
High Fat Diet-Induced Obesity Dysregulates Splenic B Cell Mitochondrial Activity

 , ,
, , 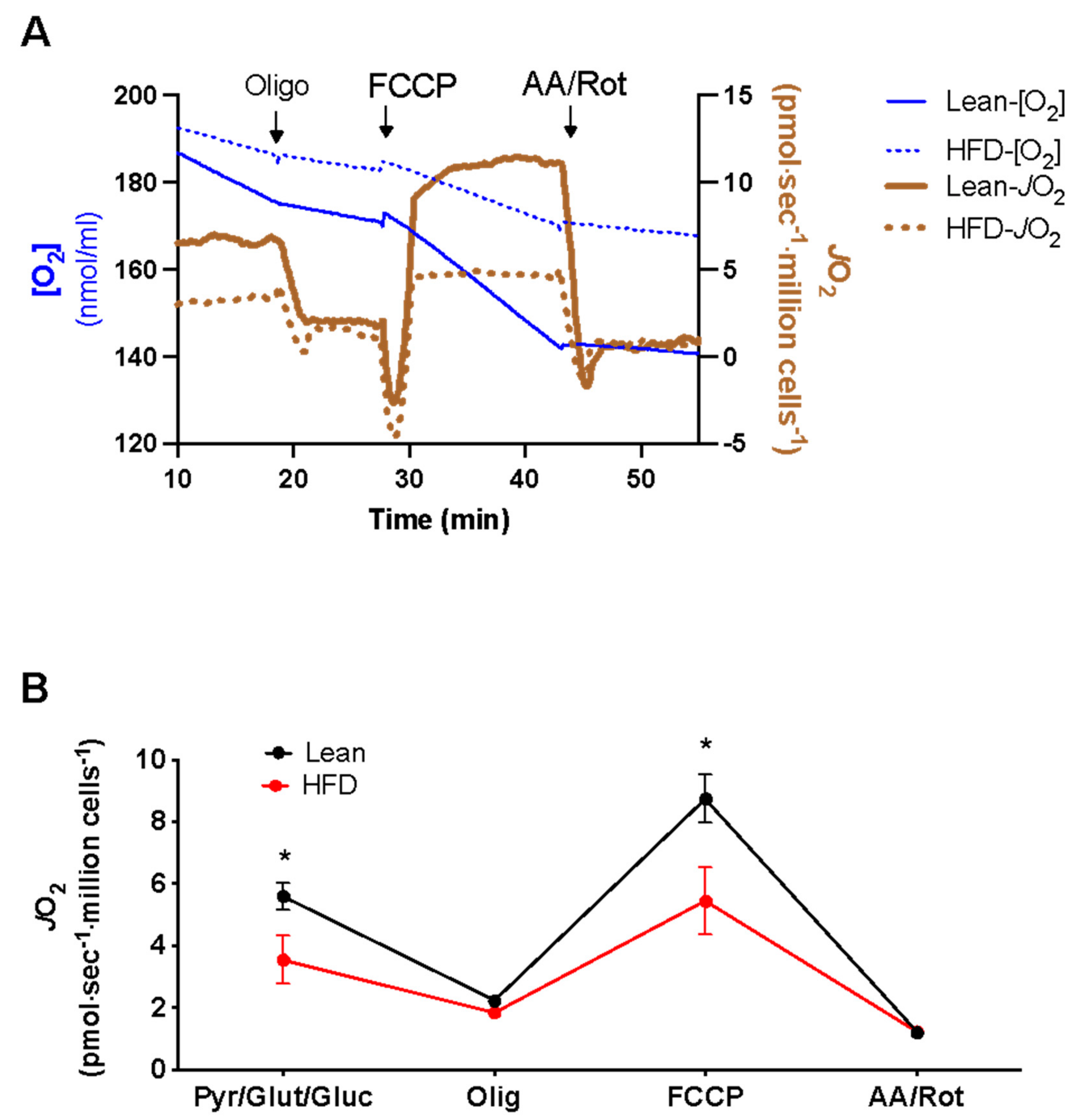{kind=link}
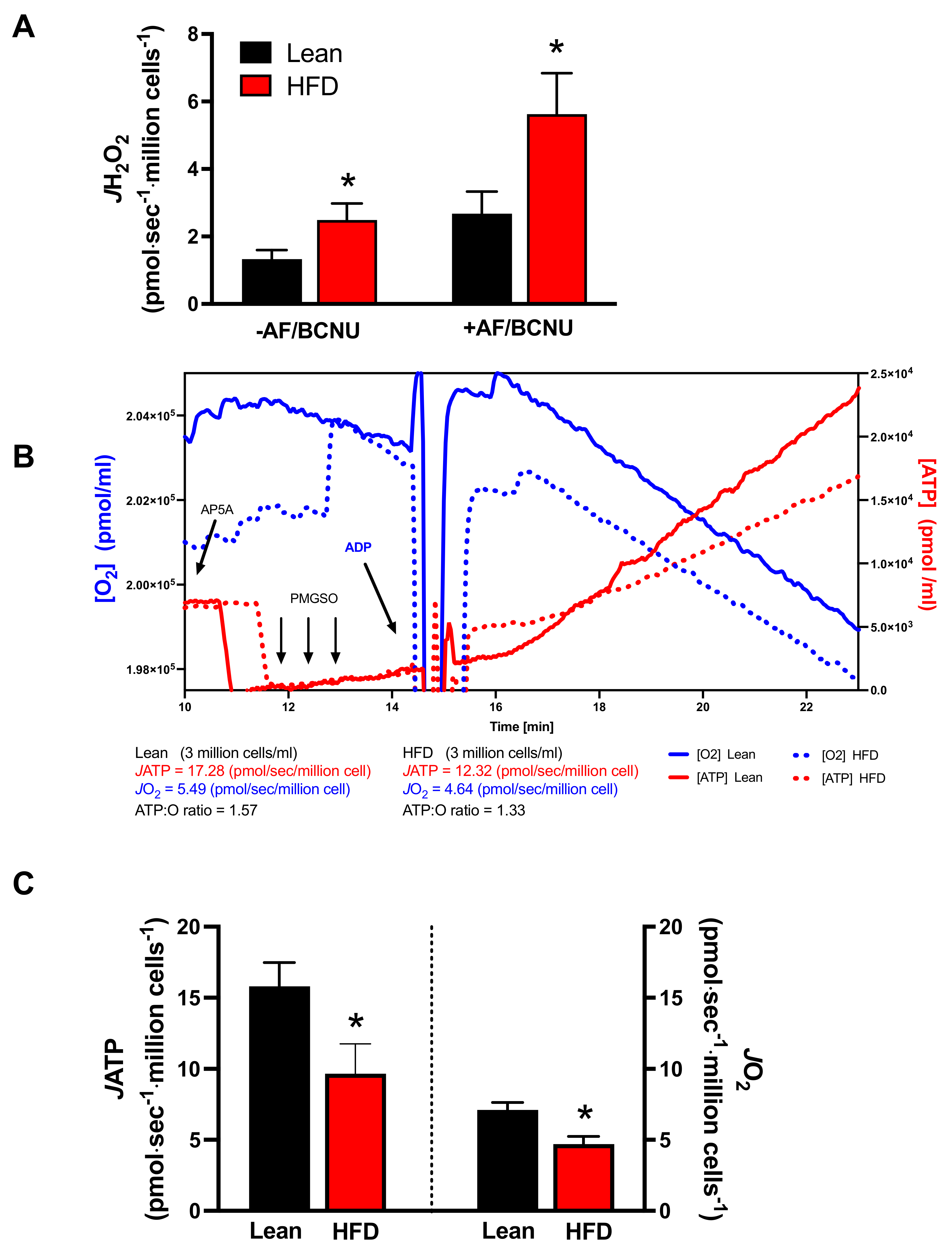{kind=link}
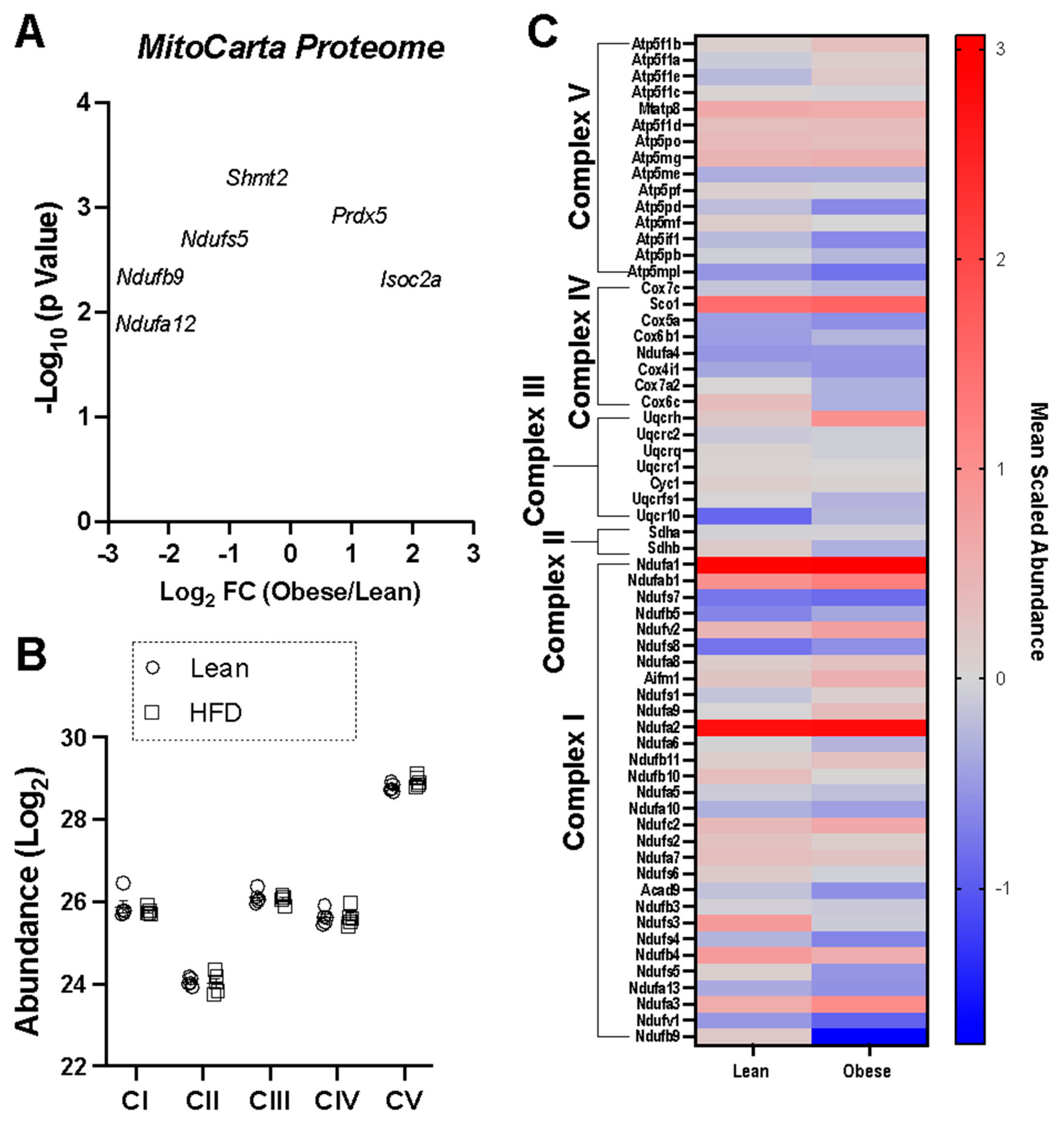{kind=link}
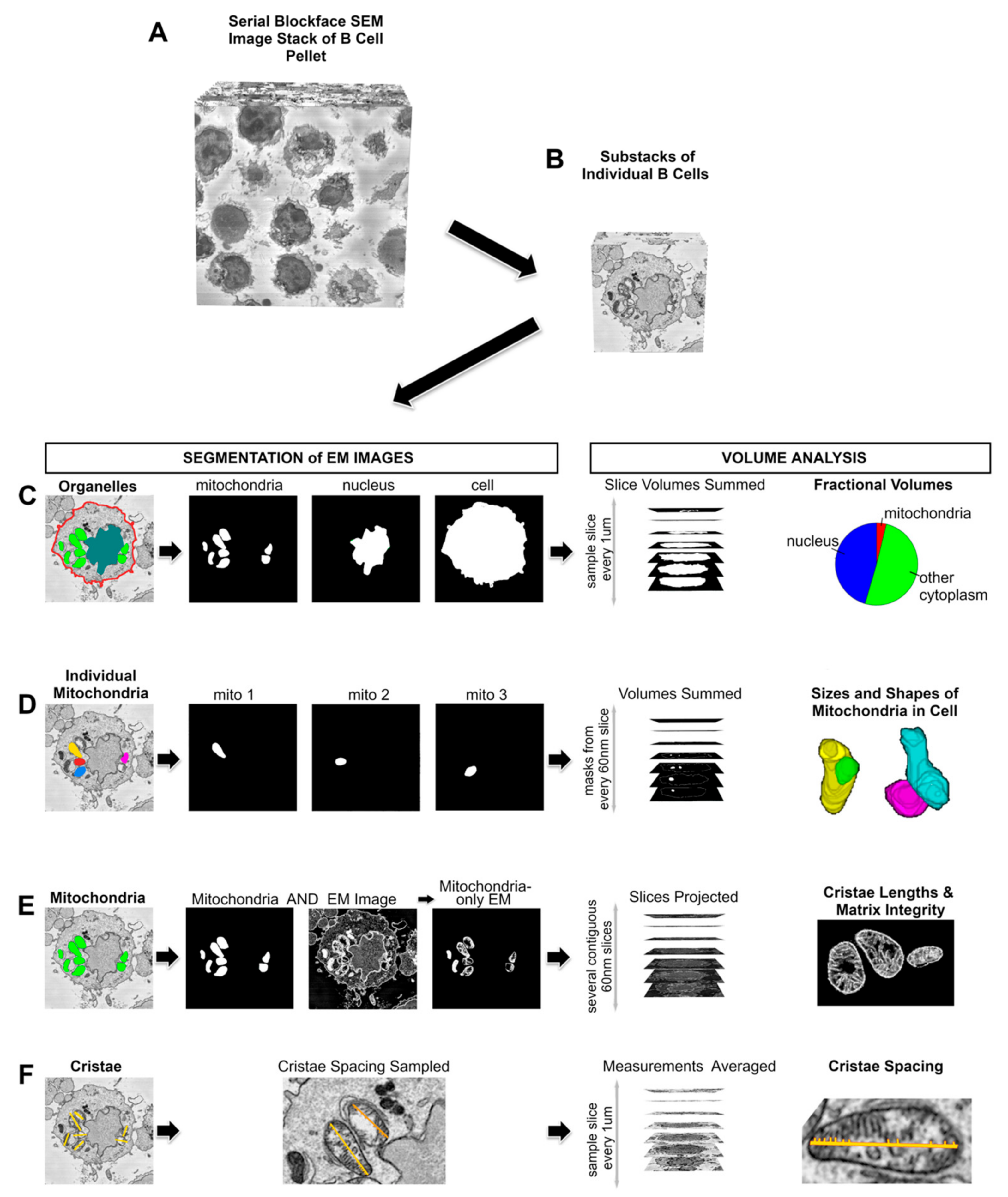{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
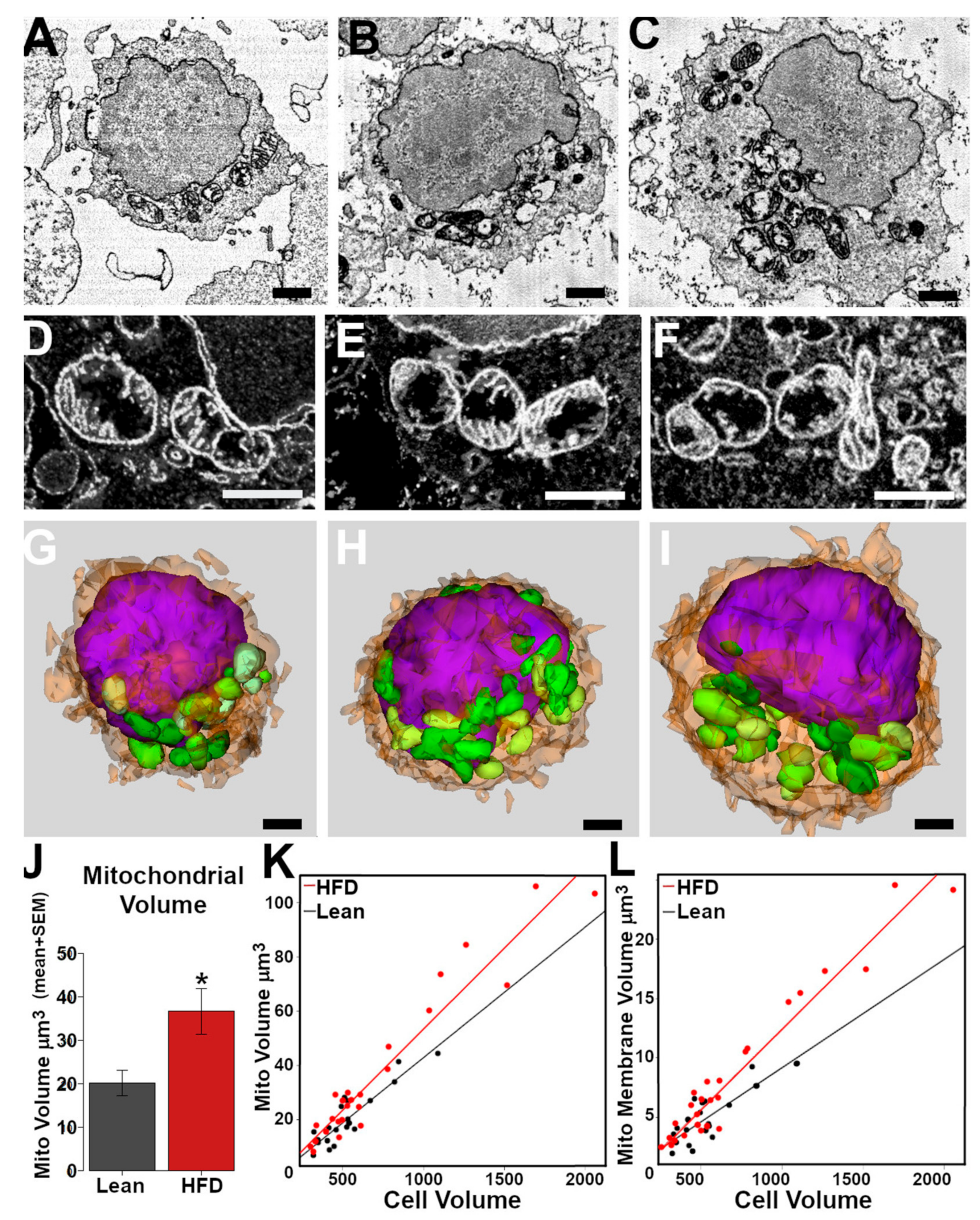
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- De Mello, A.H.; Costa, A.B.; Engel, J.D.G.; Rezin, G.T. Mitochondrial dysfunction in obesity. Life Sci. 2018, 192, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Hartley, R.C. Mitochondria as a therapeutic target for common pathologies. Nat. Rev. Drug Discov. 2018, 17, 865–886. [Google Scholar] [CrossRef] [PubMed]
- Allen, M.E.; Pennington, E.R.; Perry, J.B.; Dadoo, S.; Makrecka-Kuka, M.; Dambrova, M.; Moukdar, F.; Patel, H.D.; Han, X.; Kidd, G.K.; et al. The cardiolipin-binding peptide elamipretide mitigates fragmentation of cristae networks following cardiac ischemia reperfusion in rats. Commun. Biol. 2020, 3, 389. [Google Scholar] [CrossRef] [PubMed]
- Anderson, E.J.; Lustig, M.E.; Boyle, K.E.; Woodlief, T.L.; Kane, D.A.; Lin, C.T.; Price, J.W.; Kang, L.I.; Rabinovitch, P.S.; Szeto, H.H.; et al. Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Investig. 2009, 119, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Frasca, D.; Blomberg, B.B. The Impact of Obesity and Metabolic Syndrome on Vaccination Success. In Interdisciplinary Topics in Gerontology and Geriatrics; Karger: Basel, Switzerland, 2020; Volume 43, pp. 86–97. [Google Scholar]
- DeFuria, J.; Belkina, A.C.; Jagannathan-Bogdan, M.; Snyder-Cappione, J.; Carr, J.D.; Nersesova, Y.R.; Markham, D.; Strissel, K.J.; Watkins, A.A.; Zhu, M.; et al. B cells promote inflammation in obesity and type 2 diabetes through regulation of T-cell function and an inflammatory cytokine profile. Proc. Natl. Acad. Sci. USA 2013, 110, 5133–5138. [Google Scholar] [CrossRef] [PubMed]
- SantaCruz-Calvo, S.; Bharath, L.; Pugh, G.; SantaCruz-Calvo, L.; Lenin, R.R.; Lutshumba, J.; Liu, R.; Bachstetter, A.D.; Zhu, B.; Nikolajczyk, B.S. Adaptive immune cells shape obesity-associated type 2 diabetes mellitus and less prominent comorbidities. Nat. Rev. Endocrinol. 2022, 18, 23–42. [Google Scholar] [CrossRef]
- Frasca, D.; Diaz, A.; Romero, M.; Blomberg, B.B. Leptin induces immunosenescence in human B cells. Cell Immunol. 2020, 348, 103994. [Google Scholar] [CrossRef] [PubMed]
- Winer, D.A.; Winer, S.; Shen, L.; Wadia, P.P.; Yantha, J.; Paltser, G.; Tsui, H.; Wu, P.; Davidson, M.G.; Alonso, M.N.; et al. B cells promote insulin resistance through modulation of T cells and production of pathogenic IgG antibodies. Nat. Med. 2011, 17, 610–617. [Google Scholar] [CrossRef]
- Frasca, D.; Ferracci, F.; Diaz, A.; Romero, M.; Lechner, S.; Blomberg, B.B. Obesity decreases B cell responses in young and elderly individuals. Obesity 2016, 24, 615–625. [Google Scholar] [CrossRef]
- Sheridan, P.A.; Paich, H.A.; Handy, J.; Karlsson, E.A.; Hudgens, M.G.; Sammon, A.B.; Holland, L.A.; Weir, S.; Noah, T.L.; Beck, M.A. Obesity is associated with impaired immune response to influenza vaccination in humans. Int. J. Obes. 2012, 36, 1072–1077. [Google Scholar] [CrossRef]
- Milner, J.J.; Sheridan, P.A.; Karlsson, E.A.; Schultz-Cherry, S.; Shi, Q.; Beck, M.A. Diet-induced obese mice exhibit altered heterologous immunity during a secondary 2009 pandemic H1N1 infection. J. Immunol. 2013, 191, 2474–2485. [Google Scholar] [CrossRef] [PubMed]
- Virk, R.; Buddenbaum, N.; Al-Shaer, A.; Armstrong, M.; Manke, J.; Reisdorph, N.; Sergin, S.; Fenton, J.I.; Wallace, E.D.; Ehrmann, B.M.; et al. Obesity reprograms the pulmonary polyunsaturated fatty acid-derived lipidome, transcriptome, and gene-oxylipin networks. J. Lipid. Res. 2022, 63, 100267. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.; Guesdon, W.; Torres, M.J.; Armstrong, M.; Quinn, K.; Davis, T.; Reisdorph, N.; Neufer, P.D.; Spangenburg, E.E.; Carroll, I.; et al. Resolvin E1 derived from eicosapentaenoic acid prevents hyperinsulinemia and hyperglycemia in a host genetic manner. FASEB J. 2020, 34, 10640–10656. [Google Scholar] [CrossRef] [PubMed]
- Crouch, M.J.; Kosaraju, R.; Guesdon, W.; Armstrong, M.; Reisdorph, N.; Jain, R.; Fenton, J.; Shaikh, S.R. Frontline Science: A reduction in DHA-derived mediators in male obesity contributes toward defects in select B cell subsets and circulating antibody. J. Leukoc. Biol. 2019, 106, 241–257. [Google Scholar] [CrossRef] [PubMed]
- Lark, D.S.; Torres, M.J.; Lin, C.T.; Ryan, T.E.; Anderson, E.J.; Neufer, P.D. Direct real-time quantification of mitochondrial oxidative phosphorylation efficiency in permeabilized skeletal muscle myofibers. Am. J. Physiol. Cell Physiol. 2016, 311, C239–C245. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.J.; Kew, K.A.; Ryan, T.E.; Pennington, E.R.; Lin, C.-T.; Buddo, K.A.; Fix, A.M.; Smith, C.A.; Gilliam, L.A.; Karvinen, S.; et al. 17β-Estradiol Directly Lowers Mitochondrial Membrane Microviscosity and Improves Bioenergetic Function in Skeletal Muscle. Cell Metab. 2018, 27, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Iñigo, M.R.; Amorese, A.J.; Tarpey, M.D.; Balestrieri, N.P.; Jones, K.G.; Patteson, D.J.; Jackson, K.C.; Torres, M.; Lin, C.-T.; Smith, C.D.; et al. Estrogen receptor-alpha in female skeletal muscle is not required for regulation of muscle insulin sensitivity and mitochondrial regulation. Mol. Metab. 2020, 34, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Fisher-Wellman, K.H.; Davidson, M.T.; Narowski, T.M.; Lin, C.T.; Koves, T.R.; Muoio, D.M. Mitochondrial Diagnostics: A Multiplexed Assay Platform for Comprehensive Assessment of Mitochondrial Energy Fluxes. Cell Rep. 2018, 24, 3593–3606.e10. [Google Scholar] [CrossRef]
- Goldberg, E.J.; Buddo, K.A.; McLaughlin, K.L.; Fernandez, R.F.; Pereyra, A.S.; Psaltis, C.E.; Lin, C.-T.; Hagen, J.T.; Boykov, I.N.; Nguyen, T.K.; et al. Tissue-specific characterization of mitochondrial branched-chain keto acid oxidation using a multiplexed assay platform. Biochem. J. 2019, 476, 1521–1537, Correction in Biochem. J. 2019, 476, 2519. [Google Scholar] [CrossRef]
- McLaughlin, K.L.; Nelson, M.A.; Coalson, H.S.; Hagen, J.T.; Montgomery, M.M.; Wooten, A.R.; Zeczycki, T.N.; Vohra, N.A.; Fisher-Wellman, K.H. Bioenergetic Phenotyping of DEN-Induced Hepatocellular Carcinoma Reveals a Link Between Adenylate Kinase Isoform Expression and Reduced Complex I-Supported Respiration. Front. Oncol. 2022, 12, 919880. [Google Scholar] [CrossRef]
- Hagen, J.T.; Montgomery, M.M.; Biagioni, E.M.; Krassovskaia, P.; Jevtovic, F.; Shookster, D.; Sharma, U.; Tung, K.; Broskey, N.T.; May, L.; et al. Intrinsic adaptations in OXPHOS power output and reduced tumorigenicity characterize doxorubicin resistant ovarian cancer cells. Biochim. Biophys. Acta Bioenerg. 2022, 1863, 148915. [Google Scholar] [CrossRef] [PubMed]
- Calvo, S.E.; Clauser, K.R.; Mootha, V.K. MitoCarta2.0: An updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2015, 44, D1251–D1257. [Google Scholar] [CrossRef] [PubMed]
- McLaughlin, K.L.; Hagen, J.T.; Coalson, H.S.; Nelson, M.A.M.; Kew, K.A.; Wooten, A.R.; Fisher-Wellman, K.H. Novel approach to quantify mitochondrial content and intrinsic bioenergetic efficiency across organs. Sci. Rep. 2020, 10, 17599. [Google Scholar] [CrossRef] [PubMed]
- Benson, E.; Kidd, G.; Campbell, J.; Goodman, S. Serial Block-Face SEM of Brain Tissue Using Rapid Automated Preparation. Microsc. Microanal. 2020, 26 (Suppl. S2), 1372–1373. [Google Scholar] [CrossRef]
- Kamiński, M.M.; Sauer, S.W.; Kamiński, M.; Opp, S.; Ruppert, T.; Grigaravičius, P.; Grudnik, P.; Gröne, H.J.; Krammer, P.H.; Gülow, K. T cell activation is driven by an ADP-dependent glucokinase linking enhanced glycolysis with mitochondrial reactive oxygen species generation. Cell Rep. 2012, 2, 1300–1315. [Google Scholar] [CrossRef]
- Sandoval, H.; Kodali, S.; Wang, J. Regulation of B cell fate, survival, and function by mitochondria and autophagy. Mitochondrion 2018, 41, 58–65. [Google Scholar] [CrossRef]
- Ogura, M.; Inoue, T.; Yamaki, J.; Homma, M.K.; Kurosaki, T.; Homma, Y. Mitochondrial reactive oxygen species suppress humoral immune response through reduction of CD19 expression in B cells in mice. Eur. J. Immunol. 2017, 47, 406–418. [Google Scholar] [CrossRef]
- Jang, K.-J.; Mano, H.; Aoki, K.; Hayashi, T.; Muto, A.; Nambu, Y.; Takahashi, K.; Itoh, K.; Taketani, S.; Nutt, S.L.; et al. Mitochondrial function provides instructive signals for activation-induced B-cell fates. Nat. Commun. 2015, 6, 6750. [Google Scholar] [CrossRef]
- Frasca, D.; Romero, M.; Diaz, A.; Garcia, D.; Thaller, S.; Blomberg, B.B. B Cells with a senescent-associated secretory phenotype accumulate in the adipose tissue of individuals with obesity. Int. J. Mol. Sci. 2021, 22, 1839. [Google Scholar] [CrossRef]
- Frasca, D. Obesity accelerates age defects in human B cells and induces autoimmunity. Immunometabolism 2022, 4, e220010. [Google Scholar] [CrossRef]
- Green, W.D.; Al-Shaer, A.E.; Shi, Q.; Gowdy, K.M.; MacIver, N.J.; Milner, J.J.; Beck, M.A.; Shaikh, S.R. Metabolic and functional impairment of CD8(+) T cells from the lungs of influenza-infected obese mice. J. Leukoc. Biol. 2021, 111, 147–159. [Google Scholar] [CrossRef]
- Alwarawrah, Y.; Nichols, A.G.; Green, W.D.; Eisner, W.; Kiernan, K.; Warren, J.; Hale, L.P.; Beck, M.A.; MacIver, N.J. Targeting T-cell oxidative metabolism to improve influenza survival in a mouse model of obesity. Int. J. Obes. 2020, 44, 2419–2429. [Google Scholar] [CrossRef]
- Wu, L.; Lu, P.; Guo, X.; Song, K.; Lyu, Y.; Bothwell, J.; Wu, J.; Hawkins, O.; Clarke, S.L.; Lucas, E.A.; et al. Beta-carotene oxygenase 2 deficiency-triggered mitochondrial oxidative stress promotes low-grade inflammation and metabolic dysfunction. Free Radic. Biol. Med. 2021, 164, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Aryaman, J.; Johnston, I.G.; Jones, N.S. Mitochondrial Heterogeneity. Front. Genet. 2019, 9, 718. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, E.M.; Pennington, E.R.; Sparagna, G.C.; Torres, M.J.; Neufer, P.D.; Harris, M.; Washington, J.; Anderson, E.J.; Zeczycki, T.N.; Brown, D.A.; et al. Docosahexaenoic acid lowers cardiac mitochondrial enzyme activity by replacing linoleic acid in the phospholipidome. J. Biol. Chem. 2018, 293, 466–483. [Google Scholar] [CrossRef]
- Sullivan, E.M.; Fix, A.; Crouch, M.J.; Sparagna, G.C.; Zeczycki, T.N.; Brown, D.A.; Shaikh, S.R. Murine diet-induced obesity remodels cardiac and liver mitochondrial phospholipid acyl chains with differential effects on respiratory enzyme activity. J. Nutr. Biochem. 2017, 45, 94–103. [Google Scholar] [CrossRef] [PubMed]
- Cardenas-Perez, R.E.; Fuentes-Mera, L.; de la Garza, A.L.; Torre-Villalvazo, I.; Reyes-Castro, L.A.; Rodriguez-Rocha, H.; Garcia-Garcia, A.; Corona-Castillo, J.C.; Tovar, A.R.; Zambrano, E.; et al. Maternal overnutrition by hypercaloric diets programs hypothalamic mitochondrial fusion and metabolic dysfunction in rat male offspring. Nutr. Metab. (Lond.) 2018, 15, 38. [Google Scholar] [CrossRef]
- Kosaraju, R.; Guesdon, W.; Crouch, M.J.; Teague, H.L.; Sullivan, E.M.; Karlsson, E.A.; Schultz-Cherry, S.; Gowdy, K.; Bridges, L.C.; Reese, L.R.; et al. B Cell activity is impaired in human and mouse obesity and is responsive to an essential fatty acid upon murine influenza infection. J. Immunol. 2017, 198, 4738–4752. [Google Scholar] [CrossRef]
- Li, Z.; Zhao, M.; Li, J.; Luo, W.; Huang, J.; Huang, G.; Xie, Z.; Xiao, Y.; Huang, J.; Li, X.; et al. Elevated glucose metabolism driving pro-inflammatory response in B cells contributes to the progression of type 1 diabetes. Clin. Immunol. 2023, 255, 109729. [Google Scholar] [CrossRef] [PubMed]
- Šlisere, B.; Arisova, M.; Aizbalte, O.; Salmiņa, M.M.; Zolovs, M.; Levenšteins, M.; Mukāns, M.; Troickis, I.; Meija, L.; Lejnieks, A.; et al. Distinct B cell profiles characterise healthy weight and obesity pre- and post-bariatric surgery. Int. J. Obes. (Lond.) 2023, 47, 970–978. [Google Scholar] [CrossRef]
- Frasca, D.; Garcia, D.; Diaz, A.; Romero, M.; Thaller, S.; Blomberg, B.B. Phenotypic and functional features of B cells from two different human subcutaneous adipose depots. PLoS ONE 2023, 18, e0285025. [Google Scholar] [CrossRef]
- Frasca, D. Several areas of overlap between obesity and aging indicate obesity as a biomarker of accelerated aging of human B cell function and antibody responses. Immun. Ageing 2022, 19, 48. [Google Scholar] [CrossRef]
- Khan, S.; Winer, D.A. T-bet+ B cells exacerbate obesity-related metabolic disease. Trends Immunol. 2022, 43, 855–857. [Google Scholar] [CrossRef] [PubMed]
- Hägglöf, T.; Vanz, C.; Kumagai, A.; Dudley, E.; Ortega, V.; Siller, M.; Parthasarathy, R.; Keegan, J.; Koenigs, A.; Shute, T.; et al. T-bet+B cells accumulate in adipose tissue and exacerbate metabolic disorder during obesity. Cell Metab. 2022, 34, 1121–1136.e6. [Google Scholar] [CrossRef]
- Frasca, D.; Diaz, A.; Romero, M.; Blomberg, B.B. Phenotypic and Functional Characterization of Double Negative B Cells in the Blood of Individuals With Obesity. Front. Immunol. 2021, 12, 616650. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pal, A.; Lin, C.-T.; Boykov, I.; Benson, E.; Kidd, G.; Fisher-Wellman, K.H.; Neufer, P.D.; Shaikh, S.R. High Fat Diet-Induced Obesity Dysregulates Splenic B Cell Mitochondrial Activity. Nutrients 2023, 15, 4807. https://doi.org/10.3390/nu15224807
Pal A, Lin C-T, Boykov I, Benson E, Kidd G, Fisher-Wellman KH, Neufer PD, Shaikh SR. High Fat Diet-Induced Obesity Dysregulates Splenic B Cell Mitochondrial Activity. Nutrients. 2023; 15(22):4807. https://doi.org/10.3390/nu15224807
Chicago/Turabian StylePal, Anandita, Chien-Te Lin, Ilya Boykov, Emily Benson, Grahame Kidd, Kelsey H. Fisher-Wellman, P. Darrell Neufer, and Saame Raza Shaikh. 2023. "High Fat Diet-Induced Obesity Dysregulates Splenic B Cell Mitochondrial Activity" Nutrients 15, no. 22: 4807. https://doi.org/10.3390/nu15224807
APA StylePal, A., Lin, C.-T., Boykov, I., Benson, E., Kidd, G., Fisher-Wellman, K. H., Neufer, P. D., & Shaikh, S. R. (2023). High Fat Diet-Induced Obesity Dysregulates Splenic B Cell Mitochondrial Activity. Nutrients, 15(22), 4807. https://doi.org/10.3390/nu15224807