
Time to Consider the “Exposome Hypothesis” in the Development of the Obesity Pandemic

 ,
,  , , ,
, , ,  ,
,  and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
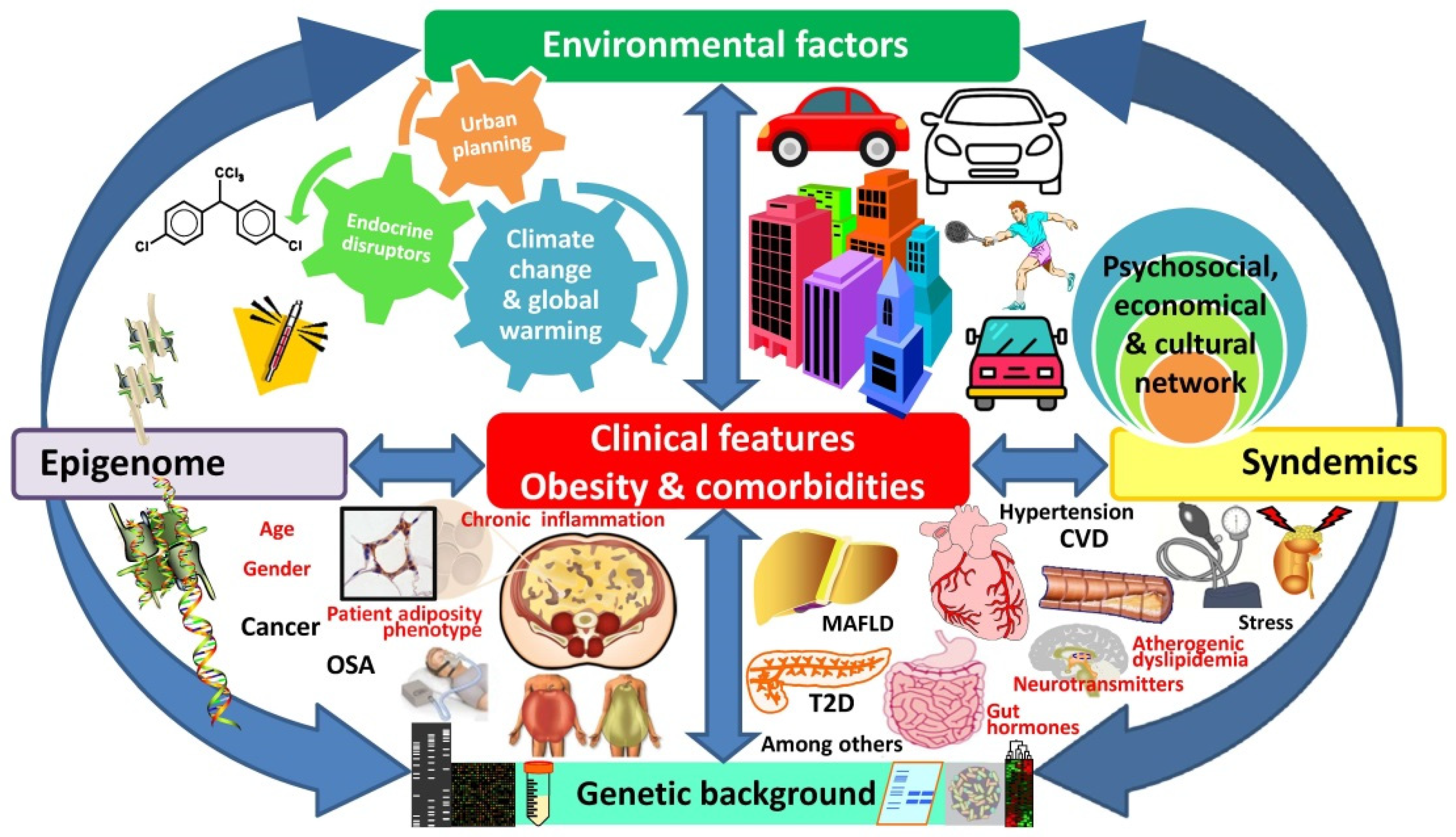{kind=link}
1. Introduction
2. Emerging Evidence Working as Warning Signs
2.1. Genetics
2.2. Microbiome
2.3. Infectobesity
2.4. Chronobiology
2.5. Endocrine Disrupters–Obesogens
2.6. Urban Planning
2.7. Climate Change
2.8. Plurality of Obesity Epidemics
3. The “Exposome” as a Plausible Underlying Mechanism of Action
Need for an Integral Consideration of the Collective Impact of Simultaneously Acting Drivers
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Finucane, M.M.; Stevens, G.A.; Cowan, M.J.; Danaei, G.; Lin, J.K.; Paciorek, C.J.; Singh, G.M.; Gutierrez, H.R.; Lu, Y.; Bahalim, A.N.; et al. National, regional, and global trends in body-mass index since 1980: Systematic analysis of health examination surveys and epidemiological studies with 960 country-years and 9.1 million participants. Lancet 2011, 377, 557–567. [Google Scholar] [CrossRef]
- NCD Risk Factor Collaboration. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: A pooled analysis of 2416 population-based measurement studies in 128.9 million children, adolescents, and adults. Lancet 2017, 390, 2627–2642. [Google Scholar] [CrossRef]
- NCD Risk Factor Collaboration. Rising rural body-mass index is the main driver of the global obesity epidemic in adults. Nature 2019, 569, 260–264. [Google Scholar] [CrossRef] [PubMed]
- Collaborators, G.R.F. Global burden of 87 risk factors in 204 countries and territories, 1990–2019: A systematic analysis for the Global Burden of Disease Study 2019. Lancet 2020, 396, 1223–1249. [Google Scholar]
- Hill, J.O. Understanding and addressing the epidemic of obesity: An energy balance perspective. Endocr. Rev. 2006, 27, 750–761. [Google Scholar] [CrossRef]
- Loos, R.J.F.; Yeo, G.S.H. The genetics of obesity: From discovery to biology. Nat. Rev. Genet. 2022, 23, 120–133. [Google Scholar] [CrossRef]
- McGee, S.L.; Hargreaves, M. Exercise adaptations: Molecular mechanisms and potential targets for therapeutic benefit. Nat. Rev. Endocrinol. 2020, 16, 495–505. [Google Scholar] [CrossRef]
- Dixon, B.N.; Ugwoaba, U.A.; Brockmann, A.N.; Ross, K.M. Associations between the built environment and dietary intake, physical activity, and obesity: A scoping review of reviews. Obes. Rev. 2021, 22, e13171. [Google Scholar] [CrossRef]
- Woessner, M.N.; Tacey, A.; Levinger-Limor, A.; Parker, A.G.; Levinger, P.; Levinger, I. The evolution of technology and physical inactivity: The good, the bad, and the way forward. Front. Public Health 2021, 9, 655491. [Google Scholar] [CrossRef]
- Rodríguez, A.; Becerril, S.; Ezquerro, S.; Méndez-Giménez, L.; Frühbeck, G. Crosstalk between adipokines and myokines in fat browning. Acta. Physiol. 2017, 219, 362–381. [Google Scholar] [CrossRef]
- Prentice, A.M. Early influences on human energy regulation: Thrifty genotypes and thrifty phenotypes. Physiol. Behav. 2005, 86, 640–645. [Google Scholar] [CrossRef] [PubMed]
- Zukiewicz-Sobczak, W.; Wroblewska, P.; Zwolinski, J.; Chmielewska-Badora, J.; Adamczuk, P.; Krasowska, E.; Zagorski, J.; Oniszczuk, A.; Piatek, J.; Silny, W. Obesity and poverty paradox in developed countries. Ann. Agric. Environ. Med. 2014, 21, 590–594. [Google Scholar] [CrossRef] [PubMed]
- Speliotes, E.K.; Willer, C.J.; Berndt, S.I.; Monda, K.L.; Thorleifsson, G.; Jackson, A.U.; Allen, H.L.; Lindgren, C.M.; Luan, J.; Magi, R.; et al. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat. Genet. 2010, 42, 937–948. [Google Scholar] [CrossRef]
- Li, S.; Zhao, J.H.; Luan, J.; Luben, R.N.; Rodwell, S.A.; Khaw, K.T.; Ong, K.K.; Wareham, N.J.; Loos, R.J. Cumulative effects and predictive value of common obesity-susceptibility variants identified by genome-wide association studies. Am. J. Clin. Nutr. 2010, 91, 184–190. [Google Scholar] [CrossRef] [PubMed]
- Bray, M.S.; Loos, R.J.; McCaffery, J.M.; Ling, C.; Franks, P.W.; Weinstock, G.M.; Snyder, M.P.; Vassy, J.L.; Agurs-Collins, T. NIH working group report-using genomic information to guide weight management: From universal to precision treatment. Obesity 2016, 24, 14–22. [Google Scholar] [CrossRef]
- Wahl, S.; Drong, A.; Lehne, B.; Loh, M.; Scott, W.R.; Kunze, S.; Tsai, P.C.; Ried, J.S.; Zhang, W.; Yang, Y.; et al. Epigenome-wide association study of body mass index, and the adverse outcomes of adiposity. Nature 2017, 541, 81–86. [Google Scholar] [CrossRef]
- Graham, S.E.; Clarke, S.L.; Wu, K.H.; Kanoni, S.; Zajac, G.J.M.; Ramdas, S.; Surakka, I.; Ntalla, I.; Vedantam, S.; Winkler, T.W.; et al. The power of genetic diversity in genome-wide association studies of lipids. Nature 2021, 600, 675–679. [Google Scholar] [CrossRef]
- Christakis, N.A.; Fowler, J.H. The spread of obesity in a large social network over 32 years. N. Engl. J. Med. 2007, 357, 370–379. [Google Scholar] [CrossRef]
- Cole, T.J.; Power, C.; Moore, G.E. Intergenerational obesity involves both the father and the mother. Am. J. Clin. Nutr. 2008, 87, 1535–1536. [Google Scholar] [CrossRef]
- Pollard, T.M.; Rousham, E.K.; Colls, R. Intergenerational and familial approaches to obesity and related conditions. Ann. Hum. Biol. 2011, 38, 385–389. [Google Scholar] [CrossRef]
- Adamo, K.B.; Ferraro, Z.M.; Brett, K.E. Can we modify the intrauterine environment to halt the intergenerational cycle of obesity? Int. J. Environ. Res. Public Health 2012, 9, 1263–1307. [Google Scholar] [CrossRef] [PubMed]
- Johnson, P.C.; Logue, J.; McConnachie, A.; Abu-Rmeileh, N.M.; Hart, C.; Upton, M.N.; Lean, M.; Sattar, N.; Watt, G. Intergenerational change and familial aggregation of body mass index. Eur. J. Epidemiol. 2012, 27, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Mohajer, N.; Joloya, E.M.; Seo, J.; Shioda, T.; Blumberg, B. Epigenetic transgenerational inheritance of the effects of obesogen exposure. Front. Endocrinol. 2021, 12, 787580. [Google Scholar] [CrossRef]
- Ling, C.; Rönn, T. Epigenetics in human obesity and type 2 diabetes. Cell Metab. 2019, 29, 1028–1044. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.-F.; Lin, R.C.Y.; Laybutt, D.R.; Barres, R.; Owens, J.A.; Morris, M.J. Chronic high-fat diet in fathers programs b-cell dysfunction in female rat offspring. Nature 2010, 467, 963–966. [Google Scholar] [CrossRef] [PubMed]
- Carone, B.R.; Fauquier, L.; Habib, N.; Shea, J.M.; Hart, C.E.; Li, R.; Bock, C.; Li, C.; Gu, H.; Zamore, P.D.; et al. Paternally induced transgenerational environmental reprogramming of metabolic gene expression in mammals. Cell 2010, 143, 1084–1096. [Google Scholar] [CrossRef] [PubMed]
- Donkin, I.; Versteyhe, S.; Ingerslev, L.R.; Qian, K.; Mechta, M.; Nordkap, L.; Mortensen, B.; Appel, E.V.; Jorgensen, N.; Kristiansen, V.B.; et al. Obesity and bariatric surgery drive epigenetic variation of spermatozoa in humans. Cell Metab. 2016, 23, 369–378. [Google Scholar] [CrossRef]
- McAllister, E.J.; Dhurandhar, N.V.; Keith, S.W.; Aronne, L.J.; Barger, J.; Baskin, M.; Benca, R.M.; Biggio, J.; Boggiano, M.M.; Eisenmann, J.C.; et al. Ten putative contributors to the obesity epidemic. Crit. Rev. Food Sci. Nutr. 2009, 49, 868–913. [Google Scholar] [CrossRef]
- Faienza, M.F.; Wang, D.Q.; Frühbeck, G.; Garruti, G.; Portincasa, P. The dangerous link between childhood and adulthood predictors of obesity and metabolic syndrome. Intern. Emerg. Med. 2016, 11, 175–182. [Google Scholar] [CrossRef]
- Heitmann, B.L.; Westerterp, K.R.; Loos, R.J.; Sørensen, T.I.; O’Dea, K.; McLean, P.; Jensen, T.K.; Eisenmann, J.; Speakman, J.R.; Simpson, S.J.; et al. Obesity: Lessons from evolution and the environment. Obes. Rev. 2012, 13, 910–922. [Google Scholar] [CrossRef]
- Speakman, J.R.; Djafarian, K.; Stewart, J.; Jackson, D.M. Assortative mating for obesity. Am. J. Clin. Nutr. 2007, 86, 316–323. [Google Scholar] [CrossRef] [PubMed]
- Ajslev, T.A.; Angquist, L.; Silventoinen, K.; Gamborg, M.; Allison, D.B.; Baker, J.L.; Sorensen, T.I. Assortative marriages by body mass index have increased simultaneously with the obesity epidemic. Front. Genet. 2012, 3, 125. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ley, R.E.; Mahowald, M.A.; Magrini, V.; Mardis, E.R.; Gordon, J.I. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006, 444, 1027–1131. [Google Scholar] [CrossRef] [PubMed]
- Pinart, M.; Dötsch, A.; Schlicht, K.; Laudes, M.; Bouwman, J.; Forslund, S.K.; Pischon, T.; Nimptsch, K. Gut microbiome composition in obese and non-obese persons: A systematic review and meta-analysis. Nutrients 2021, 14, 12. [Google Scholar] [CrossRef]
- Furet, J.P.; Kong, L.C.; Tap, J.; Poitou, C.; Basdevant, A.; Bouillot, J.L.; Mariat, D.; Corthier, G.; Dore, J.; Henegar, C.; et al. Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss. Links with metabolic and low-grade inflammation markers. Diabetes 2010, 59, 3049–3057. [Google Scholar] [CrossRef]
- Liou, A.P.; Paziuk, M.; Luevano, J.M., Jr.; Machineni, S.; Turnbaugh, P.J.; Kaplan, L.M. Conserved shifts in the gut microbiota due to gastric bypass reduce host weight and adiposity. Sci. Transl. Med. 2013, 5, 178ra141. [Google Scholar] [CrossRef]
- Ciobarca, D.; Catoi, A.F.; Copaescu, C.; Miere, D.; Crisan, G. Bariatric surgery in obesity: Effects on gut microbiota and micronutrient status. Nutrients 2020, 12, 235. [Google Scholar] [CrossRef]
- Michels, N.; Zouiouich, S.; Vanderbauwhede, B.; Vanacker, J.; Indave Ruiz, B.I.; Huybrechts, I. Human microbiome and metabolic health: An overview of systematic reviews. Obes. Rev. 2022, 3, e13409. [Google Scholar] [CrossRef]
- Le Roy, T.; Moens de Hase, E.; Van Hul, M.; Paquot, A.; Pelicaen, R.; Régnier, M.; Depommier, C.; Druart, C.; Everard, A.; Maiter, D.; et al. Dysosmobacter welbionis is a newly isolated human commensal bacterium preventing diet-induced obesity and metabolic disorders in mice. Gut 2022, 71, 534–543. [Google Scholar] [CrossRef]
- Snedeker, S.M.; Hay, A.G. Do interactions between gut ecology and environmental chemicals contribute to obesity and diabetes? Environ. Health Perspect. 2012, 120, 332–339. [Google Scholar] [CrossRef] [PubMed]
- Henao-Mejia, J.; Elinav, E.; Jin, C.; Hao, L.; Mehal, W.Z.; Strowig, T.; Thaiss, C.A.; Kau, A.L.; Eisenbarth, S.C.; Jurczak, M.J.; et al. Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity. Nature 2012, 482, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P.; Bonfrate, L.; Khalil, M.; Angelis, M.; Calabrese, F.M.; D’Amato, M.; Wang, D.Q.; Di Ciaula, A. Intestinal barrier and permeability in health, obesity and NAFLD. Biomedicines 2021, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Greenblum, S.; Turnbaugh, P.J.; Borenstein, E. Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc. Natl. Acad. Sci. USA 2012, 109, 594–599. [Google Scholar] [CrossRef] [PubMed]
- Genoni, G.; Prodam, F.; Marolda, A.; Giglione, E.; Demarchi, I.; Bellone, S.; Bona, G. Obesity and infection: Two sides of one coin. Eur. J. Pediatr. 2014, 173, 25–32. [Google Scholar] [CrossRef] [PubMed]
- Green, W.D.; Beck, M.A. Obesity altered T cell metabolism and the response to infection. Curr. Opin. Immunol. 2017, 46, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Frühbeck, G.; Baker, J.L.; Busetto, L.; Dicker, D.; Goossens, G.H.; Halford, J.C.G.; Handjieva-Darlenska, T.; Hassapidou, M.; Holm, J.C.; Lehtinen-Jacks, S.; et al. European Association for the Study of Obesity Position Statement on the Global COVID-19 Pandemic. Obes. Facts 2020, 13, 292–296. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Piernas, C.; Astbury, N.M.; Hippisley-Cox, J.; O’Rahilly, S.; Aveyard, P.; Jebb, S.A. Associations between body-mass index and COVID-19 severity in 6·9 million people in England: A prospective, community-based, cohort study. Lancet Diabetes Endocrinol. 2021, 9, 350–359. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Krawczyk, M.; Filipiak, K.J.; Geier, A.; Bonfrate, L.; Portincasa, P. Noncommunicable diseases, climate change and iniquities: What COVID-19 has taught us about syndemic. Eur. J. Clin. Investig. 2021, 51, e13682. [Google Scholar] [CrossRef]
- Dhurandhar, N.V. Infectobesity: Obesity of infectious origin. J. Nutr. 2001, 131, 2794S–2797S. [Google Scholar] [CrossRef]
- Pasarica, M.; Dhurandhar, N.V. Infectobesity: Obesity of infectious origin. Adv. Food Nutr. Res. 2007, 52, 61–102. [Google Scholar]
- van Ginneken, V.; Sitnyakowsky, L.; Jeffery, J.E. Infectobesity: Viral infections (especially with human adenovirus-36: Ad-36) may be a cause of obesity. Med. Hypotheses 2009, 72, 383–388. [Google Scholar] [CrossRef]
- Voss, J.D.; Dhurandhar, N.V. Viral infections and obesity. Curr. Obes. Rep. 2017, 6, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Kanneganti, T.D.; Dixit, V.D. Immunological complications of obesity. Nat. Immunol. 2012, 13, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Dhurandhar, N.V.; Bailey, D.; Thomas, D. Interaction of obesity and infections. Obes. Rev. 2015, 16, 1017–1029. [Google Scholar] [CrossRef] [PubMed]
- Honce, R.; Schultz-Cherry, S. Impact of Obesity on Influenza A Virus Pathogenesis, Immune Response, and Evolution. Front. Immunol. 2019, 10, 1071. [Google Scholar] [CrossRef]
- Sheridan, P.A.; Paich, H.A.; Handy, J.; Karlsson, E.A.; Hudgens, M.G.; Sammon, A.B.; Holland, L.A.; Weir, S.; Noah, T.L.; Beck, M.A. Obesity is associated with impaired immune response to influenza vaccination in humans. Int. J. Obes. 2012, 36, 1072–1077. [Google Scholar] [CrossRef] [PubMed]
- Dicker, D.; Golan, R.; Baker, J.L.; Busetto, L.; Frühbeck, G.; Goossens, G.H.; Halford, J.C.G.; Holm, J.C.; Woodward, E.; Farpour-Lambert, N.J. Vaccinating people with obesity for COVID-19: EASO call for action. Obes. Facts 2021, 14, 334–335. [Google Scholar] [CrossRef] [PubMed]
- Lajunen, T.; Bloigu, A.; Paldanius, M.; Pouta, A.; Laitinen, J.; Ruokonen, A.; Hartikainen, A.L.; Savolainen, M.; Herzig, K.H.; Leinonen, M.; et al. The association of body mass index, waist and hip circumference, and waist-hip ratio with Chlamydia pneumoniae IgG antibodies and high-sensitive C-reactive protein at 31 years of age in Northern Finland Birth Cohort 1966. Int. J. Obes. 2011, 35, 1470–1478. [Google Scholar] [CrossRef][Green Version]
- Rubicz, R.; Leach, C.T.; Kraig, E.; Dhurandhar, N.V.; Grubbs, B.; Blangero, J.; Yolken, R.; Göring, H.H. Seroprevalence of 13 common pathogens in a rapidly growing U.S. minority population: Mexican Americans from San Antonio, TX. BMC Res. Notes 2011, 4, 433. [Google Scholar] [CrossRef]
- Dhurandhar, N.V. A framework for identification of infections that contribute to human obesity. Lancet Infect. Dis. 2011, 11, 963–969. [Google Scholar] [CrossRef]
- Na, H.N.; Nam, J.H. Adenovirus 36 as an obesity agent maintains the obesity state by increasing MCP-1 and inducing inflammation. J. Infect. Dis. 2012, 205, 914–922. [Google Scholar] [CrossRef] [PubMed]
- Na, H.N.; Kim, H.; Nam, J.H. Novel genes and cellular pathways related to infection with adenovirus-36 as an obesity agent in human mesenchymal stem cells. Int. J. Obes. 2012, 36, 195–200. [Google Scholar] [CrossRef]
- Tarantino, G.; Citro, V.; Cataldi, M. Findings from studies are congruent with obesity having a viral origin, but what about obesity-related NAFLD? Viruses 2021, 13, 1285. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Calamita, G.; Shanmugam, H.; Khalil, M.; Bonfrate, L.; Wang, D.Q.; Baffy, G.; Portincasa, P. Mitochondria matter: Systemic aspects of nonalcoholic fatty liver disease (NAFLD) and diagnostic assessment of liver function by stable isotope dynamic breath tests. Int. J. Mol. Sci. 2021, 22, 7702. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Passarella, S.; Shanmugam, H.; Noviello, M.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Nonalcoholic fatty liver disease (NAFLD). Mitochondria as players and targets of therapies? Int. J. Mol. Sci. 2021, 22, 5375. [Google Scholar] [CrossRef] [PubMed]
- Baldini, F.; Fabbri, R.; Eberhagen, C.; Voci, A.; Portincasa, P.; Zischka, H.; Vergani, L. Adipocyte hypertrophy parallels alterations of mitochondrial status in a cell model for adipose tissue dysfunction in obesity. Life Sci. 2021, 15, 118812. [Google Scholar] [CrossRef]
- Santos, J.P.M.D.; Maio, M.C.; Lemes, M.A.; Laurindo, L.F.; Haber, J.F.D.S.; Bechara, M.D.; Prado, P.S.D.J.; Rauen, E.C.; Costa, F.; Pereira, B.C.A.; et al. Non-alcoholic steatohepatitis (NASH) and organokines: What is now and what will be in the future. Int. J. Mol. Sci. 2022, 23, 498. [Google Scholar] [CrossRef]
- Gallego-Escuredo, J.M.; Gómez-Ambrosi, J.; Catalán, V.; Domingo, P.; Giralt, M.; Frühbeck, G.; Villarroya, F. Opposite alterations in FGF21 and FGF19 levels and disturbed expression of the receptor machinery for endocrine FGFs in obese patients. Int. J. Obes. 2015, 39, 121–129. [Google Scholar] [CrossRef]
- Gomez-Ambrosi, J.; Gallego-Escuredo, J.M.; Catalan, V.; Rodriguez, A.; Domingo, P.; Moncada, R.; Valenti, V.; Salvador, J.; Giralt, M.; Villarroya, F.; et al. FGF19 and FGF21 serum concentrations in human obesity and type 2 diabetes behave differently after diet- or surgically-induced weight loss. Clin. Nutr. 2017, 36, 861–868. [Google Scholar] [CrossRef]
- Basolo, A.; Bechi Genzano, S.; Piaggi, P.; Krakoff, J.; Santini, F. Energy balance and control of body weight: Possible effects of meal timing and circadian rhythm dysregulation. Nutrients 2021, 13, 3276. [Google Scholar] [CrossRef]
- Fatima, N.; Rana, S. Metabolic implications of circadian disruption. Pflug. Arch. 2020, 472, 513–526. [Google Scholar] [CrossRef] [PubMed]
- Karatsoreos, I.N.; Bhagat, S.; Bloss, E.B.; Morrison, J.H.; McEwen, B.S. Disruption of circadian clocks has ramifications for metabolism, brain, and behavior. Proc. Natl. Acad. Sci. USA 2011, 108, 1657–1662. [Google Scholar] [CrossRef] [PubMed]
- Spiegel, K.; Tasali, E.; Leproult, R.; Van Cauter, E. Effects of poor and short sleep on glucose metabolism and obesity risk. Nat. Rev. Endocrinol. 2009, 9, 253–261. [Google Scholar] [CrossRef]
- Tasali, E.; Leproult, R.; Spiegel, K. Reduced sleep duration or quality: Relationships with insulin resistance and type 2 diabetes. Prog. Cardiovasc. Dis. 2009, 51, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Fonken, L.K.; Workman, J.L.; Walton, J.C.; Weil, Z.M.; Morris, J.S.; Haim, A.; Nelson, R.J. Light at night increases body mass by shifting the time of food intake. Proc. Natl. Acad. Sci. USA 2010, 107, 18664–18669. [Google Scholar] [CrossRef]
- Spiegel, K.; Tasali, E.; Leproult, R.; Scherberg, N.; Van Cauter, E. Twenty-four-hour profiles of acylated and total ghrelin: Relationship with glucose levels and impact of time of day and sleep. J. Clin. Endocrinol. Metab. 2011, 96, 486–493. [Google Scholar] [CrossRef]
- Nedeltcheva, A.V.; Kilkus, J.M.; Imperial, J.; Schoeller, D.A.; Penev, P.D. Insufficient sleep undermines dietary efforts to reduce adiposity. Ann. Intern. Med. 2010, 153, 435–441. [Google Scholar] [CrossRef]
- Vgontzas, A.N.; Zoumakis, M.; Bixler, E.O.; Lin, H.M.; Prolo, P.; Vela-Bueno, A.; Kales, A.; Chrousos, G.P. Impaired nighttime sleep in healthy old versus young adults is associated with elevated plasma interleukin-6 and cortisol levels: Physiologic and therapeutic implications. J. Clin. Endocrinol. Metab. 2003, 88, 2087–2095. [Google Scholar] [CrossRef]
- Kritikou, I.; Basta, M.; Tappouni, R.; Pejovic, S.; Fernandez-Mendoza, J.; Nazir, R.; Shaffer, M.; Liao, D.; Bixler, E.; Chrousos, G.; et al. Sleep apnoea and visceral adiposity in middle-aged males and females. Eur. Respir. J. 2013, 41, 601–609. [Google Scholar] [CrossRef]
- Lorton, D.; Lubahn, C.L.; Estus, C.; Millar, B.A.; Carter, J.L.; Wood, C.A.; Bellinger, D.L. Bidirectional communication between the brain and the immune system: Implications for physiological sleep and disorders with disrupted sleep. Neuroimmunomodulation 2006, 13, 357–374. [Google Scholar] [CrossRef]
- van der Valk, E.S.; Savas, M.; van Rossum, E.F.C. Stress and obesity: Are there more susceptible individuals? Curr. Obes. Rep. 2018, 7, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Tasali, E.; Wroblewski, K.; Kahn, E.; Kilkus, J.; Schoeller, D.A. Effect of sleep extension on objectively assessed energy intake among adults with overweight in real-life settings: A randomized clinical trial. JAMA Intern. Med. 2022, 182, 365–374. [Google Scholar] [CrossRef] [PubMed]
- Plagemann, A. Perinatal programming and functional teratogenesis: Impact on body weight regulation and obesity. Physiol. Behav. 2005, 86, 661–668. [Google Scholar] [CrossRef] [PubMed]
- Lindblom, R.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. The early life origin theory in the development of cardiovascular disease and type 2 diabetes. Mol. Biol. Rep. 2015, 42, 791–797. [Google Scholar] [CrossRef] [PubMed]
- Heindel, J.J.; Newbold, R.; Schug, T.T. Endocrine disruptors and obesity. Nat. Rev. Endocrinol. 2015, 11, 653–661. [Google Scholar] [CrossRef] [PubMed]
- Azoulay, L.; Bouvattier, C.; Christin-Maitre, S. Impact of intra-uterine life on future health. Ann. Endocrinol. 2022, 83, 54–58. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Portincasa, P. Fat, epigenome and pancreatic diseases. Interplay and common pathways from a toxic and obesogenic environment. Eur. J. Intern. Med. 2014, 25, 865–873. [Google Scholar] [CrossRef]
- Haverinen, E.; Fernandez, M.F.; Mustieles, V.; Tolonen, H. Metabolic syndrome and endocrine disrupting chemicals: An overview of exposure and health effects. Int. J. Environ. Res. Public Health 2021, 18, 13047. [Google Scholar] [CrossRef]
- Heindel, J.J.; Blumberg, B. Environmental obesogens: Mechanisms and controversies. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 89–106. [Google Scholar] [CrossRef]
- Sun, J.; Fang, R.; Wang, H.; Xu, D.X.; Yang, J.; Huang, X.; Cozzolino, D.; Fang, M.; Huang, Y. A review of environmental metabolism disrupting chemicals and effect biomarkers associating disease risks: Where exposomics meets metabolomics. Environ. Int. 2022, 158, 106941. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Portincasa, P. Diet and contaminants: Driving the rise to obesity epidemics? Curr Med Chem 2019, 26, 3471–3482. [Google Scholar] [CrossRef] [PubMed]
- Grun, F.; Blumberg, B. Endocrine disrupters as obesogens. Mol. Cell. Endocrinol. 2009, 304, 19–29. [Google Scholar] [CrossRef] [PubMed]
- Grun, F.; Blumberg, B. Minireview: The case for obesogens. Mol. Endocrinol. 2009, 23, 1127–1134. [Google Scholar] [CrossRef] [PubMed]
- Boudalia, S.; Bousbia, A.; Boumaaza, B.; Oudir, M.; Canivenc Lavier, M.C. Relationship between endocrine disruptors and obesity with a focus on bisphenol A: A narrative review. Bioimpacts 2021, 11, 289–300. [Google Scholar] [CrossRef]
- Elobeid, M.A.; Padilla, M.A.; Brock, D.W.; Ruden, D.M.; Allison, D.B. Endocrine disruptors and obesity: An examination of selected persistent organic pollutants in the NHANES 1999–2002 data. Int. J. Environ. Res. Public Health 2010, 7, 2988–3005. [Google Scholar] [CrossRef]
- Brulport, A.; Le Corre, L.; Chagnon, M.C. Chronic exposure of 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) induces an obesogenic effect in C57BL/6J mice fed a high fat diet. Toxicology 2017, 390, 43–52. [Google Scholar] [CrossRef]
- Guo, J.; Ito, S.; Nguyen, H.T.; Yamamoto, K.; Tanoue, R.; Kunisue, T.; Iwata, H. Effects of prenatal exposure to triclosan on the liver transcriptome in chicken embryos. Toxicol. Appl. Pharmacol. 2018, 347, 23–32. [Google Scholar] [CrossRef]
- Frühbeck, G.; Méndez-Giménez, L.; Fernández-Formoso, J.A.; Fernández, S.; Rodríguez, A. Regulation of adipocyte lipolysis. Nutr. Res. Rev. 2014, 27, 63–93. [Google Scholar] [CrossRef]
- Frühbeck, G.; Gómez Ambrosi, J.; Salvador, J. Leptin-induced lipolysis opposes the tonic inhibition of endogenous adenosine in white adipocytes. FASEB J. 2001, 15, 333–340. [Google Scholar] [CrossRef]
- Frühbeck, G.; Gómez-Ambrosi, J. Modulation of the leptin-induced white adipose tissue lipolysis by nitric oxide. Cell Signal. 2001, 13, 827–833. [Google Scholar] [CrossRef]
- Muruzábal, F.J.; Frühbeck, G.; Gómez-Ambrosi, J.; Archanco, M.; Burrell, M.A. Immunocytochemical detection of leptin in non-mammalian vertebrate stomach. Gen. Comp. Endocrinol. 2002, 128, 149–152. [Google Scholar] [CrossRef]
- Moreno-Navarrete, J.M.; Martínez-Barricarte, R.; Catalán, V.; Sabater, M.; Gómez-Ambrosi, J.; Ortega, F.J.; Ricart, W.; Blüher, M.; Frühbeck, G.; de Córdoba, S.R.; et al. Complement Factor H is expressed in adipose tissue in association with insulin resistance. Diabetes 2010, 59, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Pulido, M.R.; Diaz-Ruiz, A.; Jimenez-Gomez, Y.; Garcia-Navarro, S.; Gracia-Navarro, F.; Tinahones, F.; Lopez-Miranda, J.; Frühbeck, G.; Vazquez-Martinez, R.; Malagon, M.M. Rab18 dynamics in adipocytes in relation to lipogenesis, lipolysis and obesity. PLoS ONE 2011, 6, e22931. [Google Scholar] [CrossRef] [PubMed]
- Catalán, V.; Gómez-Ambrosi, J.; Rodríguez, A.; Ramírez, B.; Rotellar, F.; Valentí, V.; Silva, C.; Gil, M.J.; Fernández-Real, J.M.; Salvador, J.; et al. Increased levels of calprotectin in obesity are related to macrophage content: Impact on inflammation and effect of weight loss. Mol. Med. 2011, 17, 1157–1167. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez, A.; Gómez-Ambrosi, J.; Catalán, V.; Rotellar, F.; Valentí, V.; Silva, C.; Mugueta, C.; Pulido, M.R.; Vázquez, R.; Salvador, J.; et al. The ghrelin O-acyltransferase-ghrelin system reduces TNF-a-induced apoptosis and autophagy in human visceral adipocytes. Diabetologia 2012, 55, 3038–3050. [Google Scholar] [CrossRef] [PubMed]
- Calamita, G.; Delporte, C. Involvement of aquaglyceroporins in energy metabolism in health and disease. Biochimie 2021, 188, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Frühbeck, G. Obesity: Aquaporin enters the picture. Nature 2005, 438, 436–437. [Google Scholar] [CrossRef]
- Francis, C.E.; Allee, L.; Nguyen, H.; Grindstaff, R.D.; Miller, C.N.; Rayalam, S. Endocrine disrupting chemicals: Friend or foe to brown and beige adipose tissue? Toxicology 2021, 463, 152972. [Google Scholar] [CrossRef]
- Ohanyan, H.; Portengen, L.; Huss, A.; Traini, E.; Beulens, J.W.J.; Hoek, G.; Lakerveld, J.; Vermeulen, R. Machine learning approaches to characterize the obesogenic urban exposome. Environ. Int. 2022, 158, 107015. [Google Scholar] [CrossRef]
- Guo, B.; Guo, Y.; Nima, Q.; Feng, Y.; Wang, Z.; Lu, R.; Baimayangji; Ma, Y.; Zhou, J.; Xu, H.; et al. Exposure to air pollution is associated with an increased risk of metabolic dysfunction-associated fatty liver disease. J. Hepatol. 2022, 76, 518–525. [Google Scholar] [CrossRef]
- Koch, C.A.; Sharda, P.; Patel, J.; Gubbi, S.; Bansal, R.; Bartel, M.J. Climate change and obesity. Horm. Metab. Res. 2021, 53, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M. Reassessing human adipose tissue. N. Engl. J. Med. 2022, 386, 768–779. [Google Scholar] [CrossRef] [PubMed]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.H.; Doria, A.; et al. Identification and importance of brown adipose tissue in adult humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef] [PubMed]
- Frühbeck, G.; Becerril, S.; Sáinz, N.; Garrastachu, P.; García-Velloso, M.J. BAT: A new target for human obesity? Trends Pharmacol. Sci. 2009, 30, 387–396. [Google Scholar] [CrossRef]
- Tseng, Y.-H.; Cypess, A.M.; Kahn, C.R. Cellular bioenergetics as a target for obesity therapy. Nat. Rev. Drug. Discov. 2010, 9, 465–481. [Google Scholar] [CrossRef]
- Xue, R.; Lynes, M.D.; Dreyfuss, J.M.; Shamsi, F.; Schulz, T.J.; Zhang, H.; Huang, T.L.; Townsend, K.L.; Li, Y.; Takahashi, H.; et al. Clonal analyses and gene profiling identify genetic biomarkers of the thermogenic potential of human brown and white preadipocytes. Nat. Med. 2015, 21, 760–768. [Google Scholar] [CrossRef]
- Shinoda, K.; Luijten, I.H.; Hasegawa, Y.; Hong, H.; Sonne, S.B.; Kim, M.; Xue, R.; Chondronikola, M.; Cypess, A.M.; Tseng, Y.H.; et al. Genetic and functional characterization of clonally derived adult human brown adipocytes. Nat. Med. 2015, 21, 389–394. [Google Scholar] [CrossRef]
- Sakers, A.; De Siqueira, M.K.; Seale, P.; Villanueva, C.J. Adipose-tissue plasticity in health and disease. Cell 2022, 185, 419–446. [Google Scholar] [CrossRef]
- Shamsi, F.; Wang, C.H.; Tseng, Y.H. The evolving view of thermogenic adipocytes—ontogeny, niche and function. Nat. Rev. Endocrinol. 2021, 17, 726–744. [Google Scholar] [CrossRef]
- Symonds, M.E.; Farhat, G.; Aldiss, P.; Pope, M.; Budge, H. Brown adipose tissue and glucose homeostasis—the link between climate change and the global rise in obesity and diabetes. Adipocyte 2019, 8, 46–50. [Google Scholar] [CrossRef]
- Gaspar, R.C.; Pauli, J.R.; Shulman, G.I.; Muñoz, V.R. An update on brown adipose tissue biology: A discussion of recent findings. Am. J. Physiol. Endocrinol. Metab. 2021, 320, E488–E495. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.T.; Stanford, K.I. Batokines: Mediators of inter-tissue communication (a mini-review). Curr. Obes. Rep. 2022. online ahead of print. [Google Scholar] [CrossRef]
- Gavaldà-Navarro, A.; Villarroya, J.; Cereijo, R.; Giralt, M.; Villarroya, F. The endocrine role of brown adipose tissue: An update on actors and actions. Rev. Endocr. Metab. Disord. 2022, 23, 31–41. [Google Scholar] [CrossRef] [PubMed]
- Lynes, M.D.; Leiria, L.O.; Lundh, M.; Bartelt, A.; Shamsi, F.; Huang, T.L.; Takahashi, H.; Hirshman, M.F.; Schlein, C.; Lee, A.; et al. The cold-induced lipokine 12,13-diHOME promotes fatty acid transport into brown adipose tissue. Nat. Med. 2017, 23, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Macedo, A.P.A.; Munoz, V.R.; Cintra, D.E.; Pauli, J.R. 12,13-diHOME as a new therapeutic target for metabolic diseases. Life Sci. 2022, 290, 120229. [Google Scholar] [CrossRef] [PubMed]
- Frühbeck, G.; Gómez Ambrosi, J. Rationale for the existence of additional adipostatic hormones. FASEB J. 2001, 15, 1996–2006. [Google Scholar] [CrossRef] [PubMed]
- Landecho, M.F.; Tuero, C.; Valentí, V.; Bilbao, I.; de la Higuera, M.; Frühbeck, G. Relevance of leptin and other adipokines in obesity-associated cardiovascular risk. Nutrients 2019, 11, 2664. [Google Scholar] [CrossRef]
- Klimentidis, Y.C.; Beasley, T.M.; Lin, H.Y.; Murati, G.; Glass, G.E.; Guyton, M.; Newton, W.; Jorgensen, M.; Heymsfield, S.B.; Kemnitz, J.; et al. Canaries in the coal mine: A cross-species analysis of the plurality of obesity epidemics. Proc. Biol. Sci. 2011, 278, 1626–1632. [Google Scholar] [CrossRef]
- Rappaport, S.M.; Smith, M.T. Epidemiology. Environment and disease risks. Science 2010, 330, 460–461. [Google Scholar] [CrossRef]
- Zhang, P.; Carlsten, C.; Chaleckis, R.; Hanhineva, K.; Huang, M.; Isobe, T.; Koistinen, V.M.; Meister, I.; Papazian, S.; Sdougkou, K.; et al. Defining the scope of exposome studies and research needs from a multidisciplinary perspective. Environ. Sci. Technol. Lett. 2021, 8, 839–852. [Google Scholar] [CrossRef]
- Fang, M.; Hu, L.; Chen, D.; Guo, Y.; Liu, J.; Lan, C.; Gong, J.; Wang, B. Exposome in human health: Utopia or wonderland? Innovation 2021, 2, 100172. [Google Scholar] [CrossRef]
- Wild, C.P. Complementing the genome with an "exposome": The outstanding challenge of environmental exposure measurement in molecular epidemiology. Cancer Epidemiol. Biomarks Prev. 2005, 14, 1847–1850. [Google Scholar] [CrossRef] [PubMed]
- Wild, C.P. The exposome: From concept to utility. Int. J. Epidemiol. 2012, 41, 24–32. [Google Scholar] [CrossRef] [PubMed]
- The, N.S.; Suchindran, C.; North, K.E.; Popkin, B.M.; Gordon-Larsen, P. Association of adolescent obesity with risk of severe obesity in adulthood. JAMA 2010, 304, 2042–2047. [Google Scholar] [CrossRef] [PubMed]
- Golding, J.; Gregory, S.; Northstone, K.; Iles-Caven, Y.; Ellis, G.; Pembrey, M. Investigating possible trans/intergenerational associations with obesity in young adults using an exposome approach. Front. Genet. 2019, 10, 314. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Perng, W.; Peterson, K.E. Precision Nutrition and Childhood Obesity: A Scoping Review. Metabolites 2020, 10, 235. [Google Scholar] [CrossRef]
- Mahmoud, A.M. An overview of epigenetics in obesity: The role of lifestyle and therapeutic interventions. Int. J. Mol. Sci. 2022, 23, 1341. [Google Scholar] [CrossRef]
- Rajpurkar, P.; Chen, E.; Banerjee, O.; Topol, E.J. AI in health and medicine. Nat. Med. 2022, 28, 31–38. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Catalán, V.; Avilés-Olmos, I.; Rodríguez, A.; Becerril, S.; Fernández-Formoso, J.A.; Kiortsis, D.; Portincasa, P.; Gómez-Ambrosi, J.; Frühbeck, G. Time to Consider the “Exposome Hypothesis” in the Development of the Obesity Pandemic. Nutrients 2022, 14, 1597. https://doi.org/10.3390/nu14081597
Catalán V, Avilés-Olmos I, Rodríguez A, Becerril S, Fernández-Formoso JA, Kiortsis D, Portincasa P, Gómez-Ambrosi J, Frühbeck G. Time to Consider the “Exposome Hypothesis” in the Development of the Obesity Pandemic. Nutrients. 2022; 14(8):1597. https://doi.org/10.3390/nu14081597
Chicago/Turabian StyleCatalán, Victoria, Iciar Avilés-Olmos, Amaia Rodríguez, Sara Becerril, José Antonio Fernández-Formoso, Dimitrios Kiortsis, Piero Portincasa, Javier Gómez-Ambrosi, and Gema Frühbeck. 2022. "Time to Consider the “Exposome Hypothesis” in the Development of the Obesity Pandemic" Nutrients 14, no. 8: 1597. https://doi.org/10.3390/nu14081597
APA StyleCatalán, V., Avilés-Olmos, I., Rodríguez, A., Becerril, S., Fernández-Formoso, J. A., Kiortsis, D., Portincasa, P., Gómez-Ambrosi, J., & Frühbeck, G. (2022). Time to Consider the “Exposome Hypothesis” in the Development of the Obesity Pandemic. Nutrients, 14(8), 1597. https://doi.org/10.3390/nu14081597