
Fisetin Suppresses the Inflammatory Response and Oxidative Stress in Bronchial Epithelial Cells

 ,
,  ,
,  , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Cytotoxicity Assay
2.3. BEAS-2B Cell Culture and Fisetin Treatment
2.4. Cell Adhesion Experiment
2.5. ROS Expression
2.6. Animal Experiments
2.7. Mouse Sensitization and Administration of Fisetin
2.8. Airway Hyperresponsiveness
2.9. Bronchoalveolar Lavage Fluid
2.10. Immunohistochemistry
2.11. Glutathione Assay
2.12. Malondialdehyde Activity
2.13. Real-Time PCR
2.14. Western Blot Analysis
2.15. ELISA
2.16. Transfection and Luciferase Assays
2.17. Data Analysis
3. Results
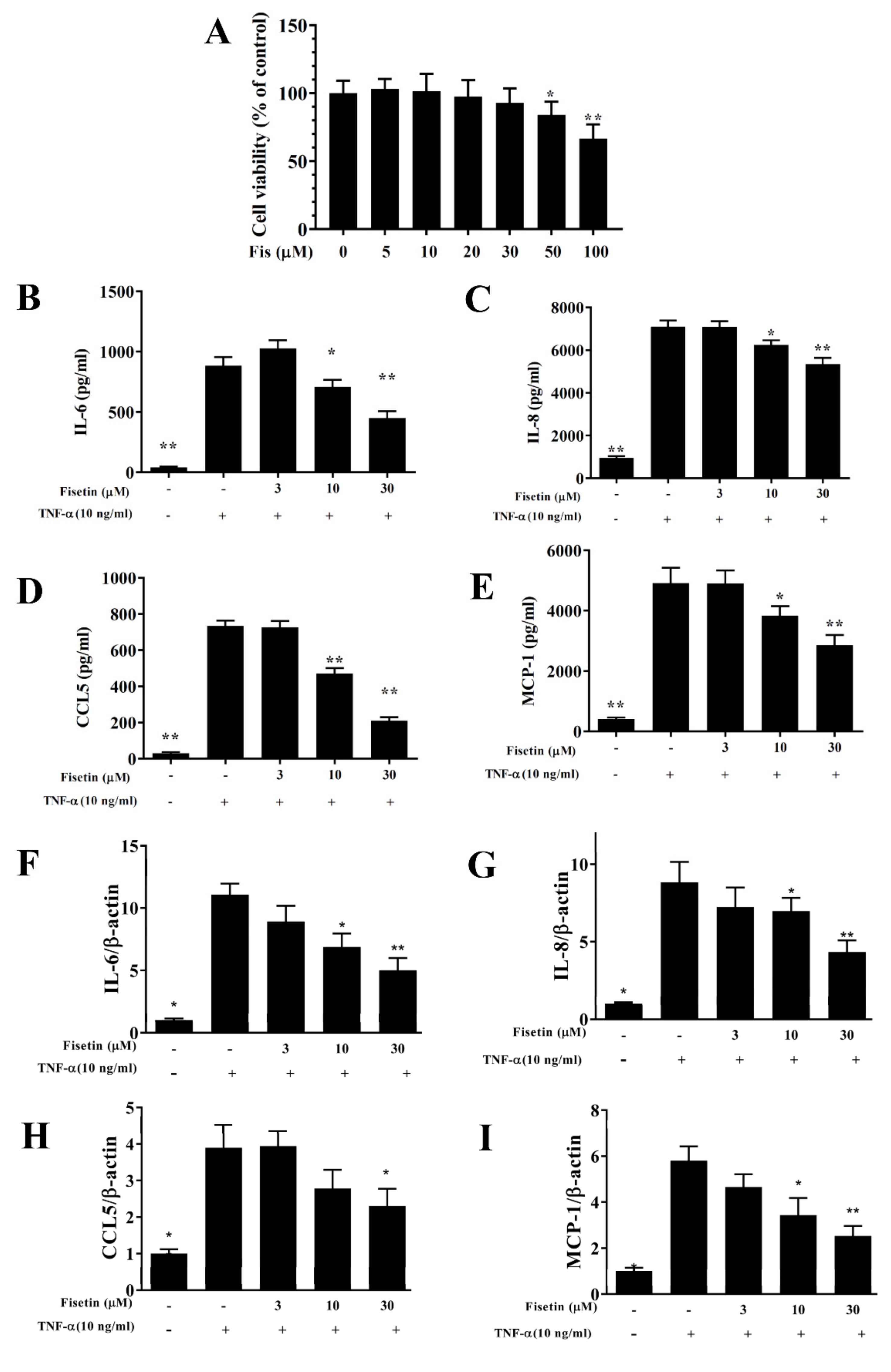
3.1. Fisetin Reduced Pro-Inflammatory Cytokine Expressions in BEAS-2B Cells
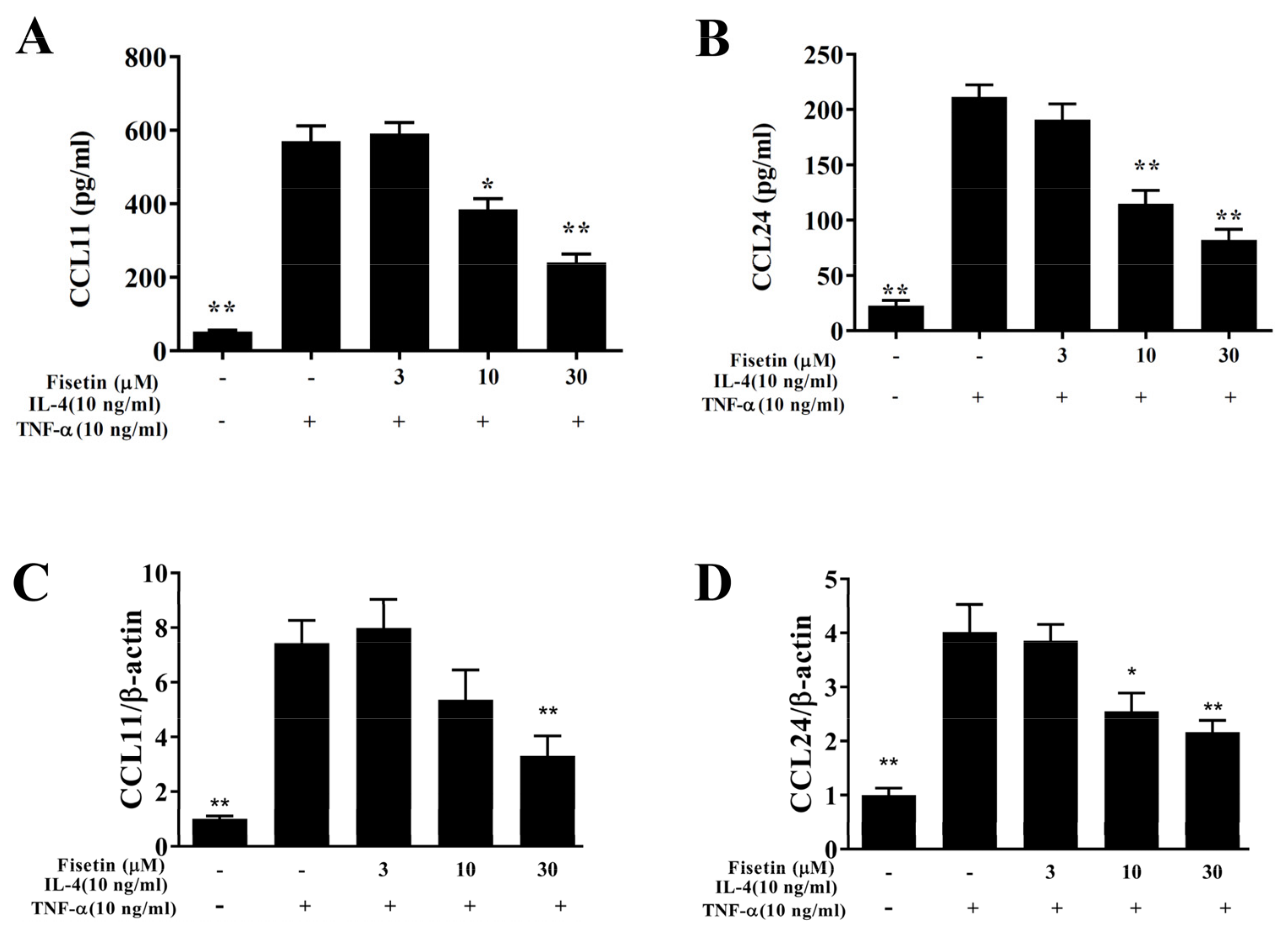
3.2. Fisetin Attenuated ICAM-1 and MUC5AC Expression
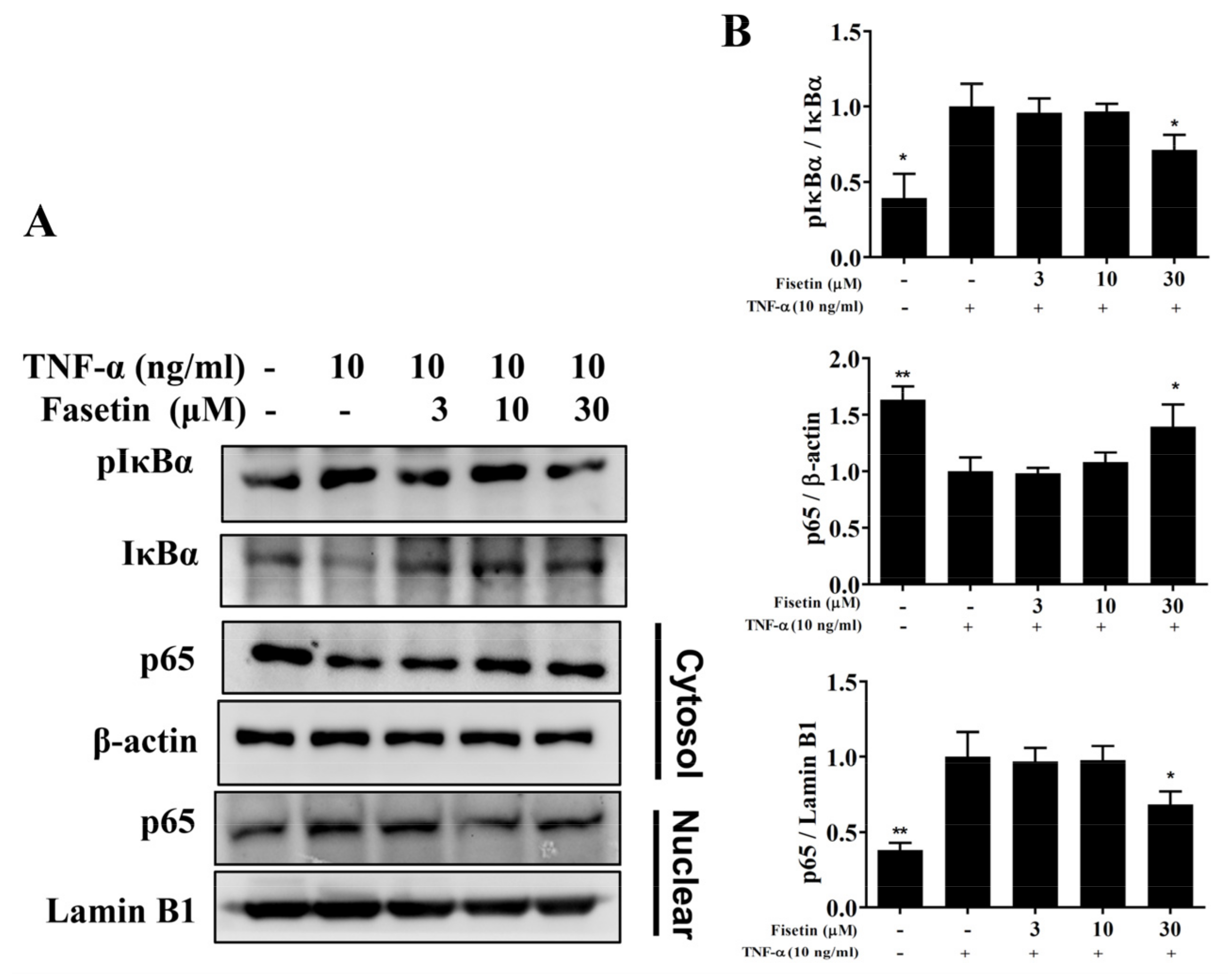
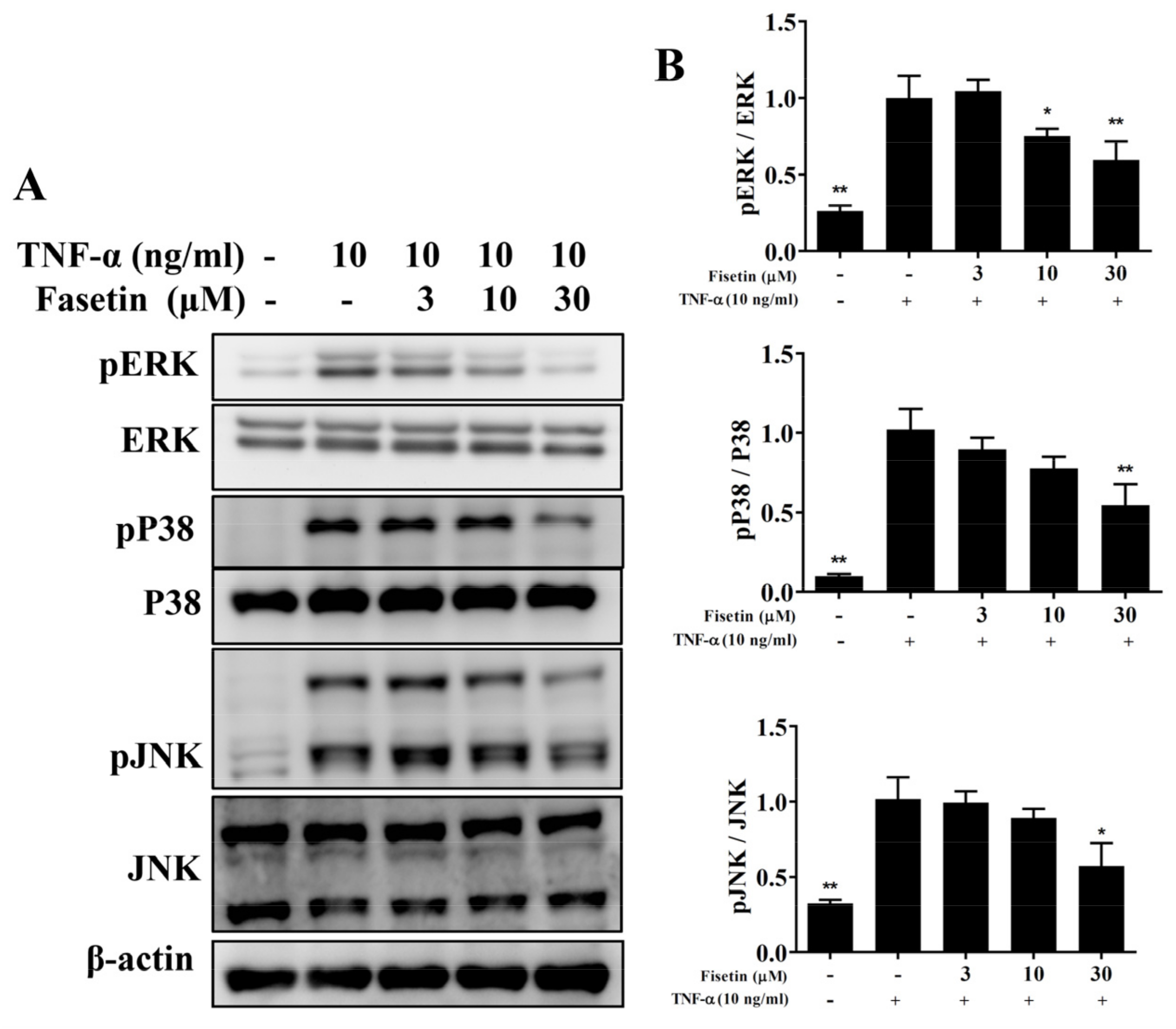
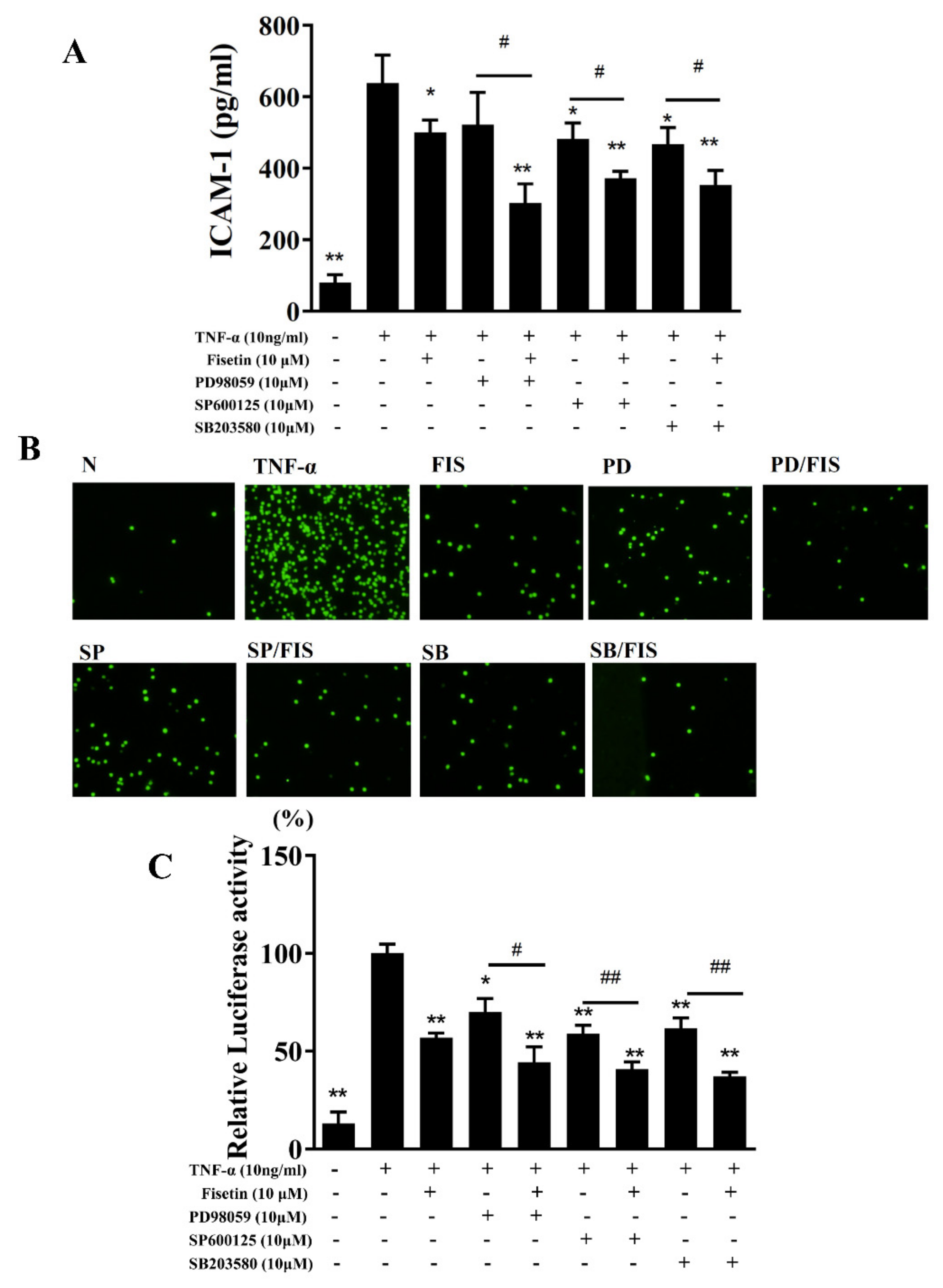
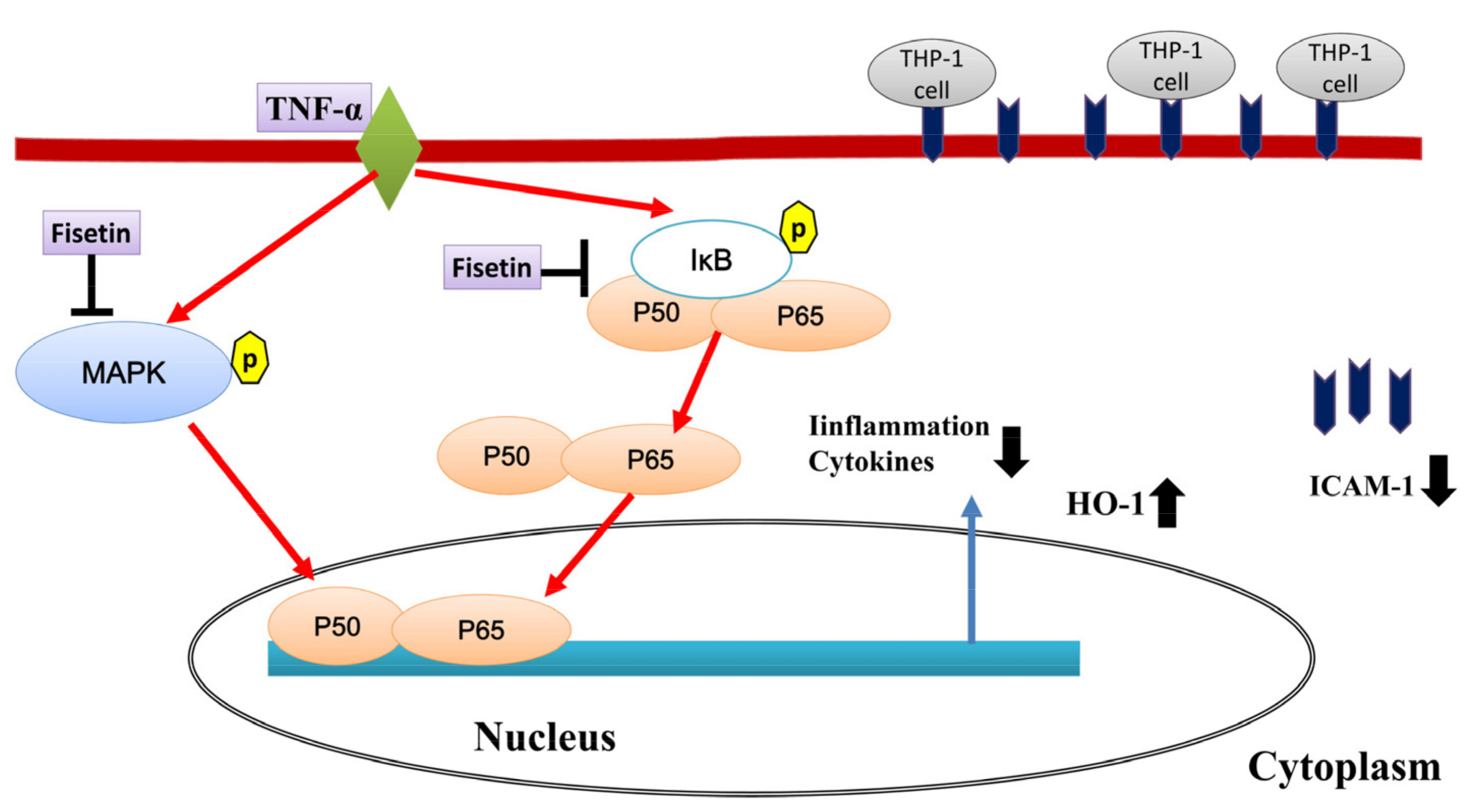
3.3. Fisetin Regulated NF-κB and MAPK Pathways in BEAS-2B Cells
3.4. Effect of Fisetin on ROS Expression
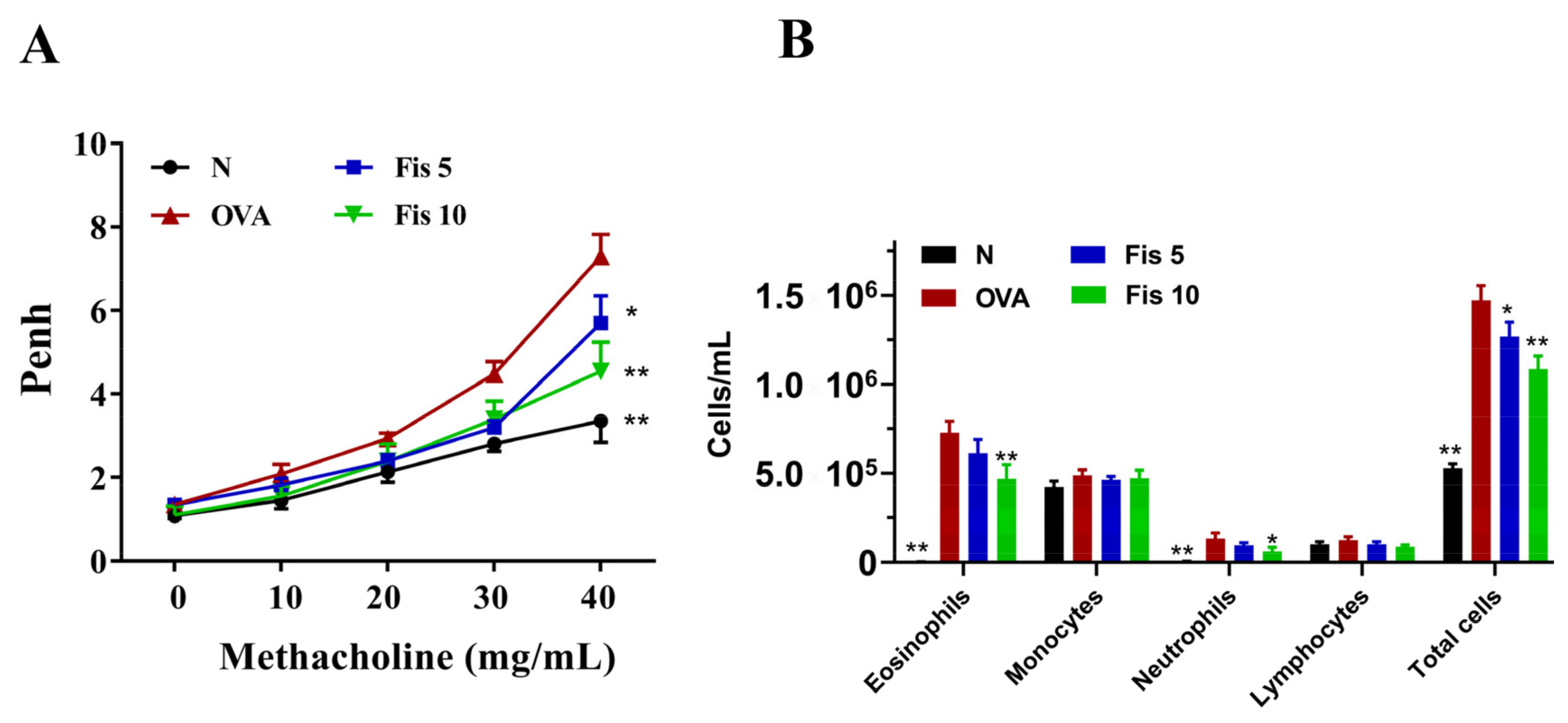
3.5. Fisetin Effects on AHR in Asthmatic Mice
3.6. Fisetin Suppressed Eosinophil Numbers in the BALF
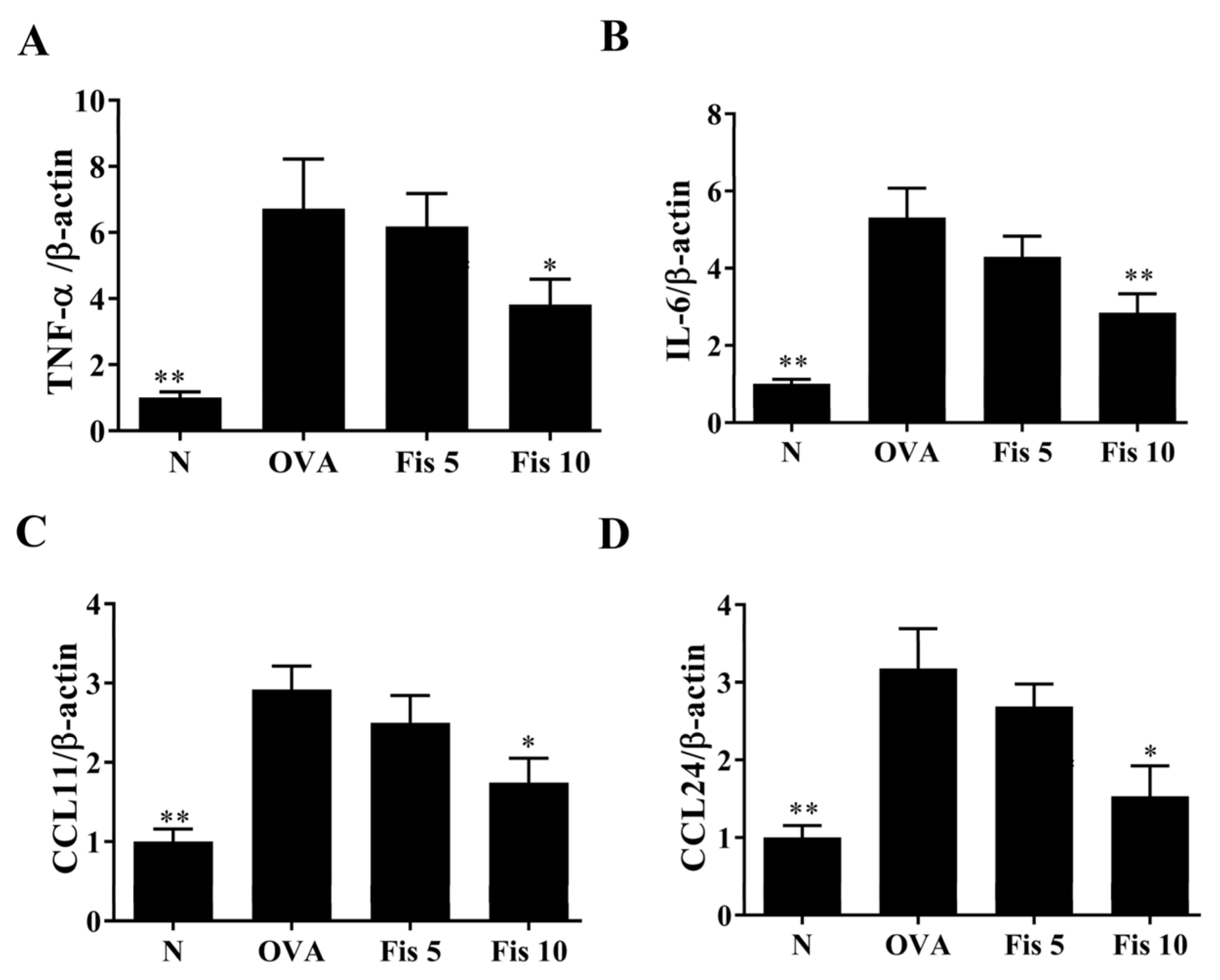
3.7. Fisetin Reduced OVA-Induced Inflammatory Mediators in the Lungs
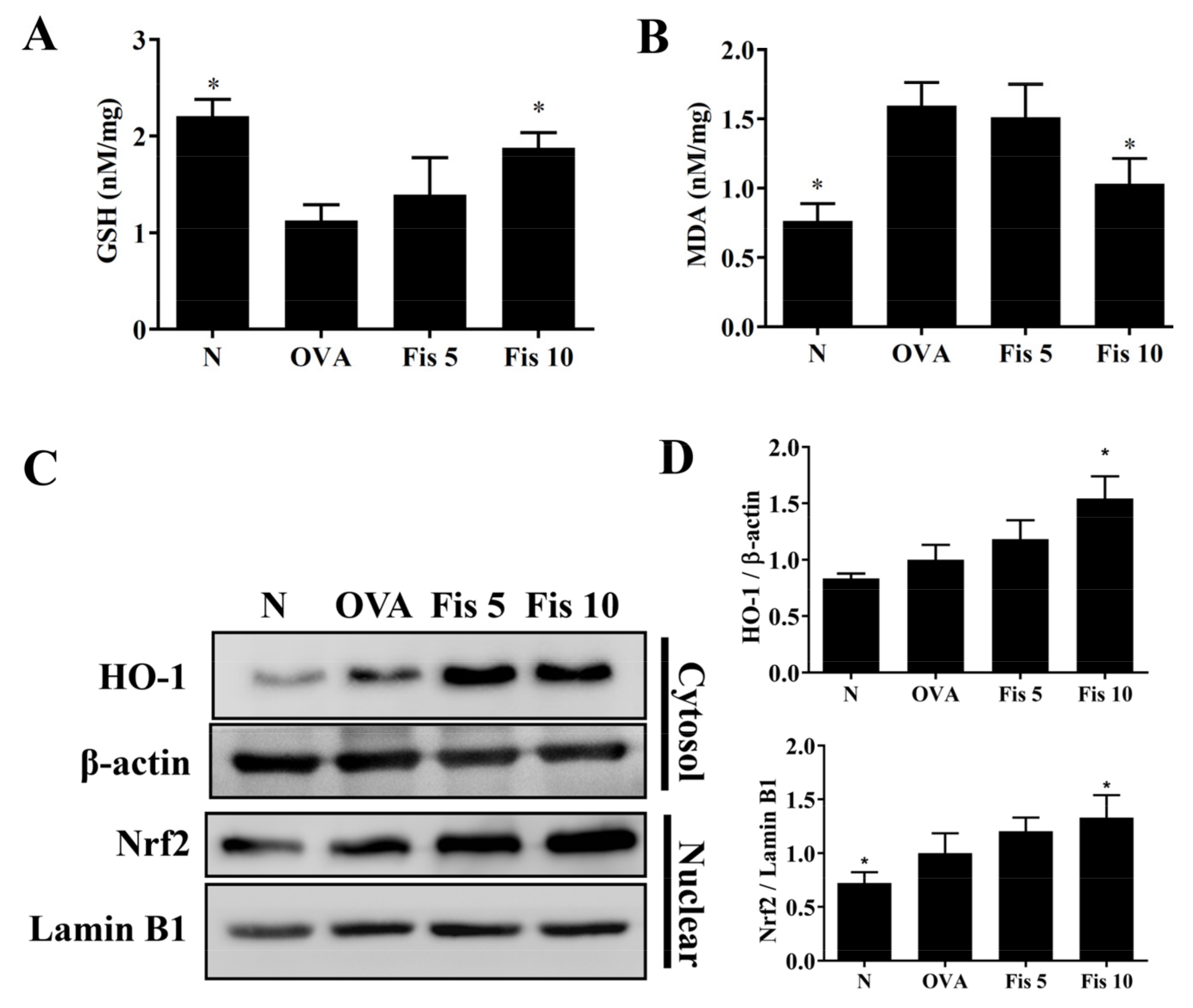
3.8. Fisetin Modulated MDA and GSH Levels in the Lungs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Guarnieri, M.; Balmes, J.R. Outdoor air pollution and asthma. Lancet 2014, 383, 1581–1592. [Google Scholar] [CrossRef] [Green Version]
- Maciag, M.C.; Phipatanakul, W. Prevention of asthma: Targets for intervention. Chest 2020, 158, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Cevhertas, L.; Ogulur, I.; Maurer, D.J.; Burla, D.; Ding, M.; Jansen, K.; Koch, J.; Liu, C.; Ma, S.; Mitamura, Y.; et al. Advances and recent developments in asthma in 2020. Allergy 2020, 75, 3124–3146. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, H.; Braubach, P.; Schilpp, C.; Lochbaum, R.; Neuland, K.; Thompson, K.; Jonigk, D.; Frick, M.; Dietl, P.; Wittekindt, O.H. IL-13 impairs tight junctions in airway epithelia. Int. J. Mol. Sci. 2019, 20, 3222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goleva, E.; Berdyshev, E.; Leung, D.Y. Epithelial barrier repair and prevention of allergy. J. Clin. Investig. 2019, 129, 1463–1474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cargnello, M.; Roux, P.P. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol. Mol. Biol. Rev. 2011, 75, 50–83. [Google Scholar] [CrossRef] [Green Version]
- Coleman, S.L.; Kruger, M.C.; Sawyer, G.M.; Hurst, R.D. Procyanidin A2 modulates IL-4-induced CCL26 production in human alveolar epithelial cells. Int. J. Mol. Sci. 2016, 17, 1888. [Google Scholar] [CrossRef] [Green Version]
- Alagha, K.; Bourdin, A.; Vernisse, C.; Garulli, C.; Tummino, C.; Charriot, J.; Vachier, I.; Suehs, C.; Chanez, P.; Gras, D. Goblet cell hyperplasia as a feature of neutrophilic asthma. Clin. Exp. Allergy 2019, 49, 781–788. [Google Scholar] [CrossRef]
- Tanabe, T.; Rubin, B.K. Airway goblet cells secrete pro-inflammatory cytokines, chemokines, and growth factors. Chest 2016, 149, 714–720. [Google Scholar] [CrossRef]
- Kanoh, S.; Tanabe, T.; Rubin, B.K. IL-13-induced Muc5AC production and goblet cell differentiation is steroid resistant in human airway cells. Clin. Exp. Allergy 2011, 41, 1747–1756. [Google Scholar] [CrossRef]
- Fatani, S.H. Biomarkers of oxidative stress in acute and chronic bronchial asthma. J. Asthma 2014, 51, 578–584. [Google Scholar] [CrossRef]
- Teskey, G.; Abrahem, R.; Cao, R.; Gyurjian, K.; Islamoglu, H.; Lucero, M.; Martinez, A.; Paredes, E.; Salaiz, O.; Robinson, B.; et al. Glutathione as a marker for human disease. Adv. Clin. Chem. 2018, 87, 141–159. [Google Scholar]
- Kleniewska, P.; Pawliczak, R. The participation of oxidative stress in the pathogenesis of bronchial asthma. Biomed. Pharmacother. 2017, 94, 100–108. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Demchuk, O.M. New perspectives for fisetin. Front. Chem. 2019, 7, 697. [Google Scholar] [CrossRef]
- Pal, H.C.; Pearlman, R.L.; Afaq, F. Fisetin and its role in chronic diseases. Adv. Exp. Med. Biol. 2016, 928, 213–244. [Google Scholar]
- Sim, H.; Choo, S.; Kim, J.; Baek, M.C.; Bae, J.S. Fisetin suppresses pulmonary inflammatory responses through heme oxygenase-1 mediated downregulation of inducible nitric oxide synthase. J. Med. Food 2020, 23, 1163–1168. [Google Scholar] [CrossRef]
- Wu, M.Y.; Hung, S.K.; Fu, S.L. Immunosuppressive effects of fisetin in ovalbumin-induced asthma through inhibition of NF-κB activity. J. Agric. Food Chem. 2011, 59, 10496–10504. [Google Scholar] [CrossRef]
- Huang, W.C.; Huang, T.H.; Yeh, K.W.; Chen, Y.L.; Shen, S.C.; Liou, C.J. Ginsenoside Rg3 ameliorates allergic airway inflammation and oxidative stress in mice. J. Ginseng Res. 2021, 45, 654–664. [Google Scholar] [CrossRef]
- Liou, C.J.; Chen, Y.L.; Yu, M.C.; Yeh, K.W.; Shen, S.C.; Huang, W.C. Sesamol alleviates airway hyperresponsiveness and oxidative stress in asthmatic mice. Antioxidants 2020, 9, 295. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.C.; Liu, C.Y.; Shen, S.C.; Chen, L.C.; Yeh, K.W.; Liu, S.H.; Liou, C.J. Protective effects of licochalcone A improve airway hyper-responsiveness and oxidative stress in a mouse model of asthma. Cells 2019, 8, 617. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.C.; Ting, N.C.; Huang, Y.L.; Chen, L.C.; Lin, C.F.; Liou, C.J. Helminthostachys zeylanica water extract ameliorates airway hyperresponsiveness and eosinophil infiltration by reducing oxidative stress and Th2 cytokine production in a mouse asthma model. Mediat. Inflamm. 2020, 2020, 1702935. [Google Scholar] [CrossRef] [PubMed]
- Liou, C.J.; Cheng, C.Y.; Yeh, K.W.; Wu, Y.H.; Huang, W.C. Protective effects of casticin from Vitex trifolia alleviate eosinophilic airway inflammation and oxidative stress in a murine asthma model. Front. Pharmacol. 2018, 9, 635. [Google Scholar] [CrossRef] [PubMed]
- Liou, C.J.; Dai, Y.W.; Wang, C.L.; Fang, L.W.; Huang, W.C. Maslinic acid protects against obesity-induced nonalcoholic fatty liver disease in mice through regulation of the sirt1/AMPK signaling pathway. FASEB J. 2019, 33, 11791–11803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, S.J.; Huang, W.C.; Yu, M.C.; Chen, Y.L.; Shen, S.C.; Yeh, K.W.; Liou, C.J. Tomatidine ameliorates obesity-induced nonalcoholic fatty liver disease in mice. J. Nutr. Biochem. 2021, 91, 108602. [Google Scholar] [CrossRef]
- Huang, W.C.; Chan, C.C.; Wu, S.J.; Chen, L.C.; Shen, J.J.; Kuo, M.L.; Chen, M.C.; Liou, C.J. Matrine attenuates allergic airway inflammation and eosinophil infiltration by suppressing eotaxin and Th2 cytokine production in asthmatic mice. J. Ethnopharmacol. 2014, 151, 470–477. [Google Scholar] [CrossRef]
- Song, W.J.; Lee, J.H.; Kang, Y.; Joung, W.J.; Chung, K.F. Future risks in patients with severe asthma. Allergy Asthma Immunol. Res. 2019, 11, 763–778. [Google Scholar] [CrossRef]
- Huang, W.; Li, M.L.; Xia, M.Y.; Shao, J.Y. Fisetin-treatment alleviates airway inflammation through inhbition of MYD88/NF-κB signaling pathway. Int. J. Mol. Med. 2018, 42, 208–218. [Google Scholar] [CrossRef] [Green Version]
- Mitra, S.; Ghosh, N.; Paul, P.; Banerjee, E.R. Orally administered fisetin reduces the symptoms of acute allergic asthma in a preclinical mouse model. Biomed. Res. Ther. 2022, 9, 4953–4970. [Google Scholar] [CrossRef]
- Paul, P.; Majhi, S.; Mitra, S.; Banerjee, E.R. Orally administered fisetin as an immuno-modulatory and therapeutic agent in a mouse model of chronic allergic airway disease. Biomed. Res. Ther. 2019, 6, 3262–3273. [Google Scholar] [CrossRef]
- Peng, H.L.; Huang, W.C.; Cheng, S.C.; Liou, C.J. Fisetin inhibits the generation of inflammatory mediators in interleukin-1β-induced human lung epithelial cells by suppressing the NF-κB and ERK1/2 pathways. Int. Immunopharmacol. 2018, 60, 202–210. [Google Scholar] [CrossRef]
- Zhang, H.; Zheng, W.; Feng, X.; Yang, F.; Qin, H.; Wu, S.; Hou, D.X.; Chen, J. Nrf2− are signaling acts as master pathway for the cellular antioxidant activity of fisetin. Molecules 2019, 24, 708. [Google Scholar] [CrossRef] [Green Version]
- Gour, N.; Wills-Karp, M. IL-4 and IL-13 signaling in allergic airway disease. Cytokine 2015, 75, 68–78. [Google Scholar] [CrossRef] [Green Version]
- Aghasafari, P.; George, U.; Pidaparti, R. A review of inflammatory mechanism in airway diseases. Inflamm. Res. 2019, 68, 59–74. [Google Scholar] [CrossRef]
- Lambrecht, B.N.; Hammad, H.; Fahy, J.V. The cytokines of asthma. Immunity 2019, 50, 975–991. [Google Scholar] [CrossRef]
- Zahiruddin, A.S.; Grant, J.A.; Sur, S. Role of epigenetics and DNA-damage in asthma. Curr. Opin. Allergy Clin. Immunol. 2018, 18, 32–37. [Google Scholar] [CrossRef]
- Bonser, L.R.; Erle, D.J. Airway mucus and asthma: The role of Muc5AC and Muc5B. J. Clin. Med. 2017, 6, 112. [Google Scholar] [CrossRef] [Green Version]
- Caminati, M.; Pham, D.L.; Bagnasco, D.; Canonica, G.W. Type 2 immunity in asthma. World Allergy Organ. J. 2018, 11, 13. [Google Scholar] [CrossRef] [Green Version]
- Persson, C. Airways exudation of plasma macromolecules: Innate defense, epithelial regeneration, and asthma. J. Allergy Clin. Immunol. 2019, 143, 1271–1286. [Google Scholar] [CrossRef] [Green Version]
- Bush, A. Cytokines and chemokines as biomarkers of future asthma. Front. Pediatr. 2019, 7, 72. [Google Scholar] [CrossRef] [Green Version]
- Goh, F.Y.; Upton, N.; Guan, S.; Cheng, C.; Shanmugam, M.K.; Sethi, G.; Leung, B.P.; Wong, W.S. Fisetin, a bioactive flavonol, attenuates allergic airway inflammation through negative regulation of NF-κB. Eur. J. Pharmacol. 2012, 679, 109–116. [Google Scholar] [CrossRef]
- Yancey, S.W.; Keene, O.N.; Albers, F.C.; Ortega, H.; Bates, S.; Bleecker, E.R.; Pavord, I. Biomarkers for severe eosinophilic asthma. J. Allergy Clin. Immunol. 2017, 140, 1509–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parulekar, A.D.; Diamant, Z.; Hanania, N.A. Role of T2 inflammation biomarkers in severe asthma. Curr. Opin. Pulm. Med. 2016, 22, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Rumzhum, N.N.; Ammit, A.J. Cyclooxygenase 2: Its regulation, role and impact in airway inflammation. Clin. Exp. Allergy 2016, 46, 397–410. [Google Scholar] [CrossRef]
- Liaras, K.; Fesatidou, M.; Geronikaki, A. Thiazoles and thiazolidinones as COX/LOX inhibitors. Molecules 2018, 23, 685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiao, W.; Kiang, J.G.; Cary, L.; Elliott, T.B.; Pellmar, T.C.; Ledney, G.D. COX-2 inhibitors are contraindicated for treatment of combined injury. Radiat. Res. 2009, 172, 686–697. [Google Scholar] [CrossRef] [PubMed]
- Weissler, J.C. Eosinophilic lung disease. Am. J. Med. Sci. 2017, 354, 339–349. [Google Scholar] [CrossRef] [PubMed]
- Athari, S.S. Targeting cell signaling in allergic asthma. Signal Transduct. Target. Ther. 2019, 4, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Wei, W.; Hong, C.; Wang, Y.; Sun, X.; Ma, J.; Zheng, F. Calreticulin induced endothelial ICAM-1 up-regulation associated with tristetraprolin expression alteration through PI3K/AKT/eNOS/p38 MAPK signaling pathway in rheumatoid arthritis. Mol. Immunol. 2019, 107, 10–20. [Google Scholar] [CrossRef]
- Huang, W.C.; Wu, L.Y.; Hu, S.; Wu, S.J. Spilanthol inhibits COX-2 and ICAM-1 expression via suppression of NF-κB and MAPK signaling in interleukin-1β-stimulated human lung epithelial cells. Inflammation 2018, 41, 1934–1944. [Google Scholar] [CrossRef]
- Menzel, M.; Ramu, S.; Calven, J.; Olejnicka, B.; Sverrild, A.; Porsbjerg, C.; Tufvesson, E.; Bjermer, L.; Akbarshahi, H.; Uller, L. Oxidative stress attenuates TLR3 responsiveness and impairs anti-viral mechanisms in bronchial epithelial cells from COPD and asthma patients. Front. Immunol. 2019, 10, 2765. [Google Scholar] [CrossRef]
- Laforgia, N.; Di Mauro, A.; Favia Guarnieri, G.; Varvara, D.; De Cosmo, L.; Panza, R.; Capozza, M.; Baldassarre, M.E.; Resta, N. The role of oxidative stress in the pathomechanism of congenital malformations. Oxid. Med. Cell. Longev. 2018, 2018, 7404082. [Google Scholar] [CrossRef]
- Drake, M.G.; Lebold, K.M.; Roth-Carter, Q.R.; Pincus, A.B.; Blum, E.D.; Proskocil, B.J.; Jacoby, D.B.; Fryer, A.D.; Nie, Z. Eosinophil and airway nerve interactions in asthma. J. Leukoc. Biol. 2018, 104, 61–67. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, S.-J.; Huang, W.-C.; Cheng, C.-Y.; Wang, M.-C.; Cheng, S.-C.; Liou, C.-J. Fisetin Suppresses the Inflammatory Response and Oxidative Stress in Bronchial Epithelial Cells. Nutrients 2022, 14, 1841. https://doi.org/10.3390/nu14091841
Wu S-J, Huang W-C, Cheng C-Y, Wang M-C, Cheng S-C, Liou C-J. Fisetin Suppresses the Inflammatory Response and Oxidative Stress in Bronchial Epithelial Cells. Nutrients. 2022; 14(9):1841. https://doi.org/10.3390/nu14091841
Chicago/Turabian StyleWu, Shu-Ju, Wen-Chung Huang, Ching-Yi Cheng, Meng-Chun Wang, Shu-Chen Cheng, and Chian-Jiun Liou. 2022. "Fisetin Suppresses the Inflammatory Response and Oxidative Stress in Bronchial Epithelial Cells" Nutrients 14, no. 9: 1841. https://doi.org/10.3390/nu14091841
APA StyleWu, S.-J., Huang, W.-C., Cheng, C.-Y., Wang, M.-C., Cheng, S.-C., & Liou, C.-J. (2022). Fisetin Suppresses the Inflammatory Response and Oxidative Stress in Bronchial Epithelial Cells. Nutrients, 14(9), 1841. https://doi.org/10.3390/nu14091841