

Iron: Not Just a Passive Bystander in AITD

 , and
, and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Material and Methods
3. Etiology of AITD
3.1. AITD Is Associated with Other Autoimmune Disorders
3.2. Impact of Genetic Factors on the Development of AITD
3.3. Impact of Viral Infections, including COVID-19, on the Development of AITD
3.4. How the Inflamamtory Infiltration Develops in AITD
3.5. ID and Production of Anti-TPO Antibodies
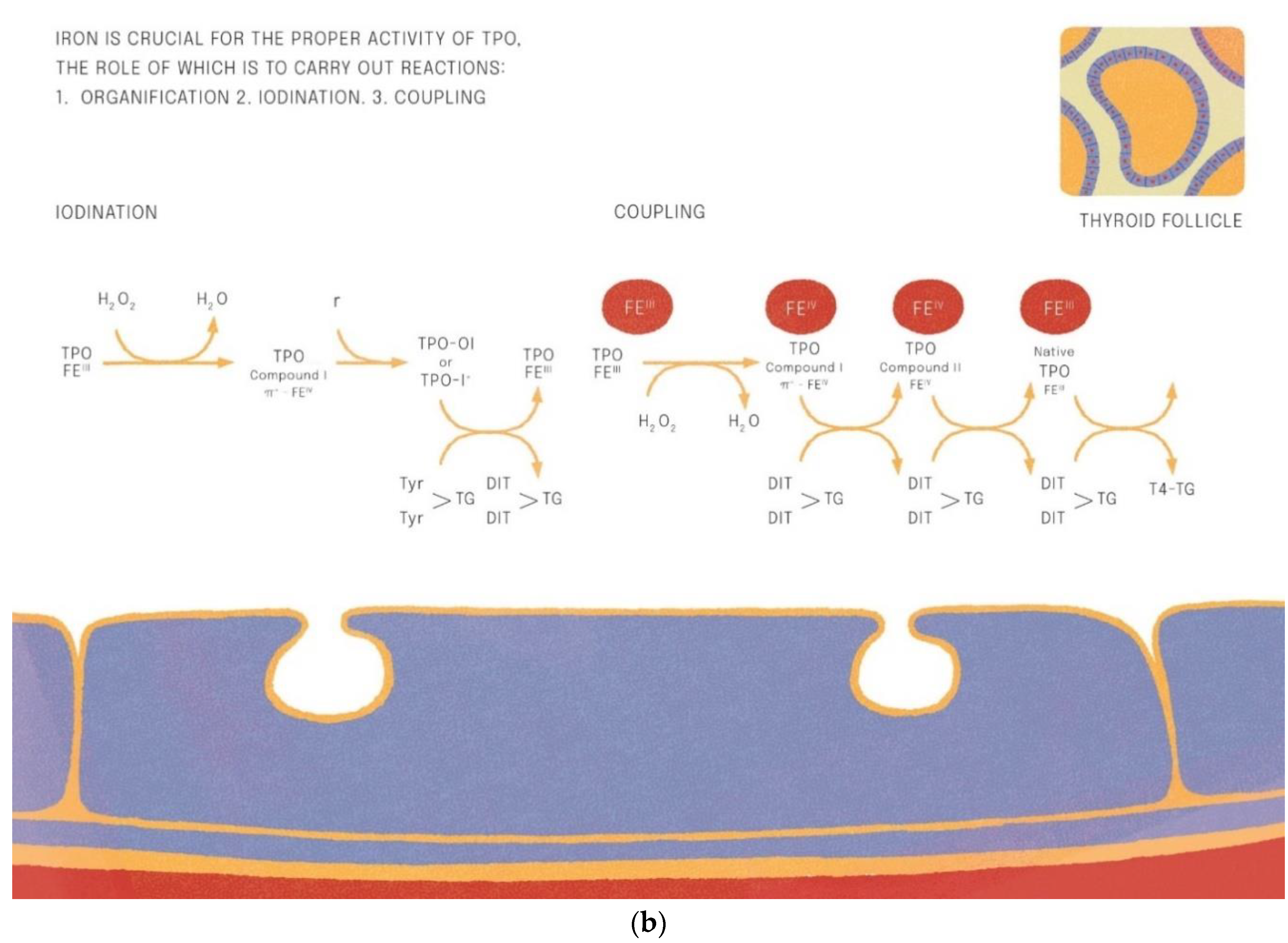
3.6. Iron Function in the Synthesis and Tissue Actions of Thyroid Hormones
4. Iron Deficiency Impairs the Body’s Immune Response and Alters the Human Microbiome
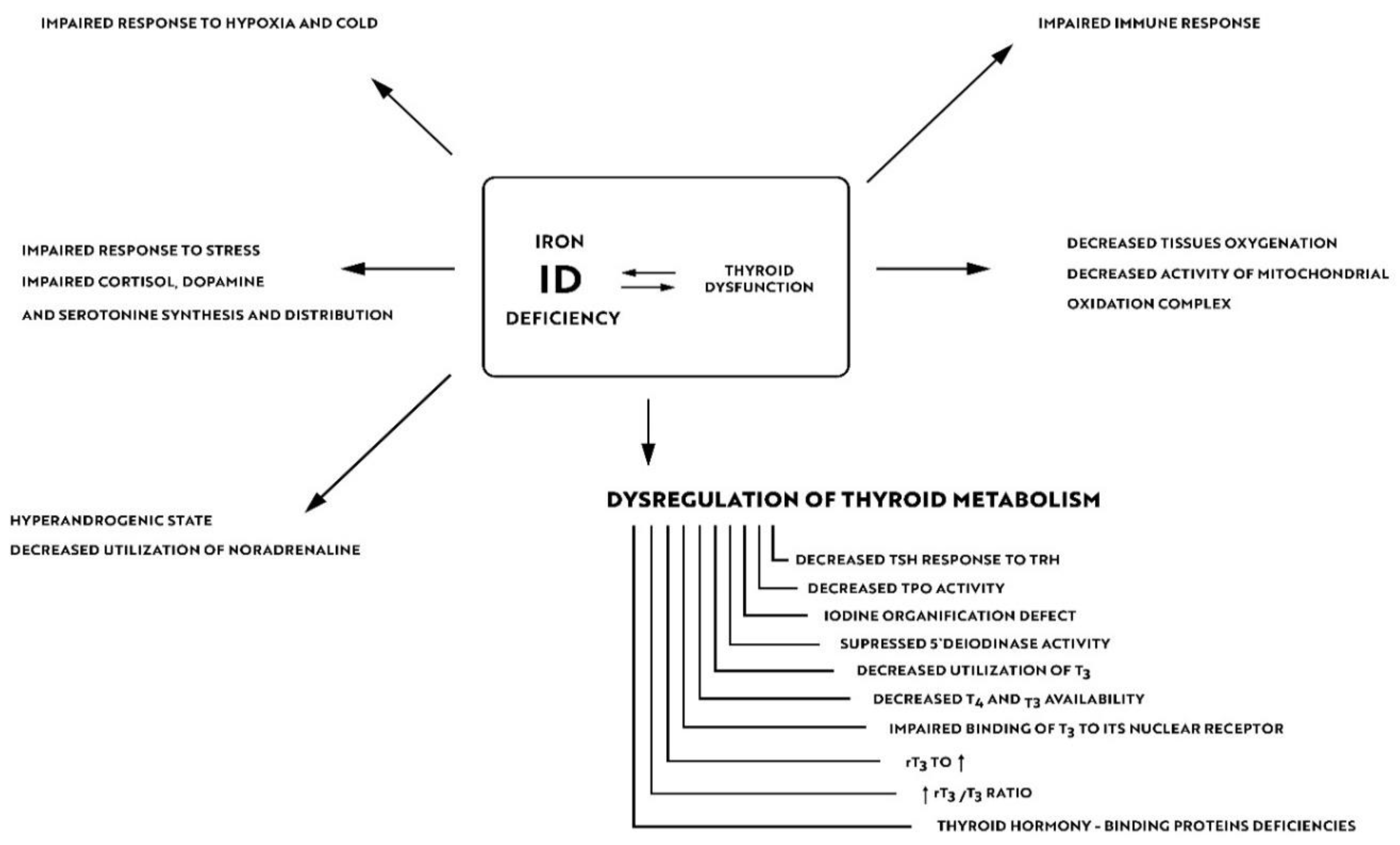
5. Iron Deficiency and Thyroid Sensitivity to Environmental Factors
6. Impact of Iron Deficiency on Residual Symptoms in Euthyroid Patients with Aitd
7. Epigenetics in AITD Development
8. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Hiromatsu, Y.; Satoh, H.; Amino, N. Hashimoto’s thyroiditis: History and future outlook. Hormones 2013, 12, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Pearce, E.N.; Farwell, A.P.; Braverman, L.E. Thyroiditis. N. Engl. J. Med. 2003, 348, 2646–2655, Erratum in N. Engl. J. Med. 2003, 349, 620. [Google Scholar] [CrossRef] [PubMed]
- Ragusa, F.; Fallahi, P.; Elia, G.; Gonnella, D.; Paparo, S.R.; Giusti, C.; Churilov, L.P.; Ferrari, S.M.; Antonelli, A. Hashimotos’ thyroiditis: Epidemiology, pathogenesis, clinic and therapy. Best Pract. Res. Clin. Endocrinol. Metab. 2019, 33, 101367. [Google Scholar] [CrossRef] [PubMed]
- McLeod, D.S.; Cooper, D.S. The incidence and prevalence of thyroid autoimmunity. Endocrine 2012, 42, 252–265. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Ferrari, S.M.; Corrado, A.; Di Domenicantonio, A.; Fallahi, P. Autoimmune thyroid disorders. Autoimmun. Rev. 2015, 14, 174–180. [Google Scholar] [CrossRef]
- Ferrari, S.M.; Fallahi, P.; Antonelli, A.; Benvenga, S. Environmental Issues in Thyroid Diseases. Front. Endocrinol. 2017, 8, 50. [Google Scholar] [CrossRef]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef]
- Shero, N.; Fiset, S.; Plamondon, H.; Thabet, M.; Rioux, F.M. Increase serum cortisol in young guinea pig offspring in response to maternal iron deficiency. Nutr. Res. 2018, 54, 69–79. [Google Scholar] [CrossRef]
- Hu, X.; Wang, R.; Shan, Z.; Dong, Y.; Zheng, H.; Jesse, F.F.; Rao, E.; Takahashi, E.; Li, W.; Teng, W.; et al. Perinatal Iron Deficiency-Induced Hypothyroxinemia Impairs Early Brain Development Regardless of Normal Iron Levels in the Neonatal Brain. Thyroid 2016, 26, 891–900. [Google Scholar] [CrossRef]
- Lopez, A.; Cacoub, P.; Macdougall, I.C.; Peyrin-Biroulet, L. Iron deficiency anaemia. Lancet 2016, 387, 907–916. [Google Scholar] [CrossRef]
- GBD 2016 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017, 390, 1211–1259, Erratum in Lancet 2017, 390, e38. [Google Scholar] [CrossRef]
- Gómez-Ramírez, S.; Bisbe, E.; Shander, A.; Spahn, D.R.; Muñoz, M. Management of Perioperative Iron Deficiency Anemia. Acta Haematol. 2019, 142, 21–29. [Google Scholar] [CrossRef]
- Jankowska, E.A.; Rozentryt, P.; Witkowska, A.; Nowak, J.; Hartmann, O.; Ponikowska, B.; Borodulin-Nadzieja, L.; Banasiak, W.; Polonski, L.; Filippatos, G.; et al. Iron deficiency: An ominous sign in patients with systolic chronic heart failure. Eur. Heart J. 2011, 31, 1872–1880, Erratum in Eur. Heart J. 2011, 32, 1054. [Google Scholar] [CrossRef]
- Beard, J.L.; Connor, J.R.; Jones, B.C. Iron in the brain. Nutr. Rev. 1993, 51, 157–170. [Google Scholar] [CrossRef]
- Pinero, D.J.; Jones, B.; Beard, J.L. Alterations in brain iron metabolism in response to dietary iron changes. J. Nutr. 2000, 130, 254–263. [Google Scholar] [CrossRef]
- Georgieff, M.K. The role of iron in neurodevelopment: Fetal iron deficiency and the developing hippocampus. Biochem. Soc. Trans. 2008, 36 Pt 6, 1267–1271. [Google Scholar] [CrossRef]
- Youdim, M.B. Nutrient deprivation and brain function: Iron. Nutrition 2000, 16, 504–508. [Google Scholar] [CrossRef]
- McEchron, M.D.; Cheng, A.Y.; Liu, H.; Connor, J.R.; Gilmartin, M.R. Perinatal nutritional iron deficiency permanently impairs hippocampus-dependent trace fear conditioning in rats. Nutr. Neurosci. 2005, 8, 195–206. [Google Scholar] [CrossRef]
- Insel, B.J.; Schaefer, C.A.; McKeague, I.W.; Susser, E.S.; Brown, A.S. Maternal iron deficiency and the risk of schizophrenia in offspring. Arch. Gen. Psychiatry 2008, 65, 1136–1144. [Google Scholar] [CrossRef]
- Min, Y.; Wang, X.; Chen, H.; Yin, G. The exploration of Hashimoto’s Thyroiditis related miscarriage for better treatment modalities. Int. J. Med. Sci. 2020, 17, 2402–2415. [Google Scholar] [CrossRef]
- Lee, H.J.; Li, C.W.; Hammerstad, S.S.; Stefan, M.; Tomer, Y. Immunogenetics of autoimmune thyroid diseases: A comprehensive review. J. Autoimmun. 2015, 64, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Kawashima, A.; Tanigawa, K.; Akama, T.; Yoshihara, A.; Ishii, N.; Suzuki, K. Innate immune activation and thyroid autoimmunity. J. Clin. Endocrinol. Metab. 2011, 96, 3661–3671. [Google Scholar] [CrossRef] [PubMed]
- Dittmar, M.; Kahaly, G.J. Polyglandular autoimmune syndromes: Immunogenetics and long-term follow-up. J. Clin. Endocrinol. Metab. 2003, 88, 2983–2992. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, S.M.; Fallahi, P.; Ruffilli, I.; Elia, G.; Ragusa, F.; Benvenga, S.; Antonelli, A. The association of other autoimmune diseases in patients with Graves’ disease (with or without ophthalmopathy): Review of the literature and report of a large series. Autoimmun. Rev. 2019, 18, 287–292. [Google Scholar] [CrossRef] [PubMed]
- Brix, T.H.; Hegedüs, L. Twin studies as a model for exploring the aetiology of autoimmune thyroid disease. Clin. Endocrinol. 2012, 76, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Tomer, Y.; Davies, T.F. Searching for the autoimmune thyroid disease susceptibility genes: From gene mapping to gene function. Endocr. Rev. 2003, 24, 694–717. [Google Scholar] [CrossRef]
- Simmonds, M.J. GWAS in autoimmune thyroid disease: Redefining our understanding of pathogenesis. Nat. Rev. Endocrinol. 2013, 9, 277–287. [Google Scholar] [CrossRef]
- Lo, M.S.; Towne, M.; VanNoy, G.E.; Brownstein, C.A.; Lane, A.A.; Chatila, T.A.; Agrawal, P.B. Monogenic Hashimoto thyroiditis associated with a variant in the thyroglobulin (TG) gene. J. Autoimmun. 2018, 86, 116–119. [Google Scholar] [CrossRef]
- Desailloud, R.; Hober, D. Viruses and thyroiditis: An update. Virol. J. 2009, 6, 5. [Google Scholar] [CrossRef]
- Morohoshi, K.; Takahashi, Y.; Mori, K. Viral infection and innate pattern recognition receptors in induction of Hashimoto’s thyroiditis. Discov. Med. 2011, 12, 505–511. [Google Scholar]
- Frost, J.N.; Tan, T.K.; Abbas, M.; Wideman, S.K.; Bonadonna, M.; Stoffel, N.U.; Wray, K.; Kronsteiner, B.; Smits, G.; Campagna, D.R.; et al. Hepcidin-Mediated Hypoferremia Disrupts Immune Responses to Vaccination and Infection. Med 2021, 2, 164–179.e12. [Google Scholar] [CrossRef]
- Bach, J.F. The effect of infections on susceptibility to autoimmune and allergic diseases. N. Engl. J. Med. 2002, 347, 911–920. [Google Scholar] [CrossRef]
- Aleksandrov, Y.; Semikov, V.; Shulutko, A.; Gogokhia, T.; Gorbacheva, A.; Mansurova, G. Subacute thyroiditis and COVID-19 (review). Georgian Med. News. 2021, 311, 98–103. (In Russian) [Google Scholar]
- Bellmann-Weiler, R.; Lanser, L.; Barket, R.; Rangger, L.; Schapfl, A.; Schaber, M.; Fritsche, G.; Wöll, E.; Weiss, G. Prevalence and Predictive Value of Anemia and Dysregulated Iron Homeostasis in Patients with COVID-19 Infection. J. Clin. Med. 2020, 9, 2429. [Google Scholar] [CrossRef]
- Drakesmith, H.; Prentice, A. Viral infection and iron metabolism. Nat. Rev. Microbiol. 2008, 6, 541–552. [Google Scholar] [CrossRef]
- Caturegli, P.; De Remigis, A.; Rose, N.R. Hashimoto thyroiditis: Clinical and diagnostic criteria. Autoimmun. Rev. 2014, 13, 391–397. [Google Scholar] [CrossRef]
- Iddah, M.A.; Macharia, B.N. Autoimmune thyroid disorders. ISRN Endocrinol. 2013, 2013, 509764. [Google Scholar] [CrossRef]
- Hu, Y.; Zhang, L.; Chen, H.; Liu, X.; Zheng, X.; Shi, H.; Jiang, L.; Cui, D. Analysis of Regulatory T Cell Subsets and Their Expression of Helios and PD-1 in Patients with Hashimoto Thyroiditis. Int. J. Endocrinol. 2019, 2019, 5368473. [Google Scholar] [CrossRef]
- Álvarez-Sierra, D.; Marín-Sánchez, A.; Ruiz-Blázquez, P.; de Jesús Gil, C.; Iglesias-Felip, C.; González, Ó.; Casteras, A.; Costa, R.F.; Nuciforo, P.; Colobran, R.; et al. Analysis of the PD-1/PD-L1 axis in human autoimmune thyroid disease: Insights into pathogenesis and clues to immunotherapy associated thyroid autoimmunity. J. Autoimmun. 2019, 103, 102285. [Google Scholar] [CrossRef]
- Nanba, T.; Watanabe, M.; Inoue, N.; Iwatani, Y. Increases of the Th1/Th2 cell ratio in severe Hashimoto’s disease and in the proportion of Th17 cells in intractable Graves’ disease. Thyroid 2009, 19, 495–501. [Google Scholar] [CrossRef]
- Xue, H.; Yu, X.; Ma, L.; Song, S.; Li, Y.; Zhang, L.; Yang, T.; Liu, H. The possible role of CD4+CD25highFoxp3+/CD4+IL-17A+ cell imbalance in the autoimmunity of patients with Hashimoto thyroiditis. Endocrine 2015, 50, 665–673. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Liu, Y.; Wang, S.; Zhao, N.; Qin, J.; Li, Y.; Fan, C.; Shan, Z.; Teng, W. Circulating Exosomes Activate Dendritic Cells and Induce Unbalanced CD4+ T Cell Differentiation in Hashimoto Thyroiditis. J. Clin. Endocrinol. Metab. 2019, 104, 4607–4618. [Google Scholar] [CrossRef] [PubMed]
- Antonelli, A.; Ferrari, S.M.; Frascerra, S.; Galetta, F.; Franzoni, F.; Corrado, A.; Miccoli, M.; Benvenga, S.; Paolicchi, A.; Ferrannini, E.; et al. Circulating chemokine (CXC motif) ligand (CXCL)9 is increased in aggressive chronic autoimmune thyroiditis, in association with CXCL10. Cytokine 2011, 55, 288–293. [Google Scholar] [CrossRef] [PubMed]
- Jogdand, G.M.; Mohanty, S.; Devadas, S. Regulators of Tfh Cell Differentiation. Front. Immunol. 2016, 7, 520. [Google Scholar] [CrossRef] [PubMed]
- Dubska, M.; Banga, J.P.; Plochocka, D.; Hoser, G.; Kemp, E.H.; Sutton, B.J.; Gardas, A.; Gora, M. Structural insights into autoreactive determinants in thyroid peroxidase composed of discontinuous and multiple key contact amino acid residues contributing to epitopes recognized by patients’ autoantibodies. Endocrinology 2006, 147, 5995–6003. [Google Scholar] [CrossRef]
- Orgiazzi, J. Thyroid autoimmunity. Presse Med. 2012, 41, e611–e625. [Google Scholar] [CrossRef]
- Chang, R.; Chu, K.A.; Lin, M.C.; Chu, Y.H.; Hung, Y.M.; Wei, J.C. Newly diagnosed iron deficiency anemia and subsequent autoimmune disease: A matched cohort study in Taiwan. Curr. Med. Res. Opin. 2020, 36, 985–992. [Google Scholar] [CrossRef]
- Luo, J.; Wang, X.; Yuan, L.; Guo, L. Iron Deficiency, a Risk Factor of Thyroid Disorders in Reproductive-Age and Pregnant Women: A Systematic Review and Meta-Analysis. Front. Endocrinol. 2021, 12, 629831. [Google Scholar] [CrossRef]
- Fayadat, L.; Niccoli-Sire, P.; Lanet, J.; Franc, J.L. Role of heme in intracellular trafficking of thyroperoxidase and involvement of H2O2 generated at the apical surface of thyroid cells in autocatalytic covalent heme binding. J. Biol. Chem. 1999, 274, 10533–10538. [Google Scholar] [CrossRef]
- Hurrell, R.F. Bioavailability of iodine. Eur. J. Clin. Nutr. 1997, 51 (Suppl. 1), S9–S12. [Google Scholar]
- Hess, S.Y.; Zimmermann, M.B.; Arnold, M.; Langhans, W.; Hurrell, R.F. Iron deficiency anemia reduces thyroid peroxidase activity in rats. J. Nutr. 2002, 132, 1951–1955. [Google Scholar] [CrossRef]
- Beard, J.; Tobin, B.; Green, W. Evidence for thyroid hormone deficiency in iron-deficient anemic rats. J. Nutr. 1989, 119, 772–778. [Google Scholar] [CrossRef]
- Dillman, E.; Gale, C.; Green, W.; Johnson, D.G.; Mackler, B.; Finch, C. Hypothermia in iron deficiency due to altered triiodothyronine metabolism. Am. J. Physiol. 1980, 239, R377–R381. [Google Scholar] [CrossRef]
- Smith, S.M.; Finley, J.; Johnson, L.K.; Lukaski, H.C. 1994. Indices of in vivo and in vitro thyroid hormone metabolism in irondeficient rats. Nutr. Res. 1994, 14, 729–739. [Google Scholar] [CrossRef]
- Jabara, H.H.; Boyden, S.E.; Chou, J.; Ramesh, N.; Massaad, M.J.; Benson, H.; Bainter, W.; Fraulino, D.; Rahimov, F.; Sieff, C.; et al. A missense mutation in TFRC, encoding transferrin receptor 1, causes combined immunodeficiency. Nat. Genet. 2016, 48, 74–78. [Google Scholar] [CrossRef]
- Hershko, C.; Peto, T.E.; Weatherall, D.J. Iron and infection. Br. Med. J. (Clin. Res. Ed.) 1988, 296, 660–664. [Google Scholar] [CrossRef]
- Jason, J.; Archibald, L.K.; Nwanyanwu, O.C.; Bell, M.; Jensen, R.J.; Gunter, E.; Buchanan, I.; Larned, J.; Kazembe, P.N.; Dobbie, H.; et al. The effects of iron deficiency on lymphocyte cytokine production and activation: Preservation of hepatic iron but not at all cost. Clin. Exp. Immunol. 2001, 126, 466–473. [Google Scholar] [CrossRef]
- Wang, L.; Harrington, L.; Trebicka, E.; Shi, H.N.; Kagan, J.C.; Hong, C.C.; Lin, H.Y.; Babitt, J.L.; Cherayil, B.J. Selective modulation of TLR4-activated inflammatory responses by altered iron homeostasis in mice. J. Clin. Investig. 2009, 119, 3322–3328. [Google Scholar] [CrossRef]
- Salahudeen, A.A.; Thompson, J.W.; Ruiz, J.C.; Ma, H.W.; Kinch, L.N.; Li, Q.; Grishin, N.V.; Bruick, R.K. An E3 ligase possessing an iron-responsive hemerythrin domain is a regulator of iron homeostasis. Science 2009, 326, 722–726. [Google Scholar] [CrossRef]
- Gomez, M.A.; Alisaraie, L.; Shio, M.T.; Berghuis, A.M.; Lebrun, C.; Gautier-Luneau, I.; Olivier, M. Protein tyrosine phosphatases are regulated by mononuclear iron dicitrate. J. Biol. Chem. 2010, 285, 24620–24628. [Google Scholar] [CrossRef]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef]
- Kochanowski, B.A.; Sherman, A.R. Cellular growth in iron-deficient rats: Effect of pre- and postweaning iron repletion. J. Nutr. 1985, 115, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Spear, A.T.; Sherman, A.R. Iron deficiency alters DMBA-induced tumor burden and natural killer cell cytotoxicity in rats. J. Nutr. 1992, 122, 46–55. [Google Scholar] [CrossRef]
- Hassan, T.H.; Badr, M.A.; Karam, N.A.; Zkaria, M.; El Saadany, H.F.; Abdel Rahman, D.M.; Shahbah, D.A.; Al Morshedy, S.M.; Fathy, M.; Esh, A.M.H.; et al. Impact of iron deficiency anemia on the function of the immune system in children. Medicine 2016, 95, e5395. [Google Scholar] [CrossRef]
- Kuvibidila, S.R.; Kitchens, D.; Baliga, B.S. In vivo and in vitro iron deficiency reduces protein kinase C activity and translocation in murine splenic and purified T cells. J. Cell Biochem. 1999, 74, 468–478. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, C.; Wu, Q.; An, P.; Huang, L.; Wang, J.; Chen, C.; Chen, X.; Zhang, F.; Ma, L.; et al. Iron-dependent histone 3 lysine 9 demethylation controls B cell proliferation and humoral immune responses. Nat. Commun. 2019, 10, 2935. [Google Scholar] [CrossRef]
- Zhu, J.; Yamane, H.; Paul, W.E. Differentiation of effector CD4 T cell populations. Annu. Rev. Immunol. 2010, 28, 445–489. [Google Scholar] [CrossRef] [PubMed]
- Miao, F.; Chen, Z.; Genuth, S.; Paterson, A.; Zhang, L.; Wu, X.; Li, S.M.; Cleary, P.; Riggs, A.; Harlan, D.M.; et al. Evaluating the role of epigenetic histone modifications in the metabolic memory of type 1 diabetes. Diabetes 2014, 63, 1748–1762. [Google Scholar] [CrossRef]
- David, L.A.; Maurice, C.F.; Carmody, R.N.; Gootenberg, D.B.; Button, J.E.; Wolfe, B.E.; Ling, A.V.; Devlin, A.S.; Varma, Y.; Fischbach, M.A.; et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature 2014, 505, 559–563. [Google Scholar] [CrossRef] [PubMed]
- Reddy, B.S.; Pleasants, J.R.; Wostmann, B.S. Effect of intestinal microflora on iron and zinc metabolism, and on activities of metalloenzymes in rats. J. Nutr. 1972, 102, 101–107. [Google Scholar] [CrossRef]
- Schaible, U.E.; Kaufmann, S.H. Iron and microbial infection. Nat. Rev. Microbiol. 2004, 2, 946–953, Erratum in Nat. Rev. Microbiol. 2005, 3, 268. [Google Scholar] [CrossRef]
- Knezevic, J.; Starchl, C.; Tmava Berisha, A.; Amrein, K. Thyroid-Gut-Axis: How Does the Microbiota Influence Thyroid Function? Nutrients 2020, 12, 1769. [Google Scholar] [CrossRef]
- Ghio, A.J.; Soukup, J.M.; Dailey, L.A. Air pollution particles and iron homeostasis. Biochim. Biophys. Acta 2016, 1860, 2816–2825. [Google Scholar] [CrossRef]
- Dugger, D.L.; Irby, B.N.; McConnell, B.L.; Cummings, W.W.; Mattman, R.W. The exchange of twenty metal ions with the weakly acidic silanol group of silica gel. J. Phys. Chem. 1964, 68, 757–760. [Google Scholar] [CrossRef]
- Koerten, H.K.; Brederoo, P.; Ginsel, L.A.; Daems, W.T. The endocytosis of asbestos by mouse peritoneal macrophages and its long-term effect on iron accumulation and labyrinth formation. Eur. J. Cell. Biol. 1986, 40, 25–36. [Google Scholar]
- Boas, M.; Feldt-Rasmussen, U.; Main, K.M. Thyroid effects of endocrine disrupting chemicals. Mol. Cell. Endocrinol. 2012, 355, 240–248. [Google Scholar] [CrossRef]
- Jancic, S.A.; Stosic, B.Z. Cadmium effects on the thyroid gland. Vitam. Horm. 2014, 94, 391–425. [Google Scholar] [CrossRef]
- Soldin, O.P.; Aschner, M. Effects of manganese on thyroid hormone homeostasis: Potential links. Neurotoxicology 2007, 28, 951–956. [Google Scholar] [CrossRef]
- Benvenga, S.; Vigo, M.T.; Metro, D.; Granese, R.; Vita, R.; Le Donne, M. Type of fish consumed and thyroid autoimmunity in pregnancy and postpartum. Endocrine 2016, 52, 120–129. [Google Scholar] [CrossRef]
- Fallahi, P.; Foddis, R.; Elia, G.; Ragusa, F.; Patrizio, A.; Frenzilli, G.; Benvenga, S.; Cristaudo, A.; Antonelli, A.; Ferrari, S.M. Differential modulation by vanadium pentoxide of the secretion of CXCL8 and CXCL11 chemokines in thyroid cells. Mol. Med. Rep. 2018, 17, 7415–7420. [Google Scholar] [CrossRef]
- Shukla, A.; Agarwal, K.N.; Shukla, G.S. Effect of latent iron deficiency on metal levels of rat brain regions. Biol. Trace Elem. Res. 1989, 22, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Liu, W.; Zhang, B.; Shen, X.; Hu, C.; Chen, X.; Jin, S.; Jiang, Y.; Liu, H.; Cao, Z.; et al. Maternal Heavy Metal Exposure, Thyroid Hormones, and Birth Outcomes: A Prospective Cohort Study. J. Clin. Endocrinol. Metab. 2019, 104, 5043–5052. [Google Scholar] [CrossRef] [PubMed]
- Ghio, A.J.; Stonehuerner, J.; Soukup, J.M.; Dailey, L.A.; Kesic, M.J.; Cohen, M.D. Iron diminishes the in vitro biological effect of vanadium. J. Inorg. Biochem. 2015, 147, 126–133. [Google Scholar] [CrossRef] [PubMed]
- Sabbioni, E.; Rade, J. Relationships between iron and vanadium metabolism: The association of vanadium with bovine lactoferrin. Toxicol. Lett. 1980, 5, 381–387. [Google Scholar] [CrossRef]
- Monteiro, H.P.; Winterbourn, C.C.; Stern, A. Tetravalent vanadium releases ferritin iron which stimulates vanadium-dependent lipid peroxidation. Free Radic. Res. Commun. 1991, 12–13 Pt 1, 125–129. [Google Scholar] [CrossRef]
- Macomber, L.; Imlay, J.A. The iron-sulfur clusters of dehydratases are primary intracellular targets of copper toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 8344–8349. [Google Scholar] [CrossRef]
- Grady, J.K.; Shao, J.; Arosio, P.; Santambrogio, P.; Chasteen, N.D. Vanadyl(IV) binding to mammalian ferritins. An EPR study aided by site-directed mutagenesis. J. Inorg. Biochem. 2000, 80, 107–113. [Google Scholar] [CrossRef]
- Arena, S.; Latina, A.; Baratta, R.; Burgio, G.; Gullo, D.; Benvenga, S. Chronic lymphocytic thyroiditis: Could it be influenced by a petrochemical complex? Data from a cytological study in South-Eastern Sicily. Eur. J. Endocrinol. 2015, 172, 383–389. [Google Scholar] [CrossRef]
- de Freitas, C.U.; Grimaldi Campos, R.A.; Rodrigues Silva, M.A.; Panachão, M.R.; de Moraes, J.C.; Waissmann, W.; Roberto Chacra, A.; Maeda, M.Y.; Minazzi Rodrigues, R.S.; Gonçalves Belchor, J.; et al. Can living in the surroundings of a petrochemical complex be a risk factor for autoimmune thyroid disease? Environ. Res. 2010, 110, 112–117. [Google Scholar] [CrossRef]
- Freire, C.; Koifman, R.J.; Sarcinelli, P.N.; Simões Rosa, A.C.; Clapauch, R.; Koifman, S. Long-term exposure to organochlorine pesticides and thyroid status in adults in a heavily contaminated area in Brazil. Environ. Res. 2013, 127, 7–15. [Google Scholar] [CrossRef]
- Langer, P.; Tajtáková, M.; Fodor, G.; Kocan, A.; Bohov, P.; Michálek, J.; Kreze, A. Increased thyroid volume and prevalence of thyroid disorders in an area heavily polluted by polychlorinated biphenyls. Eur. J. Endocrinol. 1998, 139, 402–409. [Google Scholar] [CrossRef]
- Bahn, A.K.; Mills, J.L.; Snyder, P.J.; Gann, P.H.; Houten, L.; Bialik, O.; Hollmann, L.; Utiger, R.D. Hypothyroidism in workers exposed to polybrominated biphenyls. N. Engl. J. Med. 1980, 302, 31–33. [Google Scholar] [CrossRef]
- Schell, L.M.; Gallo, M.V.; Ravenscroft, J.; DeCaprio, A.P. Persistent organic pollutants and anti-thyroid peroxidase levels in Akwesasne Mohawk young adults. Environ. Res. 2009, 109, 86–92. [Google Scholar] [CrossRef]
- Yi, S.W.; Hong, J.S.; Ohrr, H.; Yi, J.J. Agent Orange exposure and disease prevalence in Korean Vietnam veterans: The Korean veterans health study. Environ. Res. 2014, 133, 56–65. [Google Scholar] [CrossRef]
- Agate, L.; Mariotti, S.; Elisei, R.; Mossa, P.; Pacini, F.; Molinaro, E.; Grasso, L.; Masserini, L.; Mokhort, T.; Vorontsova, T.; et al. Thyroid autoantibodies and thyroid function in subjects exposed to Chernobyl fallout during childhood: Evidence for a transient radiation-induced elevation of serum thyroid antibodies without an increase in thyroid autoimmune disease. J. Clin. Endocrinol. Metab. 2008, 93, 2729–2736. [Google Scholar] [CrossRef]
- Tajtáková, M.; Semanová, Z.; Tomková, Z.; Szökeová, E.; Majoros, J.; Rádiková, Z.; Seböková, E.; Klimes, I.; Langer, P. Increased thyroid volume and frequency of thyroid disorders signs in schoolchildren from nitrate polluted area. Chemosphere 2006, 62, 559–564. [Google Scholar] [CrossRef]
- Colucci, R.; Lotti, F.; Arunachalam, M.; Lotti, T.; Dragoni, F.; Benvenga, S.; Moretti, S. Correlation of Serum Thyroid Hormones Autoantibodies with Self-Reported Exposure to Thyroid Disruptors in a Group of Nonsegmental Vitiligo Patients. Arch. Environ. Contam. Toxicol. 2015, 69, 181–190. [Google Scholar] [CrossRef]
- Sato, K.; Inoue, S.; Igarashi, A.; Tokairin, Y.; Yamauchi, K.; Kimura, T.; Nishiwaki, M.; Nemoto, T.; Nakano, H.; Sato, M.; et al. Effect of Iron Deficiency on a Murine Model of Smoke-induced Emphysema. Am. J. Respir. Cell. Mol. Biol. 2020, 62, 588–597. [Google Scholar] [CrossRef]
- Kaufman, J.D.; Adar, S.D.; Barr, R.G.; Budoff, M.; Burke, G.L.; Curl, C.L.; Daviglus, M.L.; Diez Roux, A.V.; Gassett, A.J.; Jacobs, D.R.; et al. Association between air pollution and coronary artery calcification within six metropolitan areas in the USA (the Multi-Ethnic Study of Atherosclerosis and Air Pollution): A longitudinal cohort study. Lancet 2016, 388, 696–704, Erratum in Lancet 2016, 388, 660. [Google Scholar] [CrossRef]
- Samuels, M.H.; Bernstein, L.J. Brain Fog in Hypothyroidism: What Is It, How Is It Measured, and What Can Be Done About It. Thyroid 2022, 32, 752–763. [Google Scholar] [CrossRef]
- Ettleson, M.D.; Raine, A.; Batistuzzo, A.; Batista, S.P.; McAninch, E.; Teixeira, M.C.T.V.; Jonklaas, J.; Laiteerapong, N.; Ribeiro, M.O.; Bianco, A.C. Brain Fog in Hypothyroidism: Understanding the Patient’s Perspective. Endocr. Pract. 2022, 28, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Jurado-Flores, M.; Warda, F.; Mooradian, A. Pathophysiology and Clinical Features of Neuropsychiatric Manifestations of Thyroid Disease. J. Endocr. Soc. 2022, 6, bvab194. [Google Scholar] [CrossRef] [PubMed]
- Watt, T.; Hegedüs, L.; Bjorner, J.B.; Groenvold, M.; Bonnema, S.J.; Rasmussen, A.K.; Feldt-Rasmussen, U. Is Thyroid Autoimmunity per se a Determinant of Quality of Life in Patients with Autoimmune Hypothyroidism? Eur. Thyroid J. 2012, 1, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Sun, Y.; Yang, H.; Xu, Y.; Cai, Y.; Liu, T.; Xia, Q.; Zhu, D.; Wang, F. Hashimoto’s Thyroiditis Induces Hippocampus-Dependent Cognitive Alterations by Impairing Astrocytes in Euthyroid Mice. Thyroid 2021, 31, 482–493. [Google Scholar] [CrossRef] [PubMed]
- Guldvog, I.; Reitsma, L.C.; Johnsen, L.; Lauzike, A.; Gibbs, C.; Carlsen, E.; Lende, T.H.; Narvestad, J.K.; Omdal, R.; Kvaløy, J.T.; et al. Thyroidectomy Versus Medical Management for Euthyroid Patients With Hashimoto Disease and Persisting Symptoms: A Randomized Trial. Ann. Intern. Med. 2019, 170, 453–464. [Google Scholar] [CrossRef]
- Hidese, S.; Saito, K.; Asano, S.; Kunugi, H. Association between iron-deficiency anemia and depression: A web-based Japanese investigation. Psychiatry Clin. Neurosci. 2018, 72, 513–521. [Google Scholar] [CrossRef]
- Soppi, E. Iron deficiency is the main cause of symp-tom persistence in patients treated for hypothyroidism. Thyroid 2015, 25, A74. [Google Scholar]
- Cañas, C.A.; Cañas, F.; Bonilla-Abadía, F.; Ospina, F.E.; Tobón, G.J. Epigenetics changes associated to environmental triggers in autoimmunity. Autoimmunity 2016, 49, 1–11. [Google Scholar] [CrossRef]
- Marsit, C.J. Influence of environmental exposure on human epigenetic regulation. J. Exp. Biol. 2015, 218 Pt 1, 71–79. [Google Scholar] [CrossRef]
- Cai, T.T.; Muhali, F.S.; Song, R.H.; Qin, Q.; Wang, X.; Shi, L.F.; Jiang, W.J.; Xiao, L.; Li, D.F.; Zhang, J.A. Genome-wide DNA methylation analysis in Graves’ disease. Genomics 2015, 105, 204–210. [Google Scholar] [CrossRef]
- Limbach, M.; Saare, M.; Tserel, L.; Kisand, K.; Eglit, T.; Sauer, S.; Axelsson, T.; Syvänen, A.C.; Metspalu, A.; Milani, L.; et al. Epigenetic profiling in CD4+ and CD8+ T cells from Graves’ disease patients reveals changes in genes associated with T cell receptor signaling. J. Autoimmun. 2016, 67, 46–56. [Google Scholar] [CrossRef]
- Arakawa, Y.; Watanabe, M.; Inoue, N.; Sarumaru, M.; Hidaka, Y.; Iwatani, Y. Association of polymorphisms in DNMT1, DNMT3A, DNMT3B, MTHFR and MTRR genes with global DNA methylation levels and prognosis of autoimmune thyroid disease. Clin. Exp. Immunol. 2012, 170, 194–201. [Google Scholar] [CrossRef]
- Yan, N.; Zhou, J.Z.; Zhang, J.A.; Cai, T.; Zhang, W.; Wang, Y.; Muhali, F.S.; Guan, L.; Song, R.H. Histone hypoacetylation and increased histone deacetylases in peripheral blood mononuclear cells from patients with Graves’ disease. Mol. Cell. Endocrinol. 2015, 414, 143–147. [Google Scholar] [CrossRef]
- Stefan, M.; Jacobson, E.M.; Huber, A.K.; Greenberg, D.A.; Li, C.W.; Skrabanek, L.; Conception, E.; Fadlalla, M.; Ho, K.; Tomer, Y. Novel variant of thyroglobulin promoter triggers thyroid autoimmunity through an epigenetic interferon alpha-modulated mechanism. J. Biol. Chem. 2011, 286, 31168–31179. [Google Scholar] [CrossRef]
- Stefan, M.; Wei, C.; Lombardi, A.; Li, C.W.; Concepcion, E.S.; Inabnet, W.B., 3rd; Owen, R.; Zhang, W.; Tomer, Y. Genetic-epigenetic dysregulation of thymic TSH receptor gene expression triggers thyroid autoimmunity. Proc. Natl. Acad. Sci. USA 2014, 111, 12562–12567. [Google Scholar] [CrossRef]
- Sarumaru, M.; Watanabe, M.; Inoue, N.; Hisamoto, Y.; Morita, E.; Arakawa, Y.; Hidaka, Y.; Iwatani, Y. Association between functional SIRT1 polymorphisms and the clinical characteristics of patients with autoimmune thyroid disease. Autoimmunity 2016, 49, 329–337. [Google Scholar] [CrossRef]
- Mehta, A.; Baltimore, D. MicroRNAs as regulatory elements in immune system logic. Nat. Rev. Immunol. 2016, 16, 279–294. [Google Scholar] [CrossRef]
- Vicente, R.; Noël, D.; Pers, Y.M.; Apparailly, F.; Jorgensen, C. Deregulation and therapeutic potential of microRNAs in arthritic diseases. Nat. Rev. Rheumatol. 2016, 12, 496, Erratum in Nat. Rev. Rheumatol. 2016, 12, 211–220. [Google Scholar] [CrossRef]
- Rothchild, A.C.; Sissons, J.R.; Shafiani, S.; Plaisier, C.; Min, D.; Mai, D.; Gilchrist, M.; Peschon, J.; Larson, R.P.; Bergthaler, A.; et al. MiR-155-regulated molecular network orchestrates cell fate in the innate and adaptive immune response to Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 2016, 113, E6172–E6181. [Google Scholar] [CrossRef]
- Seddiki, N.; Brezar, V.; Ruffin, N.; Lévy, Y.; Swaminathan, S. Role of miR-155 in the regulation of lymphocyte immune function and disease. Immunology 2014, 142, 32–38. [Google Scholar] [CrossRef]
- Chatzikyriakidou, A.; Voulgari, P.V.; Georgiou, I.; Drosos, A.A. The role of microRNA-146a (miR-146a) and its target IL-1R-associated kinase (IRAK1) in psoriatic arthritis susceptibility. Scand. J. Immunol. 2010, 71, 382–385. [Google Scholar] [CrossRef]
- Park, H.; Huang, X.; Lu, C.; Cairo, M.S.; Zhou, X. MicroRNA-146a and microRNA-146b regulate human dendritic cell apoptosis and cytokine production by targeting TRAF6 and IRAK1 proteins. J. Biol. Chem. 2015, 290, 2831–2841. [Google Scholar] [CrossRef]
- Bernecker, C.; Lenz, L.; Ostapczuk, M.S.; Schinner, S.; Willenberg, H.; Ehlers, M.; Vordenbäumen, S.; Feldkamp, J.; Schott, M. MicroRNAs miR-146a1, miR-155_2, and miR-200a1 are regulated in autoimmune thyroid diseases. Thyroid 2012, 22, 1294–1295. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.; Zhang, Y.; Zhang, W.; Zhang, W.; Fan, L.; Wang, L.; Liu, Y.; Liu, S.; Guo, Y.; Wang, Y.; et al. MicroRNA-142-5p contributes to Hashimoto’s thyroiditis by targeting CLDN1. J. Transl. Med. 2016, 14, 166. [Google Scholar] [CrossRef] [PubMed]
- Peng, H.; Liu, Y.; Tian, J.; Ma, J.; Tang, X.; Yang, J.; Rui, K.; Zhang, Y.; Mao, C.; Lu, L.; et al. Decreased expression of microRNA-125a-3p upregulates interleukin-23 receptor in patients with Hashimoto’s thyroiditis. Immunol. Res. 2015, 62, 129–136. [Google Scholar] [CrossRef]
- Stachurska, A.; Zorro, M.M.; van der Sijde, M.R.; Withoff, S. Small and Long Regulatory RNAs in the Immune System and Immune Diseases. Front. Immunol. 2014, 5, 513. [Google Scholar] [CrossRef] [PubMed]
- Engreitz, J.M.; Haines, J.E.; Perez, E.M.; Munson, G.; Chen, J.; Kane, M.; McDonel, P.E.; Guttman, M.; Lander, E.S. Local regulation of gene expression by lncRNA promoters, transcription and splicing. Nature 2016, 539, 452–455. [Google Scholar] [CrossRef]
- Engreitz, J.M.; Ollikainen, N.; Guttman, M. Long non-coding RNAs: Spatial amplifiers that control nuclear structure and gene expression. Nat. Rev. Mol. Cell. Biol. 2016, 17, 756–770. [Google Scholar] [CrossRef]
- Huang, W.; Thomas, B.; Flynn, R.A.; Gavzy, S.J.; Wu, L.; Kim, S.V.; Hall, J.A.; Miraldi, E.R.; Ng, C.P.; Rigo, F.; et al. Retraction Note: DDX5 and its associated lncRNA Rmrp modulate TH17 cell effector functions. Nature 2018, 562, 150. [Google Scholar] [CrossRef]
- Aune, T.M.; Crooke, P.S., 3rd; Spurlock, C.F., 3rd. Long noncoding RNAs in T lymphocytes. J. Leukoc. Biol. 2016, 99, 31–44. [Google Scholar] [CrossRef]
- Peng, H.; Liu, Y.; Tian, J.; Ma, J.; Tang, X.; Rui, K.; Tian, X.; Mao, C.; Lu, L.; Xu, H.; et al. The Long Noncoding RNA IFNG-AS1 Promotes T Helper Type 1 Cells Response in Patients with Hashimoto’s Thyroiditis. Sci. Rep. 2015, 5, 17702. [Google Scholar] [CrossRef]
- Briggs, S.F.; Reijo Pera, R.A. X chromosome inactivation: Recent advances and a look forward. Curr. Opin. Genet. Dev. 2014, 28, 78–82. [Google Scholar] [CrossRef]
- Gendrel, A.V.; Heard, E. Noncoding RNAs and epigenetic mechanisms during X-chromosome inactivation. Annu. Rev. Cell. Dev. Biol. 2014, 30, 561–580. [Google Scholar] [CrossRef]
- Minks, J.; Robinson, W.P.; Brown, C.J. A skewed view of X chromosome inactivation. J. Clin. Investig. 2008, 118, 20–23. [Google Scholar] [CrossRef]
- Seidel, M.G.; Rami, B.; Item, C.; Schober, E.; Zeitlhofer, P.; Huber, W.D.; Heitger, A.; Bodamer, O.A.; Haas, O.A. Concurrent FOXP3- and CTLA4-associated genetic predisposition and skewed X chromosome inactivation in an autoimmune disease-prone family. Eur. J. Endocrinol. 2012, 167, 131–134. [Google Scholar] [CrossRef][Green Version]
- Brix, T.H.; Knudsen, G.P.; Kristiansen, M.; Kyvik, K.O.; Orstavik, K.H.; Hegedüs, L. High frequency of skewed X-chromosome inactivation in females with autoimmune thyroid disease: A possible explanation for the female predisposition to thyroid autoimmunity. J. Clin. Endocrinol. Metab. 2005, 90, 5949–5953. [Google Scholar] [CrossRef]
- Yin, X.; Latif, R.; Tomer, Y.; Davies, T.F. Thyroid epigenetics: X chromosome inactivation in patients with autoimmune thyroid disease. Ann. N. Y. Acad. Sci. 2007, 1110, 193–200. [Google Scholar] [CrossRef]
- Aksu, B.Y.; Hasbal, C.; Himmetoglu, S.; Dincer, Y.; Koc, E.E.; Hatipoglu, S.; Akcay, T. Leukocyte DNA damage in children with iron deficiency anemia: Effect of iron supplementation. Eur. J. Pediatr. 2010, 169, 951–956. [Google Scholar] [CrossRef]
- Ames, B.N. Micronutrient deficiencies. A major cause of DNA damage. Ann. N. Y. Acad. Sci. 1999, 889, 87–106. [Google Scholar] [CrossRef]
- Aslan, M.; Horoz, M.; Kocyigit, A.; Ozgonül, S.; Celik, H.; Celik, M.; Erel, O. Lymphocyte DNA damage and oxidative stress in patients with iron deficiency anemia. Mutat. Res. 2006, 601, 144–149. [Google Scholar] [CrossRef]
- Walter, P.B.; Knutson, M.D.; Paler-Martinez, A.; Lee, S.; Xu, Y.; Viteri, F.E.; Ames, B.N. Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proc. Natl. Acad. Sci. USA 2002, 99, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
- Furukawa, T.; Naitoh, Y.; Kohno, H.; Tokunaga, R.; Taketani, S. Iron deprivation decreases ribonucleotide reductase activity and DNA synthesis. Life Sci. 1992, 50, 2059–2065. [Google Scholar] [CrossRef]
- Zohora, F.; Bidad, K.; Pourpak, Z.; Moin, M. Biological and Immunological Aspects of Iron Deficiency Anemia in Cancer Development: A Narrative Review. Nutr. Cancer 2018, 70, 546–556. [Google Scholar] [CrossRef] [PubMed]
- Inoue, H.; Kobayashi, K.; Ndong, M.; Yamamoto, Y.; Katsumata, S.; Suzuki, K.; Uehara, M. Activation of Nrf2/Keap1 signaling and autophagy induction against oxidative stress in heart in iron deficiency. Biosci. Biotechnol. Biochem. 2015, 79, 1366–1368. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.; Clarke, S. Influence of microRNA on the maintenance of human iron metabolism. Nutrients 2013, 5, 2611–2628. [Google Scholar] [CrossRef]
- Weitz, S.H.; Gong, M.; Barr, I.; Weiss, S.; Guo, F. Processing of microRNA primary transcripts requires heme in mammalian cells. Proc. Natl. Acad. Sci. USA 2014, 111, 1861–1866. [Google Scholar] [CrossRef]
- Nallamshetty, S.; Chan, S.Y.; Loscalzo, J. Hypoxia: A master regulator of microRNA biogenesis and activity. Free Radic Biol. Med. 2013, 64, 20–30. [Google Scholar] [CrossRef]
- Bao, B.; Azmi, A.S.; Li, Y.; Ahmad, A.; Ali, S.; Banerjee, S.; Kong, D.; Sarkar, F.H. Targeting CSCs in tumor microenvironment: The potential role of ROS-associated miRNAs in tumor aggressiveness. Curr. Stem. Cell. Res. Ther. 2014, 9, 22–35. [Google Scholar] [CrossRef]
- Crosby, M.E.; Kulshreshtha, R.; Ivan, M.; Glazer, P.M. MicroRNA regulation of DNA repair gene expression in hypoxic stress. Cancer Res. 2009, 69, 1221–1229, Erratum in Cancer Res. 2009, 69, 3240. [Google Scholar] [CrossRef]
- Rasmussen, K.D.; Helin, K. Role of TET enzymes in DNA methylation, development, and cancer. Genes Dev. 2016, 30, 733–750. [Google Scholar] [CrossRef]
- Barks, A.K.; Liu, S.X.; Georgieff, M.K.; Hallstrom, T.C.; Tran, P.V. Early-Life Iron Deficiency Anemia Programs the Hippocampal Epigenomic Landscape. Nutrients 2021, 13, 3857. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Szklarz, M.; Gontarz-Nowak, K.; Matuszewski, W.; Bandurska-Stankiewicz, E. Iron: Not Just a Passive Bystander in AITD. Nutrients 2022, 14, 4682. https://doi.org/10.3390/nu14214682
Szklarz M, Gontarz-Nowak K, Matuszewski W, Bandurska-Stankiewicz E. Iron: Not Just a Passive Bystander in AITD. Nutrients. 2022; 14(21):4682. https://doi.org/10.3390/nu14214682
Chicago/Turabian StyleSzklarz, Michał, Katarzyna Gontarz-Nowak, Wojciech Matuszewski, and Elżbieta Bandurska-Stankiewicz. 2022. "Iron: Not Just a Passive Bystander in AITD" Nutrients 14, no. 21: 4682. https://doi.org/10.3390/nu14214682
APA StyleSzklarz, M., Gontarz-Nowak, K., Matuszewski, W., & Bandurska-Stankiewicz, E. (2022). Iron: Not Just a Passive Bystander in AITD. Nutrients, 14(21), 4682. https://doi.org/10.3390/nu14214682