
Neurometabolic Dysfunction in SPG11 Hereditary Spastic Paraplegia

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Participants
2.2. Laboratory Workup
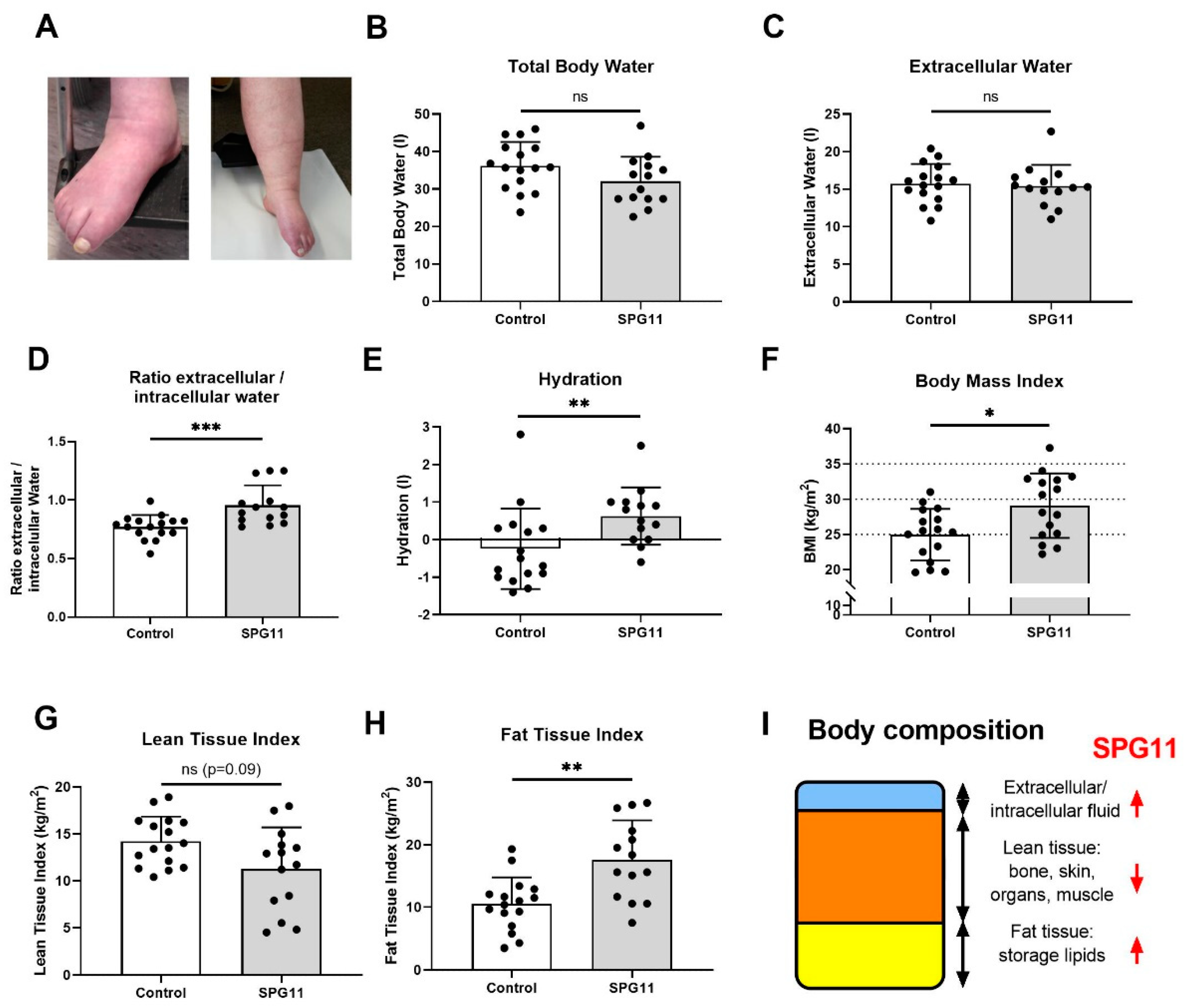
2.3. Anthropometry and Bioimpedance Spectroscopy
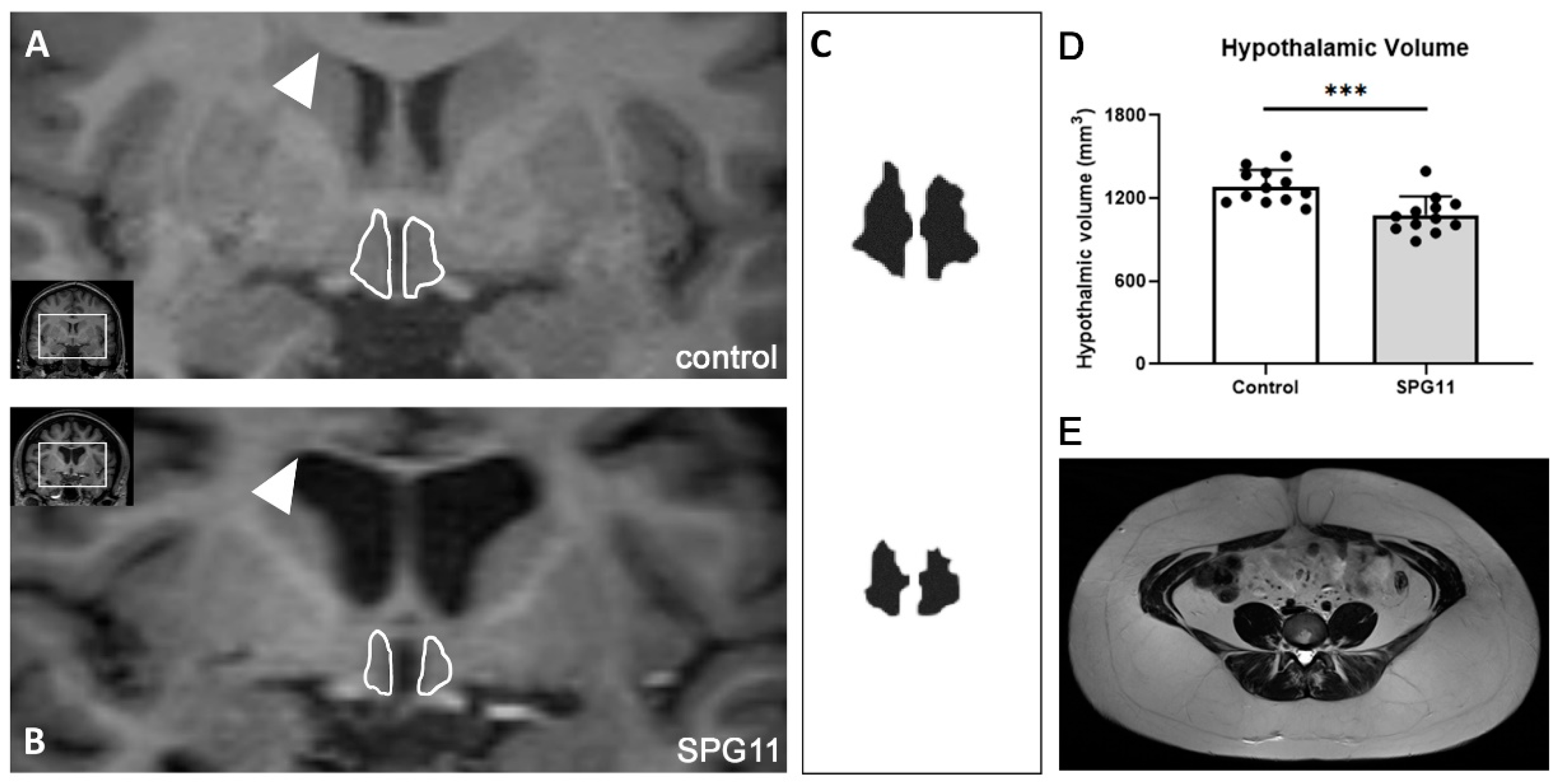
2.4. Determination of Hypothalamic Volume
2.5. Statistics
3. Results
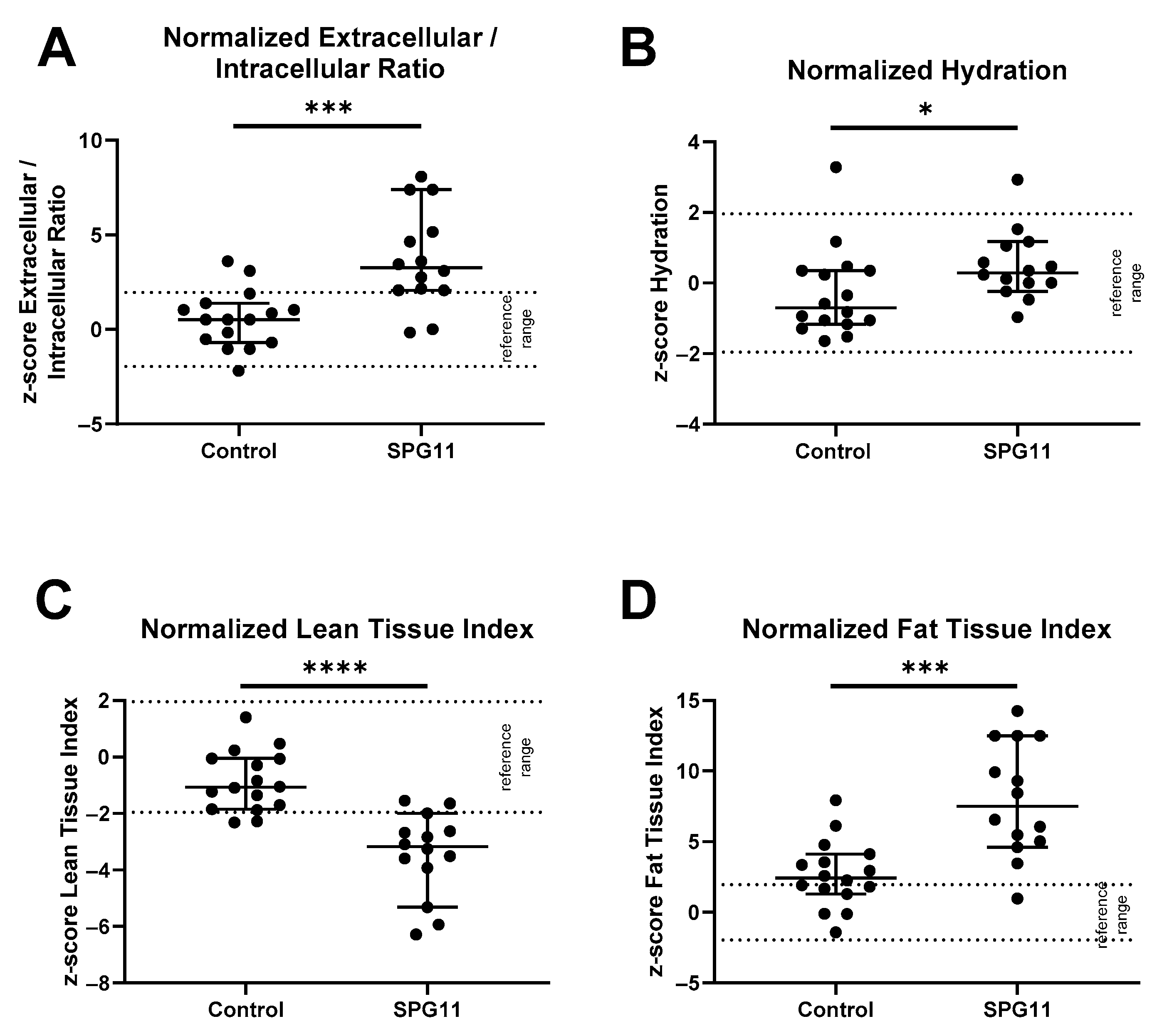
3.1. Altered Body Composition in SPG11 Patients
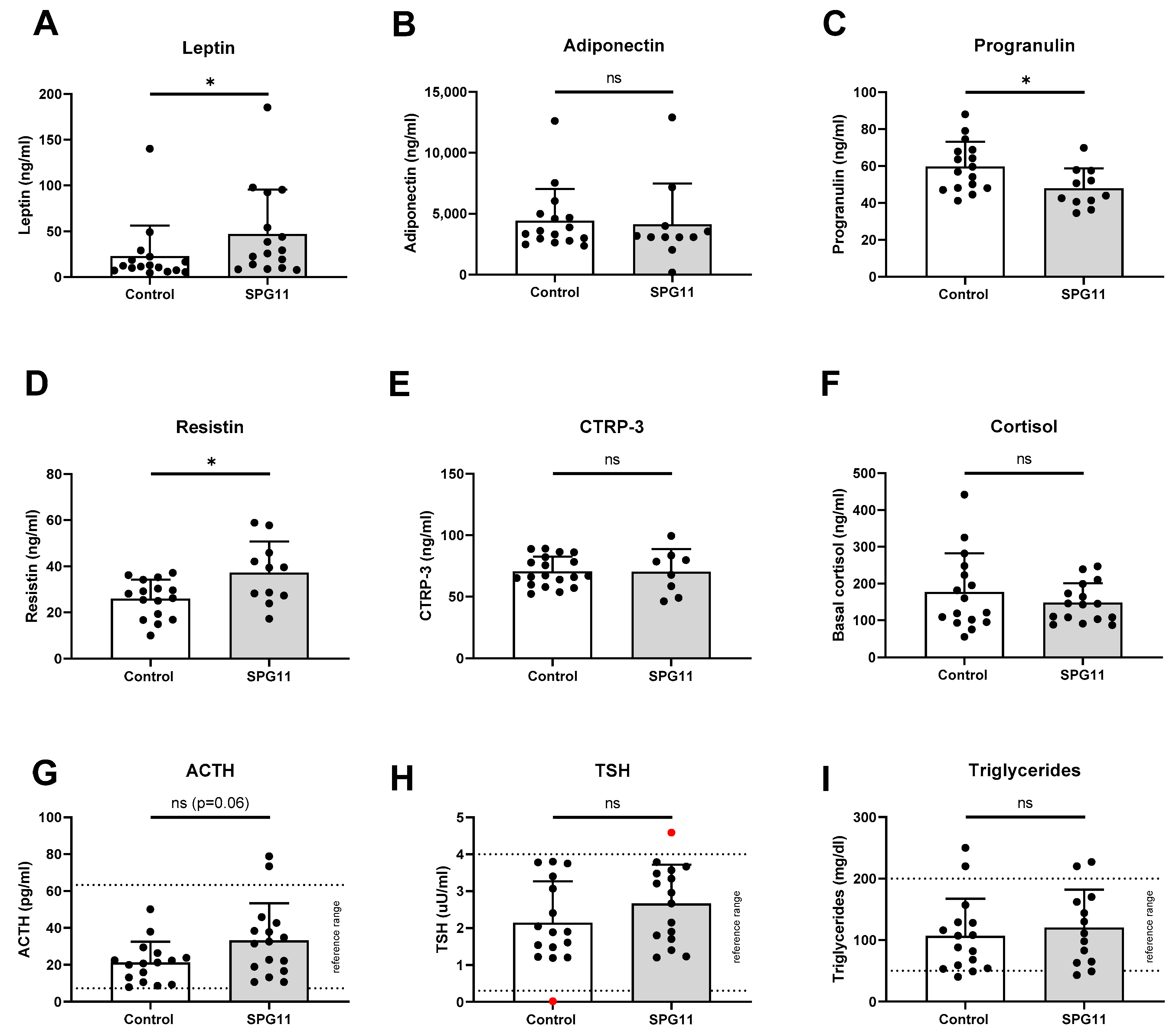
3.2. Increased Levels of Leptin in SPG11
3.3. Unchanged Pituitary Gland Hormones and Lipid Parameters in SPG11
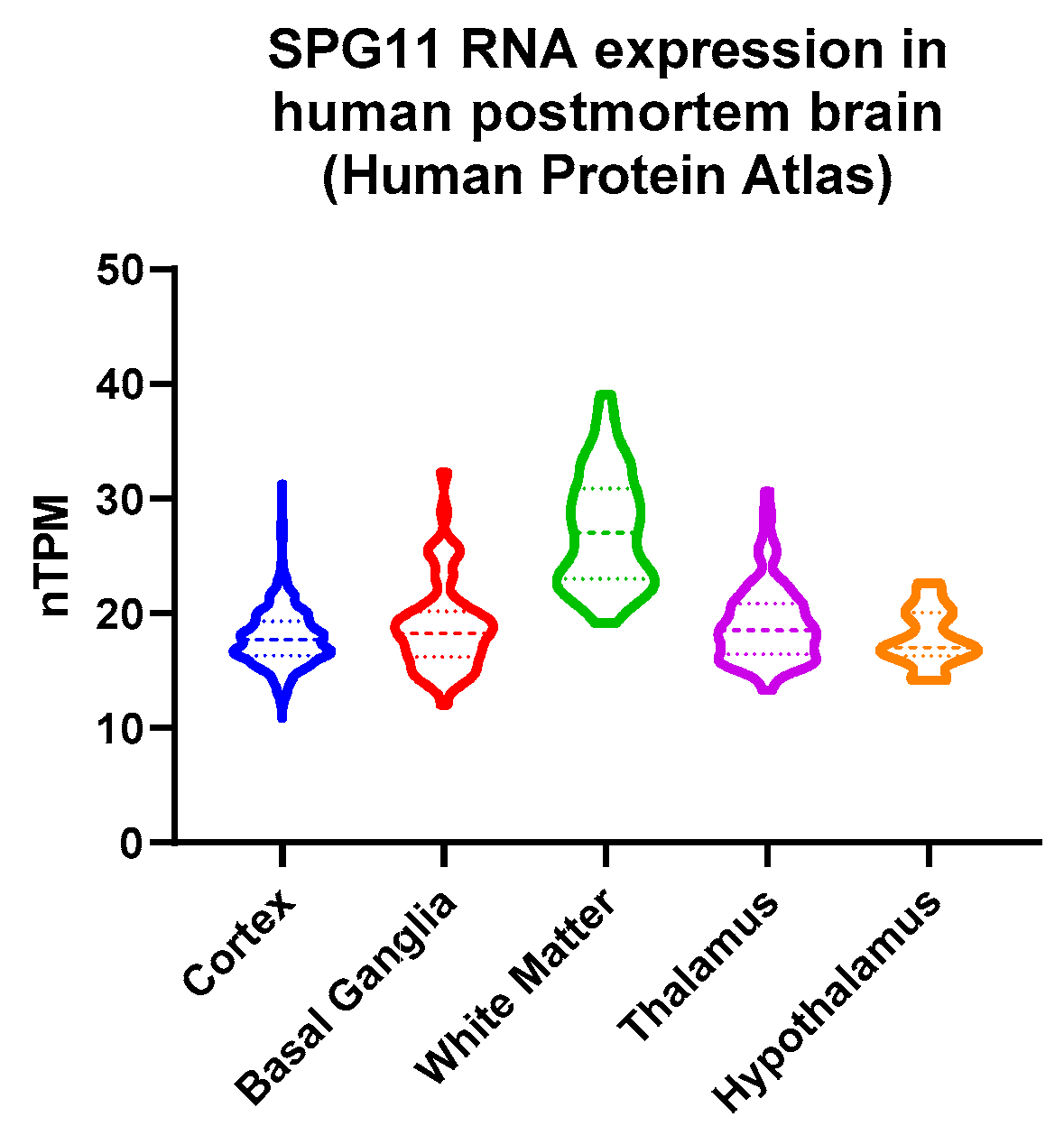
3.4. Decreased Hypothalamic Volume in SPG11
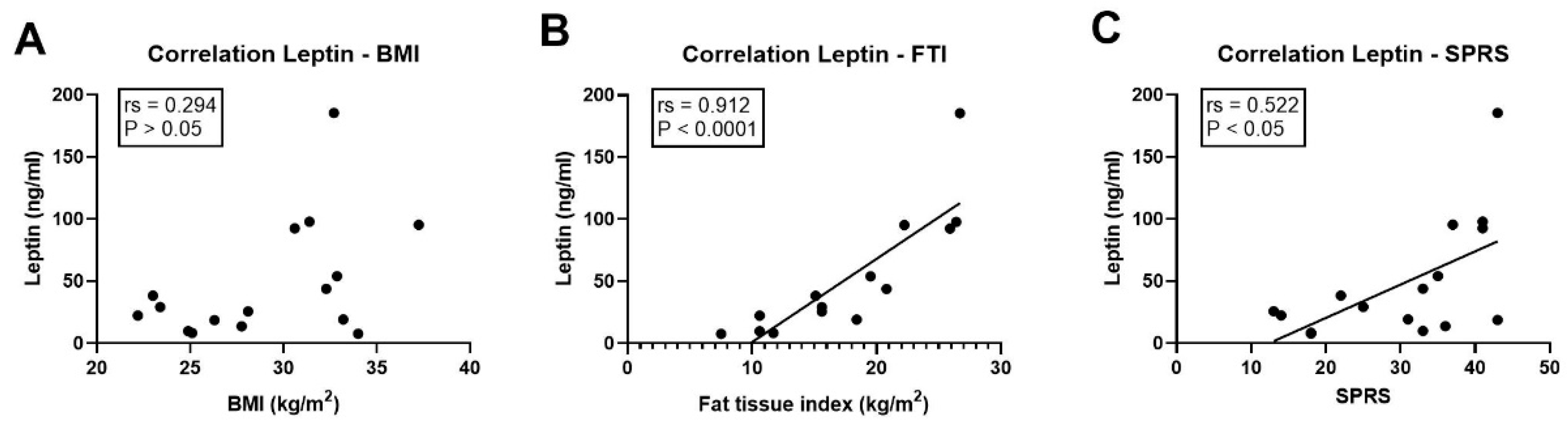
3.5. Association of Leptin with Clinical Parameters
4. Discussion
4.1. Role of Disability-Related Causes of Obesity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ref. | No. of Families | No. of SPG11 Patients | No. (%) of Obese Patients | No. (%) of Patients with Edema | Remarks |
|---|---|---|---|---|---|
| [27] | 1 | 2 | 2 (100%) | 2 (100%) | Absence of obesity in 4 unaffected siblings. |
| [28] | 2 | 4 | 2 (50%) | not reported | |
| [4] | 20 | 38 | 1 (3%) | not reported | |
| [29] | 1 | 5 | 3 (60%) | not reported | |
| [30] | 4 | 4 | 2 (50%) | not reported | Patients with Kjellin’s syndrome. |
| [31] | 3 | 4 | not reported | 1 (25%) | Congenital left leg lymphedema. |
| [5] | 9 | 14 | 18 (78%) | not reported | Five patients were still ambulatory. |
| [7] | 2 | 4 | not reported | 2 (50%) | Two members of same family affected. |
| [6] | n/a | 20 | 5 (25%) | n/a | Includes patients from [32]; increased BMI compared to FRDA individuals. |
| this report | 14 | 16 | 12 (75%) | 10 (63%) | Includes 2 patients from [27]. |
4.2. Implications of Increased Leptin Levels
4.3. Immune–Metabolic Implications of Dysregulated Progranulin and Resistin
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kara, E.; Tucci, A.; Manzoni, C.; Lynch, D.S.; Elpidorou, M.; Bettencourt, C.; Chelban, V.; Manole, A.; Hamed, S.A.; Haridy, N.A.; et al. Genetic and Phenotypic Characterization of Complex Hereditary Spastic Paraplegia. Brain 2016, 139, 1904–1918. [Google Scholar] [CrossRef] [PubMed]
- Schüle, R.; Wiethoff, S.; Martus, P.; Karle, K.N.; Otto, S.; Klebe, S.; Klimpe, S.; Gallenmüller, C.; Kurzwelly, D.; Henkel, D.; et al. Hereditary Spastic Paraplegia: Clinicogenetic Lessons from 608 Patients. Ann. Neurol. 2016, 79, 646–658. [Google Scholar] [CrossRef] [PubMed]
- Pozner, T.; Regensburger, M.; Engelhorn, T.; Winkler, J.; Winner, B. Janus-Faced Spatacsin (SPG11): Involvement in Neurodevelopment and Multisystem Neurodegeneration. Brain 2020, 143, 2369–2379. [Google Scholar] [CrossRef] [PubMed]
- Stevanin, G.; Azzedine, H.; Denora, P.; Boukhris, A.; Tazir, M.; Lossos, A.; Rosa, A.L.; Lerer, I.; Hamri, A.; Alegria, P.; et al. Mutations in SPG11 Are Frequent in Autosomal Recessive Spastic Paraplegia with Thin Corpus Callosum, Cognitive Decline and Lower Motor Neuron Degeneration. Brain 2008, 131, 772–784. [Google Scholar] [CrossRef]
- Bot, S.T.D.; Burggraaff, R.C.; Herkert, J.C.; Schelhaas, H.J.; Post, B.; Diekstra, A.; van Vliet, R.O.; van der Knaap, M.S.; Kamsteeg, E.-J.; Scheffer, H.; et al. Rapidly Deteriorating Course in Dutch Hereditary Spastic Paraplegia Type 11 Patients. Eur. J. Hum. Genet. 2013, 21, 1312–1315. [Google Scholar] [CrossRef]
- Cardozo-Hernández, A.L.D.C.; Rezende, T.J.R.; França, M.C. Hereditary Spastic Paraplegia Type 11 (SPG11) Is Associated with Obesity and Hypothalamic Damage. J. Neurol. Sci. 2020, 416, 116982. [Google Scholar] [CrossRef]
- Manole, A.; Chelban, V.; Haridy, N.A.; Hamed, S.A.; Berardo, A.; Reilly, M.M.; Houlden, H. Severe Axonal Neuropathy Is a Late Manifestation of SPG11. J. Neurol. 2016, 263, 2278–2286. [Google Scholar] [CrossRef]
- Zung, W.W. A Self-Rating Depression Scale. Arch. Gen. Psychiatry 1965, 12, 63–70. [Google Scholar] [CrossRef]
- Schmid, A.; Berghoff, M.; Hochberg, A.; Schäffler, A.; Karrasch, T. CTRP-3 Is Permeable to the Blood-Brain Barrier and Is Not Regulated by Glucose or Lipids in Vivo. Eur. J. Clin. Investig. 2017, 47, 203–212. [Google Scholar] [CrossRef]
- Berghoff, M.; Hochberg, A.; Schmid, A.; Schlegel, J.; Karrasch, T.; Kaps, M.; Schäffler, A. Quantification and Regulation of the Adipokines Resistin and Progranulin in Human Cerebrospinal Fluid. Eur. J. Clin. Investig. 2016, 46, 15–26. [Google Scholar] [CrossRef]
- Marcelli, D.; Usvyat, L.A.; Kotanko, P.; Bayh, I.; Canaud, B.; Etter, M.; Gatti, E.; Grassmann, A.; Wang, Y.; Marelli, C.; et al. Body Composition and Survival in Dialysis Patients: Results from an International Cohort Study. Clin. J. Am. Soc. Nephro. 2015, 10, 1192–1200. [Google Scholar] [CrossRef] [PubMed]
- Wabel, P.; Chamney, P.; Moissl, U.; Jirka, T. Importance of Whole-Body Bioimpedance Spectroscopy for the Management of Fluid Balance. Blood Purificat. 2009, 27, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Chamney, P.W.; Wabel, P.; Moissl, U.M.; Müller, M.J.; Bosy-Westphal, A.; Korth, O.; Fuller, N.J. A Whole-Body Model to Distinguish Excess Fluid from the Hydration of Major Body Tissues. Am. J. Clin. Nutr. 2007, 85, 80–89. [Google Scholar] [CrossRef] [PubMed]
- Moissl, U.M.; Wabel, P.; Chamney, P.W.; Bosaeus, I.; Levin, N.W.; Bosy-Westphal, A.; Korth, O.; Müller, M.J.; Ellegård, L.; Malmros, V.; et al. Body Fluid Volume Determination via Body Composition Spectroscopy in Health and Disease. Physiol. Meas. 2006, 27, 921–933. [Google Scholar] [CrossRef] [PubMed]
- Mulasi, U.; Kuchnia, A.J.; Cole, A.J.; Earthman, C.P. Bioimpedance at the Bedside. Nutr. Clin. Pract. 2015, 30, 180–193. [Google Scholar] [CrossRef]
- Passauer, J.; Petrov, H.; Schleser, A.; Leicht, J.; Pucalka, K. Evaluation of Clinical Dry Weight Assessment in Haemodialysis Patients Using Bioimpedance Spectroscopy: A Cross-Sectional Study. Nephrol. Dial. Transpl. 2010, 25, 545–551. [Google Scholar] [CrossRef]
- Tang, X.; Oishi, K.; Faria, A.V.; Hillis, A.E.; Albert, M.S.; Mori, S.; Miller, M.I. Bayesian Parameter Estimation and Segmentation in the Multi-Atlas Random Orbit Model. PLoS ONE 2013, 8, e65591. [Google Scholar] [CrossRef]
- Friedman, J.M. Causes and Control of Excess Body Fat. Nature 2009, 459, 340–342. [Google Scholar] [CrossRef]
- Nguyen, A.D.; Nguyen, T.A.; Martens, L.H.; Mitic, L.L.; Farese, R.V. Progranulin: At the Interface of Neurodegenerative and Metabolic Diseases. Trends Endocrinol. Metab. 2013, 24, 597–606. [Google Scholar] [CrossRef]
- Steppan, C.M.; Lazar, M.A. The Current Biology of Resistin. J. Intern. Med. 2004, 255, 439–447. [Google Scholar] [CrossRef]
- Sjöstedt, E.; Zhong, W.; Fagerberg, L.; Karlsson, M.; Mitsios, N.; Adori, C.; Oksvold, P.; Edfors, F.; Limiszewska, A.; Hikmet, F.; et al. An Atlas of the Protein-Coding Genes in the Human, Pig, and Mouse Brain. Science 2020, 367, eaay5947. [Google Scholar] [CrossRef] [PubMed]
- Avena, N.M.; Murray, S.; Gold, M.S. The next Generation of Obesity Treatments: Beyond Suppressing Appetite. Front. Psychol. 2013, 4, 721. [Google Scholar] [CrossRef] [PubMed]
- Luppino, F.S.; Wit, L.M.D.; Bouvy, P.F.; Stijnen, T.; Cuijpers, P.; Penninx, B.W.J.H.; Zitman, F.G. Overweight, Obesity, and Depression: A Systematic Review and Meta-Analysis of Longitudinal Studies. Arch. Gen. Psychiatry 2010, 67, 220–229. [Google Scholar] [CrossRef] [PubMed]
- Alhurani, R.E.; Vassilaki, M.; Aakre, J.A.; Mielke, M.M.; Kremers, W.K.; Machulda, M.M.; Geda, Y.E.; Knopman, D.S.; Peterson, R.C.; Roberts, R.O. Decline in Weight and Incident Mild Cognitive Impairment: Mayo Clinic Study of Aging. JAMA Neurol. 2016, 73, 439–446. [Google Scholar] [CrossRef] [PubMed]
- Voll, S.L.; Boot, E.; Butcher, N.J.; Cooper, S.; Heung, T.; Chow, E.W.C.; Silversides, C.K.; Bassett, A.S. Obesity in Adults with 22q11.2 Deletion Syndrome. Genet. Med. Off. J. Am. Coll. Med. Genet. 2017, 19, 204–208. [Google Scholar] [CrossRef]
- Butler, M.G. Prader-Willi Syndrome: Obesity Due to Genomic Imprinting. Curr. Genom. 2011, 12, 204–215. [Google Scholar] [CrossRef]
- Winner, B.; Uyanik, G.; Gross, C.; Lange, M.; Schulte-Mattler, W.; Schuierer, G.; Marienhagen, J.; Hehr, U.; Winkler, J. Clinical Progression and Genetic Analysis in Hereditary Spastic Paraplegia with Thin Corpus Callosum in Spastic Gait Gene 11 (SPG11). Arch. Neurol. 2004, 61, 117–121. [Google Scholar] [CrossRef][Green Version]
- Lossos, A.; Stevanin, G.; Meiner, V.; Argov, Z.; Bouslam, N.; Newman, J.P.; Gomori, J.M.; Klebe, S.; Lerer, I.; Elleuch, N.; et al. Hereditary Spastic Paraplegia with Thin Corpus Callosum: Reduction of the SPG11 Interval and Evidence for Further Genetic Heterogeneity. Arch. Neurol. 2006, 63, 756–760. [Google Scholar] [CrossRef]
- Orlén, H.; Melberg, A.; Raininko, R.; Kumlien, E.; Entesarian, M.; Söderberg, P.; Påhlman, M.; Darin, N.; Kyllerman, M.; Holmberg, E.; et al. SPG11 Mutations Cause Kjellin Syndrome, a Hereditary Spastic Paraplegia with Thin Corpus Callosum and Central Retinal Degeneration. Am. J. Med. Genetics. Part B Neuropsychiatr. Genet. Off. Publ. Int. Soc. Psychiatr. Genet. 2009, 150B, 984–992. [Google Scholar] [CrossRef]
- Puech, B.; Lacour, A.; Stevanin, G.; Sautiere, B.G.; Devos, D.; Depienne, C.; Denis, E.; Mundwiller, E.; Ferriby, D.; Vermersch, P.; et al. Kjellin Syndrome: Long-Term Neuro-Ophthalmologic Follow-up and Novel Mutations in the SPG11 Gene. Ophthalmology 2011, 118, 564–573. [Google Scholar] [CrossRef]
- Vanderver, A.; Tonduti, D.; Auerbach, S.; Schmidt, J.L.; Parikh, S.; Gowans, G.C.; Jackson, K.E.; Brock, P.L.; Patterson, M.; Nehrebecky, M.; et al. Neurotransmitter Abnormalities and Response to Supplementation in SPG11. Mol. Genet. Metab. 2012, 107, 229–233. [Google Scholar] [CrossRef] [PubMed]
- Faber, I.; Martinez, A.R.M.; Rezende, T.J.R.D.; Martins, C.R.; Martins, M.P.; Lourenco, C.M.; Marques, W.; Montecchiani, C.; Orlacchio, A.; Pedroso, J.L.; et al. SPG11 Mutations Cause Widespread White Matter and Basal Ganglia Abnormalities, but Restricted Cortical Damage. NeuroImage Clin. 2018, 19, 848–857. [Google Scholar] [CrossRef] [PubMed]
- Maffei, M.; Fei, H.; Lee, G.H.; Dani, C.; Leroy, P.; Zhang, Y.; Proenca, R.; Negrel, R.; Ailhaud, G.; Friedman, J.M. Increased Expression in Adipocytes of Ob RNA in Mice with Lesions of the Hypothalamus and with Mutations at the Db Locus. Proc. Natl. Acad. Sci. USA 1995, 92, 6957–6960. [Google Scholar] [CrossRef] [PubMed]
- Clément, K.; Vaisse, C.; Lahlou, N.; Cabrol, S.; Pelloux, V.; Cassuto, D.; Gourmelen, M.; Dina, C.; Chambaz, J.; Lacorte, J.-M.; et al. A Mutation in the Human Leptin Receptor Gene Causes Obesity and Pituitary Dysfunction. Nature 1998, 392, 398–401. [Google Scholar] [CrossRef]
- Denora, P.S.; Smets, K.; Zolfanelli, F.; Groote, C.C.; Casali, C.; Deconinck, T.; Sieben, A.; Gonzales, M.; Zuchner, S.; Darios, F.; et al. Motor Neuron Degeneration in Spastic Paraplegia 11 Mimics Amyotrophic Lateral Sclerosis Lesions. Brain 2016, 139, 1723–1734. [Google Scholar] [CrossRef] [PubMed]
- Kuru, S.; Sakai, M.; Konagaya, M.; Yoshida, M.; Hashizume, Y. Autopsy Case of Hereditary Spastic Paraplegia with Thin Corpus Callosum Showing Severe Gliosis in the Cerebral White Matter. Neuropathology 2005, 25, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Honda, H.; Hamasaki, H.; Sasagasako, N.; Suzuki, S.O.; Furuya, H.; Taniwaki, T.; Iwaki, T. Transactivation Response DNA-binding Protein of 43 KDa Proteinopathy and Lysosomal Abnormalities in Spastic Paraplegia Type 11. Neuropathology 2021, 41, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, M.; Matsubara, T.; Mochizuki, Y.; Takeuchi, C.; Minamitani, M.; Imai, M.; Kosaki, K.; Arai, T.; Murayama, S. An Autopsied Case Report of Spastic Paraplegia with Thin Corpus Callosum Carrying a Novel Mutation in the SPG11 Gene: Widespread Degeneration with Eosinophilic Inclusions. BMC Neurol. 2022, 22, 2. [Google Scholar] [CrossRef]
- Faber, I.; Martinez, A.R.M.; Martins, C.R.; Maia, M.L.; Souza, J.P.; Lourenço, C.M.; Marques, W.; Montecchiani, C.; Orlacchio, A.; Pedroso, J.L.; et al. SPG11-related Parkinsonism: Clinical Profile, Molecular Imaging and L-dopa Response. Mov. Disord. 2018, 33, 1650–1656. [Google Scholar] [CrossRef]
- Ueda, M.; Katayama, Y.; Kamiya, T.; Mishina, M.; Igarashi, H.; Okubo, S.; Senda, M.; Iwabuchi, K.; Terashi, A. Hereditary Spastic Paraplegia with a Thin Corpus Callosum and Thalamic Involvement in Japan. Neurology 1998, 51, 1751–1754. [Google Scholar] [CrossRef]
- Orlacchio, A.; Babalini, C.; Borreca, A.; Patrono, C.; Massa, R.; Basaran, S.; Munhoz, R.P.; Rogaeva, E.A.; George-Hyslop, P.H.S.; Bernardi, G.; et al. SPATACSIN Mutations Cause Autosomal Recessive Juvenile Amyotrophic Lateral Sclerosis. Brain 2010, 133, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Nagel, G.; Peter, R.S.; Rosenbohm, A.; Koenig, W.; Dupuis, L.; Rothenbacher, D.; Ludolph, A.C. Adipokines, C-Reactive Protein and Amyotrophic Lateral Sclerosis—Results from a Population- Based ALS Registry in Germany. Sci. Rep. 2017, 7, 4374. [Google Scholar] [CrossRef] [PubMed]
- Gorges, M.; Vercruysse, P.; Müller, H.-P.; Huppertz, H.-J.; Rosenbohm, A.; Nagel, G.; Weydt, P.; Petersén, Å.; Ludolph, A.C.; Kassubek, J.; et al. Hypothalamic Atrophy Is Related to Body Mass Index and Age at Onset in Amyotrophic Lateral Sclerosis. J. Neurol. Neurosurg. Psychiatry 2017, 88, 1033. [Google Scholar] [CrossRef] [PubMed]
- Baskin, D.G.; Schwartz, M.W.; Seeley, R.J.; Woods, S.C.; Porte, D.; Breininger, J.F.; Jonak, Z.; Schaefer, J.; Krouse, M.; Burghardt, C.; et al. Leptin Receptor Long-Form Splice-Variant Protein Expression in Neuron Cell Bodies of the Brain and Co-Localization with Neuropeptide Y MRNA in the Arcuate Nucleus. J. Histochem. Cytochem. Off. J. Histochem. Soc. 1999, 47, 353–362. [Google Scholar] [CrossRef] [PubMed]
- Couce, M.E.; Burguera, B.; Parisi, J.E.; Jensen, M.D.; Lloyd, R.V. Localization of Leptin Receptor in the Human Brain. Neuroendocrinology 1997, 66, 145–150. [Google Scholar] [CrossRef] [PubMed]
- Valerio, A.; Ghisi, V.; Dossena, M.; Tonello, C.; Giordano, A.; Frontini, A.; Ferrario, M.; Pizzi, M.; Spano, P.; Carruba, M.O.; et al. Leptin Increases Axonal Growth Cone Size in Developing Mouse Cortical Neurons by Convergent Signals Inactivating Glycogen Synthase Kinase-3β*. J. Biol. Chem. 2006, 281, 12950–12958. [Google Scholar] [CrossRef]
- Mishra, H.K.; Prots, I.; Havlicek, S.; Kohl, Z.; Perez-Branguli, F.; Boerstler, T.; Anneser, L.; Minakaki, G.; Wend, H.; Hampl, M.; et al. GSK3ß-dependent Dysregulation of Neurodevelopment in SPG11-patient Induced Pluripotent Stem Cell Model. Ann. Neurol. 2016, 79, 826–840. [Google Scholar] [CrossRef]
- Pérez-Brangulí, F.; Buchsbaum, I.Y.; Pozner, T.; Regensburger, M.; Fan, W.; Schray, A.; Börstler, T.; Mishra, H.; Gräf, D.; Kohl, Z.; et al. Human SPG11 Cerebral Organoids Reveal Cortical Neurogenesis Impairment. Hum. Mol. Genet. 2019, 28, 961–971. [Google Scholar] [CrossRef]
- Pozner, T.; Schray, A.; Regensburger, M.; Lie, D.C.; Schlötzer-Schrehardt, U.; Winkler, J.; Turan, S.; Winner, B. Tideglusib Rescues Neurite Pathology of SPG11 IPSC Derived Cortical Neurons. Front. Neurosci. 2018, 12, 914. [Google Scholar] [CrossRef]
- Kleinridders, A.; Lauritzen, H.P.M.M.; Ussar, S.; Christensen, J.H.; Mori, M.A.; Bross, P.; Kahn, C.R. Leptin Regulation of Hsp60 Impacts Hypothalamic Insulin Signaling. J. Clin. Investig. 2013, 123, 4667–4680. [Google Scholar] [CrossRef]
- Murmu, R.P.; Martin, E.; Rastetter, A.; Esteves, T.; Muriel, M.-P.; Hachimi, K.H.E.; Denora, P.S.; Dauphin, A.; Fernandez, J.C.; Duyckaerts, C.; et al. Cellular Distribution and Subcellular Localization of Spatacsin and Spastizin, Two Proteins Involved in Hereditary Spastic Paraplegia. Mol. Cell. Neurosci. 2011, 47, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Güner, F.; Pozner, T.; Krach, F.; Prots, I.; Loskarn, S.; Schlötzer-Schrehardt, U.; Winkler, J.; Winner, B.; Regensburger, M. Axon-Specific Mitochondrial Pathology in SPG11 Alpha Motor Neurons. Front. Neurosci. 2021, 15, 680572. [Google Scholar] [CrossRef] [PubMed]
- Denton, K.; Mou, Y.; Xu, C.-C.; Shah, D.; Chang, J.; Blackstone, C.; Li, X.-J. Impaired Mitochondrial Dynamics Underlie Axonal Defects in Hereditary Spastic Paraplegias. Hum. Mol. Genet. 2018, 27, 2517–2530. [Google Scholar] [CrossRef] [PubMed]
- Spadaro, O.; Youm, Y.; Shchukina, I.; Ryu, S.; Sidorov, S.; Ravussin, A.; Nguyen, K.; Aladyeva, E.; Predeus, A.N.; Smith, S.R.; et al. Caloric Restriction in Humans Reveals Immunometabolic Regulators of Health Span. Science 2022, 375, 671–677. [Google Scholar] [CrossRef] [PubMed]
- Regensburger, M.; Chaudhry, S.R.; Yasin, H.; Zhao, Y.; Stadlbauer, A.; Buchfelder, M.; Kinfe, T. Emerging Roles of Leptin in Parkinson’s Disease: Chronic Inflammation, Neuroprotection and More? Brain Behav. Immun. 2022, 107, 53–61. [Google Scholar] [CrossRef]






| Parameter (Mean ± SD) | SPG11 HSP N = 16 | Controls N = 16 | p Value |
|---|---|---|---|
| Age (y) | 31.1 ± 10.9 | 36.1 ± 11.6 | >0.05 |
| Gender (male: female) | 8:8 | 7:9 | >0.05 * |
| Body Mass Index (kg/m2) | 29.1 ± 4.6 | 25.0 ± 3.7 | 0.02 |
| Age of onset (y) | 12.7 ± 11.0 | --- | |
| Disease duration (y) | 18.4 ± 8.0 | --- | |
| SPRS total score | 30.2 ± 10.4 | --- | |
| SPRS functional measures | 18.4 ± 6.0 | --- | |
| (sum of items #1–#6) | |||
| SPRS spasticity measures (#7–#10) | 8.0 ± 3.3 | --- | |
| SPRS non-motor measures (#11–#13) | 3.8 ± 2.3 | --- |
| Disease Stage | Patient | Sex | AAO | AAE | SPRS | TCC | ID | ZUNG | Dysarthria | Trunk | Ambul. | SPG11 Genotype |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| early | SPG11-7 | f | 17 | 27 | 17 | – | – | 73.75 | – | – | – | c.5623C>T (homozyg.) |
| SPG11-9 | f | 14 | 18 | 24 | + | + | 40 | – | – | + | c.704_705del; c.6832_6833del | |
| SPG11-10 | m | 12 | 16 | 20 | + | + | 45 | + | – | + | c.1203_1203delA (homozyg.) | |
| SPG11-12 | m | 42 | 45 | 18 | – | – | 48.75 | – | – | + | c.255G>A; c.531T>C | |
| SPG11-14 | m | 3 | 27 | 18 | + | + | 50 | – | – | + | c.1951C>T (homozyg.) | |
| SPG11-16 | f | 14 | 21 | 14 | + | + | n/a | – | – | + | c.2612dupG; c.4434G>T | |
| middle | SPG11-4 | f | 15 | 23 | 21 | + | + | 45 | + | – | + | c.3075dupA; c.6204A>G |
| SPG11-5 | f | 15 | 23 | 17 | + | + | 52.5 | + | – | + | c.3075dupA; c.6204A>G | |
| SPG11-6 | m | 14 | 22 | 21 | + | + | 43.75 | + | + | – | c.733_734del; c.4306_4307del | |
| SPG11-8 | m | 12 | 35 | 40 | + | + | 66.25 | + | + | – | c.5623C>T (homozyg.) | |
| SPG11-13 | m | 9 | 20 | 33 | + | + | n/a | + | + | + | c.2990T>A; c.4877_4878delTT | |
| SPG11-15 | m | 3 | 25 | 31 | + | + | n/a | + | + | + | c.190dupC; c.704_705delAT | |
| late | SPG11-1 | f | 24 | 46 | 44 | + | ++ | n/a | + | + | – | c.3036C>A; c.5798delC |
| SPG11-2 | f | 20 | 40 | 37 | + | + | n/a | + | + | – | c.3036C>A; c.5798delC | |
| SPG11-3 | f | 31 | 50 | 36 | + | + | 35 | + | + | – | c.267G>A; c.1457-2A>G | |
| SPG11-11 | m | 14 | 47 | 46 | + | + | n/a | + | + | – | c.3076insA; del. exon 37-39 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Regensburger, M.; Krumm, L.; Schmidt, M.A.; Schmid, A.; Spatz, I.T.; Marterstock, D.C.; Kopp, C.; Kohl, Z.; Doerfler, A.; Karrasch, T.; et al. Neurometabolic Dysfunction in SPG11 Hereditary Spastic Paraplegia. Nutrients 2022, 14, 4803. https://doi.org/10.3390/nu14224803
Regensburger M, Krumm L, Schmidt MA, Schmid A, Spatz IT, Marterstock DC, Kopp C, Kohl Z, Doerfler A, Karrasch T, et al. Neurometabolic Dysfunction in SPG11 Hereditary Spastic Paraplegia. Nutrients. 2022; 14(22):4803. https://doi.org/10.3390/nu14224803
Chicago/Turabian StyleRegensburger, Martin, Laura Krumm, Manuel Alexander Schmidt, Andreas Schmid, Imke Tabea Spatz, Dominique Cornelius Marterstock, Christoph Kopp, Zacharias Kohl, Arnd Doerfler, Thomas Karrasch, and et al. 2022. "Neurometabolic Dysfunction in SPG11 Hereditary Spastic Paraplegia" Nutrients 14, no. 22: 4803. https://doi.org/10.3390/nu14224803
APA StyleRegensburger, M., Krumm, L., Schmidt, M. A., Schmid, A., Spatz, I. T., Marterstock, D. C., Kopp, C., Kohl, Z., Doerfler, A., Karrasch, T., Winner, B., & Winkler, J. (2022). Neurometabolic Dysfunction in SPG11 Hereditary Spastic Paraplegia. Nutrients, 14(22), 4803. https://doi.org/10.3390/nu14224803