
Recent Advances in the Digestive, Metabolic and Therapeutic Effects of Farnesoid X Receptor and Fibroblast Growth Factor 19: From Cholesterol to Bile Acid Signaling

 ,
,  , ,
, ,  and
and 
Abstract
1. Introduction
2. The Enterohepatic Circulation and Kinetics of BA and the FXR–FGF19 Dynamics
3. Regulation of BA Homeostasis: The Role of Gut Microbiota
4. The Microbiota–BA–FXR Axis
5. Fibroblast Growth Factors FGF15 and Human Ortholog FGF19
6. FXR–FGF19 and BA Homeostasis
7. FXR–FGF15/19 Pathway and Metabolic Effects
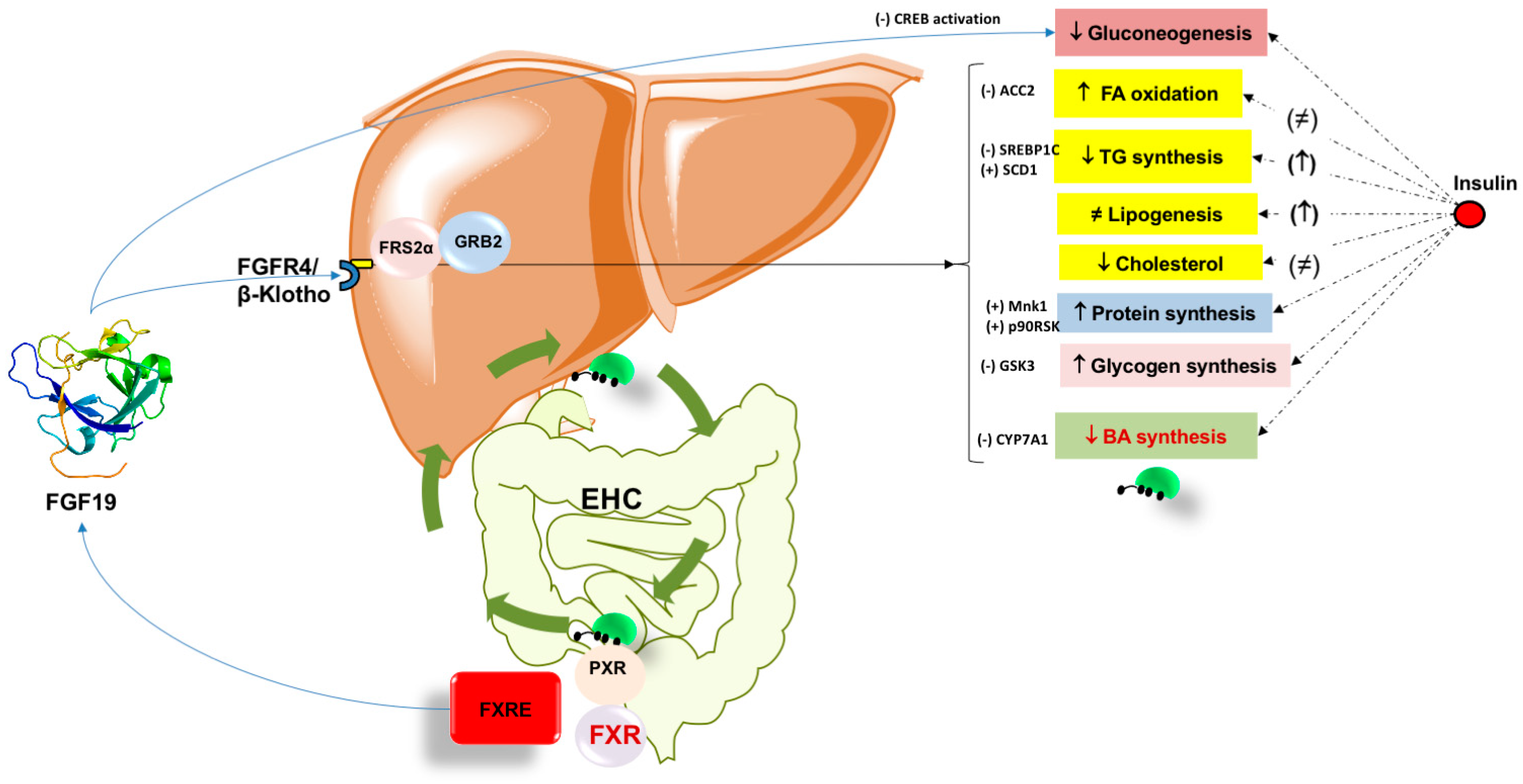
7.1. Lipid Homeostasis
7.2. Glucose Homeostasis and Gluconeogenesis
7.3. FGF19 and Role in Glycogen Synthesis
7.4. FGF19 and Role in Protein Synthesis
7.5. FGF19 and Role in Energy Expenditure
8. The Role of FXR in Liver Disease
8.1. Inflammation and Fibrosis
8.2. Cholestasis
9. FXR as a Therapeutic Target?
9.1. FGF19 and FGF21 Variants
9.2. FXR Agonists

{kind=link}
{kind=link}
| Obeticholic acid (approved for the treatment of primary biliary cholangitis) | [238,239,240,241] |
| Tropifexor (LJN452) | [242,243,244,245,246,247,248,249] |
| Cilofexor (GS-9674) | [250,251] |
| Vonafexor | [252] |
| Nidufexor | [253] |
| GW4064 | [254] |
| MET409 | [255] |
| TC-100 | [256] |
| BMS-986339 | [257] |
| HEC96719 | [258] |
| WAY-450 | [259] |
| WAY-362450 | [260] |
| Px-102 | [261] |
| Px-104 | [262] |
| TERN-101 | [263] |
| EDP-305 | [264] |
| INT-767 (FXR-TGR5 dual agonist) | [25,265,266] |
10. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 3β-HSD | 3β-hydroxy-D5-C27-steroid dehydroxylase |
| 6-ECDCA | 6a-ethyl-chenodeoxycholic acid |
| ACC2 | Acetyl CoA carboxylase |
| AGRP | Agouti-related peptide |
| AKR1C4 | 3a-hydroxysteroid dehydrogenase |
| AKR1D1 | D4-3-oxosteroid 5b-reductase |
| AKT | Protein kinase B |
| ALT | Alanine aminotransferase |
| AMPs | Antimicrobial peptides |
| ASBT | Apical sodium-dependent bile salt transporter |
| BA | Bile acids |
| BAAT | Bile acid-CoA:amino acid N-acyltransferase |
| BACS | Bile acid CoA synthase |
| ABCB11 | Bile acid export pump |
| BAT | Brown adipose tissue |
| BSEP | Bile salt export pump |
| BSH | Bile salt hydrolase |
| C4 | 7a-hydroxy-4-cholesten-3-one |
| CA | Cholic acid |
| CCK | Cholecystokinin |
| CCl4 | Carbon tetrachloride |
| CDCA | Chenodeoxycholic acid |
| CF | Cycling frequency |
| COX-2 | Cylcooxygenase-2 |
| CREB | cAMP regulatory element binding protein |
| CYP7A1 | Cholesterol-7α-hydroxylase |
| CYP27A1 | Cytochrome P450 27A1 |
| CYP2A12 | Cytochrome P450 2A12 |
| CYP2C70 | Cytochrome P450 2C70 |
| CYP7A1 | Cytochrome P450 7A1 |
| CYP7B1 | Cytochrome P450 7B1 |
| CYP8B1 | Cytochrome P450 8B1 |
| DBD | DNA binding domain |
| DCA | Deoxycholic acid |
| eIF4B | Eukaryotic initiation factor 4B |
| eIF4E | Eukaryotic initiation factor 4E |
| ER2 | Everted hexanucleotide repeat separated by 2 nucleotides |
| ERK | Extracellular-signal-regulated protein kinase |
| FAR | Fractional absorption rate |
| FFA | Free fatty acid |
| FGF | Fibroblast growth factor |
| FGF15 | Fibroblast growth factor 15 |
| FGF19 | Fibroblast growth factor 19 |
| FGFR | Fibroblast growth factor receptor |
| FGFR1 | Fibroblast growth factor receptor 1 |
| FGFR4 | Fibroblast growth factor receptor 4 |
| FLINT | Farnesoid X receptor ligand obeticholic acid in NASH treatment |
| FXR | Farnesoid X receptor |
| FXRE | Farnesoid X receptor response element |
| FTR | Fractional turnover rate |
| GGT | Gamma-glutamyltransferase |
| GLP-1 | Glucagon-like peptide 1 |
| GLUT2 | Glucose transporter 2 |
| GPBAR-1 | G-protein-coupled bile acid receptor-1 |
| GS | Glycogen synthase |
| GSK3a | Glycogen synthase kinase 3a |
| HREs | Hormone response elements |
| HSC | Hepatic stellate cell |
| IkB | Inhibitor of nuclear factor kappa B |
| IBABP | Intestinal bile acid binding protein |
| IFN-g | Interferon-gamma |
| iNOS | Inducible nitric oxide synthase |
| IR1 | Inverted hexanucleotide repeat separated by 1 nucleotide |
| KO | Knockout |
| LBD | Ligand-binding domain |
| LCA | Lithocholic acid |
| LRH-1 | Liver receptor homologue 1 |
| LPS | Lipopolysaccharide |
| JNK | JUN N-terminal kinase |
| JUN | Jun proto-oncogene |
| MAGs | Metagenome-assembled genomes |
| MCA | Muricholic acid |
| MCD | Methionine-choline deficient |
| MCP-1 | Monocyte chemoattractant protein-1 |
| MRP2 | Multidrug resistance-associated protein 2 |
| mTOR | Mammalian target of rapamycin |
| NAFLD | Non-alcoholic fatty liver disease |
| NASH | Non-alcoholic steatohepatitis |
| NF-kB | Nuclear factor kappa-light chain enhancer of activated B cell |
| NPY | Neuropeptide Y |
| NR s | Nuclear receptors |
| NLRP3 | NACHT LRR and PYD domains-containing protein 3 |
| NTCP | Sodium taurocholate co-transporting polypeptide |
| OATP | Organic anion transporting polypeptide |
| OCA | Obeticholic acid |
| OSTα | Organic solute transporter alpha |
| OSTb | Organic solute transporter beta |
| P90RSK | p90 ribosomal S6 kinase |
| PBC | Primary biliary cirrhosis |
| PEPCK | Phosphoenolpyruvate carboxykinase |
| PFIC | Progressive familial intrahepatic cholestasis |
| PGC-1α | Peroxisome proliferator-activated receptor gamma coactivator 1-alpha |
| PGC-1b | Peroxisome proliferator-activated receptor gamma coactivator 1-beta |
| PPARγ | Peroxisomal proliferator activated receptor gamma |
| PXR | Pregnane X receptor |
| RCT | Reverse cholesterol transport |
| RXR | Retinoid X receptor |
| S1PR2 | Sphingosine-1-phosphate receptor 2 |
| S6K1 | S6 kinase beta-1 |
| SCD1 | Stearoyl-CoA desaturase 1 |
| SIBO | Small intestinal bacterial overgrowth |
| SHP | Small heterodimer partner |
| SREBP1c | Sterol regulatory element-binding protein 1c |
| STAT3 | Signal transducer and activator of transcription 3 |
| SULT2A1 | Sulfotransferase 2A1 |
| TCA | Taurocholic acid |
| TDCA | Taurodeoxycholic acid |
| TGR5 | Takeda G-protein receptor 5 |
| TLCA | Taurolithocholic acid |
| TNFα | Tumor necrosis factor alpha |
| VDR | Vitamin D receptor |
| VLDL | Very low-density lipoprotein |
| WAT | White adipose tissue |
References
- Gonzalez, F.J. Nuclear receptor control of enterohepatic circulation. Compr. Physiol. 2012, 2, 2811–2828. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, T.; Li, F.; Gonzalez, F.J. FXR signaling in the enterohepatic system. Mol. Cell Endocrinol. 2013, 368, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Cai, J.; Gonzalez, F.J. The role of farnesoid X receptor in metabolic diseases, and gastrointestinal and liver cancer. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 335–347. [Google Scholar] [CrossRef]
- Nguyen, J.T.; Shaw, R.P.H.; Anakk, S. Bile Acids—A Peek Into Their History and Signaling. Endocrinology 2022, 163, bqac155. [Google Scholar] [CrossRef]
- Poland, J.C.; Flynn, C.R. Bile Acids, Their Receptors, and the Gut Microbiota. Physiology 2021, 36, 235–245. [Google Scholar] [CrossRef]
- Sorrentino, G.; Perino, A.; Yildiz, E.; El Alam, G.; Bou Sleiman, M.; Gioiello, A.; Pellicciari, R.; Schoonjans, K. Bile Acids Signal via TGR5 to Activate Intestinal Stem Cells and Epithelial Regeneration. Gastroenterology 2020, 159, 956–968.e8. [Google Scholar] [CrossRef]
- Guillot, A.; Guerri, L.; Feng, D.; Kim, S.J.; Ahmed, Y.A.; Paloczi, J.; He, Y.; Schuebel, K.; Dai, S.; Liu, F.; et al. Bile acid-activated macrophages promote biliary epithelial cell proliferation through integrin alphavbeta6 upregulation following liver injury. J. Clin. Investig. 2021, 131, e132305. [Google Scholar] [CrossRef] [PubMed]
- Hylemon, P.B.; Takabe, K.; Dozmorov, M.; Nagahashi, M.; Zhou, H. Bile acids as global regulators of hepatic nutrient metabolism. Liver Res. 2017, 1, 10–16. [Google Scholar] [CrossRef] [PubMed]
- Jiao, N.; Baker, S.S.; Chapa-Rodriguez, A.; Liu, W.; Nugent, C.A.; Tsompana, M.; Mastrandrea, L.; Buck, M.J.; Baker, R.D.; Genco, R.J.; et al. Suppressed hepatic bile acid signalling despite elevated production of primary and secondary bile acids in NAFLD. Gut 2018, 67, 1881–1891. [Google Scholar] [CrossRef]
- Pavlovic, N.; Stanimirov, B.; Mikov, M. Bile Acids as Novel Pharmacological Agents: The Interplay Between Gene Polymorphisms, Epigenetic Factors and Drug Response. Curr. Pharm. Des. 2017, 23, 187–215. [Google Scholar] [CrossRef]
- Kim, Y.C.; Jung, H.; Seok, S.; Zhang, Y.; Ma, J.; Li, T.; Kemper, B.; Kemper, J.K. MicroRNA-210 Promotes Bile Acid-Induced Cholestatic Liver Injury by Targeting Mixed-Lineage Leukemia-4 Methyltransferase in Mice. Hepatology 2020, 71, 2118–2134. [Google Scholar] [CrossRef] [PubMed]
- Zimny, S.; Koob, D.; Li, J.; Wimmer, R.; Schiergens, T.; Nagel, J.; Reiter, F.P.; Denk, G.; Hohenester, S. Hydrophobic Bile Salts Induce Pro-Fibrogenic Proliferation of Hepatic Stellate Cells through PI3K p110 Alpha Signaling. Cells 2022, 11, 2344. [Google Scholar] [CrossRef] [PubMed]
- Kumari, A.; Pal Pathak, D.; Asthana, S. Bile acids mediated potential functional interaction between FXR and FATP5 in the regulation of Lipid Metabolism. Int. J. Biol. Sci. 2020, 16, 2308–2322. [Google Scholar] [CrossRef]
- McGlone, E.R.; Bloom, S.R. Bile acids and the metabolic syndrome. Ann. Clin. Biochem. 2019, 56, 326–337. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Tang, D.; Yu, C.; Zhao, L. Progress in research on the roles of TGR5 receptor in liver diseases. Scand. J. Gastroenterol. 2021, 56, 717–726. [Google Scholar] [CrossRef]
- Wang, Y.; Aoki, H.; Yang, J.; Peng, K.; Liu, R.; Li, X.; Qiang, X.; Sun, L.; Gurley, E.C.; Lai, G.; et al. The role of sphingosine 1-phosphate receptor 2 in bile-acid-induced cholangiocyte proliferation and cholestasis-induced liver injury in mice. Hepatology 2017, 65, 2005–2018. [Google Scholar] [CrossRef]
- Nagahashi, M.; Yuza, K.; Hirose, Y.; Nakajima, M.; Ramanathan, R.; Hait, N.C.; Hylemon, P.B.; Zhou, H.; Takabe, K.; Wakai, T. The roles of bile acids and sphingosine-1-phosphate signaling in the hepatobiliary diseases. J. Lipid Res. 2016, 57, 1636–1643. [Google Scholar] [CrossRef]
- Garruti, G.; Wang, H.H.; Bonfrate, L.; de Bari, O.; Wang, D.Q.; Portincasa, P. A pleiotropic role for the orphan nuclear receptor small heterodimer partner in lipid homeostasis and metabolic pathways. J. Lipids 2012, 2012, 304292. [Google Scholar] [CrossRef]
- Portincasa, P.; Di Ciaula, A.; Garruti, G.; Vacca, M.; De Angelis, M.; Wang, D.Q. Bile Acids and GPBAR-1: Dynamic Interaction Involving Genes, Environment and Gut Microbiome. Nutrients 2020, 12, 3709. [Google Scholar] [CrossRef]
- Zollner, G.; Trauner, M. Mechanisms of cholestasis. Clin. Liver Dis. 2008, 12, 1–26. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Garruti, G.; Lunardi Baccetto, R.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Bile Acid Physiology. Ann. Hepatol. 2017, 16, s4–s14. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Wang, D.Q.; Molina-Molina, E.; Lunardi Baccetto, R.; Calamita, G.; Palmieri, V.O.; Portincasa, P. Bile Acids and Cancer: Direct and Environmental-Dependent Effects. Ann. Hepatol. 2017, 16, s87–s105. [Google Scholar] [CrossRef] [PubMed]
- Panzitt, K.; Zollner, G.; Marschall, H.U.; Wagner, M. Recent advances on FXR-targeting therapeutics. Mol. Cell. Endocrinol. 2022, 552, 111678. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Shi, A.; Wei, Y.; Luo, X.; Xi, L. Farnesoid X Receptor as a Promising Therapeutic Target for Nonalcoholic Fatty Liver Disease (NAFLD) and the Current Development of Its Agonists. Discov. Med. 2021, 32, 113–121. [Google Scholar]
- Wang, X.X.; Xie, C.; Libby, A.E.; Ranjit, S.; Levi, J.; Myakala, K.; Bhasin, K.; Jones, B.A.; Orlicky, D.J.; Takahashi, S.; et al. The Role of FXR and TGR5 in Reversing and Preventing Progression of Western Diet-induced Hepatic Steatosis, Inflammation, and Fibrosis in Mice. J. Biol. Chem. 2022, 298, 102530. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.; Yan, J.; Wu, L.; Wu, J.; Chen, Z.; Jiang, J.; Chen, Z.; He, B. Probiotics Alleviated Nonalcoholic Fatty Liver Disease in High-Fat Diet-Fed Rats via Gut Microbiota/FXR/FGF15 Signaling Pathway. J. Immunol. Res. 2021, 2021, 2264737. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Pathak, P.; Boehme, S.; Chiang, J.L. Cholesterol 7alpha-hydroxylase protects the liver from inflammation and fibrosis by maintaining cholesterol homeostasis. J. Lipid Res. 2016, 57, 1831–1844. [Google Scholar] [CrossRef] [PubMed]
- Pineda Torra, I.; Claudel, T.; Duval, C.; Kosykh, V.; Fruchart, J.C.; Staels, B. Bile acids induce the expression of the human peroxisome proliferator-activated receptor alpha gene via activation of the farnesoid X receptor. Mol. Endocrinol. 2003, 17, 259–272. [Google Scholar] [CrossRef] [PubMed]
- Sirvent, A.; Claudel, T.; Martin, G.; Brozek, J.; Kosykh, V.; Darteil, R.; Hum, D.W.; Fruchart, J.C.; Staels, B. The farnesoid X receptor induces very low density lipoprotein receptor gene expression. FEBS Lett. 2004, 566, 173–177. [Google Scholar] [CrossRef]
- Claudel, T.; Inoue, Y.; Barbier, O.; Duran-Sandoval, D.; Kosykh, V.; Fruchart, J.; Fruchart, J.C.; Gonzalez, F.J.; Staels, B. Farnesoid X receptor agonists suppress hepatic apolipoprotein CIII expression. Gastroenterology 2003, 125, 544–555. [Google Scholar] [CrossRef]
- Zhao, T.; Wang, J.; He, A.; Wang, S.; Chen, Y.; Lu, J.; Lv, J.; Li, S.; Wang, J.; Qian, M.; et al. Mebhydrolin ameliorates glucose homeostasis in type 2 diabetic mice by functioning as a selective FXR antagonist. Metabolism 2021, 119, 154771. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Wang, J.; Liu, Q.; Harnish, D.C. Farnesoid X receptor agonist WAY-362450 attenuates liver inflammation and fibrosis in murine model of non-alcoholic steatohepatitis. J. Hepatol. 2009, 51, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Roth, J.D.; Veidal, S.S.; Fensholdt, L.K.D.; Rigbolt, K.T.G.; Papazyan, R.; Nielsen, J.C.; Feigh, M.; Vrang, N.; Young, M.; Jelsing, J.; et al. Combined obeticholic acid and elafibranor treatment promotes additive liver histological improvements in a diet-induced ob/ob mouse model of biopsy-confirmed NASH. Sci. Rep. 2019, 9, 9046. [Google Scholar] [CrossRef] [PubMed]
- Stellaard, F.; Lutjohann, D. Dynamics of the enterohepatic circulation of bile acids in healthy humans. Am. J. Physiol. Gastrointest. Liver Physiol. 2021, 321, G55–G66. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.J.; Blanchard, S.G.; Bledsoe, R.K.; Chandra, G.; Consler, T.G.; Kliewer, S.A.; Stimmel, J.B.; Willson, T.M.; Zavacki, A.M.; Moore, D.D.; et al. Bile acids: Natural ligands for an orphan nuclear receptor. Science 1999, 284, 1365–1368. [Google Scholar] [CrossRef]
- Inagaki, T.; Choi, M.; Moschetta, A.; Peng, L.; Cummins, C.L.; McDonald, J.G.; Luo, G.; Jones, S.A.; Goodwin, B.; Richardson, J.A.; et al. Fibroblast growth factor 15 functions as an enterohepatic signal to regulate bile acid homeostasis. Cell Metab. 2005, 2, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Modica, S.; Gofflot, F.; Murzilli, S.; D’Orazio, A.; Salvatore, L.; Pellegrini, F.; Nicolucci, A.; Tognoni, G.; Copetti, M.; Valanzano, R.; et al. The intestinal nuclear receptor signature with epithelial localization patterns and expression modulation in tumors. Gastroenterology 2010, 138, 636–648, 648.e1-12. [Google Scholar] [CrossRef]
- Liu, H.; Hu, C.; Zhang, X.; Jia, W. Role of gut microbiota, bile acids and their cross-talk in the effects of bariatric surgery on obesity and type 2 diabetes. J. Diabetes Investig. 2018, 9, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Kemper, J.K. Regulation of FXR transcriptional activity in health and disease: Emerging roles of FXR cofactors and post-translational modifications. Biochim. Biophys. Acta 2011, 1812, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, B.; Jones, S.A.; Price, R.R.; Watson, M.A.; McKee, D.D.; Moore, L.B.; Galardi, C.; Wilson, J.G.; Lewis, M.C.; Roth, M.E.; et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol. Cell 2000, 6, 517–526. [Google Scholar] [CrossRef]
- Lu, T.T.; Makishima, M.; Repa, J.J.; Schoonjans, K.; Kerr, T.A.; Auwerx, J.; Mangelsdorf, D.J. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol. Cell 2000, 6, 507–515. [Google Scholar] [CrossRef]
- Nishimaki-Mogami, T.; Une, M.; Fujino, T.; Sato, Y.; Tamehiro, N.; Kawahara, Y.; Shudo, K.; Inoue, K. Identification of intermediates in the bile acid synthetic pathway as ligands for the farnesoid X receptor. J. Lipid Res. 2004, 45, 1538–1545. [Google Scholar] [CrossRef]
- Pircher, P.C.; Kitto, J.L.; Petrowski, M.L.; Tangirala, R.K.; Bischoff, E.D.; Schulman, I.G.; Westin, S.K. Farnesoid X receptor regulates bile acid-amino acid conjugation. J. Biol. Chem. 2003, 278, 27703–27711. [Google Scholar] [CrossRef]
- Garcia-Rodriguez, J.L.; Barbier-Torres, L.; Fernandez-Alvarez, S.; Gutierrez-de Juan, V.; Monte, M.J.; Halilbasic, E.; Herranz, D.; Alvarez, L.; Aspichueta, P.; Marin, J.J.; et al. SIRT1 controls liver regeneration by regulating bile acid metabolism through farnesoid X receptor and mammalian target of rapamycin signaling. Hepatology 2014, 59, 1972–1983. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, D.R.; Schmidt, S.; Holmstrom, S.R.; Makishima, M.; Yu, R.T.; Cummins, C.L.; Mangelsdorf, D.J.; Kliewer, S.A. AKR1B7 is induced by the farnesoid X receptor and metabolizes bile acids. J. Biol. Chem. 2011, 286, 2425–2432. [Google Scholar] [CrossRef] [PubMed]
- Modica, S.; Bellafante, E.; Moschetta, A. Master regulation of bile acid and xenobiotic metabolism via the FXR, PXR and CAR trio. Front. Biosci. (Landmark Ed.) 2008, 14, 4719–4745. [Google Scholar] [CrossRef]
- Wagner, M.; Zollner, G.; Trauner, M. New molecular insights into the mechanisms of cholestasis. J. Hepatol. 2009, 51, 565–580. [Google Scholar] [CrossRef] [PubMed]
- Gillard, J.; Clerbaux, L.A.; Nachit, M.; Sempoux, C.; Staels, B.; Bindels, L.B.; Tailleux, A.; Leclercq, I.A. Bile acids contribute to the development of non-alcoholic steatohepatitis in mice. JHEP Rep. 2022, 4, 100387. [Google Scholar] [CrossRef] [PubMed]
- Dommett, R.; Zilbauer, M.; George, J.T.; Bajaj-Elliott, M. Innate immune defence in the human gastrointestinal tract. Mol. Immunol. 2005, 42, 903–912. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Baj, J.; Garruti, G.; Celano, G.; De Angelis, M.; Wang, H.H.; Di Palo, D.M.; Bonfrate, L.; Wang, D.Q.; Portincasa, P. Liver Steatosis, Gut-Liver Axis, Microbiome and Environmental Factors. A Never-Ending Bidirectional Cross-Talk. J. Clin. Med. 2020, 9, 2648. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P.; Bonfrate, L.; Vacca, M.; De Angelis, M.; Farella, I.; Lanza, E.; Khalil, M.; Wang, D.Q.; Sperandio, M.; Di Ciaula, A. Gut Microbiota and Short Chain Fatty Acids: Implications in Glucose Homeostasis. Int. J. Mol. Sci. 2022, 23, 1105. [Google Scholar] [CrossRef]
- Biedermann, L.; Zeitz, J.; Mwinyi, J.; Sutter-Minder, E.; Rehman, A.; Ott, S.J.; Steurer-Stey, C.; Frei, A.; Frei, P.; Scharl, M.; et al. Smoking cessation induces profound changes in the composition of the intestinal microbiota in humans. PLoS ONE 2013, 8, e59260. [Google Scholar] [CrossRef]
- Mutlu, E.A.; Gillevet, P.M.; Rangwala, H.; Sikaroodi, M.; Naqvi, A.; Engen, P.A.; Kwasny, M.; Lau, C.K.; Keshavarzian, A. Colonic microbiome is altered in alcoholism. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G966–G978. [Google Scholar] [CrossRef]
- Gao, B.; Chi, L.; Mahbub, R.; Bian, X.; Tu, P.; Ru, H.; Lu, K. Multi-Omics Reveals that Lead Exposure Disturbs Gut Microbiome Development, Key Metabolites, and Metabolic Pathways. Chem. Res. Toxicol. 2017, 30, 996–1005. [Google Scholar] [CrossRef]
- Gao, B.; Bian, X.; Mahbub, R.; Lu, K. Sex-Specific Effects of Organophosphate Diazinon on the Gut Microbiome and Its Metabolic Functions. Environ. Health Perspect. 2017, 125, 198–206. [Google Scholar] [CrossRef]
- Joly, C.; Gay-Queheillard, J.; Leke, A.; Chardon, K.; Delanaud, S.; Bach, V.; Khorsi-Cauet, H. Impact of chronic exposure to low doses of chlorpyrifos on the intestinal microbiota in the Simulator of the Human Intestinal Microbial Ecosystem (SHIME) and in the rat. Environ. Sci. Pollut. Res. Int. 2013, 20, 2726–2734. [Google Scholar] [CrossRef]
- Joly Condette, C.; Bach, V.; Mayeur, C.; Gay-Queheillard, J.; Khorsi-Cauet, H. Chlorpyrifos Exposure During Perinatal Period Affects Intestinal Microbiota Associated With Delay of Maturation of Digestive Tract in Rats. J. Pediatr. Gastroenterol. Nutr. 2015, 61, 30–40. [Google Scholar] [CrossRef]
- Di Ciaula, A.; Bonfrate, L.; Portincasa, P. The role of microbiota in nonalcoholic fatty liver disease. Eur. J. Clin. Investig. 2022, 52, e13768. [Google Scholar] [CrossRef]
- Garruti, G.; Di Ciaula, A.; Wang, H.H.; Wang, D.Q.; Portincasa, P. Cross-Talk Between Bile Acids and Gastro-Intestinal and Thermogenic Hormones: Clues from Bariatric Surgery. Ann. Hepatol. 2017, 16, s68–s82. [Google Scholar] [CrossRef]
- Sayin, S.I.; Wahlstrom, A.; Felin, J.; Jantti, S.; Marschall, H.U.; Bamberg, K.; Angelin, B.; Hyotylainen, T.; Oresic, M.; Backhed, F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef]
- Long, S.L.; Gahan, C.G.M.; Joyce, S.A. Interactions between gut bacteria and bile in health and disease. Mol. Asp. Med. 2017, 56, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Parseus, A.; Sommer, N.; Sommer, F.; Caesar, R.; Molinaro, A.; Stahlman, M.; Greiner, T.U.; Perkins, R.; Backhed, F. Microbiota-induced obesity requires farnesoid X receptor. Gut 2017, 66, 429–437. [Google Scholar] [CrossRef] [PubMed]
- Yokota, A.; Fukiya, S.; Islam, K.B.; Ooka, T.; Ogura, Y.; Hayashi, T.; Hagio, M.; Ishizuka, S. Is bile acid a determinant of the gut microbiota on a high-fat diet? Gut Microbes 2012, 3, 455–459. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Hylemon, P.B. Bile acids are nutrient signaling hormones. Steroids 2014, 86, 62–68. [Google Scholar] [CrossRef]
- Ridlon, J.M.; Wolf, P.G.; Gaskins, H.R. Taurocholic acid metabolism by gut microbes and colon cancer. Gut Microbes 2016, 7, 201–215. [Google Scholar] [CrossRef] [PubMed]
- O’Keefe, S.J.; Li, J.V.; Lahti, L.; Ou, J.; Carbonero, F.; Mohammed, K.; Posma, J.M.; Kinross, J.; Wahl, E.; Ruder, E.; et al. Fat, fibre and cancer risk in African Americans and rural Africans. Nat. Commun. 2015, 6, 6342. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Li, F.; Liu, Y.; Gu, Z.; Zhang, L.; Lee, J.; He, L.; Vatsalya, V.; Zhang, H.G.; Deng, Z.; et al. Probiotic-derived nanoparticles inhibit ALD through intestinal miR194 suppression and subsequent FXR activation. Hepatology 2022. [Google Scholar] [CrossRef]
- Boursier, J.; Diehl, A.M. Implication of gut microbiota in nonalcoholic fatty liver disease. PLoS Pathog. 2015, 11, e1004559. [Google Scholar] [CrossRef]
- Lake, A.D.; Novak, P.; Shipkova, P.; Aranibar, N.; Robertson, D.; Reily, M.D.; Lu, Z.; Lehman-McKeeman, L.D.; Cherrington, N.J. Decreased hepatotoxic bile acid composition and altered synthesis in progressive human nonalcoholic fatty liver disease. Toxicol. Appl. Pharm. 2013, 268, 132–140. [Google Scholar] [CrossRef]
- Chavez-Talavera, O.; Tailleux, A.; Lefebvre, P.; Staels, B. Bile Acid Control of Metabolism and Inflammation in Obesity, Type 2 Diabetes, Dyslipidemia, and Nonalcoholic Fatty Liver Disease. Gastroenterology 2017, 152, 1679–1694.e3. [Google Scholar] [CrossRef]
- Mouzaki, M.; Wang, A.Y.; Bandsma, R.; Comelli, E.M.; Arendt, B.M.; Zhang, L.; Fung, S.; Fischer, S.E.; McGilvray, I.G.; Allard, J.P. Bile Acids and Dysbiosis in Non-Alcoholic Fatty Liver Disease. PLoS ONE 2016, 11, e0151829. [Google Scholar] [CrossRef] [PubMed]
- Cao, H.; Xu, M.; Dong, W.; Deng, B.; Wang, S.; Zhang, Y.; Wang, S.; Luo, S.; Wang, W.; Qi, Y.; et al. Secondary bile acid-induced dysbiosis promotes intestinal carcinogenesis. Int. J. Cancer 2017, 140, 2545–2556. [Google Scholar] [CrossRef] [PubMed]
- Liquori, G.E.; Mastrodonato, M.; Rossi, R.; Scillitani, G.; Gena, P.; Portincasa, P.; Calamita, G.; Ferri, D. Altered membrane glycoprotein targeting in cholestatic hepatocytes. Eur. J. Clin. Investig. 2010, 40, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Arab, J.P.; Karpen, S.J.; Dawson, P.A.; Arrese, M.; Trauner, M. Bile acids and nonalcoholic fatty liver disease: Molecular insights and therapeutic perspectives. Hepatology 2017, 65, 350–362. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.; Xie, C.; Li, F.; Zhang, L.; Nichols, R.G.; Krausz, K.W.; Cai, J.; Qi, Y.; Fang, Z.Z.; Takahashi, S.; et al. Intestinal farnesoid X receptor signaling promotes nonalcoholic fatty liver disease. J. Clin. Investig. 2015, 125, 386–402. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Jiang, C.; Krausz, K.W.; Li, Y.; Albert, I.; Hao, H.; Fabre, K.M.; Mitchell, J.B.; Patterson, A.D.; Gonzalez, F.J. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nat. Commun. 2013, 4, 2384. [Google Scholar] [CrossRef]
- Lorenzo-Zúñiga, V.; Bartolí, R.; Planas, R.; Hofmann, A.F.; Viñado, B.; Hagey, L.R.; Hernández, J.M.; Mañé, J.; Alvarez, M.A.; Ausina, V. Oral bile acids reduce bacterial overgrowth, bacterial translocation, and endotoxemia in cirrhotic rats. Hepatology 2003, 37, 551–557. [Google Scholar] [CrossRef]
- Clements, W.; Parks, R.; Erwin, P.; Halliday, M.; Barr, J.; Rowlands, B. Role of the gut in the pathophysiology of extrahepatic biliary obstruction. Gut 1996, 39, 587–593. [Google Scholar] [CrossRef]
- Inagaki, T.; Moschetta, A.; Lee, Y.K.; Peng, L.; Zhao, G.; Downes, M.; Yu, R.T.; Shelton, J.M.; Richardson, J.A.; Repa, J.J.; et al. Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc. Natl. Acad. Sci. USA 2006, 103, 3920–3925. [Google Scholar] [CrossRef]
- Sun, L.; Pang, Y.; Wang, X.; Wu, Q.; Liu, H.; Liu, B.; Liu, G.; Ye, M.; Kong, W.; Jiang, C. Ablation of gut microbiota alleviates obesity-induced hepatic steatosis and glucose intolerance by modulating bile acid metabolism in hamsters. Acta Pharm. Sin. B 2019, 9, 702–710. [Google Scholar] [CrossRef]
- Sun, L.; Xie, C.; Wang, G.; Wu, Y.; Wu, Q.; Wang, X.; Liu, J.; Deng, Y.; Xia, J.; Chen, B.; et al. Gut microbiota and intestinal FXR mediate the clinical benefits of metformin. Nat. Med. 2018, 24, 1919–1929. [Google Scholar] [CrossRef] [PubMed]
- Baron, E.J.; Summanen, P.; Downes, J.; Roberts, M.C.; Wexler, H.; Finegold, S.M. Bilophila wadsworthia, gen. nov. and sp. nov., a unique gram-negative anaerobic rod recovered from appendicitis specimens and human faeces. J. Gen. Microbiol. 1989, 135, 3405–3411. [Google Scholar] [CrossRef] [PubMed]
- Devkota, S.; Wang, Y.W.; Musch, M.W.; Leone, V.; Fehlner-Peach, H.; Nadimpalli, A.; Antonopoulos, D.A.; Jabri, B.; Chang, E.B. Dietary-fat-induced taurocholic acid promotes pathobiont expansion and colitis in Il10(−/−) mice. Nature 2012, 487, 104–108. [Google Scholar] [CrossRef] [PubMed]
- Loomba, R.; Ling, L.; Dinh, D.M.; DePaoli, A.M.; Lieu, H.D.; Harrison, S.A.; Sanyal, A.J. The Commensal Microbe Veillonella as a Marker for Response to an FGF19 Analog in NASH. Hepatology 2021, 73, 126–143. [Google Scholar] [CrossRef]
- Danneskiold-Samsøe, N.B.; Barros, H.D.d.F.Q.; Santos, R.; Bicas, J.L.; Cazarin, C.B.B.; Madsen, L.; Kristiansen, K.; Pastore, G.M.; Brix, S.; Junior, M.R.M. Interplay between food and gut microbiota in health and disease. Food Res. Int. 2019, 115, 23–31. [Google Scholar] [CrossRef]
- Liu, R.; Zhao, R.; Zhou, X.; Liang, X.; Campbell, D.J.; Zhang, X.; Zhang, L.; Shi, R.; Wang, G.; Pandak, W.M.; et al. Conjugated bile acids promote cholangiocarcinoma cell invasive growth through activation of sphingosine 1-phosphate receptor 2. Hepatology 2014, 60, 908–918. [Google Scholar] [CrossRef]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- Guo, G.L.; Lambert, G.; Negishi, M.; Ward, J.M.; Brewer, H.B., Jr.; Kliewer, S.A.; Gonzalez, F.J.; Sinal, C.J. Complementary roles of farnesoid X receptor, pregnane X receptor, and constitutive androstane receptor in protection against bile acid toxicity. J. Biol. Chem. 2003, 278, 45062–45071. [Google Scholar] [CrossRef]
- Kir, S.; Kliewer, S.A.; Mangelsdorf, D.J. Roles of FGF19 in liver metabolism. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 139–144. [Google Scholar] [CrossRef]
- Owen, B.M.; Mangelsdorf, D.J.; Kliewer, S.A. Tissue-specific actions of the metabolic hormones FGF15/19 and FGF21. Trends Endocrinol. Metab. 2015, 26, 22–29. [Google Scholar] [CrossRef]
- Zarrinpar, A.; Loomba, R. Review article: The emerging interplay among the gastrointestinal tract, bile acids and incretins in the pathogenesis of diabetes and non-alcoholic fatty liver disease. Aliment. Pharm. 2012, 36, 909–921. [Google Scholar] [CrossRef] [PubMed]
- Henkel, A.S.; Anderson, K.A.; Dewey, A.M.; Kavesh, M.H.; Green, R.M. A chronic high-cholesterol diet paradoxically suppresses hepatic CYP7A1 expression in FVB/NJ mice. J. Lipid Res. 2011, 52, 289–298. [Google Scholar] [CrossRef] [PubMed]
- Miyata, M.; Hata, T.; Yamazoe, Y.; Yoshinari, K. SREBP-2 negatively regulates FXR-dependent transcription of FGF19 in human intestinal cells. Biochem. Biophys. Res. Commun. 2014, 443, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Degirolamo, C.; Sabba, C.; Moschetta, A. Therapeutic potential of the endocrine fibroblast growth factors FGF19, FGF21 and FGF23. Nat. Rev. Drug Discov. 2016, 15, 51–69. [Google Scholar] [CrossRef]
- Beenken, A.; Mohammadi, M. The FGF family: Biology, pathophysiology and therapy. Nat. Rev. Drug Discov. 2009, 8, 235–253. [Google Scholar] [CrossRef]
- Itoh, N.; Ornitz, D.M. Functional evolutionary history of the mouse Fgf gene family. Dev. Dyn. 2008, 237, 18–27. [Google Scholar] [CrossRef]
- Itoh, N. Hormone-like (endocrine) Fgfs: Their evolutionary history and roles in development, metabolism, and disease. Cell Tissue Res 2010, 342, 1–11. [Google Scholar] [CrossRef]
- Goetz, R.; Beenken, A.; Ibrahimi, O.A.; Kalinina, J.; Olsen, S.K.; Eliseenkova, A.V.; Xu, C.; Neubert, T.A.; Zhang, F.; Linhardt, R.J.; et al. Molecular insights into the klotho-dependent, endocrine mode of action of fibroblast growth factor 19 subfamily members. Mol. Cell. Biol. 2007, 27, 3417–3428. [Google Scholar] [CrossRef]
- Nishimura, T.; Utsunomiya, Y.; Hoshikawa, M.; Ohuchi, H.; Itoh, N. Structure and expression of a novel human FGF, FGF-19, expressed in the fetal brain. Biochim. Biophys. Acta 1999, 1444, 148–151. [Google Scholar] [CrossRef]
- Fon Tacer, K.; Bookout, A.L.; Ding, X.; Kurosu, H.; John, G.B.; Wang, L.; Goetz, R.; Mohammadi, M.; Kuro-o, M.; Mangelsdorf, D.J.; et al. Research resource: Comprehensive expression atlas of the fibroblast growth factor system in adult mouse. Mol. Endocrinol. 2010, 24, 2050–2064. [Google Scholar] [CrossRef]
- Xie, M.H.; Holcomb, I.; Deuel, B.; Dowd, P.; Huang, A.; Vagts, A.; Foster, J.; Liang, J.; Brush, J.; Gu, Q.; et al. FGF-19, a novel fibroblast growth factor with unique specificity for FGFR4. Cytokine 1999, 11, 729–735. [Google Scholar] [CrossRef] [PubMed]
- Holt, J.A.; Luo, G.; Billin, A.N.; Bisi, J.; McNeill, Y.Y.; Kozarsky, K.F.; Donahee, M.; Wang, D.Y.; Mansfield, T.A.; Kliewer, S.A.; et al. Definition of a novel growth factor-dependent signal cascade for the suppression of bile acid biosynthesis. Genes Dev. 2003, 17, 1581–1591. [Google Scholar] [CrossRef] [PubMed]
- Kim, I.; Ahn, S.H.; Inagaki, T.; Choi, M.; Ito, S.; Guo, G.L.; Kliewer, S.A.; Gonzalez, F.J. Differential regulation of bile acid homeostasis by the farnesoid X receptor in liver and intestine. J. Lipid Res. 2007, 48, 2664–2672. [Google Scholar] [CrossRef] [PubMed]
- Lundasen, T.; Galman, C.; Angelin, B.; Rudling, M. Circulating intestinal fibroblast growth factor 19 has a pronounced diurnal variation and modulates hepatic bile acid synthesis in man. J. Intern. Med. 2006, 260, 530–536. [Google Scholar] [CrossRef]
- Potthoff, M.J.; Boney-Montoya, J.; Choi, M.; He, T.; Sunny, N.E.; Satapati, S.; Suino-Powell, K.; Xu, H.E.; Gerard, R.D.; Finck, B.N.; et al. FGF15/19 regulates hepatic glucose metabolism by inhibiting the CREB-PGC-1alpha pathway. Cell Metab. 2011, 13, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Potthoff, M.J.; Kliewer, S.A.; Mangelsdorf, D.J. Endocrine fibroblast growth factors 15/19 and 21: From feast to famine. Genes Dev. 2012, 26, 312–324. [Google Scholar] [CrossRef]
- Alvarez-Sola, G.; Uriarte, I.; Latasa, M.U.; Urtasun, R.; Barcena-Varela, M.; Elizalde, M.; Jimenez, M.; Rodriguez-Ortigosa, C.M.; Corrales, F.J.; Fernandez-Barrena, M.G.; et al. Fibroblast Growth Factor 15/19 in Hepatocarcinogenesis. Dig. Dis. 2017, 35, 158–165. [Google Scholar] [CrossRef]
- Uriarte, I.; Latasa, M.U.; Carotti, S.; Fernandez-Barrena, M.G.; Garcia-Irigoyen, O.; Elizalde, M.; Urtasun, R.; Vespasiani-Gentilucci, U.; Morini, S.; de Mingo, A. Ileal FGF 15 contributes to fibrosis-associated hepatocellular carcinoma development. Int. J. Cancer 2015, 136, 2469–2475. [Google Scholar] [CrossRef]
- French, D.M.; Lin, B.C.; Wang, M.; Adams, C.; Shek, T.; Hotzel, K.; Bolon, B.; Ferrando, R.; Blackmore, C.; Schroeder, K.; et al. Targeting FGFR4 inhibits hepatocellular carcinoma in preclinical mouse models. PLoS ONE 2012, 7, e36713. [Google Scholar] [CrossRef]
- Wu, X.; Lemon, B.; Li, X.; Gupte, J.; Weiszmann, J.; Stevens, J.; Hawkins, N.; Shen, W.; Lindberg, R.; Chen, J.L.; et al. C-terminal tail of FGF19 determines its specificity toward Klotho co-receptors. J. Biol. Chem. 2008, 283, 33304–33309. [Google Scholar] [CrossRef]
- Yang, C.; Jin, C.; Li, X.; Wang, F.; McKeehan, W.L.; Luo, Y. Differential specificity of endocrine FGF19 and FGF21 to FGFR1 and FGFR4 in complex with KLB. PLoS ONE 2012, 7, e33870. [Google Scholar] [CrossRef] [PubMed]
- Lin, B.C.; Wang, M.; Blackmore, C.; Desnoyers, L.R. Liver-specific activities of FGF19 require Klotho beta. J. Biol. Chem. 2007, 282, 27277–27284. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, J.D.; Guo, G.L. Pharmacologic Modulation of Bile Acid-FXR-FGF15/FGF19 Pathway for the Treatment of Nonalcoholic Steatohepatitis. Handb. Exp. Pharm. 2019, 256, 325–357. [Google Scholar] [CrossRef]
- Naugler, W.E.; Tarlow, B.D.; Fedorov, L.M.; Taylor, M.; Pelz, C.; Li, B.; Darnell, J.; Grompe, M. Fibroblast Growth Factor Signaling Controls Liver Size in Mice with Humanized Livers. Gastroenterology 2015, 149, 728–740.e15. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Yang, C.; Chang, J.Y.; You, P.; Li, Y.; Jin, C.; Luo, Y.; Li, X.; McKeehan, W.L.; Wang, F. Hepatocyte FRS2α is essential for the endocrine fibroblast growth factor to limit the amplitude of bile acid production induced by prandial activity. Curr. Mol. Med. 2014, 14, 703–711. [Google Scholar] [CrossRef]
- Kurosu, H.; Choi, M.; Ogawa, Y.; Dickson, A.S.; Goetz, R.; Eliseenkova, A.V.; Mohammadi, M.; Rosenblatt, K.P.; Kliewer, S.A.; Kuro, O.M. Tissue-specific expression of betaKlotho and fibroblast growth factor (FGF) receptor isoforms determines metabolic activity of FGF19 and FGF21. J. Biol. Chem. 2007, 282, 26687–26695. [Google Scholar] [CrossRef] [PubMed]
- Di Ciaula, A.; Molina-Molina, E.; Bonfrate, L.; Wang, D.Q.; Dumitrascu, D.L.; Portincasa, P. Gastrointestinal defects in gallstone and cholecystectomized patients. Eur. J. Clin. Investig. 2019, 49, e13066. [Google Scholar] [CrossRef]
- Molina-Molina, E.; Lunardi Baccetto, R.; Wang, D.Q.; de Bari, O.; Krawczyk, M.; Portincasa, P. Exercising the hepatobiliary-gut axis. The impact of physical activity performance. Eur. J. Clin. Investig. 2018, 48, e12958. [Google Scholar] [CrossRef]
- Diella, G.; Di Ciaula, A.; Lorusso, M.P.; Summo, C.; Caggiano, G.; Caponio, F.; Montagna, M.T.; Portincasa, P. Distinct Effects of two Almond Cultivars on Agreeability and Gastrointestinal Motility in Healthy Subjects: More than mere Nutraceuticals. J. Gastrointest. Liver Dis. JGLD 2018, 27, 31–39. [Google Scholar] [CrossRef]
- Rizzello, C.G.; Portincasa, P.; Montemurro, M.; Di Palo, D.M.; Lorusso, M.P.; De Angelis, M.; Bonfrate, L.; Genot, B.; Gobbetti, M. Sourdough Fermented Breads are More Digestible than Those Started with Baker’s Yeast Alone: An In Vivo Challenge Dissecting Distinct Gastrointestinal Responses. Nutrients 2019, 11, 2954. [Google Scholar] [CrossRef] [PubMed]
- Luiking, Y.C.; Peeters, T.L.; Stolk, M.F.; Nieuwenhuijs, V.B.; Portincasa, P.; Depoortere, I.; van Berge Henegouwen, G.P.; Akkermans, L.M. Motilin induces gall bladder emptying and antral contractions in the fasted state in humans. Gut 1998, 42, 830–835. [Google Scholar] [CrossRef] [PubMed]
- Portincasa, P.; Di Ciaula, A.; Wang, H.H.; Palasciano, G.; van Erpecum, K.J.; Moschetta, A.; Wang, D.Q. Coordinate regulation of gallbladder motor function in the gut-liver axis. Hepatology 2008, 47, 2112–2126. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.; Wang, L.; Chiang, J.Y.; Zhang, Y.; Klaassen, C.D.; Guo, G.L. Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology 2012, 56, 1034–1043. [Google Scholar] [CrossRef]
- Zollner, G.; Wagner, M.; Moustafa, T.; Fickert, P.; Silbert, D.; Gumhold, J.; Fuchsbichler, A.; Halilbasic, E.; Denk, H.; Marschall, H.U.; et al. Coordinated induction of bile acid detoxification and alternative elimination in mice: Role of FXR-regulated organic solute transporter-alpha/beta in the adaptive response to bile acids. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G923–G932. [Google Scholar] [CrossRef] [PubMed]
- Landrier, J.F.; Eloranta, J.J.; Vavricka, S.R.; Kullak-Ublick, G.A. The nuclear receptor for bile acids, FXR, transactivates human organic solute transporter-alpha and -beta genes. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G476–G485. [Google Scholar] [CrossRef]
- Chen, F.; Ma, L.; Dawson, P.A.; Sinal, C.J.; Sehayek, E.; Gonzalez, F.J.; Breslow, J.; Ananthanarayanan, M.; Shneider, B.L. Liver receptor homologue-1 mediates species- and cell line-specific bile acid-dependent negative feedback regulation of the apical sodium-dependent bile acid transporter. J. Biol. Chem. 2003, 278, 19909–19916. [Google Scholar] [CrossRef]
- Yu, C.; Wang, F.; Jin, C.; Huang, X.; McKeehan, W.L. Independent repression of bile acid synthesis and activation of c-Jun N-terminal kinase (JNK) by activated hepatocyte fibroblast growth factor receptor 4 (FGFR4) and bile acids. J. Biol. Chem. 2005, 280, 17707–17714. [Google Scholar] [CrossRef] [PubMed]
- Ito, S.; Fujimori, T.; Furuya, A.; Satoh, J.; Nabeshima, Y.; Nabeshima, Y. Impaired negative feedback suppression of bile acid synthesis in mice lacking betaKlotho. J. Clin. Investig. 2005, 115, 2202–2208. [Google Scholar] [CrossRef] [PubMed]
- Tomiyama, K.; Maeda, R.; Urakawa, I.; Yamazaki, Y.; Tanaka, T.; Ito, S.; Nabeshima, Y.; Tomita, T.; Odori, S.; Hosoda, K.; et al. Relevant use of Klotho in FGF19 subfamily signaling system in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 1666–1671. [Google Scholar] [CrossRef] [PubMed]
- Kir, S.; Zhang, Y.; Gerard, R.D.; Kliewer, S.A.; Mangelsdorf, D.J. Nuclear receptors HNF4alpha and LRH-1 cooperate in regulating Cyp7a1 in vivo. J. Biol. Chem. 2012, 287, 41334–41341. [Google Scholar] [CrossRef] [PubMed]
- Miao, J.; Choi, S.E.; Seok, S.M.; Yang, L.; Zuercher, W.J.; Xu, Y.; Willson, T.M.; Xu, H.E.; Kemper, J.K. Ligand-dependent regulation of the activity of the orphan nuclear receptor, small heterodimer partner (SHP), in the repression of bile acid biosynthetic CYP7A1 and CYP8B1 genes. Mol. Endocrinol. 2011, 25, 1159–1169. [Google Scholar] [CrossRef]
- Pols, T.W.; Noriega, L.G.; Nomura, M.; Auwerx, J.; Schoonjans, K. The bile acid membrane receptor TGR5: A valuable metabolic target. Dig. Dis. 2011, 29, 37–44. [Google Scholar] [CrossRef] [PubMed]
- Broeders, E.P.; Nascimento, E.B.; Havekes, B.; Brans, B.; Roumans, K.H.; Tailleux, A.; Schaart, G.; Kouach, M.; Charton, J.; Deprez, B.; et al. The Bile Acid Chenodeoxycholic Acid Increases Human Brown Adipose Tissue Activity. Cell Metab. 2015, 22, 418–426. [Google Scholar] [CrossRef]
- Thomas, C.; Gioiello, A.; Noriega, L.; Strehle, A.; Oury, J.; Rizzo, G.; Macchiarulo, A.; Yamamoto, H.; Mataki, C.; Pruzanski, M.; et al. TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab. 2009, 10, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Perino, A.; Schoonjans, K. TGR5 and Immunometabolism: Insights from Physiology and Pharmacology. Trends Pharm. Sci. 2015, 36, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Schaap, F.G.; Trauner, M.; Jansen, P.L. Bile acid receptors as targets for drug development. Nat. Rev. Gastroenterol. Hepatol. 2014, 11, 55–67. [Google Scholar] [CrossRef]
- Wang, D.Q.H.; Neuschwander-Tetri, B.A.; Portincasa, P. The Biliary System, Second Edition; Morgan & Claypool Life Sciences: San Rafael, CA, USA, 2017; Volume 8, p. i-178. [Google Scholar]
- Musso, G.; Gambino, R.; Cassader, M. Cholesterol metabolism and the pathogenesis of non-alcoholic steatohepatitis. Prog. Lipid Res. 2013, 52, 175–191. [Google Scholar] [CrossRef] [PubMed]
- Li, T.; Owsley, E.; Matozel, M.; Hsu, P.; Novak, C.M.; Chiang, J.Y. Transgenic expression of cholesterol 7alpha-hydroxylase in the liver prevents high-fat diet-induced obesity and insulin resistance in mice. Hepatology 2010, 52, 678–690. [Google Scholar] [CrossRef] [PubMed]
- Sinal, C.J.; Tohkin, M.; Miyata, M.; Ward, J.M.; Lambert, G.; Gonzalez, F.J. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell 2000, 102, 731–744. [Google Scholar] [CrossRef]
- Watanabe, M.; Houten, S.M.; Wang, L.; Moschetta, A.; Mangelsdorf, D.J.; Heyman, R.A.; Moore, D.D.; Auwerx, J. Bile acids lower triglyceride levels via a pathway involving FXR, SHP, and SREBP-1c. J. Clin. Investig. 2004, 113, 1408–1418. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Lee, F.Y.; Barrera, G.; Lee, H.; Vales, C.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Activation of the nuclear receptor FXR improves hyperglycemia and hyperlipidemia in diabetic mice. Proc. Natl. Acad. Sci. USA 2006, 103, 1006–1011. [Google Scholar] [CrossRef] [PubMed]
- Bhatnagar, S.; Damron, H.A.; Hillgartner, F.B. Fibroblast growth factor-19, a novel factor that inhibits hepatic fatty acid synthesis. J. Biol. Chem. 2009, 284, 10023–10033. [Google Scholar] [CrossRef]
- Kast, H.R.; Nguyen, C.M.; Sinal, C.J.; Jones, S.A.; Laffitte, B.A.; Reue, K.; Gonzalez, F.J.; Willson, T.M.; Edwards, P.A. Farnesoid X-activated receptor induces apolipoprotein C-II transcription: A molecular mechanism linking plasma triglyceride levels to bile acids. Mol. Endocrinol. 2001, 15, 1720–1728. [Google Scholar] [CrossRef] [PubMed]
- Schoenfield, L.J.; Lachin, J.M. Chenodiol (chenodeoxycholic acid) for dissolution of gallstones: The National Cooperative Gallstone Study. A controlled trial of efficacy and safety. Ann. Intern. Med. 1981, 95, 257–282. [Google Scholar] [CrossRef] [PubMed]
- Lu, D.; Liu, Y.; Luo, Y.; Zhao, J.; Feng, C.; Xue, L.; Xu, J.; Wang, Q.; Yan, T.; Xiao, P.; et al. Intestinal farnesoid X receptor signaling controls hepatic fatty acid oxidation. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2022, 1867, 159089. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.Q.H.; Neuschwander-Tetri, B.A.; Portincasa, P. The Biliary System. Colloq. Ser. Integr. Syst. Physiol. Mol. Funct. 2012, 4, 1–148. [Google Scholar] [CrossRef]
- Wang, D.Q.; Portincasa, P.; Tso, P. Transintestinal cholesterol excretion: A secondary, nonbiliary pathway contributing to reverse cholesterol transport. Hepatology 2017, 66, 1337–1340. [Google Scholar] [CrossRef] [PubMed]
- de Boer, J.F.; Schonewille, M.; Boesjes, M.; Wolters, H.; Bloks, V.W.; Bos, T.; van Dijk, T.H.; Jurdzinski, A.; Boverhof, R.; Wolters, J.C.; et al. Intestinal Farnesoid X Receptor Controls Transintestinal Cholesterol Excretion in Mice. Gastroenterology 2017, 152, 1126–1138.e6. [Google Scholar] [CrossRef]
- Deng, W.; Fan, W.; Tang, T.; Wan, H.; Zhao, S.; Tan, Y.; Oware, K.A.; Tan, J.; Li, J.; Qu, S. Farnesoid X Receptor Deficiency Induces Hepatic Lipid and Glucose Metabolism Disorder via Regulation of Pyruvate Dehydrogenase Kinase 4. Oxid. Med. Cell Longev. 2022, 2022, 3589525. [Google Scholar] [CrossRef] [PubMed]
- Kir, S.; Beddow, S.A.; Samuel, V.T.; Miller, P.; Previs, S.F.; Suino-Powell, K.; Xu, H.E.; Shulman, G.I.; Kliewer, S.A.; Mangelsdorf, D.J. FGF19 as a postprandial, insulin-independent activator of hepatic protein and glycogen synthesis. Science 2011, 331, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Lu, Y.; Li, X.Y. Farnesoid X receptor: A master regulator of hepatic triglyceride and glucose homeostasis. Acta Pharm. Sin. 2015, 36, 44–50. [Google Scholar] [CrossRef] [PubMed]
- Yamagata, K.; Daitoku, H.; Shimamoto, Y.; Matsuzaki, H.; Hirota, K.; Ishida, J.; Fukamizu, A. Bile acids regulate gluconeogenic gene expression via small heterodimer partner-mediated repression of hepatocyte nuclear factor 4 and Foxo1. J. Biol. Chem. 2004, 279, 23158–23165. [Google Scholar] [CrossRef] [PubMed]
- Thomas, A.M.; Hart, S.N.; Kong, B.; Fang, J.; Zhong, X.B.; Guo, G.L. Genome-wide tissue-specific farnesoid X receptor binding in mouse liver and intestine. Hepatology 2010, 51, 1410–1419. [Google Scholar] [CrossRef]
- Zhan, L.; Liu, H.X.; Fang, Y.; Kong, B.; He, Y.; Zhong, X.B.; Fang, J.; Wan, Y.J.; Guo, G.L. Genome-wide binding and transcriptome analysis of human farnesoid X receptor in primary human hepatocytes. PLoS ONE 2014, 9, e105930. [Google Scholar] [CrossRef]
- Ma, K.; Saha, P.K.; Chan, L.; Moore, D.D. Farnesoid X receptor is essential for normal glucose homeostasis. J. Clin. Investig. 2006, 116, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.H.; Choi, S.; Zhou, Y.; Kim, E.Y.; Lee, J.M.; Saha, P.K.; Anakk, S.; Moore, D.D. Hepatic FXR/SHP axis modulates systemic glucose and fatty acid homeostasis in aged mice. Hepatology 2017, 66, 498–509. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Wagner, M.; Xiao, R.; Kim, K.H.; Feng, D.; Lazar, M.A.; Moore, D.D. Nutrient-sensing nuclear receptors coordinate autophagy. Nature 2014, 516, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Trabelsi, M.S.; Daoudi, M.; Prawitt, J.; Ducastel, S.; Touche, V.; Sayin, S.I.; Perino, A.; Brighton, C.A.; Sebti, Y.; Kluza, J.; et al. Farnesoid X receptor inhibits glucagon-like peptide-1 production by enteroendocrine L cells. Nat. Commun. 2015, 6, 7629. [Google Scholar] [CrossRef] [PubMed]
- Pathak, P.; Xie, C.; Nichols, R.G.; Ferrell, J.M.; Boehme, S.; Krausz, K.W.; Patterson, A.D.; Gonzalez, F.J.; Chiang, J.Y.L. Intestine farnesoid X receptor agonist and the gut microbiota activate G-protein bile acid receptor-1 signaling to improve metabolism. Hepatology 2018, 68, 1574–1588. [Google Scholar] [CrossRef]
- Cully, M. FXR and JAK step up to BAT. Nat. Rev. Drug Discov. 2015, 14, 91. [Google Scholar] [CrossRef]
- Fang, S.; Suh, J.M.; Reilly, S.M.; Yu, E.; Osborn, O.; Lackey, D.; Yoshihara, E.; Perino, A.; Jacinto, S.; Lukasheva, Y.; et al. Intestinal FXR agonism promotes adipose tissue browning and reduces obesity and insulin resistance. Nat. Med. 2015, 21, 159–165. [Google Scholar] [CrossRef] [PubMed]
- Velazquez-Villegas, L.A.; Perino, A.; Lemos, V.; Zietak, M.; Nomura, M.; Pols, T.W.H.; Schoonjans, K. TGR5 signalling promotes mitochondrial fission and beige remodelling of white adipose tissue. Nat. Commun. 2018, 9, 245. [Google Scholar] [CrossRef] [PubMed]
- Stayrook, K.R.; Bramlett, K.S.; Savkur, R.S.; Ficorilli, J.; Cook, T.; Christe, M.E.; Michael, L.F.; Burris, T.P. Regulation of carbohydrate metabolism by the farnesoid X receptor. Endocrinology 2005, 146, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; John, L.M.; Adams, S.H.; Yu, X.X.; Tomlinson, E.; Renz, M.; Williams, P.M.; Soriano, R.; Corpuz, R.; Moffat, B.; et al. Fibroblast growth factor 19 increases metabolic rate and reverses dietary and leptin-deficient diabetes. Endocrinology 2004, 145, 2594–2603. [Google Scholar] [CrossRef]
- Somm, E.; Jornayvaz, F.R. Fibroblast Growth Factor 15/19: From Basic Functions to Therapeutic Perspectives. Endocr. Rev. 2018, 39, 960–989. [Google Scholar] [CrossRef]
- Morton, G.J.; Matsen, M.E.; Bracy, D.P.; Meek, T.H.; Nguyen, H.T.; Stefanovski, D.; Bergman, R.N.; Wasserman, D.H.; Schwartz, M.W. FGF19 action in the brain induces insulin-independent glucose lowering. J. Clin. Investig. 2013, 123, 4799–4808. [Google Scholar] [CrossRef]
- Taylor, R.; Magnusson, I.; Rothman, D.L.; Cline, G.W.; Caumo, A.; Cobelli, C.; Shulman, G.I. Direct assessment of liver glycogen storage by 13C nuclear magnetic resonance spectroscopy and regulation of glucose homeostasis after a mixed meal in normal subjects. J. Clin. Investig. 1996, 97, 126–132. [Google Scholar] [CrossRef]
- Benoit, B.; Meugnier, E.; Castelli, M.; Chanon, S.; Vieille-Marchiset, A.; Durand, C.; Bendridi, N.; Pesenti, S.; Monternier, P.A.; Durieux, A.C.; et al. Fibroblast growth factor 19 regulates skeletal muscle mass and ameliorates muscle wasting in mice. Nat. Med. 2017, 23, 990–996. [Google Scholar] [CrossRef]
- Massafra, V.; Milona, A.; Vos, H.R.; Ramos, R.J.J.; Gerrits, J.; Willemsen, E.C.L.; Ramos Pittol, J.M.; Ijssennagger, N.; Houweling, M.; Prinsen, H.; et al. Farnesoid X Receptor Activation Promotes Hepatic Amino Acid Catabolism and Ammonium Clearance in Mice. Gastroenterology 2017, 152, 1462–1476.e10. [Google Scholar] [CrossRef]
- Bag Soytas, R.; Suzan, V.; Arman, P.; Emiroglu Gedik, T.; Unal, D.; Cengiz, M.; Bolayirli, I.M.; Suna Erdincler, D.; Doventas, A.; Yavuzer, H. Association of FGF-19 and FGF-21 levels with primary sarcopenia. Geriatr. Gerontol. Int. 2021, 21, 959–962. [Google Scholar] [CrossRef]
- Qiu, Y.; Yu, J.; Li, Y.; Yang, F.; Yu, H.; Xue, M.; Zhang, F.; Jiang, X.; Ji, X.; Bao, Z. Depletion of gut microbiota induces skeletal muscle atrophy by FXR-FGF15/19 signalling. Ann. Med. 2021, 53, 508–522. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, E.; Fu, L.; John, L.; Hultgren, B.; Huang, X.; Renz, M.; Stephan, J.P.; Tsai, S.P.; Powell-Braxton, L.; French, D.; et al. Transgenic mice expressing human fibroblast growth factor-19 display increased metabolic rate and decreased adiposity. Endocrinology 2002, 143, 1741–1747. [Google Scholar] [CrossRef] [PubMed]
- Marcelin, G.; Jo, Y.H.; Li, X.; Schwartz, G.J.; Zhang, Y.; Dun, N.J.; Lyu, R.M.; Blouet, C.; Chang, J.K.; Chua, S., Jr. Central action of FGF19 reduces hypothalamic AGRP/NPY neuron activity and improves glucose metabolism. Mol. Metab. 2014, 3, 19–28. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Learned, R.M.; Rossi, S.J.; DePaoli, A.M.; Tian, H.; Ling, L. Engineered FGF19 eliminates bile acid toxicity and lipotoxicity leading to resolution of steatohepatitis and fibrosis in mice. Hepatol. Commun. 2017, 1, 1024–1042. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.D.; Chen, W.D.; Wang, M.; Yu, D.; Forman, B.M.; Huang, W. Farnesoid X receptor antagonizes nuclear factor kappaB in hepatic inflammatory response. Hepatology 2008, 48, 1632–1643. [Google Scholar] [CrossRef] [PubMed]
- Daly, C.; Rollins, B.J. Monocyte chemoattractant protein-1 (CCL2) in inflammatory disease and adaptive immunity: Therapeutic opportunities and controversies. Microcirculation 2003, 10, 247–257. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S. NF-ĸB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 247–257. [Google Scholar]
- Renga, B.; Mencarelli, A.; Migliorati, M.; Cipriani, S.; D’Amore, C.; Distrutti, E.; Fiorucci, S. SHP-dependent and -independent induction of peroxisome proliferator-activated receptor-gamma by the bile acid sensor farnesoid X receptor counter-regulates the pro-inflammatory phenotype of liver myofibroblasts. Inflamm. Res. 2011, 60, 577–587. [Google Scholar] [CrossRef]
- Fiorucci, S.; Rizzo, G.; Antonelli, E.; Renga, B.; Mencarelli, A.; Riccardi, L.; Morelli, A.; Pruzanski, M.; Pellicciari, R. Cross-talk between farnesoid-X-receptor (FXR) and peroxisome proliferator-activated receptor γ contributes to the antifibrotic activity of FXR ligands in rodent models of liver cirrhosis. J. Pharmacol. Exp. Ther. 2005, 315, 58–68. [Google Scholar] [CrossRef]
- El Kasmi, K.C.; Ghosh, S.; Anderson, A.L.; Devereaux, M.W.; Balasubramaniyan, N.; D’Alessandro, A.; Orlicky, D.J.; Suchy, F.J.; Shearn, C.T.; Sokol, R.J. Pharmacologic activation of hepatic farnesoid X receptor prevents parenteral nutrition-associated cholestasis in mice. Hepatology 2022, 75, 252–265. [Google Scholar] [CrossRef]
- Schumacher, J.D.; Kong, B.; Wu, J.; Rizzolo, D.; Armstrong, L.E.; Chow, M.D.; Goedken, M.; Lee, Y.H.; Guo, G.L. Direct and Indirect Effects of Fibroblast Growth Factor (FGF) 15 and FGF19 on Liver Fibrosis Development. Hepatology 2020, 71, 670–685. [Google Scholar] [CrossRef] [PubMed]
- Carulli, N.; Bertolotti, M.; Carubbi, F.; Concari, M.; Martella, P.; Carulli, L.; Loria, P. Review article: Effect of bile salt pool composition on hepatic and biliary functions. Aliment. Pharm. 2000, 14 (Suppl. 2), 14–18. [Google Scholar] [CrossRef] [PubMed]
- Merlen, G.; Ursic-Bedoya, J.; Jourdainne, V.; Kahale, N.; Glenisson, M.; Doignon, I.; Rainteau, D.; Tordjmann, T. Bile acids and their receptors during liver regeneration: “Dangerous protectors”. Mol. Asp. Med. 2017, 56, 25–33. [Google Scholar] [CrossRef]
- Donkers, J.M.; Roscam Abbing, R.L.P.; van de Graaf, S.F.J. Developments in bile salt based therapies: A critical overview. Biochem. Pharm. 2019, 161, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Dawson, P.A.; Karpen, S.J. Intestinal transport and metabolism of bile acids. J. Lipid Res. 2015, 56, 1085–1099. [Google Scholar] [CrossRef] [PubMed]
- Doignon, I.; Julien, B.; Serriere-Lanneau, V.; Garcin, I.; Alonso, G.; Nicou, A.; Monnet, F.; Gigou, M.; Humbert, L.; Rainteau, D.; et al. Immediate neuroendocrine signaling after partial hepatectomy through acute portal hyperpressure and cholestasis. J. Hepatol. 2011, 54, 481–488. [Google Scholar] [CrossRef] [PubMed]
- Fahrner, R.; Patsenker, E.; de Gottardi, A.; Stickel, F.; Montani, M.; Stroka, D.; Candinas, D.; Beldi, G. Elevated liver regeneration in response to pharmacological reduction of elevated portal venous pressure by terlipressin after partial hepatectomy. Transplantation 2014, 97, 892–900. [Google Scholar] [CrossRef]
- Miura, T.; Kimura, N.; Yamada, T.; Shimizu, T.; Nanashima, N.; Yamana, D.; Hakamada, K.; Tsuchida, S. Sustained repression and translocation of Ntcp and expression of Mrp4 for cholestasis after rat 90% partial hepatectomy. J. Hepatol. 2011, 55, 407–414. [Google Scholar] [CrossRef]
- Bhushan, B.; Borude, P.; Edwards, G.; Walesky, C.; Cleveland, J.; Li, F.; Ma, X.; Apte, U. Role of bile acids in liver injury and regeneration following acetaminophen overdose. Am. J. Pathol. 2013, 183, 1518–1526. [Google Scholar] [CrossRef]
- Meng, Z.; Wang, Y.; Wang, L.; Jin, W.; Liu, N.; Pan, H.; Liu, L.; Wagman, L.; Forman, B.M.; Huang, W. FXR regulates liver repair after CCl4-induced toxic injury. Mol. Endocrinol. 2010, 24, 886–897. [Google Scholar] [CrossRef]
- Huang, W.; Ma, K.; Zhang, J.; Qatanani, M.; Cuvillier, J.; Liu, J.; Dong, B.; Huang, X.; Moore, D.D. Nuclear receptor-dependent bile acid signaling is required for normal liver regeneration. Science 2006, 312, 233–236. [Google Scholar] [CrossRef] [PubMed]
- Pean, N.; Doignon, I.; Garcin, I.; Besnard, A.; Julien, B.; Liu, B.; Branchereau, S.; Spraul, A.; Guettier, C.; Humbert, L.; et al. The receptor TGR5 protects the liver from bile acid overload during liver regeneration in mice. Hepatology 2013, 58, 1451–1460. [Google Scholar] [CrossRef] [PubMed]
- Naugler, W.E. Bile acid flux is necessary for normal liver regeneration. PLoS ONE 2014, 9, e97426. [Google Scholar] [CrossRef] [PubMed]
- Hoekstra, L.T.; Rietkerk, M.; van Lienden, K.P.; van den Esschert, J.W.; Schaap, F.G.; van Gulik, T.M. Bile salts predict liver regeneration in rabbit model of portal vein embolization. J. Surg. Res. 2012, 178, 773–778. [Google Scholar] [CrossRef][Green Version]
- Hoekstra, L.T.; van Lienden, K.P.; Schaap, F.G.; Chamuleau, R.A.; Bennink, R.J.; van Gulik, T.M. Can plasma bile salt, triglycerides, and apoA-V levels predict liver regeneration? World J. Surg. 2012, 36, 2901–2908. [Google Scholar] [CrossRef]
- Fickert, P.; Fuchsbichler, A.; Marschall, H.U.; Wagner, M.; Zollner, G.; Krause, R.; Zatloukal, K.; Jaeschke, H.; Denk, H.; Trauner, M. Lithocholic acid feeding induces segmental bile duct obstruction and destructive cholangitis in mice. Am. J. Pathol. 2006, 168, 410–422. [Google Scholar] [CrossRef]
- Gilgenkrantz, H.; Tordjmann, T. Bile acids and FGF receptors: Orchestrators of optimal liver regeneration. Gut 2015, 64, 1351–1352. [Google Scholar] [CrossRef]
- Padrissa-Altes, S.; Bachofner, M.; Bogorad, R.L.; Pohlmeier, L.; Rossolini, T.; Bohm, F.; Liebisch, G.; Hellerbrand, C.; Koteliansky, V.; Speicher, T.; et al. Control of hepatocyte proliferation and survival by Fgf receptors is essential for liver regeneration in mice. Gut 2015, 64, 1444–1453. [Google Scholar] [CrossRef]
- Park, Y.J.; Qatanani, M.; Chua, S.S.; LaRey, J.L.; Johnson, S.A.; Watanabe, M.; Moore, D.D.; Lee, Y.K. Loss of orphan receptor small heterodimer partner sensitizes mice to liver injury from obstructive cholestasis. Hepatology 2008, 47, 1578–1586. [Google Scholar] [CrossRef]
- Modica, S.; Petruzzelli, M.; Bellafante, E.; Murzilli, S.; Salvatore, L.; Celli, N.; Di Tullio, G.; Palasciano, G.; Moustafa, T.; Halilbasic, E.; et al. Selective activation of nuclear bile acid receptor FXR in the intestine protects mice against cholestasis. Gastroenterology 2012, 142, 355–365.e4. [Google Scholar] [CrossRef]
- Honda, A.; Miyazaki, T.; Iwamoto, J.; Hirayama, T.; Morishita, Y.; Monma, T.; Ueda, H.; Mizuno, S.; Sugiyama, F.; Takahashi, S.; et al. Regulation of bile acid metabolism in mouse models with hydrophobic bile acid composition. J. Lipid Res. 2020, 61, 54–69. [Google Scholar] [CrossRef] [PubMed]
- de Boer, J.F.; Verkade, E.; Mulder, N.L.; de Vries, H.D.; Huijkman, N.; Koehorst, M.; Boer, T.; Wolters, J.C.; Bloks, V.W.; van de Sluis, B.; et al. A human-like bile acid pool induced by deletion of hepatic Cyp2c70 modulates effects of FXR activation in mice. J. Lipid Res. 2020, 61, 291–305. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, S.; Fukami, T.; Masuo, Y.; Brocker, C.N.; Xie, C.; Krausz, K.W.; Wolf, C.R.; Henderson, C.J.; Gonzalez, F.J. Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J. Lipid Res. 2016, 57, 2130–2137. [Google Scholar] [CrossRef] [PubMed]
- Hrycay, E.; Forrest, D.; Liu, L.; Wang, R.; Tai, J.; Deo, A.; Ling, V.; Bandiera, S. Hepatic bile acid metabolism and expression of cytochrome P450 and related enzymes are altered in Bsep (−/−) mice. Mol. Cell Biochem. 2014, 389, 119–132. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A. Progressive familial intrahepatic cholestasis. J. Clin. Exp. Hepatol. 2014, 4, 25–36. [Google Scholar] [CrossRef]
- Fuchs, C.D.; Paumgartner, G.; Wahlstrom, A.; Schwabl, P.; Reiberger, T.; Leditznig, N.; Stojakovic, T.; Rohr-Udilova, N.; Chiba, P.; Marschall, H.U.; et al. Metabolic preconditioning protects BSEP/ABCB11(−/−) mice against cholestatic liver injury. J. Hepatol. 2017, 66, 95–101. [Google Scholar] [CrossRef]
- Lee, C.S.; Kimura, A.; Wu, J.F.; Ni, Y.H.; Hsu, H.Y.; Chang, M.H.; Nittono, H.; Chen, H.L. Prognostic roles of tetrahydroxy bile acids in infantile intrahepatic cholestasis. J. Lipid Res. 2017, 58, 607–614. [Google Scholar] [CrossRef]
- Woolbright, B.L.; Jaeschke, H. Inflammation and Cell Death During Cholestasis: The Evolving Role of Bile Acids. Gene Expr. 2019, 19, 215–228. [Google Scholar] [CrossRef]
- Yerushalmi, B.; Dahl, R.; Devereaux, M.W.; Gumpricht, E.; Sokol, R.J. Bile acid-induced rat hepatocyte apoptosis is inhibited by antioxidants and blockers of the mitochondrial permeability transition. Hepatology 2001, 33, 616–626. [Google Scholar] [CrossRef]
- Grattagliano, I.; Montezinho, L.P.; Oliveira, P.J.; Fruhbeck, G.; Gomez-Ambrosi, J.; Montecucco, F.; Carbone, F.; Wieckowski, M.R.; Wang, D.Q.; Portincasa, P. Targeting mitochondria to oppose the progression of nonalcoholic fatty liver disease. Biochem. Pharm. 2019, 160, 34–45. [Google Scholar] [CrossRef]
- Reinehr, R.; Graf, D.; Haussinger, D. Bile salt-induced hepatocyte apoptosis involves epidermal growth factor receptor-dependent CD95 tyrosine phosphorylation. Gastroenterology 2003, 125, 839–853. [Google Scholar] [CrossRef]
- Miyoshi, H.; Rust, C.; Roberts, P.J.; Burgart, L.J.; Gores, G.J. Hepatocyte apoptosis after bile duct ligation in the mouse involves Fas. Gastroenterology 1999, 117, 669–677. [Google Scholar] [CrossRef]
- Allen, K.; Jaeschke, H.; Copple, B.L. Bile acids induce inflammatory genes in hepatocytes: A novel mechanism of inflammation during obstructive cholestasis. Am. J. Pathol. 2011, 178, 175–186. [Google Scholar] [CrossRef]
- Csanaky, I.L.; Aleksunes, L.M.; Tanaka, Y.; Klaassen, C.D. Role of hepatic transporters in prevention of bile acid toxicity after partial hepatectomy in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 297, G419–G433. [Google Scholar] [CrossRef] [PubMed]
- Geier, A.; Wagner, M.; Dietrich, C.G.; Trauner, M. Principles of hepatic organic anion transporter regulation during cholestasis, inflammation and liver regeneration. Biochim. Biophys. Acta 2007, 1773, 283–308. [Google Scholar] [CrossRef] [PubMed]
- Kong, B.; Zhang, M.; Huang, M.; Rizzolo, D.; Armstrong, L.E.; Schumacher, J.D.; Chow, M.D.; Lee, Y.H.; Guo, G.L. FXR deficiency alters bile acid pool composition and exacerbates chronic alcohol induced liver injury. Dig Liver Dis. 2019, 51, 570–576. [Google Scholar] [CrossRef]
- Kong, B.; Huang, J.; Zhu, Y.; Li, G.; Williams, J.; Shen, S.; Aleksunes, L.M.; Richardson, J.R.; Apte, U.; Rudnick, D.A.; et al. Fibroblast growth factor 15 deficiency impairs liver regeneration in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G893–G902. [Google Scholar] [CrossRef]
- Uriarte, I.; Fernandez-Barrena, M.G.; Monte, M.J.; Latasa, M.U.; Chang, H.C.; Carotti, S.; Vespasiani-Gentilucci, U.; Morini, S.; Vicente, E.; Concepcion, A.R.; et al. Identification of fibroblast growth factor 15 as a novel mediator of liver regeneration and its application in the prevention of post-resection liver failure in mice. Gut 2013, 62, 899–910. [Google Scholar] [CrossRef]
- Kong, B.; Sun, R.; Huang, M.; Chow, M.D.; Zhong, X.B.; Xie, W.; Lee, Y.H.; Guo, G.L. Fibroblast Growth Factor 15-Dependent and Bile Acid-Independent Promotion of Liver Regeneration in Mice. Hepatology 2018, 68, 1961–1976. [Google Scholar] [CrossRef]
- Cai, S.Y.; Ouyang, X.; Chen, Y.; Soroka, C.J.; Wang, J.; Mennone, A.; Wang, Y.; Mehal, W.Z.; Jain, D.; Boyer, J.L. Bile acids initiate cholestatic liver injury by triggering a hepatocyte-specific inflammatory response. JCI Insight 2017, 2, e90780. [Google Scholar] [CrossRef]
- Schumacher, J.D.; Kong, B.; Pan, Y.; Zhan, L.; Sun, R.; Aa, J.; Rizzolo, D.; Richardson, J.R.; Chen, A.; Goedken, M.; et al. The effect of fibroblast growth factor 15 deficiency on the development of high fat diet induced non-alcoholic steatohepatitis. Toxicol. Appl. Pharm. 2017, 330, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Cai, S.Y.; Boyer, J.L. Mechanisms of bile acid mediated inflammation in the liver. Mol. Asp. Med. 2017, 56, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Meadows, V.; Kennedy, L.; Ekser, B.; Kyritsi, K.; Kundu, D.; Zhou, T.; Chen, L.; Pham, L.; Wu, N.; Demieville, J.; et al. Mast Cells Regulate Ductular Reaction and Intestinal Inflammation in Cholestasis Through Farnesoid X Receptor Signaling. Hepatology 2021, 74, 2684–2698. [Google Scholar] [CrossRef] [PubMed]
- Wu, A.L.; Coulter, S.; Liddle, C.; Wong, A.; Eastham-Anderson, J.; French, D.M.; Peterson, A.S.; Sonoda, J. FGF19 regulates cell proliferation, glucose and bile acid metabolism via FGFR4-dependent and independent pathways. PLoS ONE 2011, 6, e17868. [Google Scholar] [CrossRef]
- Luo, J.; Ko, B.; Elliott, M.; Zhou, M.; Lindhout, D.A.; Phung, V.; To, C.; Learned, R.M.; Tian, H.; DePaoli, A.M.; et al. A nontumorigenic variant of FGF19 treats cholestatic liver diseases. Sci. Transl. Med. 2014, 6, 247ra100. [Google Scholar] [CrossRef] [PubMed]
- Zhou, M.; Wang, X.; Phung, V.; Lindhout, D.A.; Mondal, K.; Hsu, J.Y.; Yang, H.; Humphrey, M.; Ding, X.; Arora, T.; et al. Separating Tumorigenicity from Bile Acid Regulatory Activity for Endocrine Hormone FGF19. Cancer Res. 2014, 74, 3306–3316. [Google Scholar] [CrossRef] [PubMed]
- Gaich, G.; Chien, J.Y.; Fu, H.; Glass, L.C.; Deeg, M.A.; Holland, W.L.; Kharitonenkov, A.; Bumol, T.; Schilske, H.K.; Moller, D.E. The effects of LY2405319, an FGF21 analog, in obese human subjects with type 2 diabetes. Cell Metab. 2013, 18, 333–340. [Google Scholar] [CrossRef]
- Ogawa, Y.; Kurosu, H.; Yamamoto, M.; Nandi, A.; Rosenblatt, K.P.; Goetz, R.; Eliseenkova, A.V.; Mohammadi, M.; Kuro-o, M. BetaKlotho is required for metabolic activity of fibroblast growth factor 21. Proc. Natl. Acad. Sci. USA 2007, 104, 7432–7437. [Google Scholar] [CrossRef]
- Chen, M.M.; Hale, C.; Stanislaus, S.; Xu, J.; Veniant, M.M. FGF21 acts as a negative regulator of bile acid synthesis. J. Endocrinol. 2018, 237, 139–152. [Google Scholar] [CrossRef]
- Zhang, J.; Gupte, J.; Gong, Y.; Weiszmann, J.; Zhang, Y.; Lee, K.J.; Richards, W.G.; Li, Y. Chronic Over-expression of Fibroblast Growth Factor 21 Increases Bile Acid Biosynthesis by Opposing FGF15/19 Action. EBioMedicine 2017, 15, 173–183. [Google Scholar] [CrossRef]
- Talukdar, S.; Zhou, Y.; Li, D.; Rossulek, M.; Dong, J.; Somayaji, V.; Weng, Y.; Clark, R.; Lanba, A.; Owen, B.M.; et al. A Long-Acting FGF21 Molecule, PF-05231023, Decreases Body Weight and Improves Lipid Profile in Non-human Primates and Type 2 Diabetic Subjects. Cell Metab. 2016, 23, 427–440. [Google Scholar] [CrossRef]
- Kim, A.M.; Somayaji, V.R.; Dong, J.Q.; Rolph, T.P.; Weng, Y.; Chabot, J.R.; Gropp, K.E.; Talukdar, S.; Calle, R.A. Once-weekly administration of a long-acting fibroblast growth factor 21 analogue modulates lipids, bone turnover markers, blood pressure and body weight differently in obese people with hypertriglyceridaemia and in non-human primates. Diabetes Obes. Metab. 2017, 19, 1762–1772. [Google Scholar] [CrossRef] [PubMed]
- Charles, E.D.; Neuschwander-Tetri, B.A.; Pablo Frias, J.; Kundu, S.; Luo, Y.; Tirucherai, G.S.; Christian, R. Pegbelfermin (BMS-986036), PEGylated FGF21, in Patients with Obesity and Type 2 Diabetes: Results from a Randomized Phase 2 Study. Obesity 2019, 27, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Sanyal, A.; Charles, E.D.; Neuschwander-Tetri, B.A.; Loomba, R.; Harrison, S.A.; Abdelmalek, M.F.; Lawitz, E.J.; Halegoua-DeMarzio, D.; Kundu, S.; Noviello, S.; et al. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: A randomised, double-blind, placebo-controlled, phase 2a trial. Lancet 2019, 392, 2705–2717. [Google Scholar] [CrossRef]
- Luo, Y.; Decato, B.E.; Charles, E.D.; Shevell, D.E.; McNaney, C.; Shipkova, P.; Apfel, A.; Tirucherai, G.S.; Sanyal, A.J. Pegbelfermin selectively reduces secondary bile acid concentrations in patients with non-alcoholic steatohepatitis. JHEP Rep. 2022, 4, 100392. [Google Scholar] [CrossRef]
- Neuschwander-Tetri, B.A.; Loomba, R.; Sanyal, A.J.; Lavine, J.E.; Van Natta, M.L.; Abdelmalek, M.F.; Chalasani, N.; Dasarathy, S.; Diehl, A.M.; Hameed, B.; et al. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): A multicentre, randomised, placebo-controlled trial. Lancet 2015, 385, 956–965. [Google Scholar] [CrossRef]
- Pellicciari, R.; Costantino, G.; Camaioni, E.; Sadeghpour, B.M.; Entrena, A.; Willson, T.M.; Fiorucci, S.; Clerici, C.; Gioiello, A. Bile acid derivatives as ligands of the farnesoid X receptor. Synthesis, evaluation, and structure-activity relationship of a series of body and side chain modified analogues of chenodeoxycholic acid. J. Med. Chem. 2004, 47, 4559–4569. [Google Scholar] [CrossRef]
- Bowlus, C.L.; Pockros, P.J.; Kremer, A.E.; Pares, A.; Forman, L.M.; Drenth, J.P.H.; Ryder, S.D.; Terracciano, L.; Jin, Y.; Liberman, A.; et al. Long-Term Obeticholic Acid Therapy Improves Histological Endpoints in Patients With Primary Biliary Cholangitis. Clin. Gastroenterol. Hepatol. 2020, 18, 1170–1178.e6. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Dyson, J.K.; Alexander, G.J.M.; Chapman, M.H.; Collier, J.; Hübscher, S.; Patanwala, I.; Pereira, S.P.; Thain, C.; Thorburn, D.; et al. The British Society of Gastroenterology/UK-PBC primary biliary cholangitis treatment and management guidelines. Gut 2018, 67, 1568–1594. [Google Scholar] [CrossRef]
- Chapman, R.W.; Lynch, K.D. Obeticholic acid-a new therapy in PBC and NASH. Br. Med. Bull. 2020, 133, 95–104. [Google Scholar] [CrossRef]
- Hernandez, E.D.; Zheng, L.; Kim, Y.; Fang, B.; Liu, B.; Valdez, R.A.; Dietrich, W.F.; Rucker, P.V.; Chianelli, D.; Schmeits, J.; et al. Tropifexor-Mediated Abrogation of Steatohepatitis and Fibrosis Is Associated With the Antioxidative Gene Expression Profile in Rodents. Hepatol. Commun. 2019, 3, 1085–1097. [Google Scholar] [CrossRef] [PubMed]
- Badman, M.K.; Chen, J.; Desai, S.; Vaidya, S.; Neelakantham, S.; Zhang, J.; Gan, L.; Danis, K.; Laffitte, B.; Klickstein, L.B. Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of the Novel Non-Bile Acid FXR Agonist Tropifexor (LJN452) in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2020, 9, 395–410. [Google Scholar] [CrossRef] [PubMed]
- Camilleri, M.; Nord, S.L.; Burton, D.; Oduyebo, I.; Zhang, Y.; Chen, J.; Im, K.; Bhad, P.; Badman, M.K.; Sanders, D.S.; et al. Randomised clinical trial: Significant biochemical and colonic transit effects of the farnesoid X receptor agonist tropifexor in patients with primary bile acid diarrhoea. Aliment. Pharm. 2020, 52, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Pedrosa, M.; Seyedkazemi, S.; Francque, S.; Sanyal, A.; Rinella, M.; Charlton, M.; Loomba, R.; Ratziu, V.; Kochuparampil, J.; Fischer, L.; et al. A randomized, double-blind, multicenter, phase 2b study to evaluate the safety and efficacy of a combination of tropifexor and cenicriviroc in patients with nonalcoholic steatohepatitis and liver fibrosis: Study design of the TANDEM trial. Contemp. Clin. Trials 2020, 88, 105889. [Google Scholar] [CrossRef]
- Camilleri, M.; Nord, S.L.; Burton, D.; Oduyebo, I.; Zhang, Y.M.; Chen, J.; Bhad, P.; Badman, M.K.; Sanders, D.; Walters, J.R. A Double-Blind, Randomized, Placebo-Controlled, Crossover, Multiple-Dose Study of Tropifexor, a Non Bile Acid Fxr Agonist, in Patients with Primary Bile Acid Diarrhea. Gastroenterology 2019, 156, S204–S205. [Google Scholar] [CrossRef]
- Schramm, C.; Hirschfield, G.; Mason, A.L.; Wedemeyer, H.; Klickstein, L.; Neelakantham, S.; Koo, P.; Sanni, J.; Badman, M.; Jones, D. Early assessment of safety and efficacy of tropifexor, a potent non bile-acid FXR agonist, in patients with primary biliary cholangitis: An interim analysis of an ongoing phase 2 study. J. Hepatol. 2018, 68, S103. [Google Scholar] [CrossRef]
- Sanyal, A.; Lopez, P.; Lawitz, E.; Kim, W.; Huang, J.-F.; Andreone, P.; Goh, G.B.B.; Chen, Y.-C.; Ratziu, V.; Kim, Y.J. Tropifexor, a farnesoid X receptor agonist for the treatment of nonalcoholic steatohepatitis–Interim results based on baseline body mass index from first two parts of Phase 2b study FLIGHT-FXR. J. Hepatol. 2019, 70, E796–E797. [Google Scholar] [CrossRef]
- Schramm, C.; Wedemeyer, H.; Mason, A.; Hirschfield, G.M.; Levy, C.; Kowdley, K.V.; Milkiewicz, P.; Janczewska, E.; Malova, E.S.; Sanni, J.; et al. Farnesoid X receptor agonist tropifexor attenuates cholestasis in a randomised trial in patients with primary biliary cholangitis. JHEP Rep. 2022, 4, 100544. [Google Scholar] [CrossRef]
- Patel, K.; Harrison, S.A.; Elkhashab, M.; Trotter, J.F.; Herring, R.; Rojter, S.E.; Kayali, Z.; Wong, V.W.; Greenbloom, S.; Jayakumar, S.; et al. Cilofexor, a Nonsteroidal FXR Agonist, in Patients With Noncirrhotic NASH: A Phase 2 Randomized Controlled Trial. Hepatology 2020, 72, 58–71. [Google Scholar] [CrossRef]
- Trauner, M.; Bowlus, C.L.; Gulamhusein, A.; Hameed, B.; Caldwell, S.H.; Shiffman, M.L.; Landis, C.; Muir, A.J.; Billin, A.; Xu, J.; et al. Safety and sustained efficacy of the farnesoid X receptor (FXR) agonist cilofexor over a 96-week open-label extension in patients with PSC. Clin. Gastroenterol. Hepatol. 2022, S1542-3565(22)00720-0. [Google Scholar] [CrossRef]
- Erken, R.; Andre, P.; Roy, E.; Kootstra, N.; Barzic, N.; Girma, H.; Laveille, C.; Radreau-Pierini, P.; Darteil, R.; Vonderscher, J.; et al. Farnesoid X receptor agonist for the treatment of chronic hepatitis B: A safety study. J. Viral Hepat. 2021, 28, 1690–1698. [Google Scholar] [CrossRef] [PubMed]
- Chianelli, D.; Rucker, P.V.; Roland, J.; Tully, D.C.; Nelson, J.; Liu, X.; Bursulaya, B.; Hernandez, E.D.; Wu, J.; Prashad, M.; et al. Nidufexor (LMB763), a Novel FXR Modulator for the Treatment of Nonalcoholic Steatohepatitis. J. Med. Chem. 2020, 63, 3868–3880. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, S.K.; Singh, N.; Kumari, R.; Mishra, J.S.; Tripathi, S.; Banerjee, P.; Shah, P.; Kukshal, V.; Tyagi, A.M.; Gaikwad, A.N.; et al. Bile acid receptor agonist GW4064 regulates PPARgamma coactivator-1alpha expression through estrogen receptor-related receptor alpha. Mol. Endocrinol. 2011, 25, 922–932. [Google Scholar] [CrossRef] [PubMed]
- Harrison, S.A.; Bashir, M.R.; Lee, K.J.; Shim-Lopez, J.; Lee, J.; Wagner, B.; Smith, N.D.; Chen, H.C.; Lawitz, E.J. A structurally optimized FXR agonist, MET409, reduced liver fat content over 12 weeks in patients with non-alcoholic steatohepatitis. J. Hepatol. 2021, 75, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Marzano, M.; Fosso, B.; Colliva, C.; Notario, E.; Passeri, D.; Intranuovo, M.; Gioiello, A.; Adorini, L.; Pesole, G.; Pellicciari, R.; et al. Farnesoid X receptor activation by the novel agonist TC-100 (3alpha, 7alpha, 11beta-Trihydroxy-6alpha-ethyl-5beta-cholan-24-oic Acid) preserves the intestinal barrier integrity and promotes intestinal microbial reshaping in a mouse model of obstructed bile acid flow. Biomed. Pharm. 2022, 153, 113380. [Google Scholar] [CrossRef]
- Nara, S.J.; Jogi, S.; Cheruku, S.; Kandhasamy, S.; Jaipuri, F.; Kathi, P.K.; Reddy, S.; Sarodaya, S.; Cook, E.M.; Wang, T.; et al. Discovery of BMS-986339, a Pharmacologically Differentiated Farnesoid X Receptor Agonist for the Treatment of Nonalcoholic Steatohepatitis. J. Med. Chem. 2022, 65, 8948–8960. [Google Scholar] [CrossRef]
- Cao, S.; Yang, X.; Zhang, Z.; Wu, J.; Chi, B.; Chen, H.; Yu, J.; Feng, S.; Xu, Y.; Li, J.; et al. Discovery of a tricyclic farnesoid X receptor agonist HEC96719, a clinical candidate for treatment of non-alcoholic steatohepatitis. Eur. J. Med. Chem. 2022, 230, 114089. [Google Scholar] [CrossRef]
- Gege, C.; Hambruch, E.; Hambruch, N.; Kinzel, O.; Kremoser, C. Nonsteroidal FXR Ligands: Current Status and Clinical Applications. Handb. Exp. Pharm. 2019, 256, 167–205. [Google Scholar] [CrossRef]
- Liu, X.; Xue, R.; Ji, L.; Zhang, X.; Wu, J.; Gu, J.; Zhou, M.; Chen, S. Activation of farnesoid X receptor (FXR) protects against fructose-induced liver steatosis via inflammatory inhibition and ADRP reduction. Biochem. Biophys. Res. Commun. 2014, 450, 117–123. [Google Scholar] [CrossRef]
- Al-Khaifi, A.; Rudling, M.; Angelin, B. An FXR Agonist Reduces Bile Acid Synthesis Independently of Increases in FGF19 in Healthy Volunteers. Gastroenterology 2018, 155, 1012–1016. [Google Scholar] [CrossRef]
- Traussnigg, S.; Halilbasic, E.; Hofer, H.; Munda, P.; Stojakovic, T.; Fauler, G.; Kashofer, K.; Krssak, M.; Wolzt, M.; Trauner, M. Open-label phase II study evaluating safety and efficacy of the non-steroidal farnesoid X receptor agonist PX-104 in non-alcoholic fatty liver disease. Wien. Klin. Wochenschr. 2021, 133, 441–451. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Crittenden, D.B.; Eng, C.; Zhang, Q.; Guo, P.; Chung, D.; Fenaux, M.; Klucher, K.; Jones, C.; Jin, F.; et al. Safety, Pharmacokinetics, Pharmacodynamics, and Formulation of Liver-Distributed Farnesoid X-Receptor Agonist TERN-101 in Healthy Volunteers. Clin. Pharmacol. Drug Dev. 2021, 10, 1198–1208. [Google Scholar] [CrossRef] [PubMed]
- Ratziu, V.; Rinella, M.E.; Neuschwander-Tetri, B.A.; Lawitz, E.; Denham, D.; Kayali, Z.; Sheikh, A.; Kowdley, K.V.; Desta, T.; Elkhashab, M.; et al. EDP-305 in patients with NASH: A phase II double-blind placebo-controlled dose-ranging study. J. Hepatol. 2022, 76, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Miyata, S.; Kawashima, Y.; Sakai, M.; Matsubayashi, M.; Motoki, K.; Miyajima, Y.; Watanabe, Y.; Chikamatsu, N.; Taniguchi, T.; Tokuyama, R. Discovery, optimization, and evaluation of non-bile acid FXR/TGR5 dual agonists. Sci. Rep. 2021, 11, 9196. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.B.; Liu, X.Y.; Zhan, W. Farnesoid X receptor agonist INT-767 attenuates liver steatosis and inflammation in rat model of nonalcoholic steatohepatitis. Drug Des. Devel. 2018, 12, 2213–2221. [Google Scholar] [CrossRef]
- Cipriani, S.; Mencarelli, A.; Palladino, G.; Fiorucci, S. FXR activation reverses insulin resistance and lipid abnormalities and protects against liver steatosis in Zucker (fa/fa) obese rats. J. Lipid Res. 2010, 51, 771–784. [Google Scholar] [CrossRef]
- Renga, B.; Mencarelli, A.; Vavassori, P.; Brancaleone, V.; Fiorucci, S. The bile acid sensor FXR regulates insulin transcription and secretion. Biochim. Biophys. Acta 2010, 1802, 363–372. [Google Scholar] [CrossRef]
- Mudaliar, S.; Henry, R.R.; Sanyal, A.J.; Morrow, L.; Marschall, H.U.; Kipnes, M.; Adorini, L.; Sciacca, C.I.; Clopton, P.; Castelloe, E.; et al. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology 2013, 145, 574–582.e1. [Google Scholar] [CrossRef]
- Chalasani, N.; Younossi, Z.; Lavine, J.E.; Charlton, M.; Cusi, K.; Rinella, M.; Harrison, S.A.; Brunt, E.M.; Sanyal, A.J. The diagnosis and management of nonalcoholic fatty liver disease: Practice guidance from the American Association for the Study of Liver Diseases. Hepatology 2018, 67, 328–357. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Ratziu, V.; Loomba, R.; Rinella, M.; Anstee, Q.M.; Goodman, Z.; Bedossa, P.; Geier, A.; Beckebaum, S.; Newsome, P.N.; et al. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: Interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet 2019, 394, 2184–2196. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Stepanova, M.; Nader, F.; Loomba, R.; Anstee, Q.M.; Ratziu, V.; Harrison, S.; Sanyal, A.J.; Schattenberg, J.M.; Barritt, A.S.; et al. Obeticholic Acid Impact on Quality of Life in Patients With Nonalcoholic Steatohepatitis: REGENERATE 18-Month Interim Analysis. Clin. Gastroenterol. Hepatol. 2022, 20, 2050–2058.e12. [Google Scholar] [CrossRef] [PubMed]
- Tully, D.C.; Rucker, P.V.; Chianelli, D.; Williams, J.; Vidal, A.; Alper, P.B.; Mutnick, D.; Bursulaya, B.; Schmeits, J.; Wu, X. Discovery of tropifexor (LJN452), a Highly Potent Non-Bile Acid FXR agonist for the Treatment of Cholestatic Liver Diseases and Nonalcoholic Steatohepatitis (NASH); ACS Publications: Washington, DC, USA, 2017. [Google Scholar]
- ClinicalTrials.gov—Studies on Tropifexor. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=tropifexor&cntry=&state=&city=&dist= (accessed on 18 November 2022).
- ClinicalTrials.gov—Studies on FXF agonists. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=FXR+agonist&cntry=&state=&city=&dist=&Search=Search (accessed on 18 November 2022).
- Oduyebo, I.; Camilleri, M.; Nelson, A.D.; Khemani, D.; Nord, S.L.; Busciglio, I.; Burton, D.; Rhoten, D.; Ryks, M.; Carlson, P.; et al. Effects of NGM282, an FGF19 variant, on colonic transit and bowel function in functional constipation: A randomized phase 2 trial. Am. J. Gastroenterol. 2018, 113, 725–734. [Google Scholar] [CrossRef] [PubMed]
- Qiao, P.; Jia, Y.; Ma, A.; He, J.; Shao, C.; Li, X.; Wang, S.; Yang, B.; Zhou, H. Dapagliflozin protects against nonalcoholic steatohepatitis in db/db mice. Front. Pharm. 2022, 13, 934136. [Google Scholar] [CrossRef] [PubMed]
- Bellanti, F.; Lo Buglio, A.; Dobrakowski, M.; Kasperczyk, A.; Kasperczyk, S.; Aich, P.; Singh, S.P.; Serviddio, G.; Vendemiale, G. Impact of sodium glucose cotransporter-2 inhibitors on liver steatosis/fibrosis/inflammation and redox balance in non-alcoholic fatty liver disease. World J. Gastroenterol. 2022, 28, 3243–3257. [Google Scholar] [CrossRef]
- Wu, X.; Ge, H.; Gupte, J.; Weiszmann, J.; Shimamoto, G.; Stevens, J.; Hawkins, N.; Lemon, B.; Shen, W.; Xu, J.; et al. Co-receptor requirements for fibroblast growth factor-19 signaling. J. Biol. Chem. 2007, 282, 29069–29072. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Di Ciaula, A.; Bonfrate, L.; Baj, J.; Khalil, M.; Garruti, G.; Stellaard, F.; Wang, H.H.; Wang, D.Q.-H.; Portincasa, P. Recent Advances in the Digestive, Metabolic and Therapeutic Effects of Farnesoid X Receptor and Fibroblast Growth Factor 19: From Cholesterol to Bile Acid Signaling. Nutrients 2022, 14, 4950. https://doi.org/10.3390/nu14234950
Di Ciaula A, Bonfrate L, Baj J, Khalil M, Garruti G, Stellaard F, Wang HH, Wang DQ-H, Portincasa P. Recent Advances in the Digestive, Metabolic and Therapeutic Effects of Farnesoid X Receptor and Fibroblast Growth Factor 19: From Cholesterol to Bile Acid Signaling. Nutrients. 2022; 14(23):4950. https://doi.org/10.3390/nu14234950
Chicago/Turabian StyleDi Ciaula, Agostino, Leonilde Bonfrate, Jacek Baj, Mohamad Khalil, Gabriella Garruti, Frans Stellaard, Helen H. Wang, David Q.-H. Wang, and Piero Portincasa. 2022. "Recent Advances in the Digestive, Metabolic and Therapeutic Effects of Farnesoid X Receptor and Fibroblast Growth Factor 19: From Cholesterol to Bile Acid Signaling" Nutrients 14, no. 23: 4950. https://doi.org/10.3390/nu14234950
APA StyleDi Ciaula, A., Bonfrate, L., Baj, J., Khalil, M., Garruti, G., Stellaard, F., Wang, H. H., Wang, D. Q.-H., & Portincasa, P. (2022). Recent Advances in the Digestive, Metabolic and Therapeutic Effects of Farnesoid X Receptor and Fibroblast Growth Factor 19: From Cholesterol to Bile Acid Signaling. Nutrients, 14(23), 4950. https://doi.org/10.3390/nu14234950