
ALDH1L2 Knockout in U251 Glioblastoma Cells Reduces Tumor Sphere Formation by Increasing Oxidative Stress and Suppressing Methionine Dependency

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Background
2. Methods
2.1. Generation of ALDH1L2 CRSPR Knockouts (KO)
2.2. Whole Genome Sequencing (WGS)
2.3. Metabolites Analysis
2.4. Statistical Analysis
3. Results
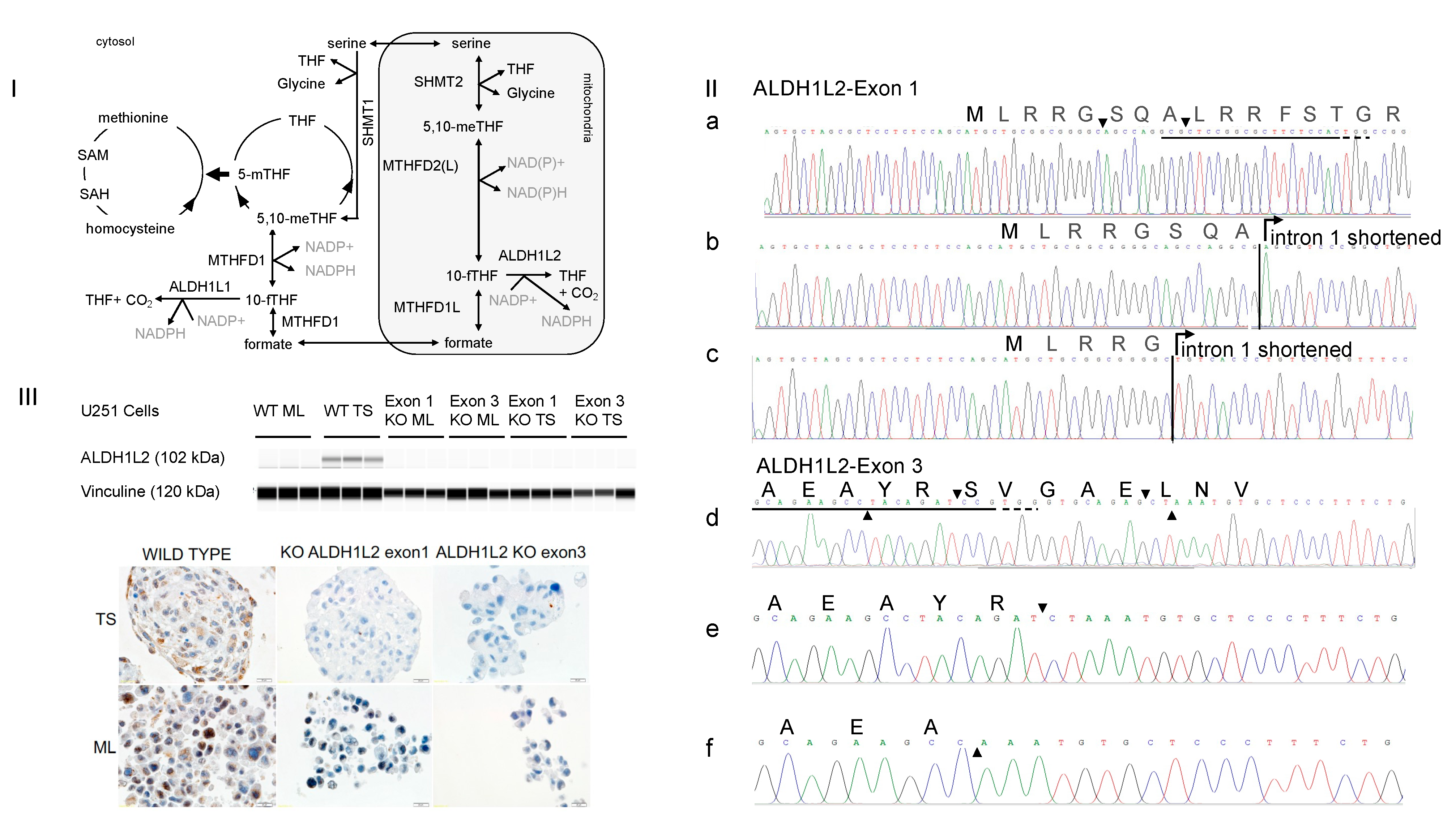
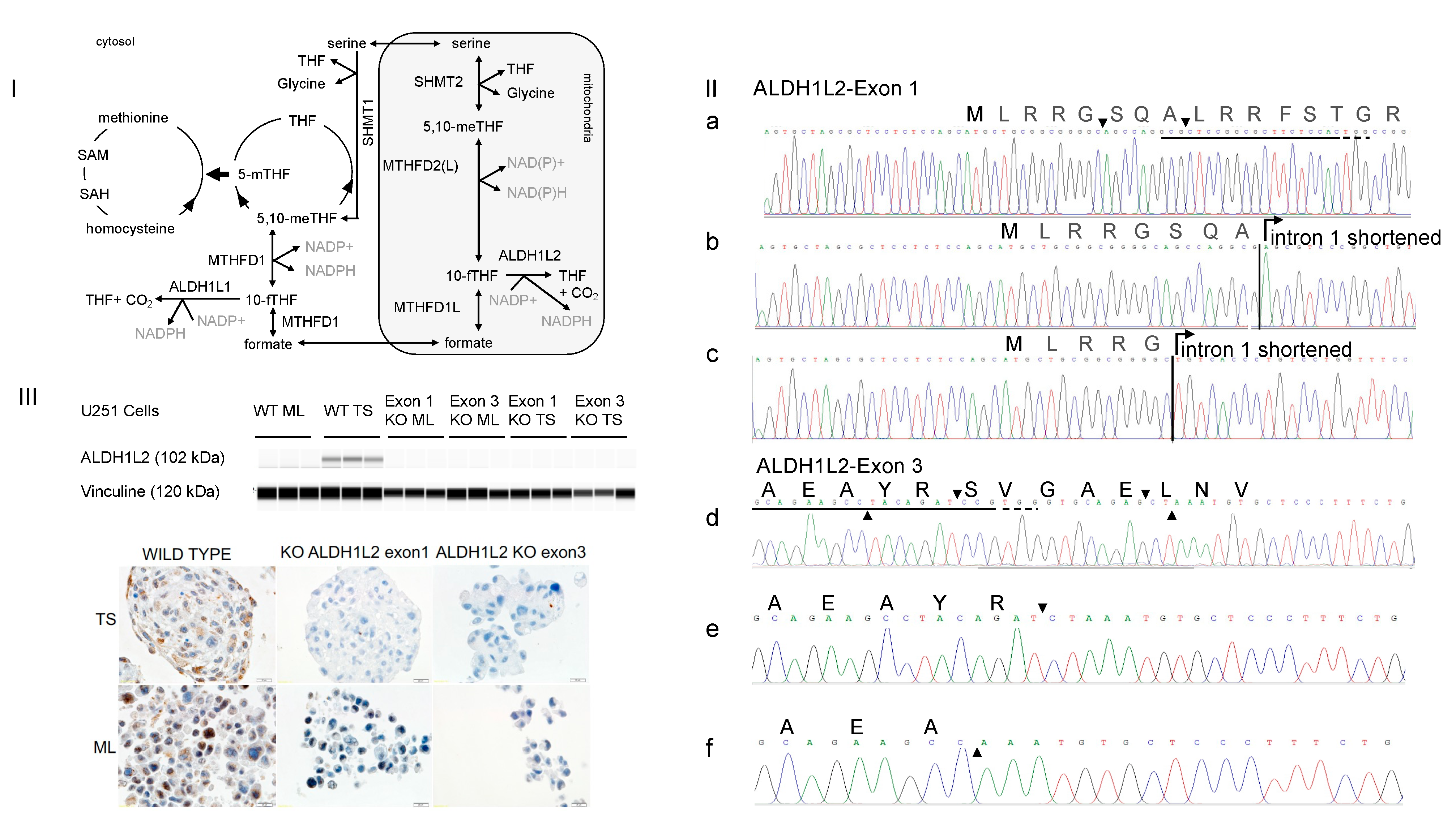
3.1. CRISPR-Cas9 Targeting Either Exon 1 or Exon 3 of ALDH1L2 Abolishes ALDH1L2 Expression
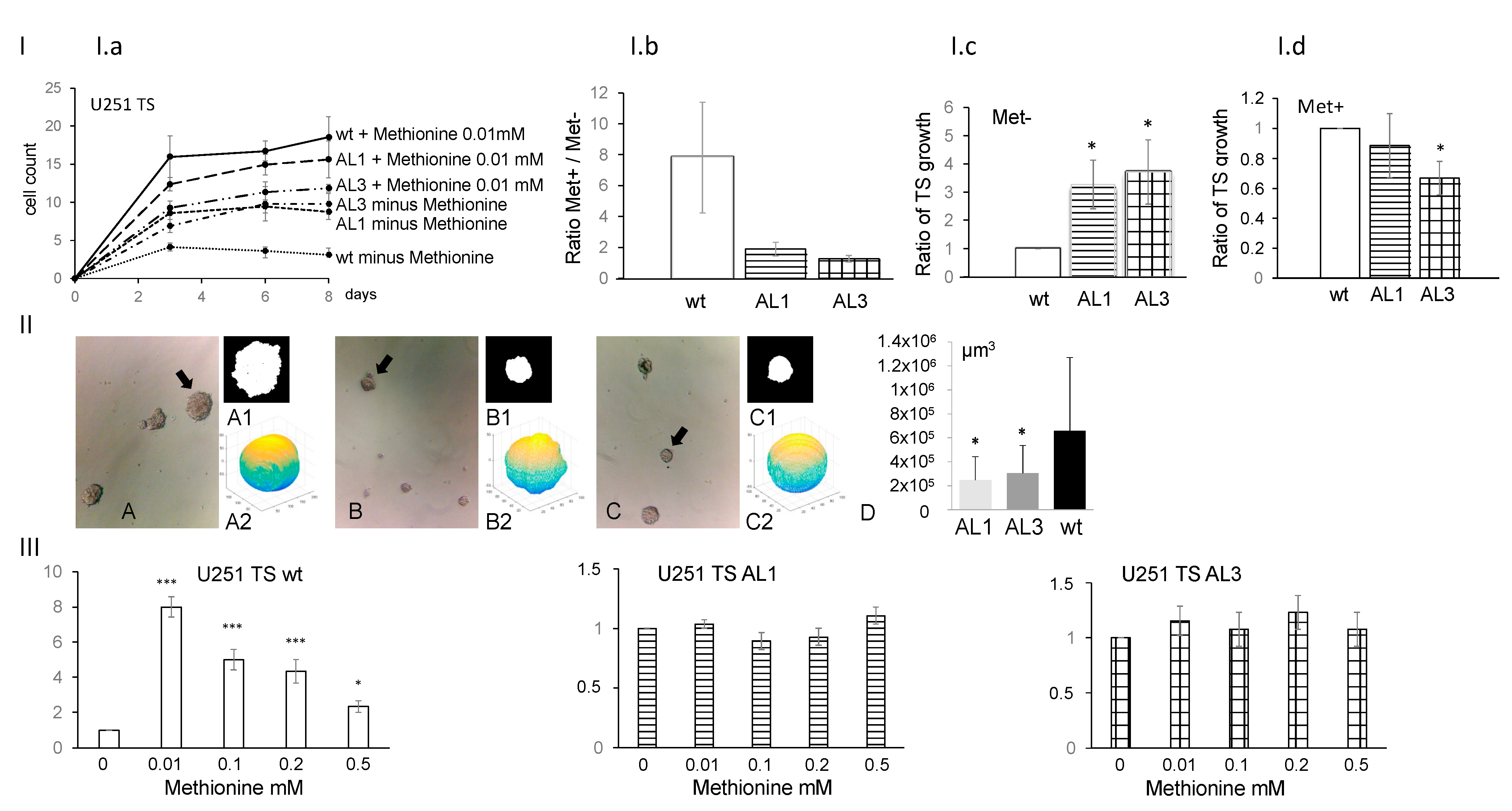
3.2. Tumor Sphere (TS) Growth of the ALDH1L2 KO Cells Are Slower and Less Methionine-Dependent
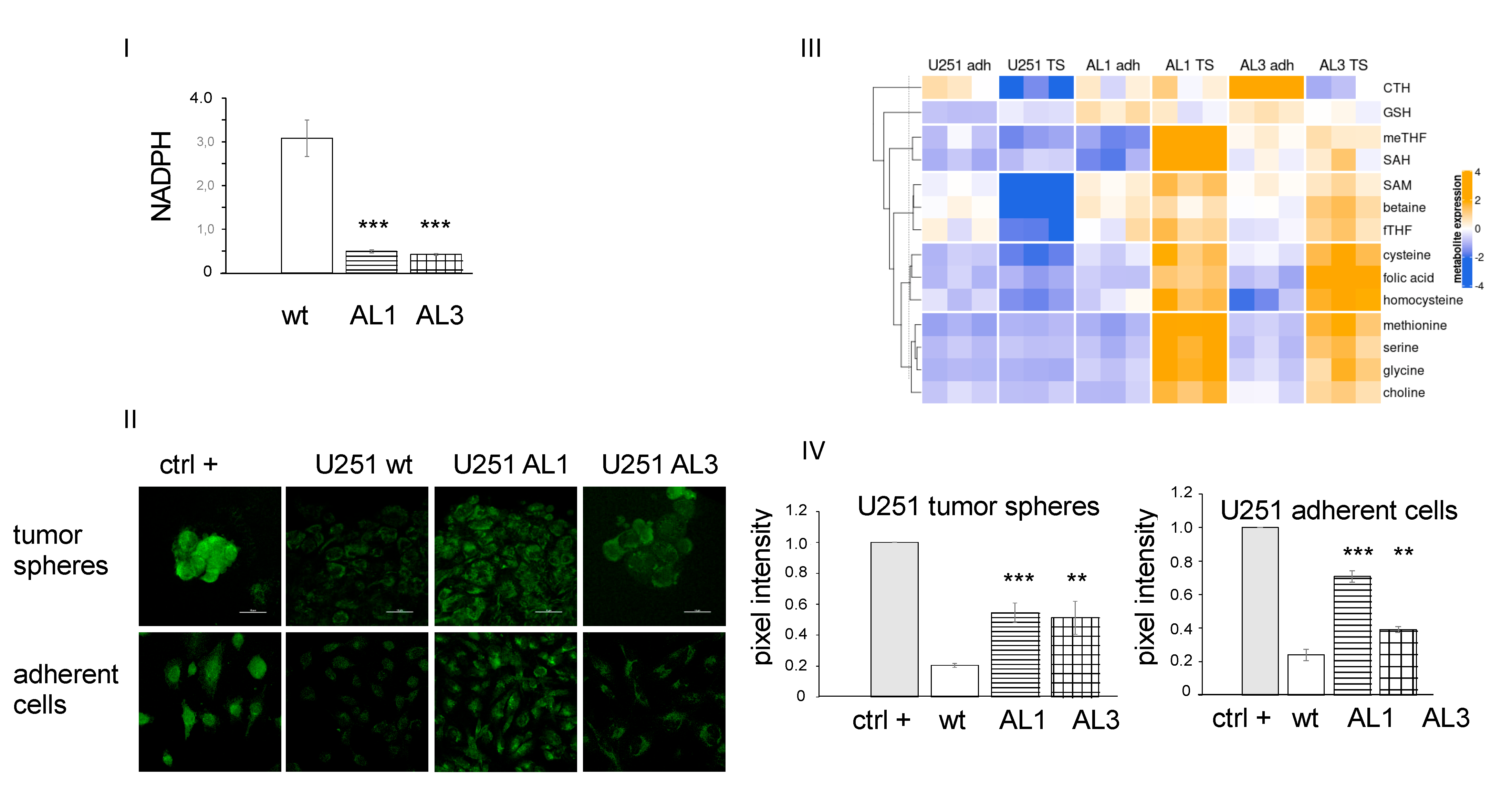
3.3. ALDHL2 Knockout U251 Cells Have Reduced NADPH but Higher Cytosolic Methyl Donors in Tumor Spheres
3.4. ALDH1L2 KO Cells Bear More ROS despite No Change in Glutathione Content
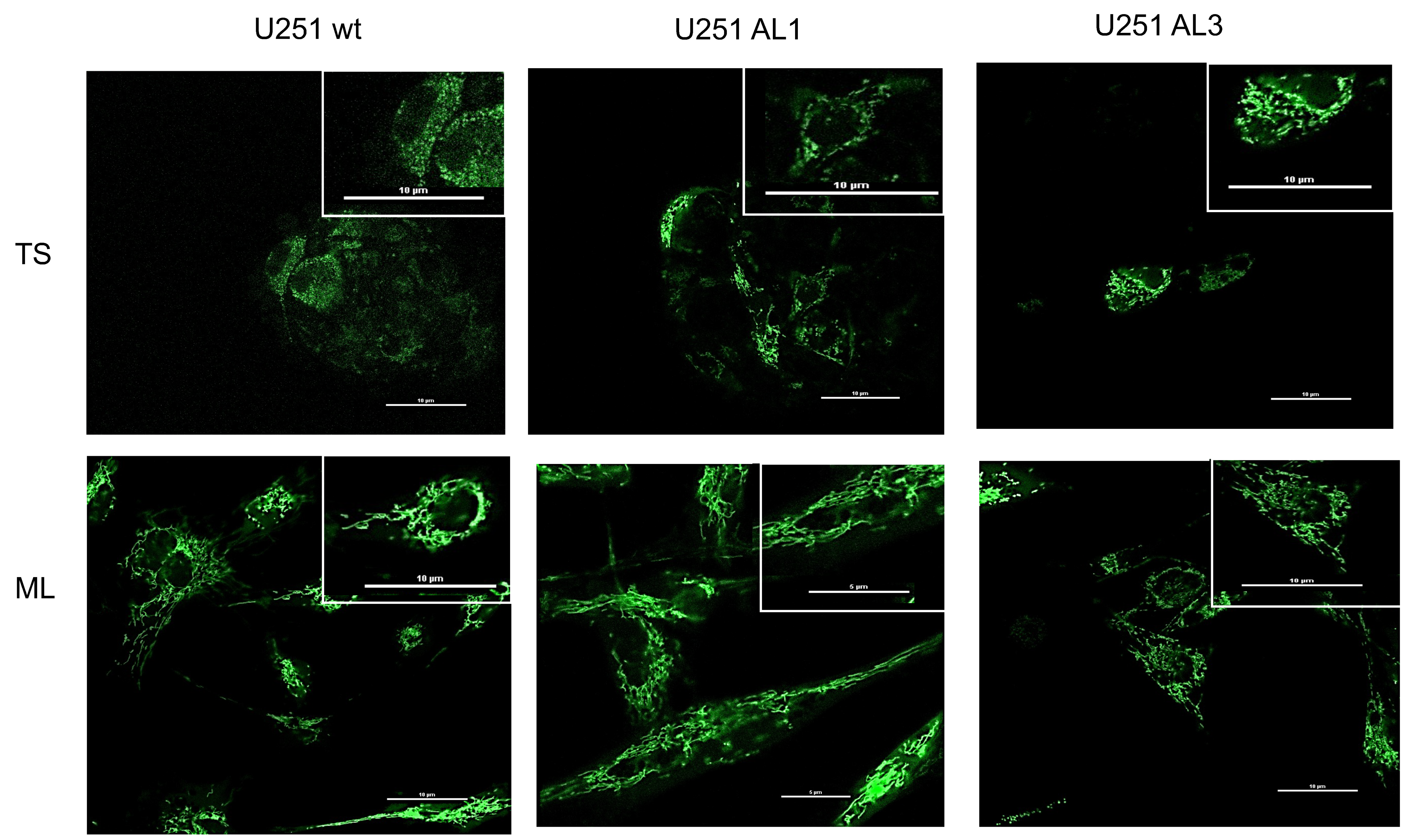
3.5. Mitochondria Morphological Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sanderson, S.M.; Mikhael, P.G.; Ramesh, V.; Dai, Z.; Locasale, J.W. Nutrient Availability Shapes Methionine Metabolism in P16/MTAP-Deleted Cells. Sci. Adv. 2019, 5, eaav7769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, M.; Vousden, K.H. Serine and One-Carbon Metabolism in Cancer. Nat. Rev. Cancer 2016, 16, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Ye, J.; Kamphorst, J.J.; Shlomi, T.; Thompson, C.B.; Rabinowitz, J.D. Quantitative Flux Analysis Reveals Folate-Dependent NADPH Production. Nature 2014, 510, 298–302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shiraki, N.; Shiraki, Y.; Tsuyama, T.; Obata, F.; Miura, M.; Nagae, G.; Aburatani, H.; Kume, K.; Endo, F.; Kume, S. Methionine Metabolism Regulates Maintenance and Differentiation of Human Pluripotent Stem Cells. Cell Metab. 2014, 19, 780–794. [Google Scholar] [CrossRef] [Green Version]
- Lamb, R.; Harrison, H.; Smith, D.L.; Townsend, P.A.; Jackson, T.; Ozsvari, B.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Howell, A.; Lisanti, M.P.; et al. Targeting Tumor-Initiating Cells: Eliminating Anabolic Cancer Stem Cells with Inhibitors of Protein Synthesis or by Mimicking Caloric Restriction. Oncotarget 2015, 6, 4585–4601. [Google Scholar] [CrossRef] [Green Version]
- Ju, H.-Q.; Lin, J.-F.; Tian, T.; Xie, D.; Xu, R.-H. NADPH Homeostasis in Cancer: Functions, Mechanisms and Therapeutic Implications. Signal Transduct. Target. Ther. 2020, 5, 231. [Google Scholar] [CrossRef]
- Bigarella, C.L.; Liang, R.; Ghaffari, S. Stem Cells and the Impact of ROS Signaling. Development 2014, 141, 4206–4218. [Google Scholar] [CrossRef] [Green Version]
- Liao, N.; Shi, Y.; Zhang, C.; Zheng, Y.; Wang, Y.; Zhao, B.; Zeng, Y.; Liu, X.; Liu, J. Antioxidants Inhibit Cell Senescence and Preserve Stemness of Adipose Tissue-Derived Stem Cells by Reducing ROS Generation during Long-Term in Vitro Expansion. Stem Cell Res. Ther. 2019, 10, 306. [Google Scholar] [CrossRef] [Green Version]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Lam, J.S.; Ailles, L.E.; Wong, M.; et al. Association of Reactive Oxygen Species Levels and Radioresistance in Cancer Stem Cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Vassalli, G. Aldehyde Dehydrogenases: Not Just Markers, but Functional Regulators of Stem Cells. Stem Cells Int. 2019, 2019, 3904645. [Google Scholar] [CrossRef] [Green Version]
- Muralikrishnan, V.; Hurley, T.D.; Nephew, K.P. Targeting Aldehyde Dehydrogenases to Eliminate Cancer Stem Cells in Gynecologic Malignancies. Cancers 2020, 12, 961. [Google Scholar] [CrossRef] [Green Version]
- Tanei, T.; Morimoto, K.; Shimazu, K.; Kim, S.J.; Tanji, Y.; Taguchi, T.; Tamaki, Y.; Noguchi, S. Association of Breast Cancer Stem Cells Identified by Aldehyde Dehydrogenase 1 Expression with Resistance to Sequential Paclitaxel and Epirubicin-Based Chemotherapy for Breast Cancers. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2009, 15, 4234–4241. [Google Scholar] [CrossRef] [Green Version]
- Duong, H.-Q.; Hwang, J.S.; Kim, H.J.; Kang, H.J.; Seong, Y.-S.; Bae, I. Aldehyde Dehydrogenase 1A1 Confers Intrinsic and Acquired Resistance to Gemcitabine in Human Pancreatic Adenocarcinoma MIA PaCa-2 Cells. Int. J. Oncol. 2012, 41, 855–861. [Google Scholar] [CrossRef] [Green Version]
- Rausch, V.; Liu, L.; Kallifatidis, G.; Baumann, B.; Mattern, J.; Gladkich, J.; Wirth, T.; Schemmer, P.; Büchler, M.W.; Zöller, M.; et al. Synergistic Activity of Sorafenib and Sulforaphane Abolishes Pancreatic Cancer Stem Cell Characteristics. Cancer Res. 2010, 70, 5004–5013. [Google Scholar] [CrossRef] [Green Version]
- Piskounova, E.; Agathocleous, M.; Murphy, M.M.; Hu, Z.; Huddlestun, S.E.; Zhao, Z.; Leitch, A.M.; Johnson, T.M.; DeBerardinis, R.J.; Morrison, S.J. Oxidative Stress Inhibits Distant Metastasis by Human Melanoma Cells. Nature 2015, 527, 186–191. [Google Scholar] [CrossRef] [Green Version]
- Sarret, C.; Ashkavand, Z.; Paules, E.; Dorboz, I.; Pediaditakis, P.; Sumner, S.; Eymard-Pierre, E.; Francannet, C.; Krupenko, N.I.; Boespflug-Tanguy, O.; et al. Deleterious Mutations in ALDH1L2 Suggest a Novel Cause for Neuro-Ichthyotic Syndrome. NPJ Genom. Med. 2019, 4, 17. [Google Scholar] [CrossRef]
- Kirsch, M.; De Groot, H. NAD(P)H, a Directly Operating Antioxidant? FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2001, 15, 1569–1574. [Google Scholar] [CrossRef] [Green Version]
- Zgheib, R.; Battaglia-Hsu, S.-F.; Hergalant, S.; Quéré, M.; Alberto, J.-M.; Chéry, C.; Rouyer, P.; Gauchotte, G.; Guéant, J.-L.; Namour, F. Folate Can Promote the Methionine-Dependent Reprogramming of Glioblastoma Cells towards Pluripotency. Cell Death Dis. 2019, 10, 596. [Google Scholar] [CrossRef] [Green Version]
- Toledano, M.B.; Delaunay-Moisan, A. Keeping Oxidative Metabolism on Time: Mitochondria as an Autonomous Redox Pacemaker Animated by H2O2 and Peroxiredoxin. Mol. Cell 2015, 59, 517–519. [Google Scholar] [CrossRef]
- Pourbagher, R.; Ghorbani, H.; Akhavan-Niaki, H.; Jorsaraei, S.G.A.; Fattahi, S.; Ghooran, S.; Abedian, Z.; Ghasemi, M.; Saeedi, F.; Jafari, N.; et al. Downregulation of Stemness Genes and Induction of Necrosis in Rat LA7 Cancer Stem Cells Induced Tumors Treated with Starved Fibroblasts Culture Supernatant. Rep. Biochem. Mol. Biol. 2021, 10, 105–118. [Google Scholar] [CrossRef]
- Buccarelli, M.; D’Alessandris, Q.G.; Matarrese, P.; Mollinari, C.; Signore, M.; Cappannini, A.; Martini, M.; D’Aliberti, P.; De Luca, G.; Pedini, F.; et al. Elesclomol-Induced Increase of Mitochondrial Reactive Oxygen Species Impairs Glioblastoma Stem-like Cell Survival and Tumor Growth. J. Exp. Clin. Cancer Res. CR 2021, 40, 228. [Google Scholar] [CrossRef]
- Mertens, J.; Herdy, J.R.; Traxler, L.; Schafer, S.T.; Schlachetzki, J.C.M.; Böhnke, L.; Reid, D.A.; Lee, H.; Zangwill, D.; Fernandes, D.P.; et al. Age-Dependent Instability of Mature Neuronal Fate in Induced Neurons from Alzheimer’s Patients. Cell Stem Cell 2021, 28, 1533–1548.e6. [Google Scholar] [CrossRef]
- Loureiro, R.; Mesquita, K.A.; Magalhães-Novais, S.; Oliveira, P.J.; Vega-Naredo, I. Mitochondrial Biology in Cancer Stem Cells. Semin. Cancer Biol. 2017, 47, 18–28. [Google Scholar] [CrossRef]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Vitale, I.; Rigoni, A.; Vacchelli, E.; Michaud, M.; Zischka, H.; Castedo, M.; Kroemer, G. Mitochondrial Gateways to Cancer. Mol. Aspects Med. 2010, 31, 1–20. [Google Scholar] [CrossRef]
- Kroemer, G.; Pouyssegur, J. Tumor Cell Metabolism: Cancer’s Achilles’ Heel. Cancer Cell 2008, 13, 472–482. [Google Scholar] [CrossRef]
- Janiszewska, M.; Suvà, M.L.; Riggi, N.; Houtkooper, R.H.; Auwerx, J.; Clément-Schatlo, V.; Radovanovic, I.; Rheinbay, E.; Provero, P.; Stamenkovic, I. Imp2 Controls Oxidative Phosphorylation and Is Crucial for Preserving Glioblastoma Cancer Stem Cells. Genes Dev. 2012, 26, 1926–1944. [Google Scholar] [CrossRef] [Green Version]
- Ye, X.-Q.; Li, Q.; Wang, G.-H.; Sun, F.-F.; Huang, G.-J.; Bian, X.-W.; Yu, S.-C.; Qian, G.-S. Mitochondrial and Energy Metabolism-Related Properties as Novel Indicators of Lung Cancer Stem Cells. Int. J. Cancer 2011, 129, 820–831. [Google Scholar] [CrossRef]
- Sancho, P.; Burgos-Ramos, E.; Tavera, A.; Bou Kheir, T.; Jagust, P.; Schoenhals, M.; Barneda, D.; Sellers, K.; Campos-Olivas, R.; Graña, O.; et al. MYC/PGC-1α Balance Determines the Metabolic Phenotype and Plasticity of Pancreatic Cancer Stem Cells. Cell Metab. 2015, 22, 590–605. [Google Scholar] [CrossRef] [Green Version]
- Ciavardelli, D.; Rossi, C.; Barcaroli, D.; Volpe, S.; Consalvo, A.; Zucchelli, M.; De Cola, A.; Scavo, E.; Carollo, R.; D’Agostino, D.; et al. Breast Cancer Stem Cells Rely on Fermentative Glycolysis and Are Sensitive to 2-Deoxyglucose Treatment. Cell Death Dis. 2014, 5, e1336. [Google Scholar] [CrossRef] [Green Version]
- Emmink, B.L.; Verheem, A.; Van Houdt, W.J.; Steller, E.J.A.; Govaert, K.M.; Pham, T.V.; Piersma, S.R.; Borel Rinkes, I.H.M.; Jimenez, C.R.; Kranenburg, O. The Secretome of Colon Cancer Stem Cells Contains Drug-Metabolizing Enzymes. J. Proteom. 2013, 91, 84–96. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, Y.; Shingu, T.; Feng, L.; Chen, Z.; Ogasawara, M.; Keating, M.J.; Kondo, S.; Huang, P. Metabolic Alterations in Highly Tumorigenic Glioblastoma Cells: Preference for Hypoxia and High Dependency on Glycolysis. J. Biol. Chem. 2011, 286, 32843–32853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiswald, L.-B.; Bellet, D.; Dangles-Marie, V. Spherical Cancer Models in Tumor Biology. Neoplasia 2015, 17, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chacko, B.K.; Kramer, P.A.; Ravi, S.; Benavides, G.A.; Mitchell, T.; Dranka, B.P.; Ferrick, D.; Singal, A.K.; Ballinger, S.W.; Bailey, S.M.; et al. The Bioenergetic Health Index: A New Concept in Mitochondrial Translational Research. Clin. Sci. 2014, 127, 367–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Quéré, M.; Alberto, J.-M.; Broly, F.; Hergalant, S.; Christov, C.; Gauchotte, G.; Guéant, J.-L.; Namour, F.; Battaglia-Hsu, S.-F. ALDH1L2 Knockout in U251 Glioblastoma Cells Reduces Tumor Sphere Formation by Increasing Oxidative Stress and Suppressing Methionine Dependency. Nutrients 2022, 14, 1887. https://doi.org/10.3390/nu14091887
Quéré M, Alberto J-M, Broly F, Hergalant S, Christov C, Gauchotte G, Guéant J-L, Namour F, Battaglia-Hsu S-F. ALDH1L2 Knockout in U251 Glioblastoma Cells Reduces Tumor Sphere Formation by Increasing Oxidative Stress and Suppressing Methionine Dependency. Nutrients. 2022; 14(9):1887. https://doi.org/10.3390/nu14091887
Chicago/Turabian StyleQuéré, Maëlle, Jean-Marc Alberto, Franck Broly, Sébastien Hergalant, Christo Christov, Guillaume Gauchotte, Jean-Louis Guéant, Farès Namour, and Shyue-Fang Battaglia-Hsu. 2022. "ALDH1L2 Knockout in U251 Glioblastoma Cells Reduces Tumor Sphere Formation by Increasing Oxidative Stress and Suppressing Methionine Dependency" Nutrients 14, no. 9: 1887. https://doi.org/10.3390/nu14091887
APA StyleQuéré, M., Alberto, J.-M., Broly, F., Hergalant, S., Christov, C., Gauchotte, G., Guéant, J.-L., Namour, F., & Battaglia-Hsu, S.-F. (2022). ALDH1L2 Knockout in U251 Glioblastoma Cells Reduces Tumor Sphere Formation by Increasing Oxidative Stress and Suppressing Methionine Dependency. Nutrients, 14(9), 1887. https://doi.org/10.3390/nu14091887