
Ursodeoxycholic Acid Exhibits Greater Inhibitory Effects on Cancerous HCT116 Colon Cells than on Noncancerous NCM460 Colon Cells


Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Cell Cycle and Apoptosis Analysis
2.3. Western Blotting Analysis
2.4. Immunofluorescent Staining
2.5. Human Apoptosis PCR Array and Gene Enrichment Functional Analyses
2.6. Analysis p21 and c-Myc Gene Expression in Human Tumor Tissues
2.7. Statistical Analysis
3. Results
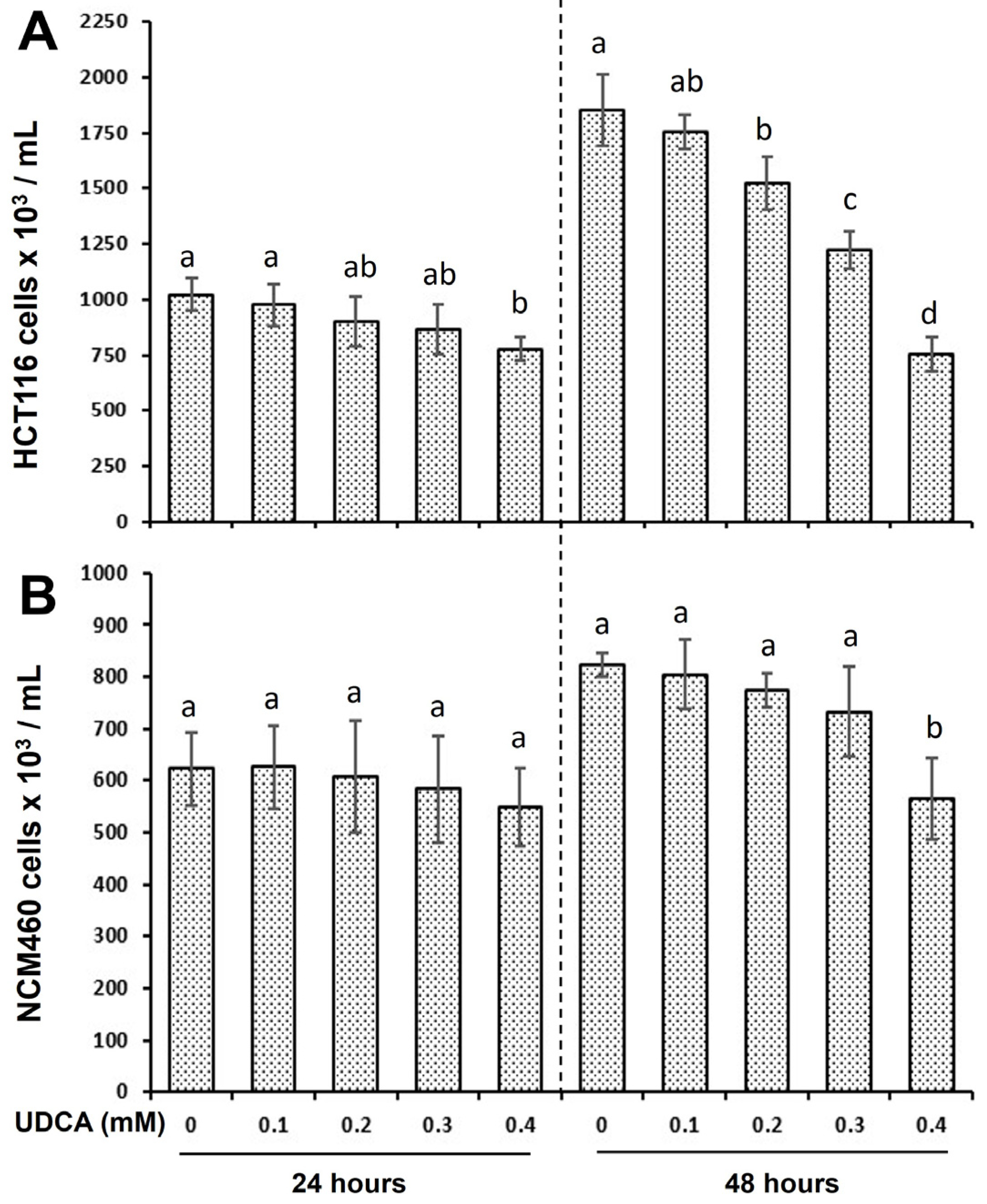
3.1. UDCA Exhibits Distinct Effects on Cell Proliferation Between HCT116 and NCM460 Cells
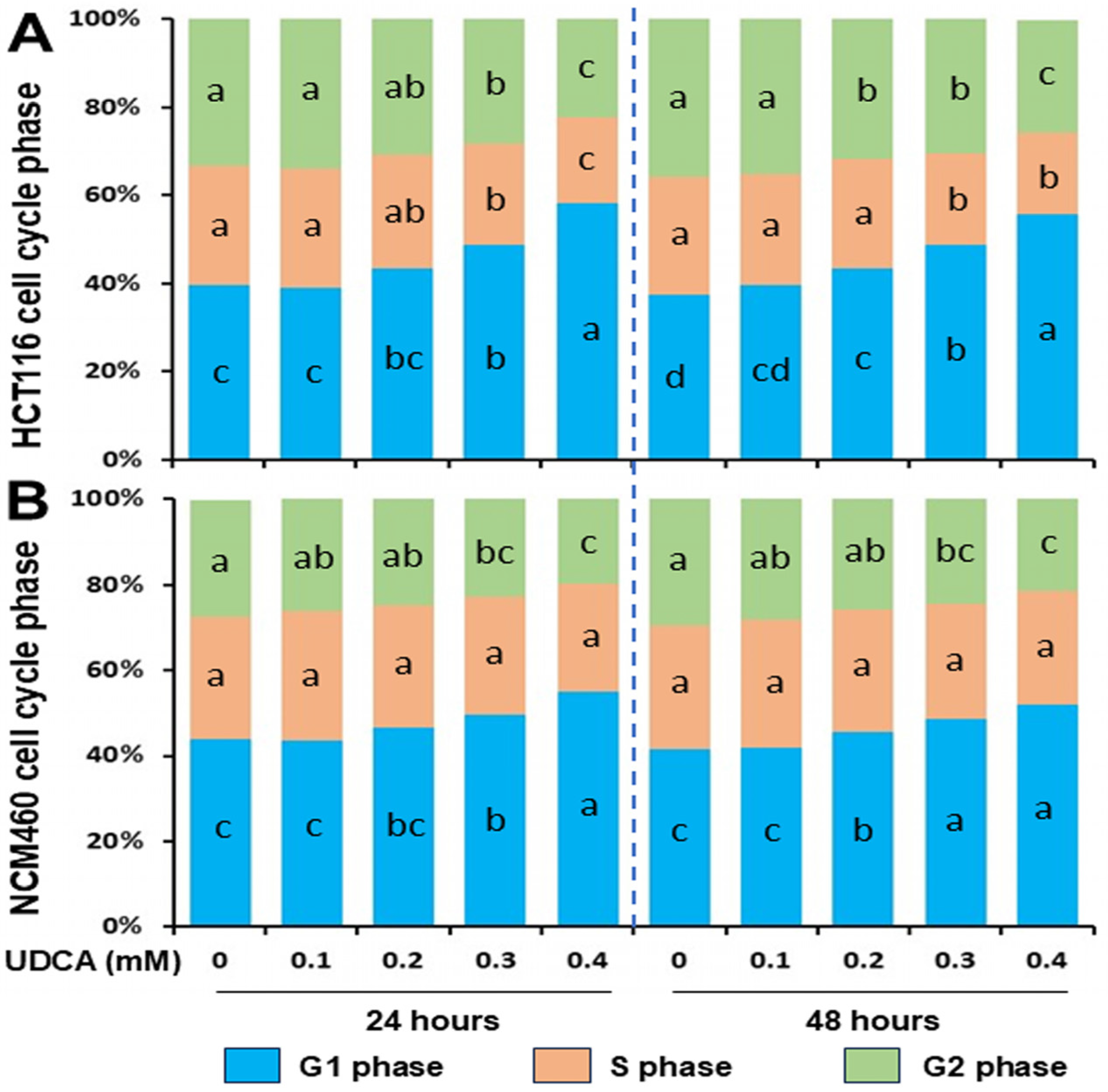
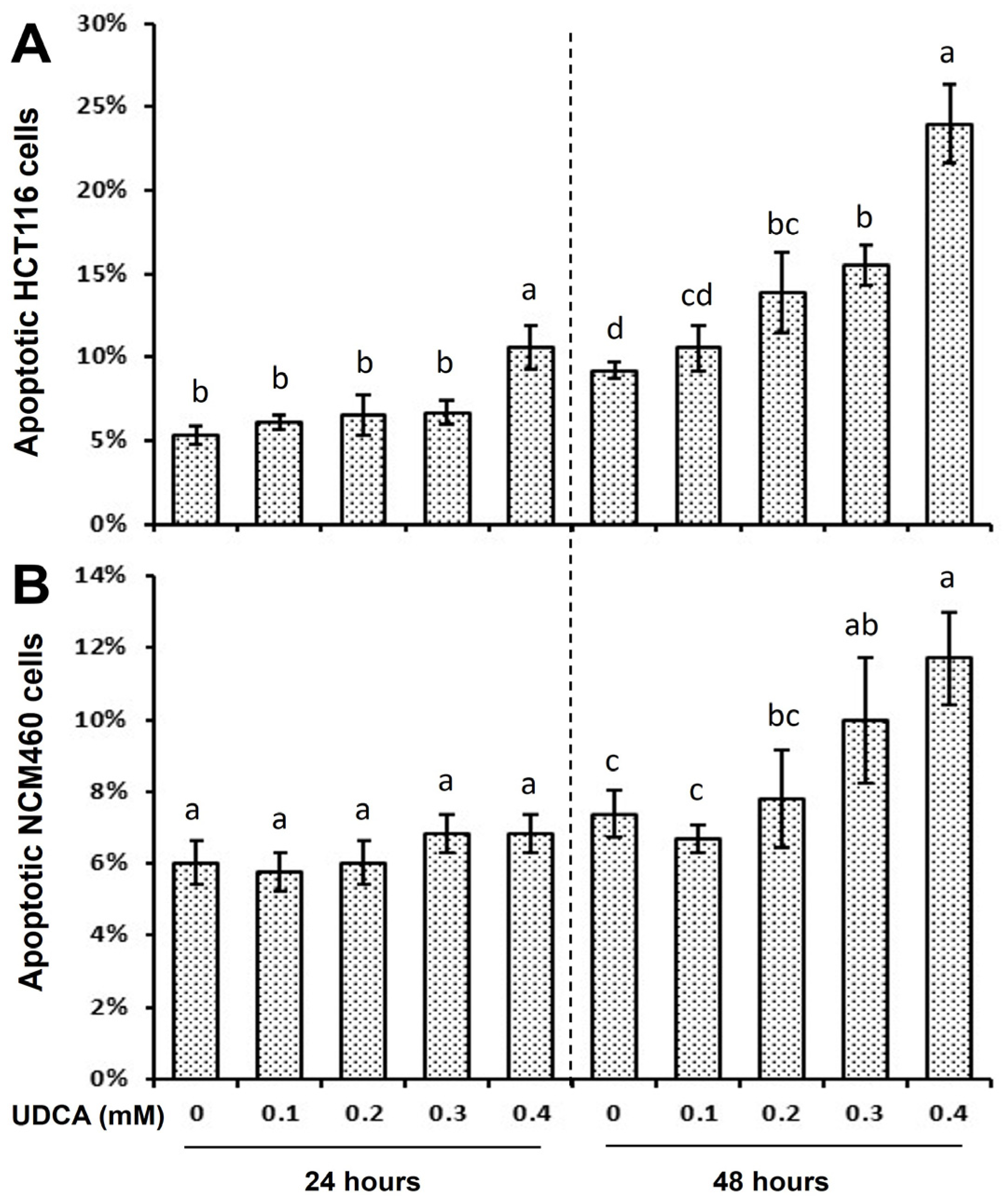
3.2. Differential Effects of UDCA on Cell Cycle Progression and Apoptosis
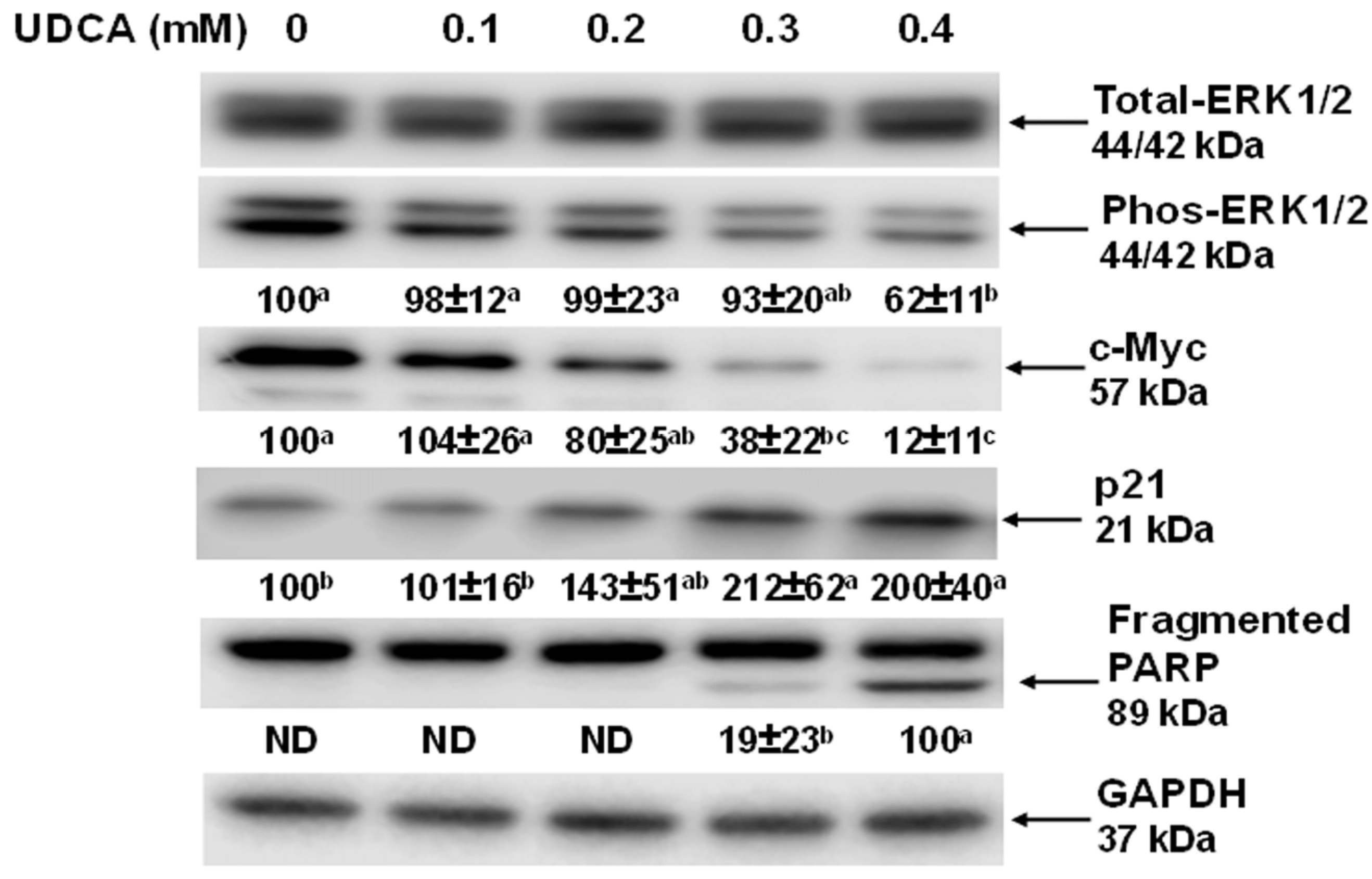
3.3. Effects of UDCA on Cell Signaling Molecules and c-Myc Cellular Localization
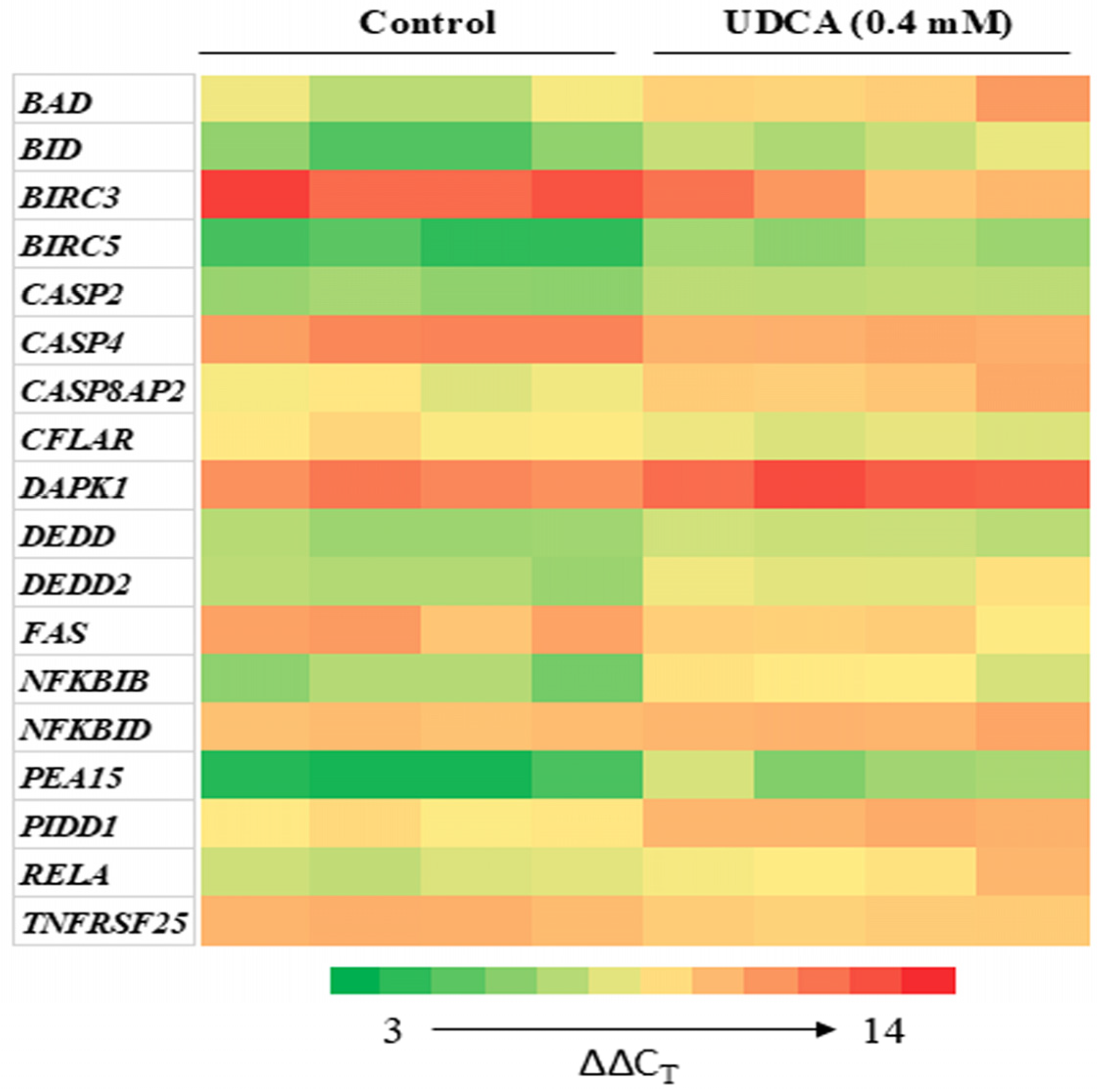
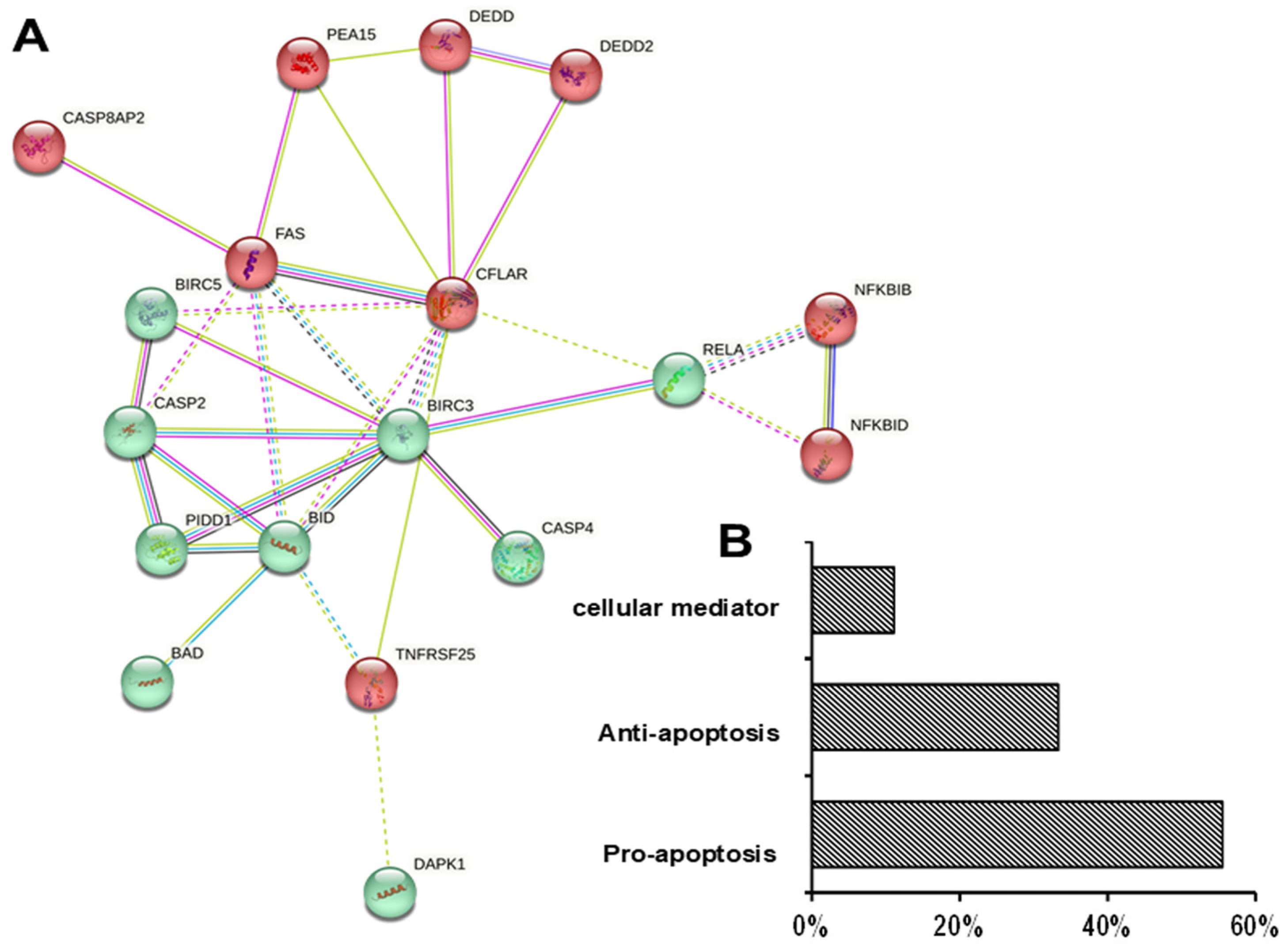
3.4. Differential Effects of UDCA on Apoptotic Gene Expression
3.5. c-Myc and p21 Expression in Human Normal and Colon Cancer Tissues
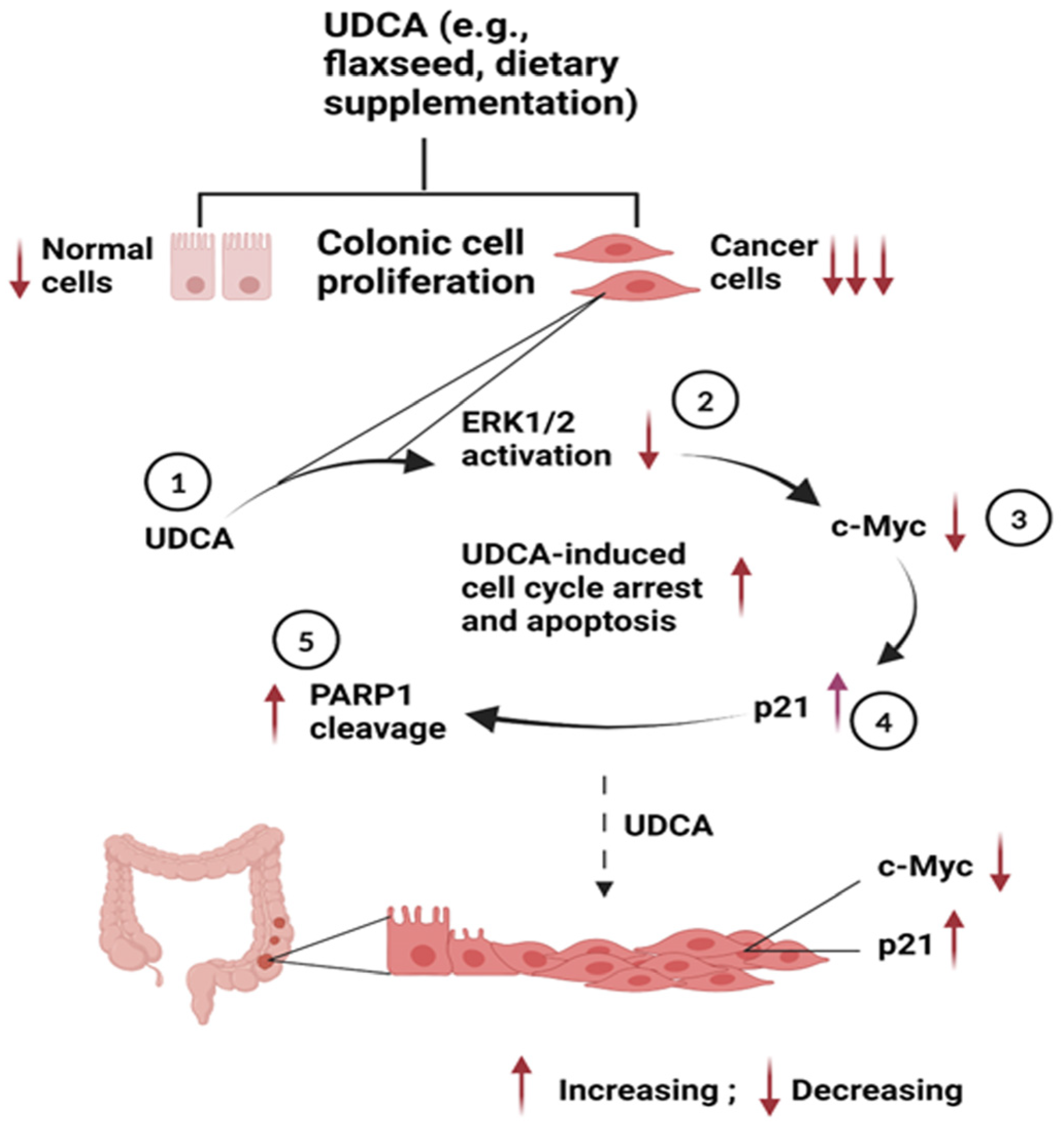
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- American Cancer Society. Cancer Facts & Figures; American Cancer Society: New York, NY, USA, 2016. [Google Scholar]
- Simon, K. Colorectal cancer development and advances in screening. Clin. Interv. Aging 2016, 11, 967–976. [Google Scholar] [PubMed]
- Carr, P.R.; Weigl, K.; Jansen, L.; Walter, V.; Erben, V.; Chang-Claude, J.; Brenner, H.; Hoffmeister, M. Healthy Lifestyle Factors Associated With Lower Risk of Colorectal Cancer Irrespective of Genetic Risk. Gastroenterology 2018, 155, 1805–1815.e5. [Google Scholar] [CrossRef]
- Ganesan, K.; Jayachandran, M.; Xu, B. Diet-Derived Phytochemicals Targeting Colon Cancer Stem Cells and Microbiota in Colorectal Cancer. Int. J. Mol. Sci. 2020, 21, 3976. [Google Scholar] [CrossRef]
- Zeng, H.; Umar, S.; Rust, B.; Lazarova, D.; Bordonaro, M. Secondary Bile Acids and Short Chain Fatty Acids in the Colon: A Focus on Colonic Microbiome, Cell Proliferation, Inflammation, and Cancer. Int. J. Mol. Sci. 2019, 20, 1214. [Google Scholar] [CrossRef] [PubMed]
- Goossens, J.F.; Bailly, C. Ursodeoxycholic acid and cancer: From chemoprevention to chemotherapy. Pharmacol. Ther. 2019, 203, 107396. [Google Scholar] [CrossRef]
- Hamilton, J.P.; Xie, G.; Raufman, J.P.; Hogan, S.; Griffin, T.L.; Packard, C.A.; Chatfield, D.A.; Hagey, L.R.; Steinbach, J.H.; Hofmann, A.F. Human cecal bile acids: Concentration and spectrum. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G256–G263. [Google Scholar] [CrossRef]
- Farhat, Z.; Sampson, J.N.; Hildesheim, A.; Safaeian, M.; Porras, C.; Cortés, B.; Herrero, R.; Romero, B.; Vogtmann, E.; Sinha, R.; et al. Reproducibility, Temporal Variability, and Concordance of Serum and Fecal Bile Acids and Short Chain Fatty Acids in a Population-Based Study. Cancer Epidemiol. Biomark. Prev. 2021, 30, 1875–1883. [Google Scholar] [CrossRef] [PubMed]
- Ndou, S.P.; Kiarie, E.; Ames, N.; Nyachoti, C.M. Flaxseed meal and oat hulls supplementation: Impact on dietary fiber digestibility, and flows of fatty acids and bile acids in growing pigs. J. Anim. Sci. 2019, 97, 291–301. [Google Scholar] [CrossRef]
- James, S.C.; Fraser, K.; Young, W.; Heenan, P.E.; Gearry, R.B.; Keenan, J.I.; Talley, N.J.; Joyce, S.A.; McNabb, W.C.; Roy, N.C. Concentrations of Fecal Bile Acids in Participants with Functional Gut Disorders and Healthy Controls. Metabolites 2021, 11, 612. [Google Scholar] [CrossRef]
- Yang, C.; Wan, M.; Xu, D.; Pan, D.; Xia, H.; Yang, L.; Sun, G. Flaxseed Powder Attenuates Non-Alcoholic Steatohepatitis via Modulation of Gut Microbiota and Bile Acid Metabolism through Gut-Liver Axis. Int. J. Mol. Sci. 2021, 22, 10858. [Google Scholar] [CrossRef]
- Yang, C.; Yang, L.; Yang, Y.; Wan, M.; Xu, D.; Pan, D.; Sun, G. Effects of flaxseed powder in improving non-alcoholic fatty liver by regulating gut microbiota-bile acids metabolic pathway through FXR/TGR5 mediating. Biomed. Pharmacother. 2023, 163, 114864. [Google Scholar] [CrossRef] [PubMed]
- Marschall, H.U.; Wagner, M.; Zollner, G.; Fickert, P.; Diczfalusy, U.; Gumhold, J.; Silbert, D.; Fuchsbichler, A.; Benthin, L.; Grundström, R.; et al. Complementary stimulation of hepatobiliary transport and detoxification systems by rifampicin and ursodeoxycholic acid in humans. Gastroenterology 2005, 129, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zheng, X.; Huang, F.; Zhao, A.; Ge, K.; Zhao, Q.; Jia, W. Ursodeoxycholic Acid Alters Bile Acid and Fatty Acid Profiles in a Mouse Model of Diet-Induced Obesity. Front. Pharmacol. 2019, 10, 842. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Harris, S.C.; Bhowmik, S.; Kang, D.J.; Hylemon, P.B. Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 2016, 7, 22–39. [Google Scholar] [CrossRef]
- Serfaty, L.; De Leusse, A.; Rosmorduc, O.; Desaint, B.; Flejou, J.F.; Chazouilleres, O.; Poupon, R.E.; Poupon, R. Ursodeoxycholic acid therapy and the risk of colorectal adenoma in patients with primary biliary cirrhosis: An observational study. Hepatology 2003, 38, 203–209. [Google Scholar] [CrossRef] [PubMed]
- Kohno, H.; Suzuki, R.; Yasui, Y.; Miyamoto, S.; Wakabayashi, K.; Tanaka, T. Ursodeoxycholic acid versus sulfasalazine in colitis-related colon carcinogenesis in mice. Clin. Cancer Res. 2007, 13, 2519–2525. [Google Scholar] [CrossRef]
- Wali, R.K.; Khare, S.; Tretiakova, M.; Cohen, G.; Nguyen, L.; Hart, J.; Wang, J.; Wen, M.; Ramaswamy, A.; Joseph, L.; et al. Ursodeoxycholic acid and F(6)-D(3) inhibit aberrant crypt proliferation in the rat azoxymethane model of colon cancer: Roles of cyclin D1 and E-cadherin. Cancer Epidemiol. Biomark. Prev. 2002, 11, 1653–1662. [Google Scholar]
- Lee, J.; Hong, E.M.; Kim, J.H.; Kim, J.H.; Jung, J.H.; Park, S.W.; Koh, D.H. Ursodeoxycholic acid inhibits epithelial-mesenchymal transition, suppressing invasiveness of bile duct cancer cells: An in vitro study. Oncol. Lett. 2022, 24, 448. [Google Scholar] [CrossRef]
- Kim, E.K.; Cho, J.H.; Kim, E.; Kim, Y.J. Ursodeoxycholic acid inhibits the proliferation of colon cancer cells by regulating oxidative stress and cancer stem-like cell growth. PLoS ONE 2017, 12, e0181183. [Google Scholar] [CrossRef]
- Wan, J.F.; Chu, S.F.; Zhou, X.; Li, Y.T.; He, W.B.; Tan, F.; Luo, P.; Ai, Q.D.; Wang, Q.; Chen, N.H. Ursodeoxycholic acid protects interstitial Cajal-like cells in the gallbladder from undergoing apoptosis by inhibiting TNF-α expression. Acta Pharmacol. Sin. 2018, 39, 1493–1500. [Google Scholar] [CrossRef]
- Moyer, M.P.; Manzano, L.A.; Merriman, R.L.; Stauffer, J.S.; Tanzer, L.R. NCM460, a normal human colon mucosal epithelial cell line. Vitr. Cell. Dev. Biol. Anim. 1996, 32, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Brattain, M.G.; Fine, W.D.; Khaled, F.M.; Thompson, J.; Brattain, D.E. Heterogeneity of malignant cells from a human colonic carcinoma. Cancer Res. 1981, 41, 1751–1756. [Google Scholar]
- Megna, B.W.; Carney, P.R.; Nukaya, M.; Geiger, P.; Kennedy, G.D. Indole-3-carbinol induces tumor cell death: Function follows form. J. Surg. Res. 2016, 204, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H. The intestinal crypt, a prototype stem cell compartment. Cell 2013, 154, 274–284. [Google Scholar] [CrossRef] [PubMed]
- van der Flier, L.G.; Clevers, H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol. 2009, 71, 241–260. [Google Scholar] [CrossRef]
- Zeng, H.; Hamlin, S.K.; Safratowich, B.D.; Cheng, W.H.; Johnson, L.K. Superior inhibitory efficacy of butyrate over propionate and acetate against human colon cancer cell proliferation via cell cycle arrest and apoptosis: Linking dietary fiber to cancer prevention. Nutr. Res. 2020, 83, 63–72. [Google Scholar] [CrossRef]
- Chen, T.R. In situ detection of mycoplasma contamination in cell cultures by fluorescent Hoechst 33258 stain. Exp. Cell Res. 1977, 104, 255–262. [Google Scholar] [CrossRef]
- Louis, K.S.; Siegel, A.C. Cell viability analysis using trypan blue: Manual and automated methods. Methods Mol. Biol. 2011, 740, 7–12. [Google Scholar]
- Zeng, H.; Safratowich, B.D.; Wang, T.T.Y.; Hamlin, S.K.; Johnson, L.K. Butyrate Inhibits Deoxycholic-Acid-Resistant Colonic Cell Proliferation via Cell Cycle Arrest and Apoptosis: A Potential Pathway Linking Dietary Fiber to Cancer Prevention. Mol. Nutr. Food Res. 2020, 64, e1901014. [Google Scholar] [CrossRef]
- Harnicarova, A.; Kozubek, S.; Pachernik, J.; Krejci, J.; Bartova, E. Distinct nuclear arrangement of active and inactive c-myc genes in control and differentiated colon carcinoma cells. Exp. Cell Res. 2006, 312, 4019–4035. [Google Scholar] [CrossRef]
- Oncel, S.; Safratowich, B.D.; Lindlauf, J.E.; Liu, Z.; Palmer, D.G.; Briske-Anderson, M.; Zeng, H. Efficacy of Butyrate to Inhibit Colonic Cancer Cell Growth Is Cell Type-Specific and Apoptosis-Dependent. Nutrients 2024, 16, 529. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef] [PubMed]
- Heid, C.A.; Stevens, J.; Livak, K.J.; Williams, P.M. Real time quantitative PCR. Genome Res. 1996, 6, 986–994. [Google Scholar] [CrossRef] [PubMed]
- Alenzi, F.Q. Links between apoptosis, proliferation and the cell cycle. Br. J. Biomed. Sci. 2004, 61, 99–102. [Google Scholar] [CrossRef]
- Squires, M.S.; Nixon, P.M.; Cook, S.J. Cell-cycle arrest by PD184352 requires inhibition of extracellular signal-regulated kinases (ERK) 1/2 but not ERK5/BMK1. Biochem. J. 2002, 366 Pt 2, 673–680. [Google Scholar] [CrossRef]
- Allison, S.J. Novel Anti-Cancer Agents and Cellular Targets and Their Mechanism(s) of Action. Biomedicines 2022, 10, 1767. [Google Scholar] [CrossRef]
- Wu, P.K.; Becker, A.; Park, J.I. Growth Inhibitory Signaling of the Raf/MEK/ERK Pathway. Int. J. Mol. Sci. 2020, 21, 5436. [Google Scholar] [CrossRef]
- Sun, Y.; Liu, W.Z.; Liu, T.; Feng, X.; Yang, N.; Zhou, H.F. Signaling pathway of MAPK/ERK in cell proliferation, differentiation, migration, senescence and apoptosis. J. Recept. Signal Transduct. Res. 2015, 35, 600–604. [Google Scholar] [CrossRef]
- Kidger, A.M.; Sipthorp, J.; Cook, S.J. ERK1/2 inhibitors: New weapons to inhibit the RAS-regulated RAF-MEK1/2-ERK1/2 pathway. Pharmacol. Ther. 2018, 187, 45–60. [Google Scholar] [CrossRef]
- Tseng, Y.S.; Wu, P.R.; Lu, J.W.; Wang, Y.F.; Yeh, K.T.; Lin, S.H. Cytoplasmic phosphorylated ERK1/2 expression in patients with melanoma is associated with tumor stage and metastasis. Biotech. Histochem. Off. Publ. Biol. Stain. Comm. 2022, 97, 118–125. [Google Scholar] [CrossRef]
- Kong, L.; Wei, Q.; Hu, X.; Chen, L.; Li, J. Ribosomal protein small subunit 15A (RPS15A) inhibits the apoptosis of breast cancer MDA-MB-231 cells via upregulating phosphorylated ERK1/2, Bad, and Chk1. J. Cell. Biochem. 2020, 121, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Bretones, G.; Delgado, M.D.; Leon, J. Myc and cell cycle control. Biochim. Biophys. Acta 2015, 1849, 506–516. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Gutierrez, L.; Delgado, M.D.; Leon, J. MYC Oncogene Contributions to Release of Cell Cycle Brakes. Genes 2019, 10, 244. [Google Scholar] [CrossRef] [PubMed]
- Madden, S.K.; de Araujo, A.D.; Gerhardt, M.; Fairlie, D.P.; Mason, J.M. Taking the Myc out of cancer: Toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer 2021, 20, 3. [Google Scholar] [CrossRef]
- Ticli, G.; Cazzalini, O.; Stivala, L.A.; Prosperi, E. Revisiting the Function of p21(CDKN1A) in DNA Repair: The Influence of Protein Interactions and Stability. Int. J. Mol. Sci. 2022, 23, 7058. [Google Scholar] [CrossRef]
- Karimian, A.; Ahmadi, Y.; Yousefi, B. Multiple functions of p21 in cell cycle, apoptosis and transcriptional regulation after DNA damage. DNA Repair 2016, 42, 63–71. [Google Scholar] [CrossRef]
- Li, X.; Li, F. p21-Activated Kinase: Role in Gastrointestinal Cancer and Beyond. Cancers 2022, 14, 4736. [Google Scholar] [CrossRef]
- Wang, Z.; Liu, M.; Zhu, H.; Zhang, W.; He, S.; Hu, C.; Quan, L.; Bai, J.; Xu, N. Suppression of p21 by c-Myc through members of miR-17 family at the post-transcriptional level. Int. J. Oncol. 2010, 37, 1315–1321. [Google Scholar]
- Gartel, A.L.; Ye, X.; Goufman, E.; Shianov, P.; Hay, N.; Najmabadi, F.; Tyner, A.L. Myc represses the p21(WAF1/CIP1) promoter and interacts with Sp1/Sp3. Proc. Natl. Acad. Sci. USA 2001, 98, 4510–4515. [Google Scholar] [CrossRef]
- Zhang, J.; Song, N.; Zang, D.; Yu, J.; Li, J.; Di, W.; Guo, R.; Zhao, W.; Wang, H. c-Myc promotes tumor proliferation and anti-apoptosis by repressing p21 in rhabdomyosarcomas. Mol. Med. Rep. 2017, 16, 4089–4094. [Google Scholar] [CrossRef]
- Bressenot, A.; Marchal, S.; Bezdetnaya, L.; Garrier, J.; Guillemin, F.; Plénat, F. Assessment of apoptosis by immunohistochemistry to active caspase-3, active caspase-7, or cleaved PARP in monolayer cells and spheroid and subcutaneous xenografts of human carcinoma. J. Histochem. Cytochem. 2009, 57, 289–300. [Google Scholar] [CrossRef] [PubMed]
- Zielińska, Z.; Ołdak, Ł.; Gorodkiewicz, E. Methods of PARP-1 Determination and its Importance in Living Organisms. Protein Pept. Lett. 2022, 29, 496–504. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| UDCA (mM) | 0 | 0.1 | 0.2 | 0.3 | 0.4 |
|---|---|---|---|---|---|
| Treatment time | 24 h | ||||
| HCT116 G1% | 39.5 ± 3.2 c | 38.9 ± 1.6 c | 43.4 ± 2.4 bc | 48.8 ± 2.3 b | 58.3 ± 2.9 a |
| HCT116 S% | 27.3 ± 2.0 a | 27.1 ± 1.5 a | 26.0 ± 1.6 ab | 23.0 ± 1.3 b | 19.5 ± 1.6 c |
| HCT116 G2% | 33.2 ± 1.8 a | 34.0 ± 0.9 a | 30.6 ± 1.4 ab | 28.2 ± 2.8 b | 22.2 ± 2.1 c |
| NCM460 G1% | 43.9 ± 2.2 c | 43.5 ± 2.4 c | 46.5 ± 3.5 bc | 49.5 ± 2.5 b | 55.1 ± 1.1 a |
| NCM460 S% | 28.7 ± 2.1 a | 30.4 ± 3.1 a | 28.6 ± 4.1 a | 27.8 ± 0.9 a | 25.2 ± 2.0 a |
| NCM460 G2% | 27.2 ± 0.9 a | 26.1 ± 3.5 ab | 24.9 ± 1.6 ab | 22.8 ± 1.6 bc | 19.7 ± 1.3 c |
| Treatment time | 48 h | ||||
| HCT116 G1% | 37.4 ± 1.2 d | 39.6 ± 1.8 cd | 43.6 ± 2.1 c | 48.7 ± 2.6 b | 55.8 ± 2.8 a |
| HCT116 S% | 26.8 ± 1.8 a | 25.2 ± 1.2 a | 24.8 ± 1.0 a | 21.0 ± 1.6 b | 18.5 ± 2.0 b |
| HCT116 G2% | 35.8 ± 2.1 a | 35.2 ± 0.6 a | 31.6 ± 1.7 b | 30.3 ± 1.7 b | 25.5 ± 1.6 c |
| NCM460 G1% | 41.6 ± 1.2 c | 41.7 ± 1.6 c | 45.4 ± 1.4 b | 48.6 ± 1.8 a | 51.8 ± 1.1 a |
| NCM460 S% | 29.0 ± 1.7 a | 30.0 ± 2.5 a | 28.6 ± 2.4 a | 26.9 ± 1.9 a | 26.6 ± 2.0 a |
| NCM460 G2% | 29.4 ± 1.5 a | 28.3 ± 1.7 ab | 26.0 ± 2.1 ab | 24.5 ± 1.9 bc | 21.5 ± 2.2 c |
| Gene | Cluster | Control, [0 mM] | UDCA, [0.4 mM] |
|---|---|---|---|
| Bad | Green | 1.00 ± 0.09 | 0.27 ± 0.14 * |
| BID | Green | 1.00 ± 0.15 | 0.28 ± 0.12 * |
| BIRC3 | Green | 1.00 ± 0.05 | 4.56 ± 2.34 * |
| BIRC5 | Green | 1.00 ± 0.10 | 0.26 ± 0.14 ** |
| CASP2 | Green | 1.00 ± 0.04 | 0.57 ± 0.12 ** |
| CASP4 | Green | 1.00 ± 0.03 | 2.02 ± 0.39 ** |
| DAPK1 | Green | 1.00 ± 0.03 | 0.44 ± 0.04 ** |
| RELA | Green | 1.00 ± 0.04 | 0.41 ± 0.14 * |
| PIDD1 | Green | 1.00 ± 0.03 | 0.38 ± 0.09 ** |
| CASP8AP2 | Red | 1.00 ± 0.05 | 0.40 ± 0.19 ** |
| CFLAR | Red | 1.00 ± 0.04 | 1.77 ± 0.63 * |
| DEDD | Red | 1.00 ± 0.04 | 0.59 ± 0.10 ** |
| DEDD2 | Red | 1.00 ± 0.05 | 0.39 ± 0.15 ** |
| FAS | Red | 1.00 ± 0.05 | 2.64 ± 1.24 * |
| NFKBIB | Red | 1.00 ± 0.11 | 0.25 ± 0.08 ** |
| NFKBID | Red | 1.00 ± 0.01 | 0.77 ± 0.10 * |
| PEA15 | Red | 1.00 ± 0.14 | 0.15 ± 0.07 ** |
| TNFRSF25 | Red | 1.00 ± 0.02 | 1.66 ± 0.27 ** |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zeng, H.; Safratowich, B.D.; Cheng, W.-H.; Briske-Anderson, M. Ursodeoxycholic Acid Exhibits Greater Inhibitory Effects on Cancerous HCT116 Colon Cells than on Noncancerous NCM460 Colon Cells. Nutrients 2025, 17, 1072. https://doi.org/10.3390/nu17061072
Zeng H, Safratowich BD, Cheng W-H, Briske-Anderson M. Ursodeoxycholic Acid Exhibits Greater Inhibitory Effects on Cancerous HCT116 Colon Cells than on Noncancerous NCM460 Colon Cells. Nutrients. 2025; 17(6):1072. https://doi.org/10.3390/nu17061072
Chicago/Turabian StyleZeng, Huawei, Bryan D. Safratowich, Wen-Hsing Cheng, and Mary Briske-Anderson. 2025. "Ursodeoxycholic Acid Exhibits Greater Inhibitory Effects on Cancerous HCT116 Colon Cells than on Noncancerous NCM460 Colon Cells" Nutrients 17, no. 6: 1072. https://doi.org/10.3390/nu17061072
APA StyleZeng, H., Safratowich, B. D., Cheng, W.-H., & Briske-Anderson, M. (2025). Ursodeoxycholic Acid Exhibits Greater Inhibitory Effects on Cancerous HCT116 Colon Cells than on Noncancerous NCM460 Colon Cells. Nutrients, 17(6), 1072. https://doi.org/10.3390/nu17061072