaki, Y.; Kohane, I.; Costello, M.; Saccone, R.; et al. Coordinated reduction of genes of oxidative metabolism in humans with insulin resistance and diabetes: Potential role of PGC1 and NRF1. Proc. Natl. Acad. Sci. USA 2003, 100, 8466–8471. [Google Scholar]
- Barres, R.; Osler, M.E.; Yan, J.; Rune, A.; Fritz, T.; Caidahl, K.; Krook, A.; Zierath, J.R. Non-CpG methylation of the PGC-1alpha promoter through DNMT3B controls mitochondrial density. Cell Metab. 2009, 10, 189–198. [Google Scholar] [CrossRef]
- Barres, R.; Yan, J.; Egan, B.; Treebak, J.T.; Rasmussen, M.; Fritz, T.; Caidahl, K.; Krook, A.; O’Gorman, D.J.; Zierath, J.R. Acute exercise remodels promoter methylation in human skeletal muscle. Cell Metab. 2012, 15, 405–411. [Google Scholar]
- Katari, S.; Turan, N.; Bibikova, M.; Erinle, O.; Chalian, R.; Foster, M.; Gaughan, J.P.; Coutifaris, C.; Sapienza, C. DNA methylation and gene expression differences in children conceived in vitro or in vivo. Hum. Mol. Genet. 2009, 18, 3769–3778. [Google Scholar] [CrossRef]
- Morgan, H.D.; Sutherland, H.G.; Martin, D.I.; Whitelaw, E. Epigenetic inheritance at the agouti locus in the mouse. Nat. Genet. 1999, 23, 314–318. [Google Scholar]
- Wolff, G.L.; Kodell, R.L.; Moore, S.R.; Cooney, C.A. Maternal epigenetics and methyl supplements affect agouti gene expression in Avy/a mice. FASEB J. 1998, 12, 949–957. [Google Scholar]
- Ng, S.F.; Lin, R.C.; Laybutt, D.R.; Barres, R.; Owens, J.A.; Morris, M.J. Chronic high-fat diet in fathers programs beta-cell dysfunction in female rat offspring. Nature 2010, 467, 963–966. [Google Scholar] [CrossRef]
- Godfrey, K.M.; Sheppard, A.; Gluckman, P.D.; Lillycrop, K.A.; Burdge, G.C.; McLean, C.; Rodford, J.; Slater-Jefferies, J.L.; Garratt, E.; Crozier, S.R.; et al. Epigenetic gene promoter methylation at birth is associated with child’s later adiposity. Diabetes 2011, 60, 1528–1534. [Google Scholar] [CrossRef]
- Bartel, D.P. MicroRNAs: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Kallio, P.; Kolehmainen, M.; Laaksonen, D.E.; Kekalainen, J.; Salopuro, T.; Sivenius, K.; Pulkkinen, L.; Mykkanen, H.M.; Niskanen, L.; Uusitupa, M.; Poutanen, K.S. Dietary carbohydrate modification induces alterations in gene expression in abdominal subcutaneous adipose tissue in persons with the metabolic syndrome: The FUNGENUT Study. Am. J. Clin. Nutr. 2007, 85, 1417–1427. [Google Scholar]
- Zhang, L.; Hou, D.; Chen, X.; Li, D.; Zhu, L.; Zhang, Y.; Li, J.; Bian, Z.; Liang, X.; Cai, X.; et al. Exogenous plant MIR168a specifically targets mammalian LDLRAP1: Evidence of cross-kingdom regulation by microRNA. Cell Res. 2012, 22, 107–126. [Google Scholar] [CrossRef]
- Panagiotou, G.; Nielsen, J. Nutritional systems biology: Definitions and approaches. Ann. Rev. Nutr. 2009, 29, 329–339. [Google Scholar]
- Crujeiras, A.B.; Parra, D.; Milagro, F.I.; Goyenechea, E.; Larrarte, E.; Margareto, J.; Martinez, J.A. Differential expression of oxidative stress and inflammation related genes in peripheral blood mononuclear cells in response to a low-calorie diet: A nutrigenomics study. OMICS 2008, 12, 251–261. [Google Scholar] [CrossRef]
- Caesar, R.; Manieri, M.; Kelder, T.; Boekschoten, M.; Evelo, C.; Muller, M.; Kooistra, T.; Cinti, S.; Kleemann, R.; Drevon, C.A. A combined transcriptomics and lipidomics analysis of subcutaneous, epididymal and mesenteric adipose tissue reveals marked functional differences. PLoS One 2010, 5, e11525. [Google Scholar]
- Henze, A.; Rohn, S.; Gericke, B.; Raila, J.; Schweigert, F.J. Structural modifications of serum transthyretin in rats during protein-energy malnutrition. Rapid Commun. Mass Spectrom. 2008, 22, 3270–3274. [Google Scholar]
- Romagnolo, D.F.; Milner, J.A. Opportunities and challenges for nutritional proteomics in cancer prevention. J. Nutr. 2012, 142, 1360S–1369S. [Google Scholar] [CrossRef]
- Kussmann, M.; Panchaud, A.; Affolter, M. Proteomics in nutrition: Status quo and outlook for biomarkers and bioactives. J. Proteome Res. 2010, 9, 4876–4887. [Google Scholar]
- Moore, J.B.; Weeks, M.E. Proteomics and systems biology: Current and future applications in the nutritional sciences. Adv. Nutr. 2011, 2, 355–364. [Google Scholar]
- Rabilloud, T. Two-dimensional gel electrophoresis in proteomics: Old, old fashioned, but it still climbs up the mountains. Proteomics 2002, 2, 3–10. [Google Scholar] [CrossRef]
- Walther, T.C.; Mann, M. Mass spectrometry-based proteomics in cell biology. J. Cell Biol. 2010, 190, 491–500. [Google Scholar]
- Domon, B.; Aebersold, R. Options and considerations when selecting a quantitative proteomics strategy. Nat. Biotechnol. 2010, 28, 710–721. [Google Scholar] [CrossRef]
- Norheim, F.; Raastad, T.; Thiede, B.; Rustan, A.C.; Drevon, C.A.; Haugen, F. Proteomic identification of secreted proteins from human skeletal muscle cells and expression in response to strength training. Am. J. Physiol. Endocrinol. Metab. 2011, 301, E1013–E1021. [Google Scholar]
- Unwin, R.D.; Griffiths, J.R.; Whetton, A.D. Simultaneous analysis of relative protein expression levels across multiple samples using iTRAQ isobaric tags with 2D nano LC-MS/MS. Nat. Protoc. 2010, 5, 1574–1582. [Google Scholar]
- Ong, S.E.; Blagoev, B.; Kratchmarova, I.; Kristensen, D.B.; Steen, H.; Pandey, A.; Mann, M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol. Cell. Proteomics 2002, 1, 376–386. [Google Scholar] [CrossRef]
- Gouw, J.W.; Krijgsveld, J.; Heck, A.J. Quantitative proteomics by metabolic labeling of model organisms. Mol. Cell. Proteomics 2010, 9, 11–24. [Google Scholar]
- Forner, F.; Kumar, C.; Luber, C.A.; Fromme, T.; Klingenspor, M.; Mann, M. Proteome differences between brown and white fat mitochondria reveal specialized metabolic functions. Cell Metab. 2009, 10, 324–335. [Google Scholar]
- Hwang, H.; Bowen, B.P.; Lefort, N.; Flynn, C.R.; de Filippis, E.A.; Roberts, C.; Smoke, C.C.; Meyer, C.; Hojlund, K.; Yi, Z.; Mandarino, L.J. Proteomics analysis of human skeletal muscle reveals novel abnormalities in obesity and type 2 diabetes. Diabetes 2010, 59, 33–42. [Google Scholar] [CrossRef]
- Schwer, B.; Eckersdorff, M.; Li, Y.; Silva, J.C.; Fermin, D.; Kurtev, M.V.; Giallourakis, C.; Comb, M.J.; Alt, F.W.; Lombard, D.B. Calorie restriction alters mitochondrial protein acetylation. Aging Cell 2009, 8, 604–606. [Google Scholar]
- Picotti, P.; Aebersold, R. Selected reaction monitoring-based proteomics: Workflows, potential, pitfalls and future directions. Nat. Methods 2012, 9, 555–566. [Google Scholar] [CrossRef]
- Patti, G.J.; Yanes, O.; Siuzdak, G. Innovation: Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 2012, 13, 263–269. [Google Scholar]
- Aune, D.; Chan, D.S.; Vieira, A.R.; Navarro Rosenblatt, D.A.; Vieira, R.; Greenwood, D.C.; Norat, T. Dietary compared with blood concentrations of carotenoids and breast cancer risk: A systematic review and meta-analysis of prospective studies. Am. J. Clin. Nutr. 2012, 96, 356–373. [Google Scholar]
- Gibney, M.J.; Walsh, M.; Brennan, L.; Roche, H.M.; German, B.; van Ommen, B. Metabolomics in human nutrition: Opportunities and challenges. Am. J. Clin. Nutr. 2005, 82, 497–503. [Google Scholar] [Green Version]
- Lodge, J.K. Symposium 2: Modern approaches to nutritional research challenges: Targeted and non-targeted approaches for metabolite profiling in nutritional research. Proc. Nutr. Soc 2010, 69, 95–102. [Google Scholar]
- Zhang, A.; Sun, H.; Wang, P.; Han, Y.; Wang, X. Modern analytical techniques in metabolomics analysis. Analyst 2012, 137, 293–300. [Google Scholar] [CrossRef]
- Griffiths, W.J.; Koal, T.; Wang, Y.; Kohl, M.; Enot, D.P.; Deigner, H.P. Targeted metabolomics for biomarker discovery. Angew. Chem. Int. Ed. Engl. 2010, 49, 5426–5445. [Google Scholar]
- Karlsen, A.; Svendsen, M.; Seljeflot, I.; Sommernes, M.A.; Sexton, J.; Brevik, A.; Erlund, I.; Serafini, M.; Bastani, N.; Remberg, S.F.; et al. Compliance, tolerability and safety of two antioxidant-rich diets: A randomised controlled trial in male smokers. Br. J. Nutr. 2011, 106, 557–571. [Google Scholar] [CrossRef]
- German, J.B.; Gillies, L.A.; Smilowitz, J.T.; Zivkovic, A.M.; Watkins, S.M. Lipidomics and lipid profiling in metabolomics. Curr. Opin. Lipidol. 2007, 18, 66–71. [Google Scholar]
- Karlsen, A.; Retterstol, L.; Laake, P.; Paur, I.; Bohn, S.K.; Sandvik, L.; Blomhoff, R. Anthocyanins inhibit nuclear factor-kappaB activation in monocytes and reduce plasma concentrations of pro-inflammatory mediators in healthy adults. J. Nutr. 2007, 137, 1951–1954. [Google Scholar]
- Karlsen, A.; Paur, I.; Bohn, S.K.; Sakhi, A.K.; Borge, G.I.; Serafini, M.; Erlund, I.; Laake, P.; Tonstad, S.; Blomhoff, R. Bilberry juice modulates plasma concentration of NF-kappaB related inflammatory markers in subjects at increased risk of CVD. Eur. J. Nutr. 2010, 49, 345–355. [Google Scholar] [CrossRef]
- Brevik, A.; Karlsen, A.; Azqueta, A.; Tirado, A.E.; Blomhoff, R.; Collins, A. Both base excision repair and nucleotide excision repair in humans are influenced by nutritional factors. Cell Biochem. Funct. 2011, 29, 36–42. [Google Scholar]
- Brevik, A.; Gaivao, I.; Medin, T.; Jorgenesen, A.; Piasek, A.; Elilasson, J.; Karlsen, A.; Blomhoff, R.; Veggan, T.; Duttaroy, A.K.; Collins, A.R. Supplementation of a western diet with golden kiwifruits (Actinidia chinensis var.‘Hort 16A’:) effects on biomarkers of oxidation damage and antioxidant protection. Nutr. J. 2011, 10, 54. [Google Scholar] [CrossRef]
- Puri, R.; Duong, M.; Uno, K.; Kataoka, Y.; Nicholls, S.J. The emerging role of plasma lipidomics in cardiovascular drug discovery. Expert Opin. Drug Discov. 2012, 7, 63–72. [Google Scholar]
- Wood, P.L. Lipidomics of Alzheimer’s disease: Current status. Alzheimers Res. Ther. 2012, 4, 5. [Google Scholar] [CrossRef]
- Wittwer, J.; Rubio-Aliaga, I.; Hoeft, B.; Bendik, I.; Weber, P.; Daniel, H. Nutrigenomics in human intervention studies: Current status, lessons learned and future perspectives. Mol. Nutr. Food Res. 2011, 55, 341–358. [Google Scholar]
- Bocci, V. The neglected organ: Bacterial flora has a crucial immunostimulatory role. Perspect. Biol. Med. 1992, 35, 251–260. [Google Scholar]
- Backhed, F.; Ley, R.E.; Sonnenburg, J.L.; Peterson, D.A.; Gordon, J.I. Host-bacterial mutualism in the human intestine. Science 2005, 307, 1915–1920. [Google Scholar]
- Nicholson, J.K.; Holmes, E.; Kinross, J.; Burcelin, R.; Gibson, G.; Jia, W.; Pettersson, S. Host-gut microbiota metabolic interactions. Science 2012, 336, 1262–1267. [Google Scholar]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef]
- Suau, A.; Bonnet, R.; Sutren, M.; Godon, J.J.; Gibson, G.R.; Collins, M.D.; Dore, J. Direct analysis of genes encoding 16S rRNA from complex communities reveals many novel molecular species within the human gut. Appl. Environ. Microbiol. 1999, 65, 4799–4807. [Google Scholar]
- Zoetendal, E.G.; Rajilic-Stojanovic, M.; de Vos, W.M. High-throughput diversity and functionality analysis of the gastrointestinal tract microbiota. Gut 2008, 57, 1605–1615. [Google Scholar]
- The Human Microbiome Project Consortium. Structure, function and diversity of the healthy human microbiome. Nature 2012, 486, 207–214.
- Frank, D.N.; St Amand, A.L.; Feldman, R.A.; Boedeker, E.C.; Harpaz, N.; Pace, N.R. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. USA 2007, 104, 13780–13785. [Google Scholar]
- Tap, J.; Mondot, S.; Levenez, F.; Pelletier, E.; Caron, C.; Furet, J.P.; Ugarte, E.; Munoz-Tamayo, R.; Paslier, D.L.; Nalin, R.; et al. Towards the human intestinal microbiota phylogenetic core. Environ. Microbiol. 2009, 11, 2574–2584. [Google Scholar] [CrossRef]
- Gill, S.R.; Pop, M.; Deboy, R.T.; Eckburg, P.B.; Turnbaugh, P.J.; Samuel, B.S.; Gordon, J.I.; Relman, D.A.; Fraser-Liggett, C.M.; Nelson, K.E. Metagenomic analysis of the human distal gut microbiome. Science 2006, 312, 1355–1359. [Google Scholar]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar]
- Dutton, R.J.; Turnbaugh, P.J. Taking a metagenomic view of human nutrition. Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 448–454. [Google Scholar]
- Wu, G.D.; Chen, J.; Hoffmann, C.; Bittinger, K.; Chen, Y.Y.; Keilbaugh, S.A.; Bewtra, M.; Knights, D.; Walters, W.A.; Knight, R.; et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011, 334, 105–108. [Google Scholar] [CrossRef]
- Walker, A.W.; Ince, J.; Duncan, S.H.; Webster, L.M.; Holtrop, G.; Ze, X.; Brown, D.; Stares, M.D.; Scott, P.; Bergerat, A.; et al. Dominant and diet-responsive groups of bacteria within the human colonic microbiota. ISME J. 2011, 5, 220–230. [Google Scholar] [CrossRef]
- Davis, L.M.; Martinez, I.; Walter, J.; Goin, C.; Hutkins, R.W. Barcoded pyrosequencing reveals that consumption of galactooligosaccharides results in a highly specific bifidogenic response in humans. PLoS One 2011, 6, e25200. [Google Scholar]
- Ramirez-Farias, C.; Slezak, K.; Fuller, Z.; Duncan, A.; Holtrop, G.; Louis, P. Effect of inulin on the human gut microbiota: Stimulation of Bifidobacterium adolescentis and Faecalibacterium prausnitzii. Br. J. Nutr. 2009, 101, 541–550. [Google Scholar]
- Flint, H.J.; Scott, K.P.; Louis, P.; Duncan, S.H. The role of the gut microbiota in nutrition and health. Nat. Rev. Gastroenterol. Hepatol. 2012, 9, 577–589. [Google Scholar]
- Weissleder, R.; Pittet, M.J. Imaging in the era of molecular oncology. Nature 2008, 452, 580–589. [Google Scholar]
- Heymsfield, S.B. Development of imaging methods to assess adiposity and metabolism. Int. J. Obes. (Lond.) 2008, 32, S76–S82. [Google Scholar] [CrossRef]
- Rokling-Andersen, M.H.; Rustan, A.C.; Wensaas, A.J.; Kaalhus, O.; Wergedahl, H.; Rost, T.H.; Jensen, J.; Graff, B.A.; Caesar, R.; Drevon, C.A. Marine n-3 fatty acids promote size reduction of visceral adipose depots, without altering body weight and composition, in male Wistar rats fed a high-fat diet. Br. J. Nutr. 2009, 102, 995–1006. [Google Scholar] [CrossRef]
- Wensaas, A.J.; Rustan, A.C.; Rokling-Andersen, M.H.; Caesar, R.; Jensen, J.; Kaalhus, O.; Graff, B.A.; Gudbrandsen, O.A.; Berge, R.K.; Drevon, C.A. Dietary supplementation of tetradecylthioacetic acid increases feed intake but reduces body weight gain and adipose depot sizes in rats fed on high-fat diets. Diabetes Obes. Metab. 2009, 11, 1034–1049. [Google Scholar]
- Seidell, J.C.; Bakker, C.J.; van der Kooy, K. Imaging techniques for measuring adipose-tissue distribution—A comparison between computed tomography and 1.5-T magnetic resonance. Am. J. Clin. Nutr. 1990, 51, 953–957. [Google Scholar]
- Klopfenstein, B.J.; Kim, M.S.; Krisky, C.M.; Szumowski, J.; Rooney, W.D.; Purnell, J.Q. Comparison of 3-T MRI and CT for the measurement of visceral and subcutaneous adipose tissue in humans. Br. J. Radiol. 2012, 85, e826–e830. [Google Scholar]
- Gray, C.; MacGillivray, T.J.; Eeley, C.; Stephens, N.A.; Beggs, I.; Fearon, K.C.; Greig, C.A. Magnetic resonance imaging with k-means clustering objectively measures whole muscle volume compartments in sarcopenia/cancer cachexia. Clin. Nutr. 2011, 30, 106–111. [Google Scholar] [CrossRef]
- Kielland, A.; Carlsen, H. Molecular imaging of transcriptional regulation during inflammation. J. Inflamm. (Lond.) 2010, 7, 20. [Google Scholar] [CrossRef]
- Paur, I.; Balstad, T.R.; Kolberg, M.; Pedersen, M.K.; Austenaa, L.M.; Jacobs, D.R., Jr.; Blomhoff, R. Extract of oregano, coffee, thyme, clove, and walnuts inhibits NF-kappaB in monocytes and in transgenic reporter mice. Cancer Prev. Res. (Phila.) 2010, 3, 653–663. [Google Scholar] [CrossRef]
- Zhang, Y.; Proenca, R.; Maffei, M.; Barone, M.; Leopold, L.; Friedman, J.M. Positional cloning of the mouse obese gene and its human homologue. Nature 1994, 372, 425–432. [Google Scholar]
- Kaiyala, K.J.; Ramsay, D.S. Direct animal calorimetry, the underused gold standard for quantifying the fire of life. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2011, 158, 252–264. [Google Scholar] [CrossRef]
- Levine, J.A. Measurement of energy expenditure. Public Health Nutr. 2005, 8, 1123–1132. [Google Scholar]
- Tschop, M.H.; Speakman, J.R.; Arch, J.R.; Auwerx, J.; Bruning, J.C.; Chan, L.; Eckel, R.H.; Farese, R.V., Jr.; Galgani, J.E.; Hambly, C.; et al. A guide to analysis of mouse energy metabolism. Nat. Methods 2012, 9, 57–63. [Google Scholar]
- Butler, A.A.; Kozak, L.P. A recurring problem with the analysis of energy expenditure in genetic models expressing lean and obese phenotypes. Diabetes 2010, 59, 323–329. [Google Scholar] [CrossRef]
- Azuaje, F.; Devaux, Y.; Wagner, D. Computational biology for cardiovascular biomarker discovery. Brief. Bioinform. 2009, 10, 367–377. [Google Scholar]
- Ng, A.; Bursteinas, B.; Gao, Q.; Mollison, E.; Zvelebil, M. Resources for integrative systems biology: From data through databases to networks and dynamic system models. Brief. Bioinform. 2006, 7, 318–330. [Google Scholar]
- Evelo, C.T.; van Bochove, K.; Saito, J.T. Answering biological questions: Querying a systems biology database for nutrigenomics. Genes Nutr. 2011, 6, 81–87. [Google Scholar]
- Hester, R.L.; Iliescu, R.; Summers, R.; Coleman, T.G. Systems biology and integrative physiological modelling. J. Physiol. 2011, 589, 1053–1060. [Google Scholar] [CrossRef]
- Frayn, K.N. Metabolic Regulation: A Human Perspective. In Metabolic Regulation: A Human Perspective; Wiley-Blackwell: Oxford, UK, 2010. [Google Scholar]
- Rosen, E.D.; Spiegelman, B.M. Adipocytes as regulators of energy balance and glucose homeostasis. Nature 2006, 444, 847–853. [Google Scholar] [CrossRef]
- Matsuzawa, Y.; Funahashi, T.; Nakamura, T. The concept of metabolic syndrome: Contribution of visceral fat accumulation and its molecular mechanism. J. Atheroscler. Thromb. 2011, 18, 629–639. [Google Scholar]
- Fox, C.S.; Liu, Y.; White, C.C.; Feitosa, M.; Smith, A.V.; Heard-Costa, N.; Lohman, K.; Johnson, A.D.; Foster, M.C.; Greenawalt, D.M.; et al. Genome-wide association for abdominal subcutaneous and visceral adipose reveals a novel locus for visceral fat in women. PLoS Genet. 2012, 8, e1002695. [Google Scholar] [CrossRef]
- Vinknes, K.J.; Elshorbagy, A.K.; Drevon, C.A.; Gjesdal, C.G.; Tell, G.S.; Nygård, O.; Vollset, S.E.; Refsum, H. Evaluation of the Body Adiposity Index in a Caucasian Population. The Hordaland Health Study. Am. J. Epidemiol. 2012, in press.. [Google Scholar]
- Canoy, D. Coronary heart disease and body fat distribution. Curr. Atheroscler. Rep. 2010, 12, 125–133. [Google Scholar] [CrossRef]
- Heitmann, B.L.; Lissner, L. Hip Hip Hurrah! Hip size inversely related to heart disease and total mortality. Obes. Rev. 2011, 12, 478–481. [Google Scholar]
- Gesta, S.; Tseng, Y.H.; Kahn, C.R. Developmental origin of fat: Tracking obesity to its source. Cell 2007, 131, 242–256. [Google Scholar] [CrossRef]
- Cinti, S. Transdifferentiation properties of adipocytes in the adipose organ. Am. J. Physiol. Endocrinol. Metab. 2009, 297, E977–E986. [Google Scholar]
- Bjorneboe, A.; Bjorneboe, G.E.; Drevon, C.A. Absorption, transport and distribution of vitamin E. J. Nutr. 1990, 120, 233–242. [Google Scholar]
- Pond, C.M. Adipose tissue and the immune system. Prostaglandins Leukot. Essent. Fatty Acids 2005, 73, 17–30. [Google Scholar]
- Knittle, J.L.; Timmers, K.; Ginsberg-Fellner, F.; Brown, R.E.; Katz, D.P. The growth of adipose tissue in children and adolescents. Cross-sectional and longitudinal studies of adipose cell number and size. J. Clin. Invest. 1979, 63, 239–246. [Google Scholar] [CrossRef]
- Seale, P.; Bjork, B.; Yang, W.; Kajimura, S.; Chin, S.; Kuang, S.; Scime, A.; Devarakonda, S.; Conroe, H.M.; Erdjument-Bromage, H.; et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature 2008, 454, 961–967. [Google Scholar] [CrossRef]
- Virtanen, K.A.; Lidell, M.E.; Orava, J.; Heglind, M.; Westergren, R.; Niemi, T.; Taittonen, M.; Laine, J.; Savisto, N.J.; Enerback, S.; Nuutila, P. Functional brown adipose tissue in healthy adults. N. Engl. J. Med. 2009, 360, 1518–1525. [Google Scholar]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef]
- Van Marken Lichtenbelt, W.D.; Vanhommerig, J.W.; Smulders, N.M.; Drossaerts, J.M.; Kemerink, G.J.; Bouvy, N.D.; Schrauwen, P.; Teule, G.J. Cold-activated brown adipose tissue in healthy men. N. Engl. J. Med. 2009, 360, 1500–1508. [Google Scholar]
- Saito, M.; Okamatsu-Ogura, Y.; Matsushita, M.; Watanabe, K.; Yoneshiro, T.; Nio-Kobayashi, J.; Iwanaga, T.; Miyagawa, M.; Kameya, T.; Nakada, K.; et al. High incidence of metabolically active brown adipose tissue in healthy adult humans: Effects of cold exposure and adiposity. Diabetes 2009, 58, 1526–1531. [Google Scholar] [CrossRef]
- Kajimura, S.; Seale, P.; Kubota, K.; Lunsford, E.; Frangioni, J.V.; Gygi, S.P.; Spiegelman, B.M. Initiation of myoblast to brown fat switch by a PRDM16-C/EBP-beta transcriptional complex. Nature 2009, 460, 1154–1158. [Google Scholar]
- Ahfeldt, T.; Schinzel, R.T.; Lee, Y.K.; Hendrickson, D.; Kaplan, A.; Lum, D.H.; Camahort, R.; Xia, F.; Shay, J.; Rhee, E.P.; et al. Programming human pluripotent stem cells into white and brown adipocytes. Nat. Cell Biol. 2012, 14, 209–219. [Google Scholar] [CrossRef]
- Spalding, K.L.; Arner, E.; Westermark, P.O.; Bernard, S.; Buchholz, B.A.; Bergmann, O.; Blomqvist, L.; Hoffstedt, J.; Naslund, E.; Britton, T.; et al. Dynamics of fat cell turnover in humans. Nature 2008, 453, 783–787. [Google Scholar]
- Arner, P.; Bernard, S.; Salehpour, M.; Possnert, G.; Liebl, J.; Steier, P.; Buchholz, B.A.; Eriksson, M.; Arner, E.; Hauner, H.; et al. Dynamics of human adipose lipid turnover in health and metabolic disease. Nature 2011, 478, 110–113. [Google Scholar]
- Simon, G. Histogenesis, Section 5: Adipose Tissue; American Physiological Society: Washington, DC, USA, 1965. [Google Scholar]
- Tang, W.; Zeve, D.; Suh, J.M.; Bosnakovski, D.; Kyba, M.; Hammer, R.E.; Tallquist, M.D.; Graff, J.M. White fat progenitor cells reside in the adipose vasculature. Science 2008, 322, 583–586. [Google Scholar] [CrossRef]
- Rodeheffer, M.S.; Birsoy, K.; Friedman, J.M. Identification of white adipocyte progenitor cells in vivo. Cell 2008, 135, 240–249. [Google Scholar] [CrossRef]
- Tran, K.V.; Gealekman, O.; Frontini, A.; Zingaretti, M.C.; Morroni, M.; Giordano, A.; Smorlesi, A.; Perugini, J.; de Matteis, R.; Sbarbati, A.; et al. The vascular endothelium of the adipose tissue gives rise to both white and brown fat cells. Cell Metab. 2012, 15, 222–229. [Google Scholar] [CrossRef]
- Gupta, R.K.; Mepani, R.J.; Kleiner, S.; Lo, J.C.; Khandekar, M.J.; Cohen, P.; Frontini, A.; Bhowmick, D.C.; Ye, L.; Cinti, S.; Spiegelman, B.M. Zfp423 expression identifies committed preadipocytes and localizes to adipose endothelial and perivascular cells. Cell Metab. 2012, 15, 230–239. [Google Scholar]
- Drevon, C.A. Fatty acids and expression of adipokines. Biochim. Biophys. Acta 2005, 1740, 287–292. [Google Scholar] [CrossRef]
- Skurk, T.; Alberti-Huber, C.; Herder, C.; Hauner, H. Relationship between adipocyte size and adipokine expression and secretion. J. Clin. Endocrinol. Metab. 2007, 92, 1023–1033. [Google Scholar]
- Haugen, F.; Zahid, N.; Dalen, K.T.; Hollung, K.; Nebb, H.I.; Drevon, C.A. Resistin expression in 3T3-L1 adipocytes is reduced by arachidonic acid. J. Lipid Res. 2005, 46, 143–153. [Google Scholar]
- DeFronzo, R.A.; Jacot, E.; Jequier, E.; Maeder, E.; Wahren, J.; Felber, J.P. The effect of insulin on the disposal of intravenous glucose. Results from indirect calorimetry and hepatic and femoral venous catheterization. Diabetes 1981, 30, 1000–1007. [Google Scholar]
- Jensen, J.; Rustad, P.I.; Kolnes, A.J.; Lai, Y.C. The role of skeletal muscle glycogen breakdown for regulation of insulin sensitivity by exercise. Front. Physiol. 2011, 2, 112. [Google Scholar]
- Jensen, J.; Ruge, T.; Lai, Y.C.; Svensson, M.K.; Eriksson, J.W. Effects of adrenaline on whole-body glucose metabolism and insulin-mediated regulation of glycogen synthase and PKB phosphorylation in human skeletal muscle. Metabolism 2011, 60, 215–226. [Google Scholar]
- Ruderman, N.B.; Xu, X.J.; Nelson, L.; Cacicedo, J.M.; Saha, A.K.; Lan, F.; Ido, Y. AMPK and SIRT1: A long-standing partnership? Am. J. Physiol. Endocrinol. Metab. 2010, 298, E751–E760. [Google Scholar] [CrossRef]
- Narkar, V.A.; Downes, M.; Yu, R.T.; Embler, E.; Wang, Y.X.; Banayo, E.; Mihaylova, M.M.; Nelson, M.C.; Zou, Y.; Juguilon, H.; et al. AMPK and PPARdelta agonists are exercise mimetics. Cell 2008, 134, 405–415. [Google Scholar] [CrossRef]
- Alibegovic, A.C.; Sonne, M.P.; Hojbjerre, L.; Bork-Jensen, J.; Jacobsen, S.; Nilsson, E.; Faerch, K.; Hiscock, N.; Mortensen, B.; Friedrichsen, M.; et al. Insulin resistance induced by physical inactivity is associated with multiple transcriptional changes in skeletal muscle in young men. Am. J. Physiol. Endocrinol. Metab. 2010, 299, E752–E763. [Google Scholar] [CrossRef]
- Giebelstein, J.; Poschmann, G.; Hojlund, K.; Schechinger, W.; Dietrich, J.W.; Levin, K.; Beck-Nielsen, H.; Podwojski, K.; Stuhler, K.; Meyer, H.E.; Klein, H.H. The proteomic signature of insulin-resistant human skeletal muscle reveals increased glycolytic and decreased mitochondrial enzymes. Diabetologia 2012, 55, 1114–1127. [Google Scholar]
- Rowlands, D.S.; Thomson, J.S.; Timmons, B.W.; Raymond, F.; Fuerholz, A.; Mansourian, R.; Zwahlen, M.C.; Metairon, S.; Glover, E.; Stellingwerff, T.; et al. Transcriptome and translational signaling following endurance exercise in trained skeletal muscle: Impact of dietary protein. Physiol. Genomics 2011, 43, 1004–1020. [Google Scholar] [CrossRef]
- Van Loon, L.J.; Greenhaff, P.L.; Constantin-Teodosiu, D.; Saris, W.H.; Wagenmakers, A.J. The effects of increasing exercise intensity on muscle fuel utilisation in humans. J. Physiol. 2001, 536, 295–304. [Google Scholar]
- Aas, V.; Kase, E.T.; Solberg, R.; Jensen, J.; Rustan, A.C. Chronic hyperglycaemia promotes lipogenesis and triacylglycerol accumulation in human skeletal muscle cells. Diabetologia 2004, 47, 1452–1461. [Google Scholar]
- Dresner, A.; Laurent, D.; Marcucci, M.; Griffin, M.E.; Dufour, S.; Cline, G.W.; Slezak, L.A.; Andersen, D.K.; Hundal, R.S.; Rothman, D.L.; et al. Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J. Clin. Invest. 1999, 103, 253–259. [Google Scholar] [CrossRef]
- Helge, J.W.; Kiens, B. Muscle enzyme activity in humans: Role of substrate availability and training. Am. J. Physiol. 1997, 272, R1620–R1624. [Google Scholar]
- Pedersen, B.K.; Febbraio, M.A. Muscle as an endocrine organ: Focus on muscle-derived interleukin-6. Physiol. Rev. 2008, 88, 1379–1406. [Google Scholar] [CrossRef]
- McPherron, A.C.; Lawler, A.M.; Lee, S.J. Regulation of skeletal muscle mass in mice by a new TGF-beta superfamily member. Nature 1997, 387, 83–90. [Google Scholar]
- Ostrowski, K.; Rohde, T.; Zacho, M.; Asp, S.; Pedersen, B.K. Evidence that interleukin-6 is produced in human skeletal muscle during prolonged running. J. Physiol. 1998, 508, 949–953. [Google Scholar] [CrossRef]
- Haugen, F.; Norheim, F.; Lian, H.; Wensaas, A.J.; Dueland, S.; Berg, O.; Funderud, A.; Skalhegg, B.S.; Raastad, T.; Drevon, C.A. IL-7 is expressed and secreted by human skeletal muscle cells. Am. J. Physiol. Cell Physiol. 2010, 298, C807–C816. [Google Scholar]
- Nielsen, A.R.; Hojman, P.; Erikstrup, C.; Fischer, C.P.; Plomgaard, P.; Mounier, R.; Mortensen, O.H.; Broholm, C.; Taudorf, S.; Krogh-Madsen, R.; et al. Association between interleukin-15 and obesity: Interleukin-15 as a potential regulator of fat mass. J. Clin. Endocrinol. Metab. 2008, 93, 4486–4493. [Google Scholar] [CrossRef]
- Nitert, M.D.; Dayeh, T.; Volkov, P.; Elgzyri, T.; Hall, E.; Nilsson, E.; Yang, B.T.; Lang, S.; Parikh, H.; Wessman, Y.; et al. Impact of an exercise intervention on DNA methylation in skeletal muscle from first-degree relatives of patients with type 2 diabetes. Diabetes 2012. [Google Scholar]
- Bostrom, P.; Wu, J.; Jedrychowski, M.P.; Korde, A.; Ye, L.; Lo, J.C.; Rasbach, K.A.; Bostrom, E.A.; Choi, J.H.; Long, J.Z.; et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 2012, 481, 463–468. [Google Scholar] [CrossRef]
- Vallim, T.; Salter, A.M. Regulation of hepatic gene expression by saturated fatty acids. Prostaglandins Leukot. Essent. Fatty Acids 2010, 82, 211–218. [Google Scholar]
- Krawczyk, M.; Bonfrate, L.; Portincasa, P. Nonalcoholic fatty liver disease. Best Pract. Res. Clin. Gastroenterol. 2010, 24, 695–708. [Google Scholar]
- Takamura, T.; Misu, H.; Ota, T.; Kaneko, S. Fatty liver as a consequence and cause of insulin resistance: Lessons from type 2 diabetic liver. Endocr. J. 2012, 59, 745–763. [Google Scholar] [CrossRef]
- Thomas, E.L.; Parkinson, J.R.; Frost, G.S.; Goldstone, A.P.; Dore, C.J.; McCarthy, J.P.; Collins, A.L.; Fitzpatrick, J.A.; Durighel, G.; Taylor-Robinson, S.D.; Bell, J.D. The missing risk: MRI and MRS phenotyping of abdominal adiposity and ectopic fat. Obesity (Silver Spring) 2012, 20, 76–87. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Diehl, A.M.; Brunt, E.M.; Cusi, K.; Charlton, M.; Sanyal, A.J. The diagnosis and management of non-alcoholic fatty liver disease: Practice Guideline by the American Association for the Study of Liver Diseases, American College of Gastroenterology, and the American Gastroenterological Association. Hepatology 2012, 55, 2005–2023. [Google Scholar] [CrossRef]
- Valenzuela, R.; Videla, L.A. The importance of the long-chain polyunsaturated fatty acid n-6/n-3 ratio in development of non-alcoholic fatty liver associated with obesity. Food Funct. 2011, 2, 644–648. [Google Scholar]
- Bjermo, H.; Iggman, D.; Kullberg, J.; Dahlman, I.; Johansson, L.; Persson, L.; Berglund, J.; Pulkki, K.; Basu, S.; Uusitupa, M.; et al. Effects of n-6 PUFAs compared with SFAs on liver fat, lipoproteins, and inflammation in abdominal obesity: A randomized controlled trial. Am. J. Clin. Nutr. 2012, 95, 1003–1012. [Google Scholar] [CrossRef]
- Farese, R.V., Jr.; Zechner, R.; Newgard, C.B.; Walther, T.C. The problem of establishing relationships between hepatic steatosis and hepatic insulin resistance. Cell Metab. 2012, 15, 570–573. [Google Scholar]
- Misu, H.; Takamura, T.; Takayama, H.; Hayashi, H.; Matsuzawa-Nagata, N.; Kurita, S.; Ishikura, K.; Ando, H.; Takeshita, Y.; Ota, T.; et al. A liver-derived secretory protein, selenoprotein P, causes insulin resistance. Cell Metab. 2010, 12, 483–495. [Google Scholar] [CrossRef]
- Tierney, A.C.; McMonagle, J.; Shaw, D.I.; Gulseth, H.L.; Helal, O.; Saris, W.H.; Paniagua, J.A.; Golabek-Leszczynska, I.; Defoort, C.; Williams, C.M.; et al. Effects of dietary fat modification on insulin sensitivity and on other risk factors of the metabolic syndrome—LIPGENE: A European randomized dietary intervention study. Int. J. Obes. (Lond.) 2011, 35, 800–809. [Google Scholar] [CrossRef]
- Brevik, A.; Andersen, L.F.; Karlsen, A.; Trygg, K.U.; Blomhoff, R.; Drevon, C.A. Six carotenoids in plasma used to assess recommended intake of fruits and vegetables in a controlled feeding study. Eur. J. Clin. Nutr. 2004, 58, 1166–1173. [Google Scholar]
- Andersen, L.F.; Solvoll, K.; Drevon, C.A. Very-long-chain n-3 fatty acids as biomarkers for intake of fish and n-3 fatty acid concentrates. Am. J. Clin. Nutr. 1996, 64, 305–311. [Google Scholar]
- Singh, B.; Saxena, A. Surrogate markers of insulin resistance: A review. World J. Diabetes 2010, 1, 36–47. [Google Scholar]
- World Health OrganizationInternational Diabetes FederationDefinition and Diagnosis of Diabetes Mellitus and Intermediate Hyperglycemia: Report of a WHO/IDF Consultation; World Health Organization: Geneva, Switerzland, 2006; p. 21.
- Monzillo, L.U.; Hamdy, O. Evaluation of insulin sensitivity in clinical practice and in research settings. Nutr. Rev. 2003, 61, 397–412. [Google Scholar] [CrossRef]
- Matsuda, M. Measuring and estimating insulin resistance in clinical and research settings. Nutr. Metab. Cardiovasc. Dis. 2010, 20, 79–86. [Google Scholar]
- Lorenzo, C.; Hazuda, H.P.; Haffner, S.M. Insulin resistance and excess risk of diabetes in Mexican-Americans: The San Antonio Heart Study. J. Clin. Endocrinol. Metab. 2012, 97, 793–799. [Google Scholar] [CrossRef]
- Shaham, O.; Wei, R.; Wang, T.J.; Ricciardi, C.; Lewis, G.D.; Vasan, R.S.; Carr, S.A.; Thadhani, R.; Gerszten, R.E.; Mootha, V.K. Metabolic profiling of the human response to a glucose challenge reveals distinct axes of insulin sensitivity. Mol. Syst. Biol. 2008, 4, 214. [Google Scholar]
- Wopereis, S.; Rubingh, C.M.; van Erk, M.J.; Verheij, E.R.; van Vliet, T.; Cnubben, N.H.; Smilde, A.K.; van der Greef, J.; van Ommen, B.; Hendriks, H.F. Metabolic profiling of the response to an oral glucose tolerance test detects subtle metabolic changes. PLoS One 2009, 4, e4525. [Google Scholar]
- Rubio-Aliaga, I.; Roos, B.; Duthie, S.; Crosley, L.; Mayer, C.; Horgan, G.; Colquhoun, I.; Gall, G.; Huber, F.; Kremer, W.; et al. Metabolomics of prolonged fasting in humans reveals new catabolic markers. Metabolomics 2010, 7, 375–387. [Google Scholar]
- Krug, S.; Kastenmuller, G.; Stuckler, F.; Rist, M.J.; Skurk, T.; Sailer, M.; Raffler, J.; Romisch-Margl, W.; Adamski, J.; Prehn, C.; et al. The dynamic range of the human metabolome revealed by challenges. FASEB J. 2012, 26, 2607–2619. [Google Scholar] [CrossRef]
- Speakman, J.R.; Levitsky, D.A.; Allison, D.B.; Bray, M.S.; de Castro, J.M.; Clegg, D.J.; Clapham, J.C.; Dulloo, A.G.; Gruer, L.; Haw, S.; et al. Set points, settling points and some alternative models: Theoretical options to understand how genes and environments combine to regulate body adiposity. Dis. Model. Mech. 2011, 4, 733–745. [Google Scholar] [CrossRef]
- Hession, M.; Rolland, C.; Kulkarni, U.; Wise, A.; Broom, J. Systematic review of randomized controlled trials of low-carbohydrate vs. low-fat/low-calorie diets in the management of obesity and its comorbidities. Obes. Rev. 2009, 10, 36–50. [Google Scholar] [CrossRef]
- Sacks, F.M.; Bray, G.A.; Carey, V.J.; Smith, S.R.; Ryan, D.H.; Anton, S.D.; McManus, K.; Champagne, C.M.; Bishop, L.M.; Laranjo, N.; et al. Comparison of weight-loss diets with different compositions of fat, protein, and carbohydrates. N. Engl. J. Med. 2009, 360, 859–873. [Google Scholar]
- Lagiou, P.; Sandin, S.; Lof, M.; Trichopoulos, D.; Adami, H.O.; Weiderpass, E. Low carbohydrate-high protein diet and incidence of cardiovascular diseases in Swedish women: Prospective cohort study. BMJ 2012, 344, e4026. [Google Scholar]
- Ericson, U.; Sonestedt, E.; Gullberg, B.; Hellstrand, S.; Hindy, G.; Wirfalt, E.; Orho-Melander, M. High intakes of protein and processed meat associate with increased incidence of type 2 diabetes. Br. J. Nutr. 2012, in press.. [Google Scholar]
- Knowler, W.C.; Fowler, S.E.; Hamman, R.F.; Christophi, C.A.; Hoffman, H.J.; Brenneman, A.T.; Brown-Friday, J.O.; Goldberg, R.; Venditti, E.; Nathan, D.M. 10-Year follow-up of diabetes incidence and weight loss in the Diabetes Prevention Program Outcomes Study. Lancet 2009, 374, 1677–1686. [Google Scholar]
- Li, G.; Zhang, P.; Wang, J.; Gregg, E.W.; Yang, W.; Gong, Q.; Li, H.; Li, H.; Jiang, Y.; An, Y.; et al. The long-term effect of lifestyle interventions to prevent diabetes in the China Da Qing Diabetes Prevention Study: A 20-year follow-up study. Lancet 2008, 371, 1783–1789. [Google Scholar]
- Lindstrom, J.; Ilanne-Parikka, P.; Peltonen, M.; Aunola, S.; Eriksson, J.G.; Hemio, K.; Hamalainen, H.; Harkonen, P.; Keinanen-Kiukaanniemi, S.; Laakso, M.; et al. Sustained reduction in the incidence of type 2 diabetes by lifestyle intervention: Follow-up of the Finnish Diabetes Prevention Study. Lancet 2006, 368, 1673–1679. [Google Scholar]
- Helland, I.B.; Smith, L.; Saarem, K.; Saugstad, O.D.; Drevon, C.A. Maternal supplementation with very-long-chain n-3 fatty acids during pregnancy and lactation augments children’s IQ at 4 years of age. Pediatrics 2003, 111, e39–e44. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Norheim, F.; Gjelstad, I.M.F.; Hjorth, M.; Vinknes, K.J.; Langleite, T.M.; Holen, T.; Jensen, J.; Dalen, K.T.; Karlsen, A.S.; Kielland, A.; et al. Molecular Nutrition Research—The Modern Way Of Performing Nutritional Science. Nutrients 2012, 4, 1898-1944. https://doi.org/10.3390/nu4121898
Norheim F, Gjelstad IMF, Hjorth M, Vinknes KJ, Langleite TM, Holen T, Jensen J, Dalen KT, Karlsen AS, Kielland A, et al. Molecular Nutrition Research—The Modern Way Of Performing Nutritional Science. Nutrients. 2012; 4(12):1898-1944. https://doi.org/10.3390/nu4121898
Chicago/Turabian StyleNorheim, Frode, Ingrid M. F. Gjelstad, Marit Hjorth, Kathrine J. Vinknes, Torgrim M. Langleite, Torgeir Holen, Jørgen Jensen, Knut Tomas Dalen, Anette S. Karlsen, Anders Kielland, and et al. 2012. "Molecular Nutrition Research—The Modern Way Of Performing Nutritional Science" Nutrients 4, no. 12: 1898-1944. https://doi.org/10.3390/nu4121898
APA StyleNorheim, F., Gjelstad, I. M. F., Hjorth, M., Vinknes, K. J., Langleite, T. M., Holen, T., Jensen, J., Dalen, K. T., Karlsen, A. S., Kielland, A., Rustan, A. C., & Drevon, C. A. (2012). Molecular Nutrition Research—The Modern Way Of Performing Nutritional Science. Nutrients, 4(12), 1898-1944. https://doi.org/10.3390/nu4121898