
The Light and the Dark Side of Maternal PKU: Single-Centre Experience of Dietary Management and Emergency Treatment Protocol of Unplanned Pregnancies

 , ,
, ,  ,
,
Abstract
1. Introduction
2. Materials and Methods
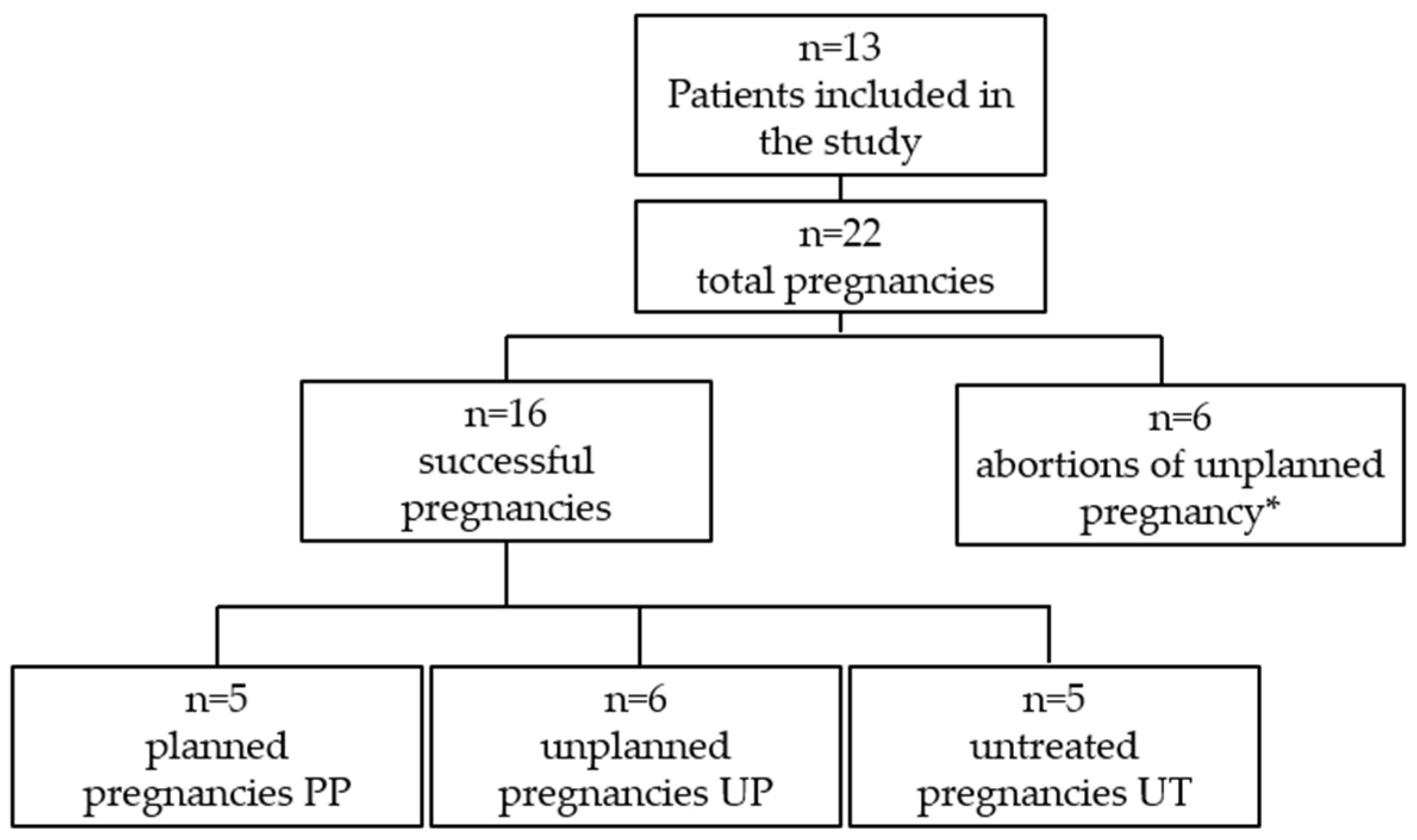
2.1. Study Sample
2.2. Amino Acid Assay
2.3. Anthropometric Parameters
2.4. Dietary Management During Pregnancy
2.5. Statistical Analysis
3. Results
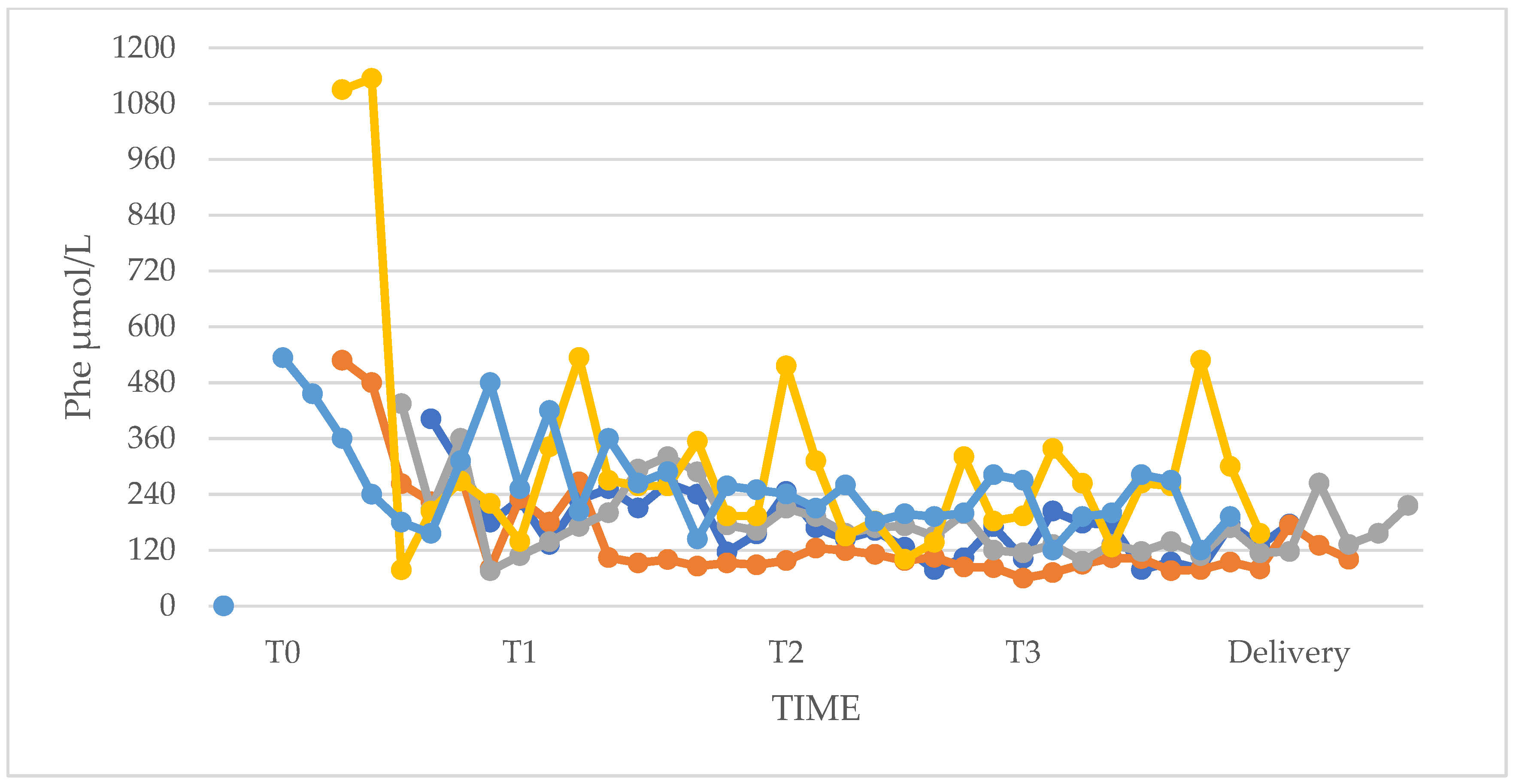
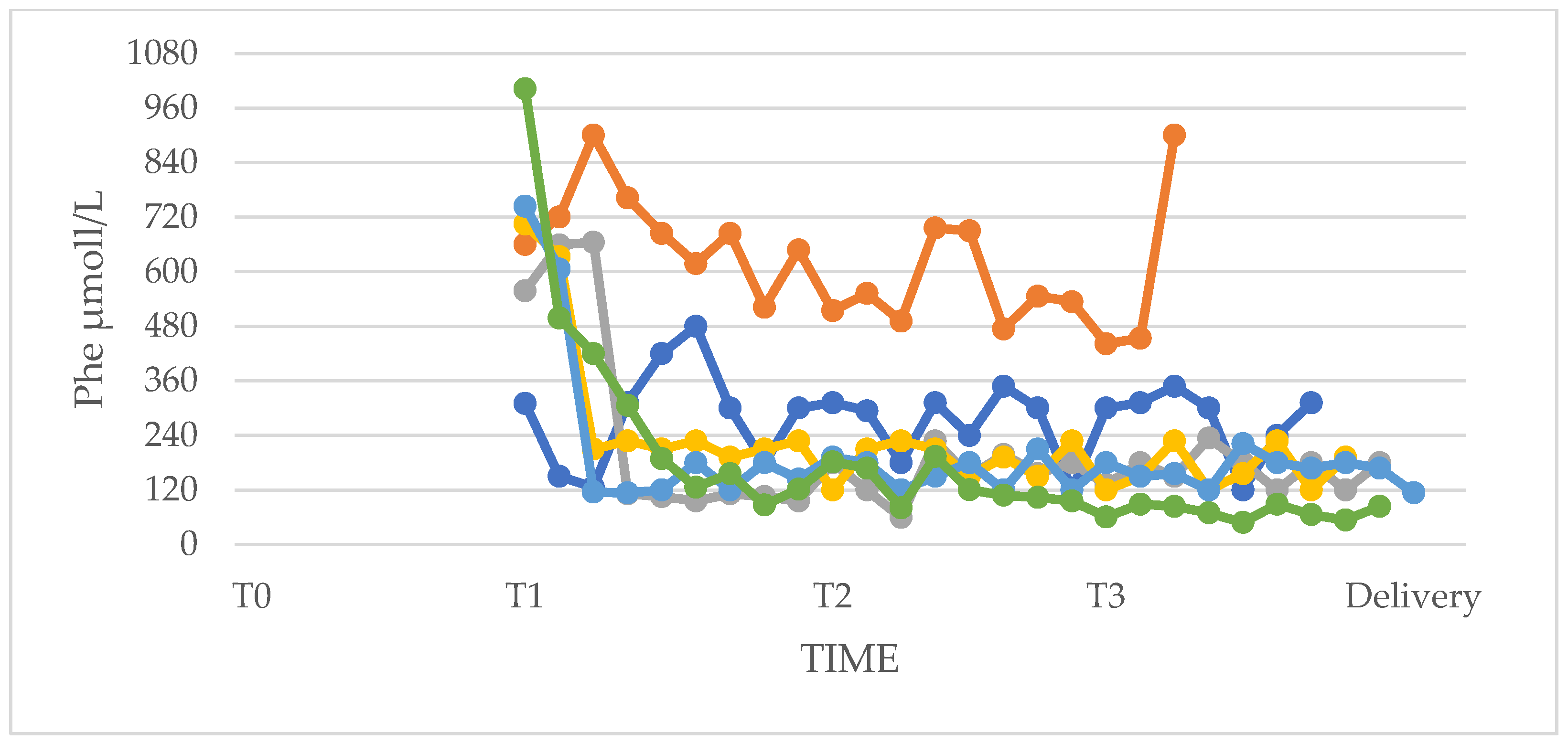
3.1. Phenylalanine and Tyrosine Levels and Phe/Tyr Ratio (PP and UP Groups)
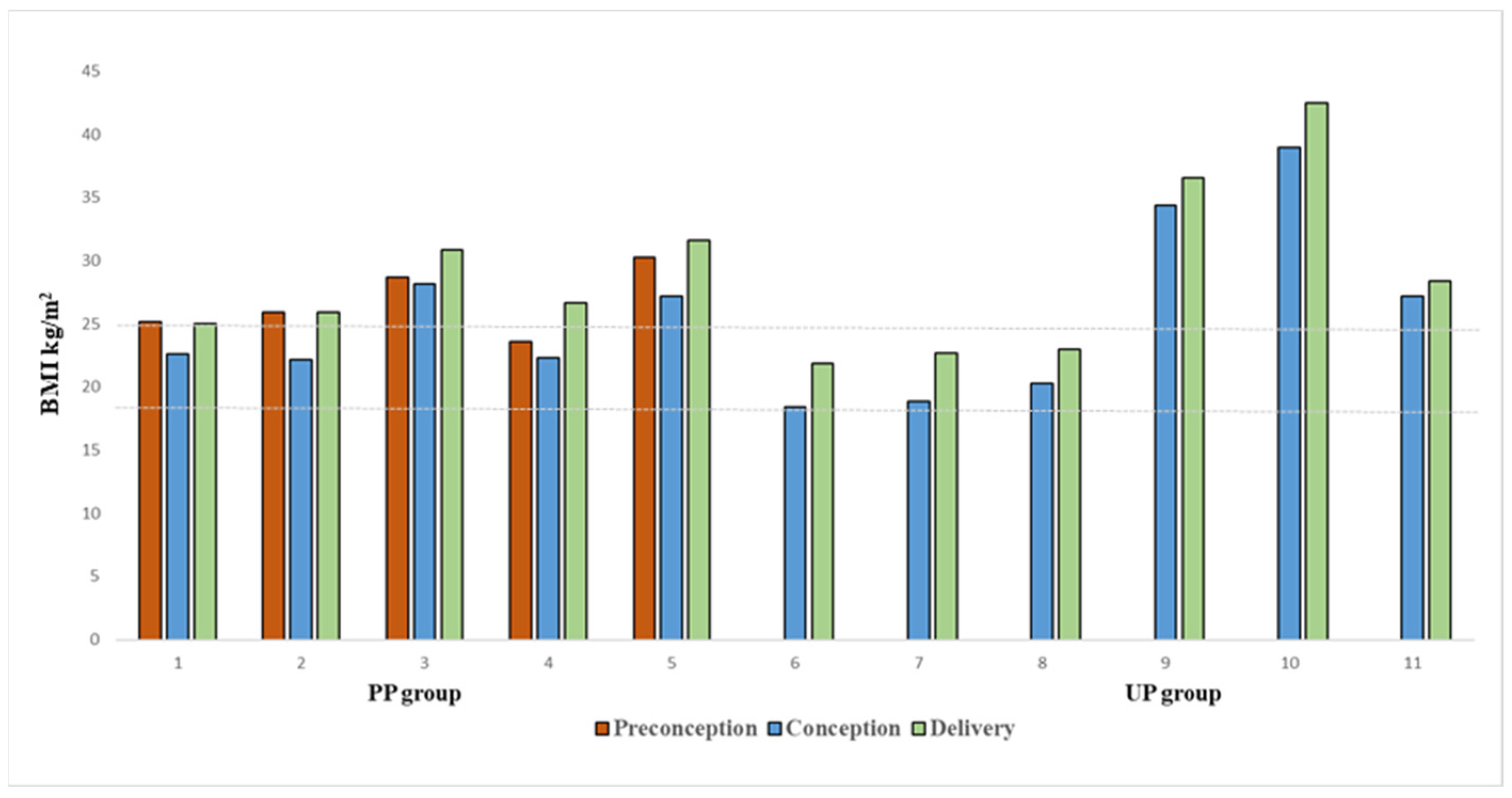
3.2. Anthropometric Characteristics of Women
3.3. Dietary Therapy
3.4. Phenylalanine Tolerance
3.5. Adherence to Dietary Therapy
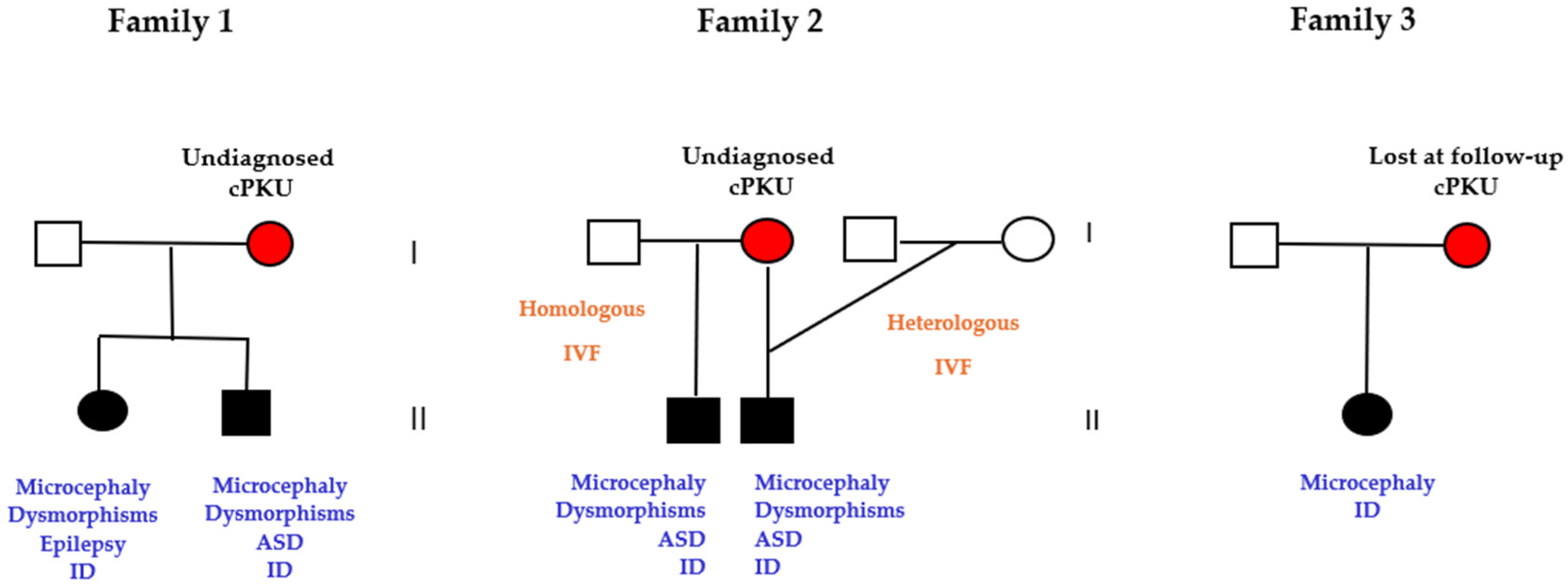
3.6. Pregnancies of Previously Undiagnosed PKU Women (UT n = 5)
3.7. Abortions of Unplanned Pregnancies
3.8. Offspring Neonatal Characteristics
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- American Academy of Pediatrics, Committee on Genetics. American Academy of Pediatrics: Maternal phenylketonuria. Pediatrics 2001, 107, 427–428. [Google Scholar] [CrossRef] [PubMed]
- Burgard, P.; Bremer, H.J.; Bührdel, P.; Clemens, P.C.; Mönch, E.; Przyrembel, H.; Trefz, F.K.; Ullrich, K. Rationale for the German recommendations for phenylalanine level control in phenylketonuria 1997. Eur. J. Pediatr. 1999, 158, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Trefz, F.K.; Ullrich, K.; Cipcic-Schmidt, S.; Fünders-Bücker, B.; van Teeffelen-Heithoff, A.; Przyrembel, H. Prophylaxis and treatment of maternal phenylketonuria (MPKU). Statement of the Working Group for Pediatric Metabolic Diseases (APS). Monatsschrift Kinderheilkd. 1995, 143, 898–899. [Google Scholar]
- Hanley, W.B.; Clarke, J.T.; Schoonheyt, W. Maternal phenylketonuria (PKU)—A review. Clin. Biochem. 1987, 20, 149–156. [Google Scholar] [CrossRef] [PubMed]
- Dent, C.E. Discussion of Armstrong MD: The relation of biochemical abnormality to the development of mental defect in phenylketonuria. In Etiologic Factors in Mental Retardation: Report of the 23rd Ross Pediatric Research Conference; Ross Laboratories: Columbus, OH, USA, 1957; pp. 32–33. [Google Scholar]
- Mabry, C.C.; Denniston, J.C.; Nelson, T.L.; Son, C.D. Maternal phenylketonuria: A cause of mental retardation in children without the metabolic defect. N. Engl. J. Med. 1963, 269, 1404–1408. [Google Scholar] [CrossRef]
- Lenke, R.R.; Levy, H.L. Maternal phenylketonuria and hyperphenylalaninemia. An international survey of the outcome of untreated and treated pregnancies. N. Engl. J. Med. 1980, 303, 1202–1208. [Google Scholar] [CrossRef]
- Koch, R.; Hanley, W.; Levy, H.; Matalon, K.; Matalon, R.; Rouse, B.; Trefz, F.; Guttler, F.; Azen, C.; Platt, L.; et al. The Maternal Phenylketonuria Collaborative Study: 1984–2002. Pediatrics 2003, 112 Pt 2, 1523–1529. [Google Scholar] [CrossRef]
- Lee, P.J.; Ridout, D.; Walter, J.H.; Cockburn, F. Maternal Phenylketonuria: Report from the United Kingdom Registry 1978–97. Arch. Dis. Child. 2005, 90, 143–146. [Google Scholar] [CrossRef]
- Medical Research Council Working Party on Phenylketonuria. Phenylketonuria due to phenylalanine hydroxylase deficiency: An unfolding story. BMJ 1993, 306, 115–119. [Google Scholar] [CrossRef]
- Matalon, K.M.; Acosta, P.B.; Azen, C. Role of nutrition in pregnancy with phenylketonuria and birth defects. Pediatrics 2003, 112 Pt 2, 1534–1536. [Google Scholar] [CrossRef] [PubMed]
- Widaman, K.F.; Azen, C. Relation of prenatal phenylalanine exposure to infant and childhood cognitive outcomes: Results from the International Maternal PKU Collaborative Study. Pediatrics 2003, 112 Pt 2, 1537–1543. [Google Scholar] [CrossRef]
- Waisbren, S.E.; Azen, C. Cognitive and behavioral development in maternal phenylketonuria offspring. Pediatrics 2003, 112, 1544–1547. [Google Scholar] [CrossRef] [PubMed]
- Vockley, J.; Andersson, H.C.; Antshel, K.M.; Braverman, N.E.; Burton, B.K.; Frazier, D.M.; Mitchell, J.; Smith, W.E.; Thompson, B.H.; Berry, S.A. Phenylalanine hydroxylase deficiency: Diagnosis and management guideline. Genet. Med. 2014, 16, 188–200. [Google Scholar] [CrossRef] [PubMed]
- ACOG Committee Opinion No. 449: Maternal phenylketonuria. Obstet. Gynecol. 2009, 114, 1432–1433. [CrossRef] [PubMed]
- Adams, A.D.; Fiesco-Roa, M.Ó.; Wong, L.; Jenkins, G.P.; Malinowski, J.; Demarest, O.M.; Rothberg, P.G.; Hobert, J.A. ACMG Therapeutics Committee. Electronic address: Documents@acmg.net. Phenylalanine hydroxylase deficiency treatment and management: A systematic evidence review of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 2023, 25, 100358. [Google Scholar] [CrossRef] [PubMed]
- Rouse, B.; Matalon, R.; Koch, R.; Azen, C.; Levy, H.; Hanley, W.; Trefz, F.; de la Cruz, F. Maternal phenylketonuria syndrome: Congenital heart defects, microcephaly, and developmental outcomes. J. Pediatr. 2000, 136, 57–61. [Google Scholar] [CrossRef] [PubMed]
- Platt, L.D.; Koch, R.; Hanley, W.B.; Levy, H.L.; Matalon, R.; Rouse, B.; Trefz, F.; de la Cruz, F.; Güttler, F.; Azen, C.; et al. The international study of pregnancy outcome in women with maternal phenylketonuria: Report of a 12-year study. Am. J. Obstet. Gynecol. 2000, 182, 326–333. [Google Scholar] [CrossRef] [PubMed]
- Grohmann-Held, K.; Burgard, P.; Baerwald, C.G.O.; Beblo, S.; Vom Dahl, S.; Das, A.; Dokoupil, K.; Fleissner, S.; Freisinger, P.; Heddrich-Ellerbrok, M.; et al. Impact of pregnancy planning and preconceptual dietary training on metabolic control and offspring’s outcome in phenylketonuria. J. Inherit. Metab. Dis. 2022, 45, 1070–1081. [Google Scholar] [CrossRef] [PubMed]
- van Vliet, D.; van Wegberg, A.M.J.; Ahring, K.; Bik-Multanowski, M.; Blau, N.; Bulut, F.D.; Casas, K.; Didycz, B.; Djordjevic, M.; Federico, A.; et al. Can untreated PKU patients escape from intellectual disability? A systematic review. Orphanet J. Rare Dis. 2018, 13, 149. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Maillot, F.; Cook, P.; Lilburn, M.; Lee, P.J. A practical approach to maternal phenylketonuria management. J. Inherit. Metab. Dis. 2007, 30, 198–201. [Google Scholar] [CrossRef] [PubMed]
- Scala, I.; Riccio, M.P.; Marino, M.; Bravaccio, C.; Parenti, G.; Strisciuglio, P. Large Neutral Amino Acids (LNAAs) Supplementation Improves Neuropsychological Performances in Adult Patients with Phenylketonuria. Nutrients 2020, 12, 1092. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Bertino, E.; Spada, E.; Occhi, L.; Coscia, A.; Giuliani, F.; Gagliardi, L.; Gilli, G.; Bona, G.; Fabris, C.; De Curtis, M.; et al. Neonatal anthropometric charts: The Italian neonatal study compared with other European studies. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 353–361. [Google Scholar] [CrossRef] [PubMed]
- Waisbren, S.E.; Hamilton, B.D.; St. James, P.; Shilo, S.; Levy, H.L. Psychological factors in maternal phenylketonuria: Women’s adherence to medical recommendations. Am. J. Public Health 1995, 12, 1636–1641. [Google Scholar] [CrossRef] [PubMed]
- Galan, H.L.; Marconi, A.M.; Paolini, C.L.; Cheung, A.; Battaglia, F.C. The transplacental transport of essential amino acids in uncomplicated human pregnancies. Am. J. Obstet. Gynecol. 2009, 200, 91.e1–91.e7. [Google Scholar] [CrossRef] [PubMed]
- van Wegberg, A.M.J.; MacDonald, A.; Ahring, K.; Bélanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Giżewska, M.; et al. The complete European guidelines on phenylketonuria: Diagnosis and treatment. Orphanet J. Rare Dis. 2017, 12, 162. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Chen, Y.; Wan, K.; Gong, Y.; Zhang, X.; Liang, Y.; Wang, X.; Feng, P.; He, F.; Zhou, R.; Yang, D.; et al. Assessing the relationship between pregravid body mass index and risk of adverse maternal pregnancy and neonatal outcomes: Prospective data in Southwest China. Sci. Rep. 2021, 11, 7591. [Google Scholar] [CrossRef] [PubMed] [PubMed Central]
- Teissier, R.; Nowak, E.; Assoun, M.; Mention, K.; Cano, A.; Fouilhoux, A.; Feillet, F.; Ogier, H.; Oger, E.; de Parscau, L.; et al. Maternal phenylketonuria: Low phenylalaninemia might increase the risk of intra uterine growth retardation. J. Inherit. Metab. Dis. 2012, 35, 993–999. [Google Scholar] [CrossRef]
- Bhasin, K.K.; van Nas, A.; Martin, L.J.; Davis, R.C.; Devaska, S.U.; Lusis, A.J. Maternal low-protein diet or hypercholesterolemia reduces circulating essential amino acids and leads to intrauterine growth restriction. Diabetes 2009, 58, 559–566. [Google Scholar] [CrossRef]
- van Spronsen, F.J.; van Wegberg, A.M.; Ahring, K.; Bélanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Giżewska, M.; et al. Key European guidelines for the diagnosis and management of patients with phenylketonuria. Lancet Diabetes Endocrinol. 2017, 5, 743–756. [Google Scholar] [CrossRef] [PubMed]
- Feillet, F.; Ficicioglu, C.; Lagler, F.B.; Longo, N.; Muntau, A.C.; Burlina, A.; Trefz, F.K.; van Spronsen, F.J.; Arnoux, J.B.; Lindstrom, K.; et al. KAMPER and PKUDOS investigators. Efficacy and safety of sapropterin before and during pregnancy: Final analysis of the Kuvan® Adult Maternal Paediatric European Registry (KAMPER) maternal and Phenylketonuria Developmental Outcomes and Safety (PKUDOS) PKU-MOMs sub-registries. J. Inherit. Metab. Dis. 2024, 47, 636–650. [Google Scholar] [CrossRef] [PubMed]
- Wiedemann, A.; Leheup, B.; Battaglia-Hsu, S.F.; Jonveaux, P.; Jeannesson, E.; Feillet, F. Undiagnosed phenylketonuria in parents of phenylketonuric patients, is it worthwhile to be checked? Mol. Genet. Metab. 2013, 110, S62–S65. [Google Scholar] [CrossRef] [PubMed]
- Yıldız, Y.; Sivri, H.S. Maternal phenylketonuria in Turkey: Outcomes of 71 pregnancies and issues in management. Eur. J. Pediatr. 2019, 178, 1005–1011. [Google Scholar] [CrossRef] [PubMed]
- Bouchlariotou, S.; Tsikouras, P.; Maroulis, G. Undiagnosed maternal phenylketonuria: Own clinical experience and literature review. J. Matern.-Fetal Neonatal Med. 2009, 22, 943–948. [Google Scholar] [CrossRef] [PubMed]
- Gokmen, T.; Oguz, S.S.; Altug, N.; Akar, M.; Erdeve, O.; Dilmen, U. A case of maternal phenylketonuria syndrome presenting with unilateral renal agenesis. J. Trop. Pediatr. 2011, 57, 138–140. [Google Scholar] [CrossRef] [PubMed]
- Hanley, W.B. Finding the fertile woman with phenylketonuria. Eur. J. Obstet. Gynecol. Reprod. Biol. 2008, 137, 131–135. [Google Scholar] [CrossRef] [PubMed]
- SINU, Italian Society of Human Nutrition. LARN-Reference Intake Levels of Nutrients and Energy for the Italian Population; IV Revision; Editorial coordination SINU-INRAN; SICS: Milan, Italy, 2014. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pregnant women with PKU | n = 13 |
| Pregnancies (including multiple pregnancies and abortions) | n = 22 |
| PKU classification | Classic PKU = 8 women; 14 pregnancies (PP = 2; UP = 2; UT = 5; AB = 5) Mild PKU = 3 women; 5 pregnancies (PP = 2; UP = 2; AB = 1) Mild HPA = 2 women; 3 pregnancies (PP = 1; UP = 2) |
| Historical tolerance (dietary Phe; mg/day) | Range 340–2100 |
| Maternal age at conception (years) | Mean (SD) 30.2 ± 4.8 Range 24–39 |
| Time | Median Phenylalanine Concentration (μmol/L) | |||||
|---|---|---|---|---|---|---|
| p-Value | ||||||
| Median Min (IQR) | Median Max (IQR) | on Min Phe Values | on Max Phe Values | |||
| PP | UP | PP | UP | |||
| I trimester (T1) | 132 (108–138) | 354 (78–630) | 324 (306–534 | 726 (594–864) | 1 | 0.030 |
| II trimester (T2) | 114 (102–132) | 156 (114–192) | 258 (210–468) | 450 (228–666) | 0.329 | 0.429 |
| III trimester (T3) | 102 (96–126) | 108 (96–114) | 264 (198–342) | 276 (138–582) | 0.931 | 1 |
| Offspring PP (N = 5) Males = 1, Females = 4 | Offspring UP (N = 5) Male = 5, Female = 0 | p Value | |
|---|---|---|---|
| Birth weight (Kg) Median (IQR) | 2.79 (2.74–3.33) | 2.40 (2.30–2.85) | 0.690 |
| Weight percentiles Median (IQR) | 21 (19–74) | 5 (3–10) | 0.151 |
| Length (cm) Median (IQR) | 49 (47–49) | 45 (45–50) | 0.600 |
| Length percentiles Median (IQR) | 48 (19–56) | 8 (4–21) | 0.310 |
| Head circumference (cm) Median (IQR) | 33 (32.5–34) | 32.5 (32–33) | 0.800 |
| Head circumference percentiles Median (IQR) | 40(40–47) | 23 (2–27) | 0.548 |
| Apgar index Median (IQR) | 8.0 (8.0–9.0) | 7.0 (7.0–8.0) | 0.400 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gautiero, C.; Scala, I.; Esposito, G.; Coppola, M.R.; Cacciapuoti, N.; Fisco, M.; Ruoppolo, M.; Strisciuglio, P.; Parenti, G.; Guida, B. The Light and the Dark Side of Maternal PKU: Single-Centre Experience of Dietary Management and Emergency Treatment Protocol of Unplanned Pregnancies. Nutrients 2025, 17, 1048. https://doi.org/10.3390/nu17061048
Gautiero C, Scala I, Esposito G, Coppola MR, Cacciapuoti N, Fisco M, Ruoppolo M, Strisciuglio P, Parenti G, Guida B. The Light and the Dark Side of Maternal PKU: Single-Centre Experience of Dietary Management and Emergency Treatment Protocol of Unplanned Pregnancies. Nutrients. 2025; 17(6):1048. https://doi.org/10.3390/nu17061048
Chicago/Turabian StyleGautiero, Claudia, Iris Scala, Giulia Esposito, Maria Rosaria Coppola, Nunzia Cacciapuoti, Mariagrazia Fisco, Margherita Ruoppolo, Pietro Strisciuglio, Giancarlo Parenti, and Bruna Guida. 2025. "The Light and the Dark Side of Maternal PKU: Single-Centre Experience of Dietary Management and Emergency Treatment Protocol of Unplanned Pregnancies" Nutrients 17, no. 6: 1048. https://doi.org/10.3390/nu17061048
APA StyleGautiero, C., Scala, I., Esposito, G., Coppola, M. R., Cacciapuoti, N., Fisco, M., Ruoppolo, M., Strisciuglio, P., Parenti, G., & Guida, B. (2025). The Light and the Dark Side of Maternal PKU: Single-Centre Experience of Dietary Management and Emergency Treatment Protocol of Unplanned Pregnancies. Nutrients, 17(6), 1048. https://doi.org/10.3390/nu17061048