
Nuclear Receptor PPARα as a Therapeutic Target in Diseases Associated with Lipid Metabolism Disorders
 ,
, {kind=link}
{kind=link}
Abstract
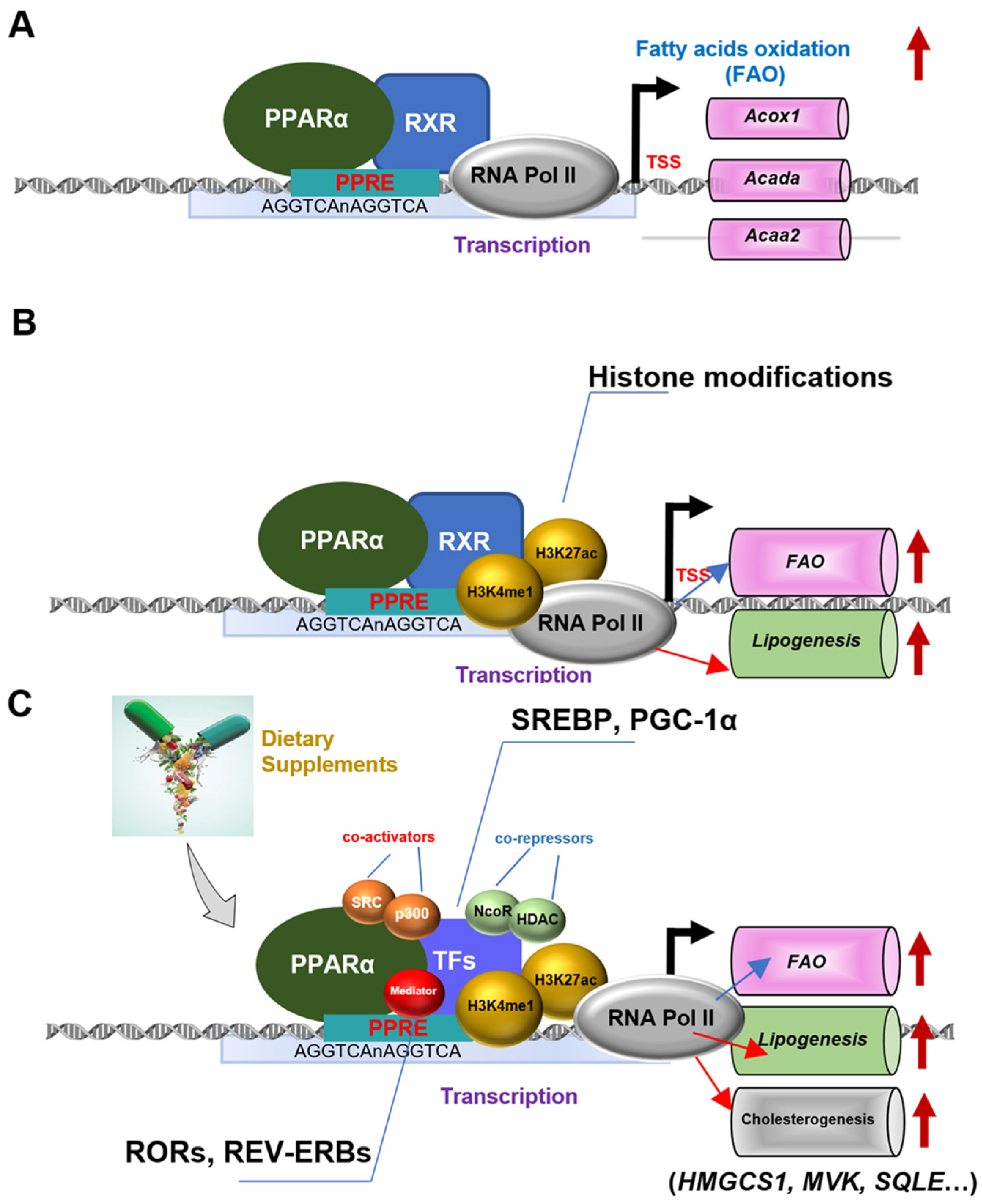
:1. Introduction
2. PPARα Is a Critical Player in Nonalcoholic and Alcoholic Liver Diseases
2.1. Nonalcoholic Fatty Liver Disease
2.2. Alcohol-Related Liver Disease
3. Therapeutic Approaches to Diabetes-Related Diseases via PPARα-Dependent Pathways
3.1. Heart Disease
3.2. Diabetic Retinopathy
3.3. Diabetic Keratopathy
4. PPARα Drives Cell Senescence in Alzheimer’s and Chronic Kidney Disease
4.1. Alzheimer’s Disease
4.2. Chronic Kidney Disease
5. PPARα Is a Potential Therapeutic Target for Cancer Treatment
5.1. Colorectal Cancer
5.2. Kidney Cancer
5.3. Breast Cancer
5.4. Liver Cancer
6. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dewidar, B.; Kahl, S.; Pafili, K.; Roden, M. Metabolic liver disease in diabetes—From mechanisms to clinical trials. Metabolism 2020, 111, 154299. [Google Scholar] [CrossRef] [PubMed]
- Amorim, J.A.; Coppotelli, G.; Rolo, A.P.; Palmeira, C.M.; Ross, J.M.; Sinclair, D.A. Mitochondrial and metabolic dysfunction in ageing and age-related diseases. Nat. Rev. Endocrinol. 2022, 18, 243–258. [Google Scholar] [CrossRef]
- Leone, R.D.; Powell, J.D. Metabolism of immune cells in cancer. Nat. Rev. Cancer 2020, 20, 516–531. [Google Scholar] [CrossRef]
- Faubert, B.; Solmonson, A.; DeBerardinis, R.J. Metabolic reprogramming and cancer progression. Science 2020, 368, eaaw5473. [Google Scholar] [CrossRef]
- Martinez-Reyes, I.; Chandel, N.S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 2021, 21, 669–680. [Google Scholar] [CrossRef] [PubMed]
- Gyamfi, J.; Kim, J.; Choi, J. Cancer as a Metabolic Disorder. Int. J. Mol. Sci. 2022, 23, 1155. [Google Scholar] [CrossRef]
- Montaigne, D.; Butruille, L.; Staels, B. PPAR control of metabolism and cardiovascular functions. Nat. Rev. Cardiol. 2021, 18, 809–823. [Google Scholar] [CrossRef]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as metabolic regulators in the liver: Lessons from liver-specific PPAR-null mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef]
- Suh, J.H.; Kim, K.H.; Conner, M.E.; Moore, D.D.; Preidis, G.A. Hepatic PPARalpha Is Destabilized by SIRT1 Deacetylase in Undernourished Male Mice. Front. Nutr. 2022, 9, 831879. [Google Scholar] [CrossRef]
- Kimura, A.; Kamimura, K.; Ohkoshi-Yamada, M.; Shinagawa-Kobayashi, Y.; Goto, R.; Owaki, T.; Oda, C.; Shibata, O.; Morita, S.; Sakai, N.; et al. Effects of a novel selective PPAR alpha modulator, statin, sodium-glucose cotransporter 2 inhibitor, and combinatorial therapy on the liver and vasculature of medaka nonalcoholic steatohepatitis model. Biochem. Biophys. Res. Commun. 2022, 596, 76–82. [Google Scholar] [CrossRef]
- Yao, H.Y.; Wang, Y.Q.; Zhang, X.; Li, P.; Shang, L.; Chen, X.C.; Zeng, J. Targeting peroxisomal fatty acid oxidation improves hepatic steatosis and insulin resistance in obese mice. J. Biol. Chem. 2023, 299, 102845. [Google Scholar] [CrossRef]
- Yang, P.; Qin, H.; Li, Y.; Xiao, A.; Zheng, E.; Zeng, H.; Su, C.; Luo, X.; Lu, Q.; Liao, M.; et al. CD36-mediated metabolic crosstalk between tumor cells and macrophages affects liver metastasis. Nat. Commun. 2022, 13, 5782. [Google Scholar] [CrossRef]
- Vega, R.B.; Kelly, D.P. Cardiac nuclear receptors: Architects of mitochondrial structure and function. J. Clin. Investig. 2017, 127, 1155–1164. [Google Scholar] [CrossRef] [PubMed]
- Cai, D.; Li, Y.; Zhang, K.; Zhou, B.; Guo, F.; Holm, L.; Liu, H.Y. Co-option of PPARalpha in the regulation of lipogenesis and fatty acid oxidation in CLA-induced hepatic steatosis. J. Cell Physiol. 2021, 236, 4387–4402. [Google Scholar] [CrossRef]
- Cai, D.; Liu, H.; Zhao, R. Nuclear Receptors in Hepatic Glucose and Lipid Metabolism During Neonatal and Adult Life. Curr. Protein Pept. Sci. 2017, 18, 548–561. [Google Scholar] [CrossRef]
- Mizukawa, Y.; Amagase, Y.; Urushidani, T. Extraction of peroxisome proliferator-activated receptor alpha agonist-induced lipid metabolism-related and unrelated genes in rat liver and analysis of their genomic location. J. Toxicol. Sci. 2020, 45, 449–473. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.; Tacke, F.; Arrese, M.; Chander Sharma, B.; Mostafa, I.; Bugianesi, E.; Wai-Sun Wong, V.; Yilmaz, Y.; George, J.; Fan, J.; et al. Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 2019, 69, 2672–2682. [Google Scholar] [CrossRef] [PubMed]
- Samuel, V.T.; Shulman, G.I. Nonalcoholic Fatty Liver Disease as a Nexus of Metabolic and Hepatic Diseases. Cell Metab. 2018, 27, 22–41. [Google Scholar] [CrossRef]
- Avila, M.A.; Dufour, J.-F.; Gerbes, A.L.; Zoulim, F.; Bataller, R.; Burra, P.; Cortez-Pinto, H.; Gao, B.; Gilmore, I.; Mathurin, P. Recent advances in alcohol-related liver disease (ALD): Summary of a Gut round table meeting. Gut 2020, 69, 764–780. [Google Scholar] [CrossRef]
- Rotman, Y.; Sanyal, A.J. Current and upcoming pharmacotherapy for non-alcoholic fatty liver disease. Gut 2017, 66, 180–190. [Google Scholar] [CrossRef]
- Yue, R.; Chen, G.-Y.; Xie, G.; Hao, L.; Guo, W.; Sun, X.; Jia, W.; Zhang, Q.; Zhou, Z.; Zhong, W. Activation of PPARα-catalase pathway reverses alcoholic liver injury via upregulating NAD synthesis and accelerating alcohol clearance. Free Radic. Biol. Med. 2021, 174, 249–263. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Hu, P.; Li, Y.; Sun, M.A.; Qu, H.; Zong, Q.; Gu, H.; Chen, X.; Bao, W.; Cai, D. Targeted inhibition of PPARalpha ameliorates CLA-induced hypercholesterolemia via hepatic cholesterol biosynthesis reprogramming. Liver Int. 2022, 42, 1449–1466. [Google Scholar] [CrossRef]
- Liu, H.Y.; Gu, H.; Li, Y.; Hu, P.; Yang, Y.; Li, K.; Li, H.; Zhang, K.; Zhou, B.; Wu, H.; et al. Dietary Conjugated Linoleic Acid Modulates the Hepatic Circadian Clock Program via PPARalpha/REV-ERBalpha-Mediated Chromatin Modification in Mice. Front. Nutr. 2021, 8, 711398. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Targher, G.; Bernardi, M.; Bonino, F.; Bugianesi, E.; Casini, A.; Gastaldelli, A.; Marchesini, G.; Marra, F.; et al. AISF position paper on nonalcoholic fatty liver disease (NAFLD): Updates and future directions. Dig. Liver Dis. 2017, 49, 471–483. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Luo, Y.; Yan, N.; Hamada, K.; Zhao, N.; Xia, Y.; Wang, P.; Zhao, C.; Qi, D.; Yang, S.; et al. Intestinal peroxisome proliferator-activated receptor alpha-fatty acid-binding protein 1 axis modulates nonalcoholic steatohepatitis. Hepatology 2023, 77, 239–255. [Google Scholar] [CrossRef]
- Régnier, M.; Polizzi, A.; Smati, S.; Lukowicz, C.; Fougerat, A.; Lippi, Y.; Fouché, E.; Lasserre, F.; Naylies, C.; Bétoulières, C. Hepatocyte-specific deletion of Pparα promotes NAFLD in the context of obesity. Sci. Rep. 2020, 10, 6489. [Google Scholar]
- Zhang, J.; Feng, Q. Pharmacological Effects and Molecular Protective Mechanisms of Astragalus Polysaccharides on Nonalcoholic Fatty Liver Disease. Front. Pharmacol. 2022, 13, 854674. [Google Scholar] [CrossRef]
- Liang, N.; Damdimopoulos, A.; Goñi, S.; Huang, Z.; Vedin, L.-L.; Jakobsson, T.; Giudici, M.; Ahmed, O.; Pedrelli, M.; Barilla, S. Hepatocyte-specific loss of GPS2 in mice reduces non-alcoholic steatohepatitis via activation of PPARα. Nat. Commun. 2019, 10, 1684. [Google Scholar]
- Karagoz, G.E.; Aragon, T.; Acosta-Alvear, D. Recent advances in signal integration mechanisms in the unfolded protein response. F1000Research 2019, 8, F1000 Faculty Rev-1840. [Google Scholar] [CrossRef]
- Zhang, N.; Wang, Y.; Zhang, J.; Liu, B.; Deng, X.; Xin, S.; Xu, K. N-glycosylation of CREBH improves lipid metabolism and attenuates lipotoxicity in NAFLD by modulating PPARalpha and SCD-1. FASEB J. 2020, 34, 15338–15363. [Google Scholar] [CrossRef]
- Musso, G.; Cassader, M.; Gambino, R. Non-alcoholic steatohepatitis: Emerging molecular targets and therapeutic strategies. Nat. Rev. Drug Discov. 2016, 15, 249–274. [Google Scholar] [CrossRef]
- Lan, T.; Hu, Y.; Hu, F.; Li, H.; Chen, Y.; Zhang, J.; Yu, Y.; Jiang, S.; Weng, Q.; Tian, S.; et al. Hepatocyte glutathione S-transferase mu 2 prevents non-alcoholic steatohepatitis by suppressing ASK1 signaling. J. Hepatol. 2022, 76, 407–419. [Google Scholar] [CrossRef]
- Moore, D.D. Nuclear receptors reverse McGarry’s vicious cycle to insulin resistance. Cell Metab. 2012, 15, 615–622. [Google Scholar] [CrossRef] [PubMed]
- Wan, C.W.; Wong, C.N.; Pin, W.K.; Wong, M.H.; Kwok, C.Y.; Chan, R.Y.; Yu, P.H.; Chan, S.W. Chlorogenic acid exhibits cholesterol lowering and fatty liver attenuating properties by up-regulating the gene expression of PPAR-alpha in hypercholesterolemic rats induced with a high-cholesterol diet. Phytother. Res. 2013, 27, 545–551. [Google Scholar] [CrossRef] [PubMed]
- Tahri-Joutey, M.; Andreoletti, P.; Surapureddi, S.; Nasser, B.; Cherkaoui-Malki, M.; Latruffe, N. Mechanisms Mediating the Regulation of Peroxisomal Fatty Acid Beta-Oxidation by PPARalpha. Int. J. Mol. Sci. 2021, 22, 8969. [Google Scholar] [CrossRef] [PubMed]
- Rowe, I.A. Lessons from Epidemiology: The Burden of Liver Disease. Dig. Dis. 2017, 35, 304–309. [Google Scholar] [CrossRef]
- Du, D.Y.; Liu, C.; Qin, M.Y.; Zhang, X.; Xi, T.; Yuan, S.T.; Hao, H.P.; Xiong, J. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharm. Sin. B 2022, 12, 558–580. [Google Scholar] [CrossRef]
- Gao, B.; Bataller, R. Alcoholic liver disease: Pathogenesis and new therapeutic targets. Gastroenterology 2011, 141, 1572–1585. [Google Scholar] [CrossRef]
- Jiang, Y.; Zhang, T.; Kusumanchi, P.; Han, S.; Yang, Z.; Liangpunsakul, S. Alcohol Metabolizing Enzymes, Microsomal Ethanol Oxidizing System, Cytochrome P450 2E1, Catalase, and Aldehyde Dehydrogenase in Alcohol-Associated Liver Disease. Biomedicines 2020, 8, 50. [Google Scholar] [CrossRef]
- Shin, M.H.; Lee, S.R.; Kim, M.K.; Shin, C.Y.; Lee, D.H.; Chung, J.H. Activation of Peroxisome Proliferator-Activated Receptor Alpha Improves Aged and UV-Irradiated Skin by Catalase Induction. PLoS ONE 2016, 11, e0162628. [Google Scholar] [CrossRef] [PubMed]
- Koo, S.H.; Satoh, H.; Herzig, S.; Lee, C.H.; Hedrick, S.; Kulkarni, R.; Evans, R.M.; Olefsky, J.; Montminy, M. PGC-1 promotes insulin resistance in liver through PPAR-alpha-dependent induction of TRB-3. Nat. Med. 2004, 10, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Zimmet, P.; Alberti, K.; Shaw, J. Global and societal implications of the diabetes epidemic. Nature 2001, 414, 782–787. [Google Scholar]
- Cole, J.B.; Florez, J.C. Genetics of diabetes mellitus and diabetes complications. Nat. Rev. Nephrol. 2020, 16, 377–390. [Google Scholar] [CrossRef]
- Feng, X.; Gao, X.; Wang, S.; Huang, M.; Sun, Z.; Dong, H.; Yu, H.; Wang, G. PPAR-alpha Agonist Fenofibrate Prevented Diabetic Nephropathy by Inhibiting M1 Macrophages via Improving Endothelial Cell Function in db/db Mice. Front. Med. Lausanne 2021, 8, 652558. [Google Scholar] [CrossRef] [PubMed]
- Wagner, N.; Wagner, K.D. The Role of PPARs in Disease. Cells 2020, 9, 2367. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Juarez, J.; Suarez, J.A.; Dillmann, W.H.; Suarez, J. Mitochondrial calcium handling and heart disease in diabetes mellitus. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2021, 1867, 165984. [Google Scholar]
- Tang, W.; Zhang, B.; Wang, H.; Li, M.; Wang, H.; Liu, F.; Zhu, D.; Bi, Y. Improved skeletal muscle energy metabolism relates to the recovery of beta cell function by intensive insulin therapy in drug naive type 2 diabetes. Diabetes Metab. Res. Rev. 2019, 35, e3177. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Liu, R.; Huang, Y.; Yang, Z.; Xian, J.; Huang, J.; Qiu, Z.; Lin, X.; Zhang, M.; Chen, H. Reactivation of PPARα alleviates myocardial lipid accumulation and cardiac dysfunction by improving fatty acid β-oxidation in Dsg2-deficient arrhythmogenic cardiomyopathy. Acta Pharm. Sin. B 2023, 13, 192–203. [Google Scholar]
- Makrecka-Kuka, M.; Liepinsh, E.; Murray, A.J.; Lemieux, H.; Dambrova, M.; Tepp, K.; Puurand, M.; Käämbre, T.; Han, W.H.; de Goede, P. Altered mitochondrial metabolism in the insulin-resistant heart. Acta Physiol. 2020, 228, e13430. [Google Scholar]
- Lopaschuk, G.D.; Karwi, Q.G.; Tian, R.; Wende, A.R.; Abel, E.D. Cardiac Energy Metabolism in Heart Failure. Circ. Res. 2021, 128, 1487–1513. [Google Scholar] [CrossRef] [PubMed]
- Ting, D.S.; Cheung, G.C.; Wong, T.Y. Diabetic retinopathy: Global prevalence, major risk factors, screening practices and public health challenges: A review. Clin. Exp. Ophthalmol. 2016, 44, 260–277. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Wang, L.; He, J.; Bi, Y.; Li, M.; Wang, T.; Wang, L.; Jiang, Y.; Dai, M.; Lu, J.; et al. Prevalence and control of diabetes in Chinese adults. JAMA 2013, 310, 948–959. [Google Scholar] [CrossRef]
- Hammes, H.P. Pericytes and the pathogenesis of diabetic retinopathy. Horm. Metab. Res. 2005, 37 (Suppl. S1), 39–43. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Kim, D.; Hernandez, C.; Simo, R.; Roy, S. Beneficial effects of fenofibric acid on overexpression of extracellular matrix components, COX-2, and impairment of endothelial permeability associated with diabetic retinopathy. Exp. Eye Res. 2015, 140, 124–129. [Google Scholar] [CrossRef]
- Deng, G.; Moran, E.P.; Cheng, R.; Matlock, G.; Zhou, K.; Moran, D.; Chen, D.; Yu, Q.; Ma, J.X. Therapeutic Effects of a Novel Agonist of Peroxisome Proliferator-Activated Receptor Alpha for the Treatment of Diabetic Retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5030–5042. [Google Scholar] [CrossRef]
- Cai, X.; Biswas, I.; Panicker, S.R.; Giri, H.; Rezaie, A.R. Activated protein C inhibits lipopolysaccharide-mediated acetylation and secretion of high-mobility group box 1 in endothelial cells. J. Thromb. Haemost. 2019, 17, 803–817. [Google Scholar] [CrossRef]
- Shiono, A.; Sasaki, H.; Sekine, R.; Abe, Y.; Matsumura, Y.; Inagaki, T.; Tanaka, T.; Kodama, T.; Aburatani, H.; Sakai, J.; et al. PPARalpha activation directly upregulates thrombomodulin in the diabetic retina. Sci. Rep. 2020, 10, 10837. [Google Scholar] [CrossRef]
- Qiu, F.; Matlock, G.; Chen, Q.; Zhou, K.; Du, Y.; Wang, X.; Ma, J.-X. Therapeutic effects of PPARα agonist on ocular neovascularization in models recapitulating neovascular age-related macular degeneration. Investig. Ophthalmol. Vis. Sci. 2017, 58, 5065–5075. [Google Scholar] [CrossRef]
- Ma, X.; Wu, W.; Liang, W.; Takahashi, Y.; Cai, J.; Ma, J.X. Modulation of cGAS-STING signaling by PPARalpha in a mouse model of ischemia-induced retinopathy. Proc. Natl. Acad. Sci. USA 2022, 119, e2208934119. [Google Scholar] [CrossRef]
- He, Y.; Yang, W.; Gan, L.; Liu, S.; Ni, Q.; Bi, Y.; Han, T.; Liu, Q.; Chen, H.; Hu, Y.; et al. Silencing HIF-1alpha aggravates non-alcoholic fatty liver disease in vitro through inhibiting PPAR-alpha/ANGPTL4 singling pathway. Gastroenterol. Hepatol. 2021, 44, 355–365. [Google Scholar] [CrossRef]
- Han, S.B.; Yang, H.K.; Hyon, J.Y. Influence of diabetes mellitus on anterior segment of the eye. Clin. Interv. Aging 2019, 14, 53–63. [Google Scholar] [CrossRef]
- Yu, F.X.; Lee, P.S.Y.; Yang, L.; Gao, N.; Zhang, Y.; Ljubimov, A.V.; Yang, E.; Zhou, Q.; Xie, L. The impact of sensory neuropathy and inflammation on epithelial wound healing in diabetic corneas. Prog. Retin. Eye Res. 2022, 89, 101039. [Google Scholar] [CrossRef]
- Matlock, H.G.; Qiu, F.; Malechka, V.; Zhou, K.; Cheng, R.; Benyajati, S.; Whelchel, A.; Karamichos, D.; Ma, J.X. Pathogenic Role of PPARalpha Downregulation in Corneal Nerve Degeneration and Impaired Corneal Sensitivity in Diabetes. Diabetes 2020, 69, 1279–1291. [Google Scholar] [CrossRef] [PubMed]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Song, W.; Li, J.; Jing, Y.; Liang, C.; Zhang, L.; Zhang, X.; Zhang, W.; Liu, B.; An, Y.; et al. The landscape of aging. Sci. China Life Sci. 2022, 65, 2354–2454. [Google Scholar] [CrossRef]
- Kim, M.J.; Kim, D.H.; Bang, E.; Noh, S.G.; Chun, P.; Yokozawa, T.; Moon, H.R.; Chung, H.Y. PPARalpha Agonist, MHY3200, Alleviates Renal Inflammation during Aging via Regulating ROS/Akt/FoxO1 Signaling. Molecules 2021, 26, 3197. [Google Scholar] [CrossRef]
- Soria Lopez, J.A.; Gonzalez, H.M.; Leger, G.C. Alzheimer’s disease. Handb. Clin. Neurol. 2019, 167, 231–255. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, A.; De Strooper, B.; Arancibia-Cárcamo, I.L. Cellular senescence at the crossroads of inflammation and Alzheimer’s disease. Trends Neurosci. 2021, 44, 714–727. [Google Scholar] [CrossRef]
- Picard, C.; Julien, C.; Frappier, J.; Miron, J.; Theroux, L.; Dea, D.; Breitner, J.C.; Poirier, J.; United Kingdom Brain Expression Consortium; Alzheimer’s Disease Neuroimaging Initiative. Alterations in cholesterol metabolism-related genes in sporadic Alzheimer’s disease. Neurobiol. Aging 2018, 66, 180.e1–180.e9. [Google Scholar] [CrossRef]
- Wojtowicz, S.; Strosznajder, A.K.; Jezyna, M.; Strosznajder, J.B. The Novel Role of PPAR Alpha in the Brain: Promising Target in Therapy of Alzheimer’s Disease and Other Neurodegenerative Disorders. Neurochem. Res. 2020, 45, 972–988. [Google Scholar] [CrossRef] [PubMed]
- D’Agostino, G.; Cristiano, C.; Lyons, D.J.; Citraro, R.; Russo, E.; Avagliano, C.; Russo, R.; Raso, G.M.; Meli, R.; De Sarro, G.; et al. Peroxisome proliferator-activated receptor alpha plays a crucial role in behavioral repetition and cognitive flexibility in mice. Mol. Metab. 2015, 4, 528–536. [Google Scholar] [CrossRef] [PubMed]
- Pierrot, N.; Ris, L.; Stancu, I.C.; Doshina, A.; Ribeiro, F.; Tyteca, D.; Bauge, E.; Lalloyer, F.; Malong, L.; Schakman, O.; et al. Sex-regulated gene dosage effect of PPARalpha on synaptic plasticity. Life Sci. Alliance 2019, 2, e201800262. [Google Scholar] [CrossRef]
- Saez-Orellana, F.; Leroy, T.; Ribeiro, F.; Kreis, A.; Leroy, K.; Lalloyer, F.; Bauge, E.; Staels, B.; Duyckaerts, C.; Brion, J.P.; et al. Regulation of PPARalpha by APP in Alzheimer disease affects the pharmacological modulation of synaptic activity. JCI Insight 2021, 6, e150099. [Google Scholar] [CrossRef] [PubMed]
- Luo, R.; Su, L.Y.; Li, G.; Yang, J.; Liu, Q.; Yang, L.X.; Zhang, D.F.; Zhou, H.; Xu, M.; Fan, Y.; et al. Activation of PPARA-mediated autophagy reduces Alzheimer disease-like pathology and cognitive decline in a murine model. Autophagy 2020, 16, 52–69. [Google Scholar] [CrossRef] [PubMed]
- Sobamowo, H.; Prabhakar, S.S. The Kidney in Aging: Physiological Changes and Pathological Implications. Prog. Mol. Biol. Transl. Sci. 2017, 146, 303–340. [Google Scholar] [CrossRef]
- Stevens, L.A.; Viswanathan, G.; Weiner, D.E. Chronic kidney disease and end-stage renal disease in the elderly population: Current prevalence, future projections, and clinical significance. Adv. Chronic Kidney Dis. 2010, 17, 293–301. [Google Scholar] [CrossRef]
- Xie, J.; Ye, Z.; Li, L.; Xia, Y.; Yuan, R.; Ruan, Y.; Zhou, X. Ferrostatin-1 alleviates oxalate-induced renal tubular epithelial cell injury, fibrosis and calcium oxalate stone formation by inhibiting ferroptosis. Mol. Med. Rep. 2022, 26, 256. [Google Scholar] [CrossRef]
- Simon, N.; Hertig, A. Alteration of Fatty Acid Oxidation in Tubular Epithelial Cells: From Acute Kidney Injury to Renal Fibrogenesis. Front. Med. Lausanne 2015, 2, 52. [Google Scholar] [CrossRef]
- Kang, H.M.; Ahn, S.H.; Choi, P.; Ko, Y.A.; Han, S.H.; Chinga, F.; Park, A.S.; Tao, J.; Sharma, K.; Pullman, J.; et al. Defective fatty acid oxidation in renal tubular epithelial cells has a key role in kidney fibrosis development. Nat. Med. 2015, 21, 37–46. [Google Scholar] [CrossRef]
- Chung, K.W.; Ha, S.; Kim, S.M.; Kim, D.H.; An, H.J.; Lee, E.K.; Moon, H.R.; Chung, H.Y. PPARα/β activation alleviates age-associated renal fibrosis in Sprague Dawley rats. J. Gerontol. Ser. A 2020, 75, 452–458. [Google Scholar] [CrossRef]
- Chung, K.W.; Lee, E.K.; Lee, M.K.; Oh, G.T.; Yu, B.P.; Chung, H.Y. Impairment of PPARalpha and the Fatty Acid Oxidation Pathway Aggravates Renal Fibrosis during Aging. J. Am. Soc. Nephrol. 2018, 29, 1223–1237. [Google Scholar] [CrossRef]
- Snaebjornsson, M.T.; Janaki-Raman, S.; Schulze, A. Greasing the wheels of the cancer machine: The role of lipid metabolism in cancer. Cell Metab. 2020, 31, 62–76. [Google Scholar] [CrossRef]
- Zeng, W.; Yin, X.; Jiang, Y.; Jin, L.; Liang, W. PPARα at the crossroad of metabolic–immune regulation in cancer. FEBS J. 2022, 289, 7726–7739. [Google Scholar] [CrossRef] [PubMed]
- Font-Díaz, J.; Jiménez-Panizo, A.; Caelles, C.; dM Vivanco, M.; Pérez, P.; Aranda, A.; Estébanez-Perpiñá, E.; Castrillo, A.; Ricote, M.; Valledor, A.F. Nuclear receptors: Lipid and hormone sensors with essential roles in the control of cancer development. In Seminars in Cancer Biology; Academic Press: Cambridge, MA, USA, 2021; pp. 58–75. [Google Scholar]
- Mirza, A.Z.; Althagafi, I.I.; Shamshad, H. Role of PPAR receptor in different diseases and their ligands: Physiological importance and clinical implications. Eur. J. Med. Chem. 2019, 166, 502–513. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Zhao, L.; Zhu, X.; Ni, Y.; You, J.; Li, A. Xiaoyaosan, a traditional Chinese medicine, inhibits the chronic restraint stress-induced liver metastasis of colon cancer in vivo. Pharm. Biol. 2020, 58, 1085–1091. [Google Scholar] [CrossRef]
- Lee, J.; Lee, K.S.; Kim, H.; Jeong, H.; Choi, M.-J.; Yoo, H.-W.; Han, T.-H.; Lee, H. The relationship between metabolic syndrome and the incidence of colorectal cancer. Environ. Health Prev. Med. 2020, 25, 6. [Google Scholar] [CrossRef]
- Lu, B.; Qian, J.M.; Li, J.N. The metabolic syndrome and its components as prognostic factors in colorectal cancer: A meta-analysis and systematic review. J. Gastroenterol. Hepatol. 2023, 38, 187–196. [Google Scholar] [CrossRef]
- Luo, Y.; Xie, C.; Brocker, C.N.; Fan, J.; Wu, X.; Feng, L.; Wang, Q.; Zhao, J.; Lu, D.; Tandon, M.; et al. Intestinal PPARalpha Protects Against Colon Carcinogenesis via Regulation of Methyltransferases DNMT1 and PRMT6. Gastroenterology 2019, 157, 744–759.e4. [Google Scholar] [CrossRef] [PubMed]
- Grau, R.; Punzón, C.; Fresno, M.; Iñiguez, M.A. Peroxisome-proliferator-activated receptor α agonists inhibit cyclo-oxygenase 2 and vascular endothelial growth factor transcriptional activation in human colorectal carcinoma cells via inhibition of activator protein-1. Biochem. J. 2006, 395, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Devchand, P.R.; Keller, H.; Peters, J.M.; Vazquez, M.; Gonzalez, F.J.; Wahli, W. The PPARalpha-leukotriene B4 pathway to inflammation control. Nature 1996, 384, 39–43. [Google Scholar] [CrossRef] [PubMed]
- You, M.; Gao, J.; Jin, J.; Hou, Y. PPARα enhances cancer cell chemotherapy sensitivity by autophagy induction. J. Oncol. 2018, 2018, 6458537. [Google Scholar] [CrossRef] [PubMed]
- Padala, S.A.; Barsouk, A.; Thandra, K.C.; Saginala, K.; Mohammed, A.; Vakiti, A.; Rawla, P.; Barsouk, A. Epidemiology of renal cell carcinoma. World J. Oncol. 2020, 11, 79–87. [Google Scholar] [CrossRef]
- Linehan, W.M.; Schmidt, L.S.; Crooks, D.R.; Wei, D.; Srinivasan, R.; Lang, M.; Ricketts, C.J. The Metabolic Basis of Kidney Cancer. Cancer Discov. 2019, 9, 1006–1021. [Google Scholar] [CrossRef]
- Hsieh, J.J.; Purdue, M.P.; Signoretti, S.; Swanton, C.; Albiges, L.; Schmidinger, M.; Heng, D.Y.; Larkin, J.; Ficarra, V. Renal cell carcinoma. Nat. Rev. Dis. Primers 2017, 3, 17009. [Google Scholar] [CrossRef] [PubMed]
- Abu Aboud, O.; Donohoe, D.; Bultman, S.; Fitch, M.; Riiff, T.; Hellerstein, M.; Weiss, R.H. PPARα inhibition modulates multiple reprogrammed metabolic pathways in kidney cancer and attenuates tumor growth. Am. J. Physiol.-Cell Physiol. 2015, 308, C890–C898. [Google Scholar] [CrossRef]
- Abu Aboud, O.; Wettersten, H.I.; Weiss, R.H. Inhibition of PPARalpha induces cell cycle arrest and apoptosis, and synergizes with glycolysis inhibition in kidney cancer cells. PLoS ONE 2013, 8, e71115. [Google Scholar] [CrossRef]
- Cohen, I.J.; Blasberg, R. Impact of the tumor microenvironment on tumor-infiltrating lymphocytes: Focus on breast cancer. Breast Cancer Basic Clin. Res. 2017, 11, 1178223417731565. [Google Scholar] [CrossRef]
- Mandard, S.; Müller, M.; Kersten, S. Peroxisome proliferator-activated receptor α target genes. Cell. Mol. Life Sci. CMLS 2004, 61, 393–416. [Google Scholar] [CrossRef]
- Qian, Z.; Chen, L.; Liu, J.; Jiang, Y.; Zhang, Y. The emerging role of PPAR-alpha in breast cancer. Biomed. Pharmacother. 2023, 161, 114420. [Google Scholar] [CrossRef] [PubMed]
- Ferraro, G.B.; Ali, A.; Luengo, A.; Kodack, D.P.; Deik, A.; Abbott, K.L.; Bezwada, D.; Blanc, L.; Prideaux, B.; Jin, X. Fatty acid synthesis is required for breast cancer brain metastasis. Nat. Cancer 2021, 2, 414–428. [Google Scholar] [CrossRef]
- Chandran, K.; Goswami, S.; Sharma-Walia, N. Implications of a peroxisome proliferator-activated receptor alpha (PPARα) ligand clofibrate in breast cancer. Oncotarget 2016, 7, 15577. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, W.-T.; Su, H.-M.; Su, K.-P.; Chen, S.-H.; Wu, H.-P.; You, Y.-L.; Fu, R.-H.; Chao, P.-M. Deficiency or activation of peroxisome proliferator-activated receptor α reduces the tissue concentrations of endogenously synthesized docosahexaenoic acid in C57BL/6J mice. Nutr. Res. Pract. 2019, 13, 286–294. [Google Scholar] [CrossRef]
- Castelli, V.; Catanesi, M.; Alfonsetti, M.; Laezza, C.; Lombardi, F.; Cinque, B.; Cifone, M.G.; Ippoliti, R.; Benedetti, E.; Cimini, A.; et al. PPARalpha-Selective Antagonist GW6471 Inhibits Cell Growth in Breast Cancer Stem Cells Inducing Energy Imbalance and Metabolic Stress. Biomedicines 2021, 9, 127. [Google Scholar] [CrossRef]
- Sun, J.; Zheng, Z.; Chen, Q.; Pan, Y.; Quan, M.; Dai, Y. Fenofibrate potentiates chemosensitivity to human breast cancer cells by modulating apoptosis via AKT/NF-κB pathway. OncoTargets Ther. 2019, 12, 773. [Google Scholar] [CrossRef] [PubMed]
- Das, U.N.; Madhavi, N. Effect of polyunsaturated fatty acids on drug-sensitive and resistant tumor cells in vitro. Lipids Health Dis. 2011, 10, 159. [Google Scholar] [CrossRef] [PubMed]
- Geng, L.; Zhou, W.; Liu, B.; Wang, X.; Chen, B. DHA induces apoptosis of human malignant breast cancer tissues by the TLR-4/PPAR-alpha pathways. Oncol. Lett. 2018, 15, 2967–2977. [Google Scholar] [CrossRef]
- Zhang, Q.; Kong, X.; Yuan, H.; Guan, H.; Li, Y.; Niu, Y. Mangiferin Improved Palmitate-Induced-Insulin Resistance by Promoting Free Fatty Acid Metabolism in HepG2 and C2C12 Cells via PPARalpha: Mangiferin Improved Insulin Resistance. J. Diabetes Res. 2019, 2019, 2052675. [Google Scholar] [CrossRef]
- Yavarow, Z.A.; Kang, H.R.; Waskowicz, L.R.; Bay, B.H.; Young, S.P.; Yen, P.M.; Koeberl, D.D. Fenofibrate rapidly decreases hepatic lipid and glycogen storage in neonatal mice with glycogen storage disease type Ia. Hum. Mol. Genet. 2020, 29, 286–294. [Google Scholar] [CrossRef]
- Shehata, A.H.F.; Ahmed, A.F.; Abdelrehim, A.B.; Heeba, G.H. The impact of single and combined PPAR-alpha and PPAR-gamma activation on the neurological outcomes following cerebral ischemia reperfusion. Life Sci. 2020, 252, 117679. [Google Scholar] [CrossRef]
- Christofides, A.; Konstantinidou, E.; Jani, C.; Boussiotis, V.A. The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metabolism 2021, 114, 154338. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, J.; Wang, G. Fenofibrate decreased microalbuminuria in the type 2 diabetes patients with hypertriglyceridemia. Lipids Health Dis. 2020, 19, 103. [Google Scholar] [CrossRef] [PubMed]
- Kataoka, S.Y.; Lois, N.; Kawano, S.; Kataoka, Y.; Inoue, K.; Watanabe, N. Fenofibrate for diabetic retinopathy. Cochrane Database Syst. Rev. 2023, 6, CD013318. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hu, P.; Li, K.; Peng, X.; Kan, Y.; Li, H.; Zhu, Y.; Wang, Z.; Li, Z.; Liu, H.-Y.; Cai, D. Nuclear Receptor PPARα as a Therapeutic Target in Diseases Associated with Lipid Metabolism Disorders. Nutrients 2023, 15, 4772. https://doi.org/10.3390/nu15224772
Hu P, Li K, Peng X, Kan Y, Li H, Zhu Y, Wang Z, Li Z, Liu H-Y, Cai D. Nuclear Receptor PPARα as a Therapeutic Target in Diseases Associated with Lipid Metabolism Disorders. Nutrients. 2023; 15(22):4772. https://doi.org/10.3390/nu15224772
Chicago/Turabian StyleHu, Ping, Kaiqi Li, Xiaoxu Peng, Yufei Kan, Hao Li, Yanli Zhu, Ziyu Wang, Zhaojian Li, Hao-Yu Liu, and Demin Cai. 2023. "Nuclear Receptor PPARα as a Therapeutic Target in Diseases Associated with Lipid Metabolism Disorders" Nutrients 15, no. 22: 4772. https://doi.org/10.3390/nu15224772
APA StyleHu, P., Li, K., Peng, X., Kan, Y., Li, H., Zhu, Y., Wang, Z., Li, Z., Liu, H.-Y., & Cai, D. (2023). Nuclear Receptor PPARα as a Therapeutic Target in Diseases Associated with Lipid Metabolism Disorders. Nutrients, 15(22), 4772. https://doi.org/10.3390/nu15224772