
Nutritional Approach in Selected Inherited Metabolic Cardiac Disorders—A Concise Summary of Available Scientific Evidence
 ,
,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
3. Overview of Cellular Energy Generation
4. Nutritional Approaches in Primary Mitochondrial Diseases
5. Nutritional Approaches in Lysosomal Storage Diseases
6. Impact of Nutritional Intervention on Cardiac Function in Patients with Inherited Metabolic Disorders
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Luca, A.C.; Miron, I.C.; Mîndru, D.E.; Curpan, A.S.; Stan, R.C.; Tarca, E.; Luca, F.A.; Paduret, A.I. Optimal Nutrition Parameters for Neonates and Infants with Congenital Heart Disease. Nutrients 2022, 14, 1671. [Google Scholar] [CrossRef]
- Trang, N.N.; Lee, T.I.; Kao, Y.H.; Chao, T.-F.; Chen, Y.-J. Ketogenic diet modulates cardiac metabolic dysregulation in streptozocin-induced diabetic rats. J. Nutr. Biochem. 2023, 111, 109161. [Google Scholar] [CrossRef]
- Valayannopoulos, V.; Bajolle, F.; Arnoux, J.-B.; Dubois, S.; Sannier, N.; Baussan, C.; Petit, F.; Labrune, P.; Rabier, D.; Ottolenghi, C.; et al. Successful treatment of severe cardiomyopathy in glycogen storage disease type III With D,L-3-hydroxybutyrate, ketogenic and high-protein diet. Pediatr. Res. 2011, 70, 638–641. [Google Scholar] [CrossRef]
- Villamizar-Schiller, I.T.; Pabón, L.A.; Hufnagel, S.B.; Serrano, N.C.; Karl, G.; Jefferies, J.L.; Hopkin, R.J.; Prada, C.E. Neurological and cardiac responses after treatment with miglustat and a ketogenic diet in a patient with Sandhoff disease. Eur. J. Med. Genet. 2015, 58, 180–183. [Google Scholar] [CrossRef]
- Marusic, T.; Tansek, M.Z.; Campa, A.S.; Mezek, A.; Berden, P.; Battelino, T.; Groselj, U. Data highlighting effects of Ketogenic diet on cardiomyopathy and hepatopathy in Glycogen storage disease Type IIIA. Data Brief. 2020, 32, 106205. [Google Scholar] [CrossRef]
- Marusic, T.; Tansek, M.Z.; Campa, A.S.; Mezek, A.; Berden, P.; Battelino, T.; Groselj, U. Normalization of obstructive cardiomyopathy and improvement of hepatopathy on ketogenic diet in patient with glycogen storage disease (GSD) type IIIa. Mol. Genet. Metab. Rep. 2020, 24, 100628. [Google Scholar] [CrossRef]
- Olgac, A.; İnci, A.; Okur, I.; Biberoğlu, G.; Oğuz, D.; Ezgü, F.S.; Kasapkara, S.; Aktaş, E.; Tümer, L. Beneficial Effects of Modified Atkins Diet in Glycogen Storage Disease Type IIIa. Ann. Nutr. Metab. 2020, 76, 233–241. [Google Scholar] [CrossRef]
- Ravaglia, S.; Pichiecchio, A.; Rossi, M.; De Filippi, P.; Minelli, A.; Moglia, A.; Danesino, C. Dietary treatment in adult-onset type II glycogenosis. J. Inherit. Metab. Dis. 2006, 29, 590. [Google Scholar] [CrossRef]
- Sofou, K.; Dahlin, M.; Hallböök, T.; Lindefeldt, M.; Viggedal, G.; Darin, N. Ketogenic diet in pyruvate dehydrogenase complex deficiency: Short- and long-term outcomes. J. Inherit. Metab. Dis. 2017, 40, 237–245. [Google Scholar] [CrossRef]
- Woloszynek, J.C.; Ko8acs, A.; Ohlemiller, K.K.; Roberts, M.S.; Sands, M.S. Metabolic adaptations to interrupted glycosaminoglycan recycling. J. Biol. Chem. 2009, 284, 29684–29691. [Google Scholar] [CrossRef]
- Ng, Y.S.; Turnbull, D.M.; Doug, M. Turnbull. Mitochondrial disease: Genetics and management. J. Neurol. 2016, 263, 179–191. [Google Scholar] [CrossRef]
- Numata-Uematsu, Y.; Uematsu, M.; Yamamoto, T.; Saitsu, H.; Katata, Y.; Oikawa, Y.; Saijyo, N.; Inui, T.; Murayama, K.; Ohtake, A.; et al. Leigh syndrome-like MRI changes in a patient with biallelic HPDL variants treated with ketogenic diet. Mol. Genet. Metab. Rep. 2021, 29, 100800. [Google Scholar] [CrossRef] [PubMed]
- Parikh, S.; Goldstein, A.; Koenig, M.K.; Scaglia, F.; Enns, G.M.; Saneto, R.; Anselm, I.; Cohen, B.H.; Falk, M.J.; Greene, C.; et al. Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet. Med. 2015, 17, 689–701. [Google Scholar] [CrossRef] [PubMed]
- Cooper, G.M. The Cell: A Molecular Approach, 2nd ed.; Sinauer Associates: Sunderland, MA, USA, 2000. [Google Scholar]
- Brambilla, A.; Olivotto, I.; Favilli, S.; Spaziani, G.; Passantino, S.; Procopio, E.; Morrone, A.; Donati, M.A. Impact of cardiovascular involvement on the clinical course of paediatric mitochondrial disorders. Orphanet J. Rare Dis. 2020, 15, 1–12. [Google Scholar] [CrossRef]
- Bray, A.W.; Ballinger, S.W. Mitochondrial DNA mutations and cardiovascular disease. Curr. Opin. Cardiol. 2017, 32, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Camp, K.M.; Krotoski, D.; Parisi, M.A.; Gwinn, K.A.; Cohen, B.H.; Cox, C.S.; Enns, G.M.; Falk, M.J.; Goldstein, A.C.; Gopal-Srivastava, R.; et al. Nutritional interventions in primary mitochondrial disorders: Developing an evidence base. Mol. Genet. Metab. 2016, 119, 187–206. [Google Scholar] [CrossRef]
- Jackson, A.; Forsyth, C.B.; Shaikh, M.; Voigt, R.M.; Engen, P.A.; Ramirez, V.; Keshavarzian, A. Diet in Parkinson’s Disease: Critical Role for the Microbiome. Front. Neurol. 2019, 10, 1245. [Google Scholar] [CrossRef]
- Janssen, M.C.; Koene, S.; Laat, P.; Hemelaar, P.; Pickkers, P.; Spaans, E.; Beukema, R.; Beyrath, J.; Groothuis, J.; Verhaak, C.; et al. The KHENERGY Study: Safety and Efficacy of KH176 in Mitochondrial m.3243A>G Spectrum Disorders. Clin. Pharmacol. Ther. 2019, 105, 101–111. [Google Scholar] [CrossRef]
- Huang, L.; Li, H.; Zhong, J.; Yang, L.; Chen, G.; Wang, D.; Zheng, G.; Han, H.; Han, X.; Long, Y.; et al. Efficacy and Safety of the Ketogenic Diet for Mitochondrial Disease With Epilepsy: A Prospective, Open-labeled, Controlled Study. Front. Neurol. 2022, 13, 880944. [Google Scholar] [CrossRef]
- Mahoney, D.J.; Parise, G.; Tarnopolsky, M.A. Nutritional and exercise-based therapies in the treatment of mitochondrial disease. Curr. Opin. Clin. Nutr. Metab. Care 2002, 5, 619–629. [Google Scholar] [CrossRef]
- Marriage, B.J.; Clandinin, M.T.; Glerum, D.M. Nutritional cofactor treatment in mitochondrial disorders. J. Am. Diet. Assoc. 2003, 103, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Meena, N.K.; Raben, N. Pompe Disease: New Developments in an Old Lysosomal Storage Disorder. Biomolecules 2020, 10, 1339. [Google Scholar] [CrossRef]
- Alraddadi, E.A.; Khojah, A.M.; Alamri, F.F.; Kecheck, H.K.; Altaf, W.F.; Khouqeer, Y. Potential role of creatine as an anticonvulsant agent: Evidence from preclinical studies. Front. Neurosci. 2023, 17, 1201971. [Google Scholar] [CrossRef]
- Arauna, D.; Furrianca, M.; Espinosa-Parrilla, Y.; Fuentes, E.; Alarcón, M.; Palomo, I. Natural Bioactive Compounds As Protectors Of Mitochondrial Dysfunction In Cardiovascular Diseases And Aging. Molecules 2019, 24, 4259. [Google Scholar] [CrossRef] [PubMed]
- Ayer, A.; Macdonald, P.S.; Stocker, R. CoQ₁₀ Function and Role in Heart Failure and Ischemic Heart Disease. Annu. Rev. Nutr. 2015, 35, 175–213. [Google Scholar] [CrossRef] [PubMed]
- Bordugo, A.; Salvetti, E.; Rodella, G.; Piazza, M.; Dianin, A.; Amoruso, A.; Piacentini, G.; Pane, M.; Torriani, S.; Vitulo, N.; et al. Assessing Gut Microbiota in an Infant with Congenital Propionic Acidemia before and after Probiotic Supplementation. Microorganisms 2021, 9, 2599. [Google Scholar] [CrossRef]
- Rahman, S. Mitochondrial disease in children. J. Intern. Med. 2020, 287, 609–633. [Google Scholar] [CrossRef]
- Magner, M.; Kolářová, H.; Honzik, T.; Švandová, I.; Zeman, J.; Zeman, J. Clinical manifestation of mitochondrial diseases. Dev. Period Med. 2015, 19, 441–449. [Google Scholar]
- Lioncino, M.; Monda, E.; Caiazza, M.; Fusco, A.; Cirillo, A.; Dongiglio, F.; Simonelli, V.; Sampaolo, S.; Ruggiero, L.; Scarano, G. Cardiovascular Involvement in mtDNA Disease. Heart Fail. Clin. 2022, 18, 51–60. [Google Scholar] [CrossRef]
- Meyers, D.E.; Basha, H.I.; Koenig, M.K. Mitochondrial cardiomyopathy: Pathophysiology, diagnosis, and management. Tex. Heart Inst. J. 2013, 40, 385–394. [Google Scholar]
- Di Lorenzo, A.; Iannuzzo, G.; Parlato, A.; Cuomo, G.; Testa, C.; Coppola, M.; D’ambrosio, G.; Oliviero, D.A.; Sarullo, S.; Vitale, G.; et al. Clinical Evidence for Q10 Coenzyme Supplementation in Heart Failure: From Energetics to Functional Improvement. J. Clin. Med. 2020, 9, 1266. [Google Scholar] [CrossRef] [PubMed]
- Madden, R.F.; Lalonde-Bester, S.; Parnell, J.A.; Trudeau, M.S.; Martin, J.M.; Khan, A.; Shearer, J. Assessment of Dietary Patterns and Supplement Use in Mitochondrial Disease. Clin. Nutr. ESPEN 2022, 51, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Orsucci, D.; Filosto, M.; Siciliano, G.; Mancuso, M. Electron transfer mediators and other metabolites and cofactors in the treatment of mitochondrial dysfunction. Nutr. Rev. 2009, 67, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Pagliaro, B.; Santolamazza, C.; Simonelli, F.; Rubattu, S. Phytochemical Compounds and Protection from Cardiovascular Diseases: A State of the Art. BioMed Res. Int. 2015, 2015, 918069. [Google Scholar] [CrossRef]
- Hey, T.M.; Nielsen, S.K.; Eriksen, U.; Hansen, F.; Mogensen, J. Leber’s Hereditary Optic Neuropathy and Hypertrophic Cardiomyopathy. CJC Open 2022, 4, 813–815. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.C.; Chiang, K.-L. Clinical Diagnosis and Treatment of Leigh Syndrome Based on SURF1: Genotype and Phenotype. Antioxidants 2021, 10, 1950. [Google Scholar] [CrossRef]
- Wesół-Kucharska, D.; Greczan, M.; Witulska, K.; Piekutowska-Abramczuk, D.; Ciara, E.; Kowalski, P.; Rokicki, D. Improvement of cardiomyopathy after ketogenic diet in a patient with Leigh syndrome caused by MTND5 mutation. Res. Sq. 2021. [Google Scholar] [CrossRef]
- Belal, S.; Goudenège, D.; Bocca, C.; Dumont, F.; De La Barca, J.M.C.; Desquiret-Dumas, V.; Gueguen, N.; Geffroy, G.; Benyahia, R.; Kane, S.; et al. Glutamate-Induced Deregulation of Krebs Cycle in Mitochondrial Encephalopathy Lactic Acidosis Syndrome Stroke-like Episodes (MELAS) Syndrome Is Alleviated by Ketone Body Exposure. Biomedicines 2022, 10, 1665. [Google Scholar] [CrossRef]
- He, F.; Ye, L.; Miao, P.; Zhou, J.; Ding, Y.; Wang, S. Long-term ketogenic diet therapy improves mitochondrial encephalopathy with lactic acidosis and stroke-like episodes (MELAS): A case report. CNS Neurosci. Ther. 2023, 29, 2717–2720. [Google Scholar] [CrossRef]
- Steriade, C.; Andrade, D.M.; Faghfoury, H.; Tarnopolsky, M.A.; Tai, P. Mitochondrial Encephalopathy With Lactic Acidosis and Stroke-like Episodes (MELAS) May Respond to Adjunctive Ketogenic Diet. Pediatr. Neurol. 2014, 50, 498–502. [Google Scholar] [CrossRef]
- Caplliure-Llopis, J.; Peralta-Chamba, T.; Carrera-Juliá, S.; Cuerda-Ballester, M.; Drehmer-Rieger, E.; López-Rodriguez, M.M.; Ortí, J.E.d.l.R. Therapeutic alternative of the ketogenic Mediterranean diet to improve mitochondrial activity in Amyotrophic Lateral Sclerosis (ALS): A Comprehensive Review. Food Sci. Nutr. 2020, 8, 23–35. [Google Scholar] [CrossRef]
- Lim, J.-M.; Letchumanan, V.; Tan, L.T.-H.; Hong, K.-W.; Wong, S.-H.; Ab Mutalib, N.-S.; Lee, L.-H.; Law, J.W.-F. Ketogenic Diet: A Dietary Intervention via Gut Microbiome Modulation for the Treatment of Neurological and Nutritional Disorders (a Narrative Review). Nutrients 2022, 14, 3566. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, R.; Neth, B.J.; Wang, S.; Craft, S.; Yadav, H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer’s disease markers in subjects with mild cognitive impairment. EBioMedicine 2019, 47, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Nicolson, G.L.; Rosenblatt, S.; Ferreira de Mattos, G.; Settineri, R.; Breeding, P.C.; Ellithorpe, R.R.; Ash, M.E. Clinical Uses of Membrane Lipid Replacement Supplements in Restoring Membrane Function and Reducing Fatigue in Chronic Diseases and Cancer. Discoveries 2016, 4, e54. [Google Scholar] [CrossRef] [PubMed]
- Pollicino, F.; Veronese, N.; Dominguez, L.J.; Barbagallo, M. Mediterranean diet and mitochondria: New findings. Exp. Gerontol. 2023, 176, 112165. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.C.; Lee, Y.M.; Lee, Y.M.; Kim, H.D.; Lee, J.S.; Slama, A. Safe and Effective Use of the Ketogenic Diet in Children with Epilepsy and Mitochondrial Respiratory Chain Complex Defects. Epilepsia 2007, 48, 82–88. [Google Scholar] [CrossRef] [PubMed]
- Kyriazis, I.D.; Vassi, E.; Alvanou, M.; Angelakis, C.; Skaperda, Z.; Tekos, F.; Garikipati, V.N.S.; Spandidos, D.A.; Kouretas, D. The impact of diet upon mitochondrial physiology (Review). Int. J. Mol. Med. 2022, 50, 135. [Google Scholar] [CrossRef]
- Qu, C.; Keijer, J.; Adjobo-Hermans, M.J.; van de Wal, M.; Schirris, T.; van Karnebeek, C.; Pan, Y.; Koopman, W.J. The ketogenic diet as a therapeutic intervention strategy in mitochondrial disease. Int. J. Biochem. Cell Biol. 2021, 138, 106050. [Google Scholar] [CrossRef]
- Butnariu, L.I.; Florea, L.; Badescu, M.C.; Țarcă, E.; Costache, I.I.; Gorduza, E.V. Etiologic Puzzle of Coronary Artery Disease: How Important Is Genetic Component? Life 2022, 12, 865. [Google Scholar] [CrossRef]
- Augustin, K.; Khabbush, A.; Williams, S.; Eaton, S.; Orford, M.; Cross, J.H.; Heales, S.J.R.; Walker, M.C.; Williams, R.S.B. Mechanisms of action for the medium-chain triglyceride ketogenic diet in neurological and metabolic disorders. Lancet Neurol. 2018, 17, 84–93. [Google Scholar] [CrossRef]
- Zweers, H.; van Wegberg, A.M.J.; Janssen, M.C.H.; Wortmann, S.B. Ketogenic diet for mitochondrial disease: A systematic review on efficacy and safety. Orphanet J. Rare Dis. 2021, 16, 295. [Google Scholar] [CrossRef] [PubMed]
- Deberles, E.; Maragnes, P.; Penniello-Valette, M.J.; Allouche, S.; Joubert, M. Reversal of Cardiac Hypertrophy With a Ketogenic Diet in a Child With Mitochondrial Disease and Hypertrophic Cardiomyopathy. Can. J. Cardiol. 2020, 36, 1690.e1–1690.e3. [Google Scholar] [CrossRef]
- Carubbi, F.; Barbato, A.; Burlina, A.B.; Francini, F.; Mignani, R.; Pegoraro, E.; Landini, L.; De Danieli, G.; Bruni, S.; Strazzullo, P. Nutrition in adult patients with selected lysosomal storage diseases. Nutr. Metab. Cardiovasc. Dis. 2020, 31, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Luong, T.V.; Abild, C.B.; Bangshaab, M.; Gormsen, L.C.; Søndergaard, E. Ketogenic Diet and Cardiac Substrate Metabolism. Nutrients 2022, 14, 1322. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Chen, H.; Wang, Y.-J.; Qiu, J.-X.; Meng, Q.-Q.; Zou, R.-J.; Li, L.; Huang, J.-G.; Zhao, Z.-K.; Huang, Y.-L.; et al. Ketogenic Diet Suppressed T-Regulatory Cells and Promoted Cardiac Fibrosis via Reducing Mitochondria-Associated Membranes and Inhibiting Mitochondrial Function. Oxid. Med. Cell. Longev. 2021, 2021, 5512322. [Google Scholar] [CrossRef]
- Ruocco, C.; Agnese Segala Segala, A.; Agnese Segala Valerio, A.; Nisoli, E. Essential amino acid formulations to prevent mitochondrial dysfunction and oxidative stress. Curr. Opin. Clin. Nutr. Metab. Care 2020, 24, 88–95. [Google Scholar] [CrossRef]
- Rusek, M.; Pluta, R.; Ułamek-Kozioł, M.; Czuczwar, S.J. Ketogenic Diet in Alzheimer’s Disease. Int. J. Mol. Sci. 2019, 20, 3892. [Google Scholar] [CrossRef]
- Ortiz, A.; Germain, D.P.; Desnick, R.J.; Politei, J.; Mauer, M.; Burlina, A.; Eng, C.; Hopkin, R.J.; Laney, D.; Linhart, A.; et al. Fabry disease revisited: Management and treatment recommendations for adult patients. Mol. Genet. Metab. 2018, 123, 416–427. [Google Scholar] [CrossRef]
- Duceac, L.D.; Marcu, C.; Ichim, D.L.; Ciomaga, I.M.; Tarca, E.; Iordache, A.C.; Ciuhodaru, M.I.; Florescu, L.; Tutunaru, D.; Luca, A.C.; et al. Antibiotic Molecules Involved in Increasing Microbial Resistance. Rev. Chim. 2019, 70, 2622–2626. [Google Scholar] [CrossRef]
- Cingolani, A.; Paduano, D.; Vecchiarelli, V.; Demelas, M.; Corrias, P.T.; Casula, L.; Usai, P. Feasibility of Low Fermentable Oligosaccharide, Disaccharide, Monosaccharide, and Polyol Diet and Its Effects on Quality of Life in an Italian Cohort. Nutrients 2020, 12, 716. [Google Scholar] [CrossRef]
- Vervier, K.; Moss, S.; Kumar, N.; Adoum, A.; Barne, M.; Browne, H.; Kaser, A.; Kiely, C.J.; Neville, B.A.; Powell, N.; et al. Two microbiota subtypes identified in irritable bowel syndrome with distinct responses to the low FODMAP diet. Gut 2022, 71, 1821–1830. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, M.I.; Crozier, M.; Di Carlo, A.; Xhuti, D.; Manta, K.; Roik, L.J.; Bujak, A.L.; Nederveen, J.P.; Tarnopolsky, M.G.; Hettinga, B.; et al. Nutritional co-therapy with 1,3-butanediol and multi-ingredient antioxidants enhances autophagic clearance in Pompe disease. Mol. Genet. Metab. 2022, 137, 228–240. [Google Scholar] [CrossRef] [PubMed]
- Sechi, A.; Zuccarelli, L.; Grassi, B.; Frangiamore, R.; De Amicis, R.; Marzorati, M.; Porcelli, S.; Tullio, A.; Bacco, A.; Bertoli, S.; et al. Exercise training alone or in combination with high-protein diet in patients with late onset Pompe disease: Results of a cross over study. Orphanet J. Rare Dis. 2020, 15, 1–11. [Google Scholar] [CrossRef]
- Scheffers, L.E.; Somers, O.C.; Dulfer, K.; Dieleman, G.C.; Walet, S.; van der Giessen, L.J.; van der Ploeg, A.T.; Hout, J.M.P.v.D.; Berg, L.E.v.D. Physical training and high-protein diet improved muscle strength, parent-reported fatigue, and physical quality of life in children with Pompe disease. J. Inherit. Metab. Dis. 2023, 46, 605–617. [Google Scholar] [CrossRef] [PubMed]
- Viana dos Santos, R.; das Neves Ferreira, T.; Oliveira de Almeida, D.; Brito da Silva Fatal, L.; Queiroz Araujo, E.M. Nutrition management of Niemann Pick disease type C: A case report. Endocr. Regul. 2021, 55, 238–241. [Google Scholar] [CrossRef] [PubMed]
- Gupta, P.; Pastores, G. Pharmacological treatment of pediatric Gaucher disease. Expert Rev. Clin. Pharmacol. 2018, 11, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Bellotti, A.S.; Andreoli, L.; Ronchi, D.; Bresolin, N.; Comi, G.P.; Corti, S. Molecular Approaches for the Treatment of Pompe Disease. Mol. Neurobiol. 2019, 10, 1665. [Google Scholar] [CrossRef]
- Borie-Guichot, M.; Lan Tran, M.; Génisson, Y.; Ballereau, S.; Dehoux, C. Phamacological Chaperone Therapy for Pompe Disease. Molecules 2021, 26, 7223. [Google Scholar] [CrossRef]
- Sitarska, D.; Tylki-Szymańska, A.; Ługowska, A. Treatment trials in Niemann-Pick type C disease. Metab. Brain Dis. 2021, 36, 2215–2221. [Google Scholar] [CrossRef]
- Höller, A.; Albrecht, U.; Sigl, S.B.; Zöggeler, T.; Ramoser, G.; Bernar, B.; Karall, D.; Scholl-Bürgi, S. Successful implementation of classical ketogenic dietary therapy in a patient with Niemann-Pick disease type C. Mol. Genet. Metab. Rep. 2021, 27, 100723. [Google Scholar] [CrossRef]
- Aguilera-Correa, J.-J.; Madrazo-Clemente, P.; Martínez-Cuesta, M.d.C.; Peláez, C.; Ortiz, A.; Sánchez-Niño, M.D.; Esteban, J.; Requena, T. Lyso-Gb3 modulates the gut microbiota and decreases butyrate production. Sci. Rep. 2019, 9, 12010. [Google Scholar] [CrossRef] [PubMed]
- Mehta, A.; Beck, M.; Sunder-Plassmann, G. (Eds.) Fabry Disease: Perspectives from 5 Years of FOS; Oxford PharmaGenesis: Oxford, UK, 2006. [Google Scholar]
- Luca, F.A.; Ioan, C.A.; Sasu, C.; Luca, A.C. The Impact of Public Health Care Services on the Patients’ Perception as Regards the Health Institutions Brand on the Background of the Health Reform in Romania. Rev. Cercet. Interv. Soc. 2015, 49, 80–97. [Google Scholar]
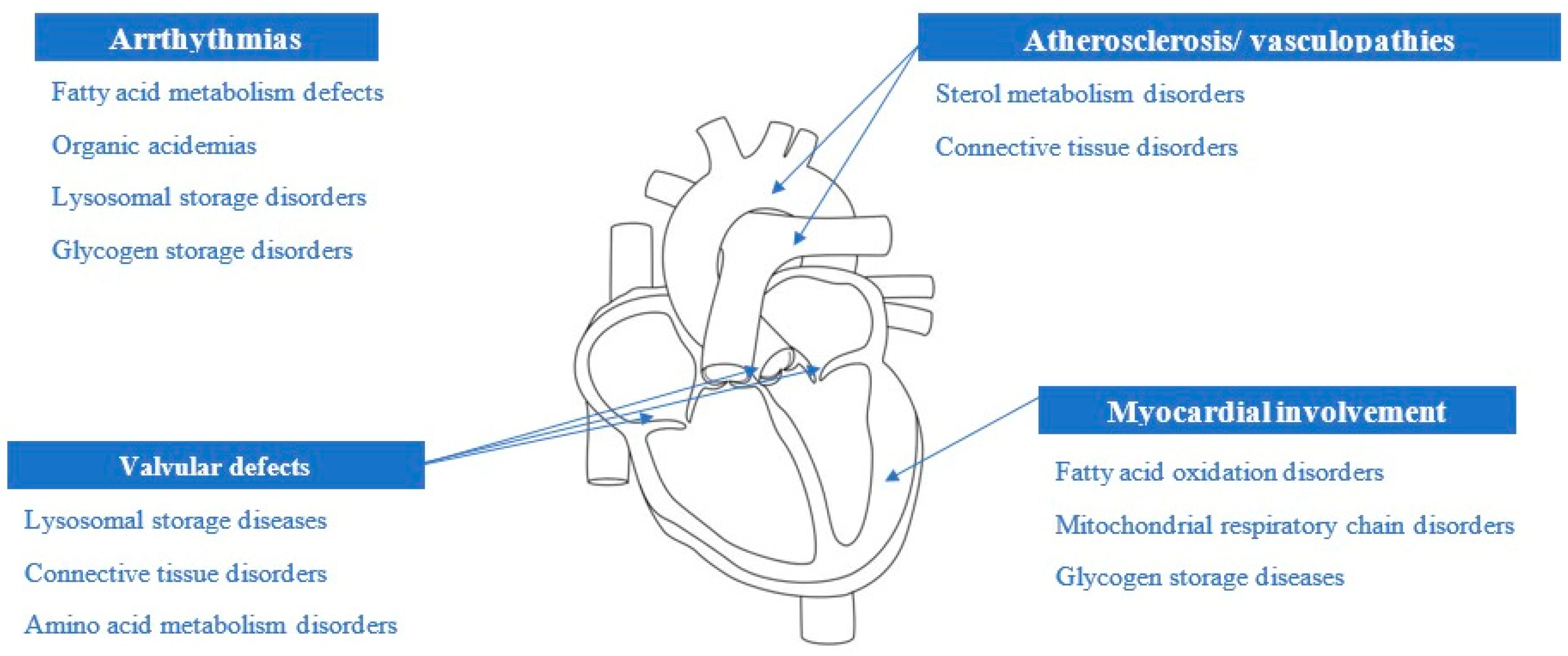

{kind=link}
| Disease Category | Cardiac Manifestations | Red Flags | Dietary Intervention [11,12,13] |
|---|---|---|---|
| Glycogen storage disorders | Hypertrophic cardiomyopathy Conduction abnormalities | Hepatomegaly Hypoglycemia | High-protein diet Modified Atkins diet [7] |
| Fatty acid oxidation disorders | Hypertrophic/dilated cardiomyopathy | Hypoketotic hypoglycemia; Episodic rhabdomyolysis | Avoidance of fasting Carnitine supplementation Fat-restricted diet and DHA supplementation for long-chain FAMD |
| Primary mitochondrial diseases | Hypertrophic/dilated cardiomyopathy Arrhythmias Conduction abnormalities | Skeletal muscle symptoms Encephalopathy Seizures Episodic vomiting/ketoacidosis | Dietary supplements Ketogenic diet |
| Organic acidemias [9] | Cardiomyopathy Arrhythmias | Metabolic decompensation Hearing impairment Renal failure | Avoid catabolism Carnitine supplementation |
| Lysosomal storage disorders | Valvular abnormalities Conduction abnormalities Large vessel anomalies | Skeletal abnormalities Dysmorphic features Hepatosplenomegaly Corneal clouding Hearing impairment | Vitamin supplementation Ketogenic diet High-protein diet |
| Disease Name [28] | Red Flags [29] | Cardiovascular Manifestations [30,31] | Dietary Intervention [21,22,32,33,34,35] |
|---|---|---|---|
| Chronic progressive external ophthalmoplegia | Ptosis Ophthalmoparesis Proximal myopathy | Prolonged intraventricular conduction time Bundle branch blocks Complete AV block | Alpha-lipoic acid Carnitine Coenzyme Q10 Creatine |
| Kearns–Sayre syndrome (KSS) | Ataxia Hearing loss Myopathy Pigmentary retinopathy Elevated CSF proteins | Atrioventricular block which requires pacemaker implantation | Carnitine Coenzyme Q10 Creatine Folinic acid |
| Leber’s hereditary optic neuropathy (LHON) [36] | Progressive bilateral visual failure | Hypertrophic cardiomyopathy Atrioventricular conduction abnormalities (long QT, WPW) Left ventricle hypertrabeculation | Coenzyme Q10 |
| Leigh syndrome [37] | Encephalopathy Seizures Hypotonia/ataxia Oculomotor dysfunction Respiratory dysfunction | Hypertrophic cardiomyopathy Conduction abnormalities | Carnitine Coenzyme Q10 Ketogenic diet [12,38] |
| Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) [39,40,41] | Stroke-like symptoms Lactic acidemia Myopathy | Hypertrophic cardiomyopathy Dilated cardiomyopathy Conduction abnormalities | Arginine Carnitine Citrulline Coenzyme Q10 Creatine Niacin |
| Mitochondrial neurogastrointestinal encephalopathy (MNGIE) | Gastrointestinal dysmotility Polyneuropathy Leukodystrophy | Conduction abnormalities Sudden cardiac death | Coenzyme Q10 |
| Myoclonic epilepsy with ragged-red fibers (MERRF) | Ataxia Myoclonus Generalized seizures | Dilated cardiomyopathy Hypertrophic cardiomyopathy Conduction abnormalities | Coenzyme Q10 Creatine |
| Disease Name | Red Flags | Cardiac Manifestations [5] | Dietary Intervention [6] |
|---|---|---|---|
| Gaucher | Hepatosplenomegaly Peripheral blood cytopenia Bone lesions Different degrees of neurologic impairment | Pulmonary hypertension Aortic and mitral valve calcifications Myocardial infiltrative damage | Vitamin D Calcium Avoid fruits and vegetables that affect cytochrome P450 |
| Fabry | Renal failure or hematuria/proteinuria Angiokeratomas Acroparesthesias | Left ventricular hypertrophy Arrhythmias | Low fermentable oligosaccharide, disaccharide, monosaccharide and polyol (FODMAP) diet [62] Low-protein + keto analogs in patients with CKD |
| Pompe [23,63,64] | Muscle weakness Motor delay Feeding difficulties Recurrent respiratory infections | Hypertrophic cardiomyopathy Conduction abnormalities | Low-carbohydrate, high-protein diet [23] L-Alanine supplementation Aerobic exercise [65] |
| Niemann–Pick | Hepatosplenomegaly Ataxia Hypotonia Progressive severe neurologic impairment Difficulty with swallowing Severe liver disease | Cardiomegaly Endocardial fibroelastosis Valvular stenosis | Low-carbohydrate diet [66] Disaccharides restriction Ketogenic diet |
| MPS | Coarse features Skeletal abnormalities Neurologic impairment | Cardiac valve thickening Valvular dysfunction | Vitamins B1, B2, B3 Vitamin C Iron No specific diet indicated |
| Diet Type | Cardiac Effect | Global Effects |
|---|---|---|
| Ketogenic | Decreases ventricular hypertrophy Improves ejection fraction | Seizure control Improves muscular function Reverses movement disorders Improves verbal response, memory, social interaction |
| FODMAP [62] | Potentially decreases cardiovascular disease risk (nonspecific impact on IMDs) | Ameliorates bowel function Decreases frequency of diarrhea, constipation, nausea |
| Low carbohydrate/high protein [3,64] | Inhibits cardiac remodeling caused by pressure overload (nonspecific impact on IMDs) | Improves muscle strength Increases exercise capacity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luca, A.C.; Pădureț, I.-A.; Țarcă, V.; David, S.G.; Mîndru, D.E.; Roșu, S.T.; Roșu, E.V.; Adumitrăchioaiei, H.; Bernic, J.; Cojocaru, E.; et al. Nutritional Approach in Selected Inherited Metabolic Cardiac Disorders—A Concise Summary of Available Scientific Evidence. Nutrients 2023, 15, 4795. https://doi.org/10.3390/nu15224795
Luca AC, Pădureț I-A, Țarcă V, David SG, Mîndru DE, Roșu ST, Roșu EV, Adumitrăchioaiei H, Bernic J, Cojocaru E, et al. Nutritional Approach in Selected Inherited Metabolic Cardiac Disorders—A Concise Summary of Available Scientific Evidence. Nutrients. 2023; 15(22):4795. https://doi.org/10.3390/nu15224795
Chicago/Turabian StyleLuca, Alina Costina, Ioana-Alexandra Pădureț, Viorel Țarcă, Simona Georgiana David, Dana Elena Mîndru, Solange Tamara Roșu, Eduard Vasile Roșu, Heidrun Adumitrăchioaiei, Jana Bernic, Elena Cojocaru, and et al. 2023. "Nutritional Approach in Selected Inherited Metabolic Cardiac Disorders—A Concise Summary of Available Scientific Evidence" Nutrients 15, no. 22: 4795. https://doi.org/10.3390/nu15224795
APA StyleLuca, A. C., Pădureț, I.-A., Țarcă, V., David, S. G., Mîndru, D. E., Roșu, S. T., Roșu, E. V., Adumitrăchioaiei, H., Bernic, J., Cojocaru, E., & Țarcă, E. (2023). Nutritional Approach in Selected Inherited Metabolic Cardiac Disorders—A Concise Summary of Available Scientific Evidence. Nutrients, 15(22), 4795. https://doi.org/10.3390/nu15224795