1. Introduction
Stimulator of interferon genes (STING,
Tmem173), a classical nucleic acid sensor of DNA, is located on the membrane of the endoplasmic reticulum. Its C-terminal tail is located in the cell cytosol and allows STING to translocate to the Golgi when activated. STING has been linked to both exogenous pathogens, such as DNA viruses, but also endogenous DNA, such as mtDNA, sensing in the body [
1]. In the liver, specifically, STING has been linked to both sterile and non-sterile inflammation, as caused by either ethanol or hepatitis virus, respectively. STING has a complex downstream molecular pathway that is dependent on the activating stimuli. The simplest pathway includes STING activation and oligomerization, phosphorylation of TBK1 (tank binding kinase 1), subsequent phosphorylation of IRF3 (IFN-regulatory factor 3), translocation of IRF3 to the nucleus, and transcription of numerous interferon type-I genes such as interferon-β (
Ifnb). However, STING can also lead to cell death. This cell death signaling cascade also begins with the phosphorylation of TBK1 but leads to the uncovering of a BH3-only domain on IRF3, which permits the interaction of IRF3 with BAX/BAK (BCL2 associated X, apoptosis regulator and BCL2 antagonist/Killer 1) on the mitochondria, leading to mitochondrial permeability transition pore opening and activation of the mitochondrial-dependent cell death cascade [
2,
3,
4].
Recently, STING has been connected to hepatic sterile inflammation by several groups. For instance, STING has been shown to worsen liver outcome following toxic liver injury such as induced by ethanol or CCl
4 [
5,
6,
7,
8]. The role of STING during metabolic liver injury as induced by high fat diets has also been briefly studied, but results have been inconsistent [
9,
10,
11]. Certain studies have demonstrated a detrimental role for STING during high fat diet (HFD) feeding or methionine- and choline-deficient (MCD) diet feeding [
10,
11], while others have demonstrated a potential benefit to STING during HFD feeding [
9].
We thus set out to determine the exact role that STING plays during different liver injury models. Notably, we administered a toxic liver injury model—tunicamycin (TM), which leads to acute injury. We then decided to further pursue the role of STING in dietary-induced NASH models: high fructose diet and the Fructose–Palmitate–Cholesterol (FPC) diet. We chose these models as TM mimics a toxic liver injury while the high fructose and FPC diets mimic NAFLD. In order to study the role of STING, Goldenticket (Tmem173gt) mice, which carry a missense point mutation in the STING encoding gene Tmem173, were used. This missense mutation leads to a complete absence of STING protein production in the whole body.
Herein, we demonstrate that, in a toxic liver injury model such as tunicamycin, the absence of STING is protective conferring a detrimental role to STING, while in both dietary models, the absence of STING did not influence the overall outcome of liver injury or NAFLD progression. In fact, the only difference we demonstrate between WT and STING-deficient mice is a significant increase in insulin resistance in STING-deficient mice fed FPC.
2. Materials and Methods
2.1. Animals
For TM studies, 8–11 wk old male wild-type control mice (C57BL/6 or XBP1
fl/fl on a C57BL/6 background) and STING-deficient C57BL/6J-Sting1
gt/J mice (
Tmem137gt, Jackson Laboratory, Bar Harbor, ME, USA) were used. All mice received one intraperitoneal (i.p.) injection of either TM (2 mg/Kg) in a DMSO/saline mixture or the equivalent DMSO/saline dose alone [
12]. The mice were then sacrificed either 48 or 72 h post-injection. For the high fructose dietary studies, 7 wk old male C57BL/6 mice were purchased from the Jackson Laboratory (Bar Harbor, Maine) and allowed to acclimatize for 1 wk;
Tmem137gt were bred in-house and used at 8 wk of age. The animals (n = 4–5 mice/group) were placed on a 60% fructose diet (Fructose, TD.89247, Envigo) for 4 wk. Finally, for the NAFLD studies, another set of C57BL/6 and
Tmem173gt mice were randomly assigned (n = 4–6 mice/group) to either control chow (Chow, #5053, PicoLab, Forth Worth, TX, USA) or a Fructose–Palmitate–Cholesterol diet (FPC, TD.160785, Envigo, Indianapolis, IN, USA) for 1–16 wk. The mice on the FPC diet also received drinking water containing a 55:45 glucose-to-fructose mixture. Prior to euthanasia, the mice were fasted for 4 h. All mouse experiments were conducted in accordance with the guidelines set by the American Veterinary Medical Association. All mouse studies were reviewed and approved by the Committee on Animal Research at the University of California, San Francisco.
2.2. Histology and Immunohistochemistry
Formalin-fixed liver sections were stained with hematoxylin and eosin. Liver inflammation was quantified by immunohistochemistry for the pan-macrophage marker CD68, and fibrosis was assessed by staining with Sirius Red. Images were taken with a Leica DM6 B upright microscope and LAS X software (Leica Microsystems, Wetzlar, Germany), and percent staining was assessed using Image J software (Bethesda, MD, USA).
2.3. Gene Expression
RNA was isolated from whole liver using TRIzol (Invitrogen, Carlsbad, CA, USA) and purified with a Direct-zol RNA miniprep kit (ZymoResearch, Irvine, CA, USA). cDNA was synthesized using an iScript kit (Bio-Rad, Hercules, CA, USA), and gene expression was measured with TaqMan probe sets (Life Technologies, Carlsbad, CA, USA). TaqMan sets used were as follows: Ifnb (Mm00439552_s1), Irf3 (Mm00516784_m1), Tbk1 (Mm00451150_m1), Ifit1 (Mm00515153_m1), Ifit3 (Mm01704846_s1), and Gusb (Mm01197698_m1).
2.4. Serum Testing
Alanine aminotransferase (ALT) and total cholesterol were measured in mouse serum using an ADVIA 1800 autoanalyzer (Siemens Healthcare Diagnostics, Deerfield, IL, USA) in the clinical chemistry laboratory at the Zuckerberg San Francisco General Hospital.
2.5. Glucose Tolerance Testing
At 15 wk of experimental FPC feeding, a glucose tolerance test was performed. Mice were fasted for 4 h prior to testing. Blood glucose was measured at time 0 followed by a glucose bolus of 20 mg/kg i.p. Subsequent glucose levels were monitored at 15, 30, 60, and 120 min.
2.6. Lipid Quantitation
Lipids were extracted from frozen liver tissue using the Folch method [
13]. Total liver triglycerides were measured spectrophotometrically as previously described (TR0100, Millipore-Sigma, Burlington, MA, USA) [
14].
2.7. Statistics
All results are reported as mean ± SEM. Statistical analysis was performed using Prism 9.1.1 software (GraphPad Software, San Diego, CA, USA). When two groups were compared, a Student’s t-test was applied. When more than two groups were compared, a one-way analysis of variance (ANOVA) was employed followed by a Tukey’s multiple comparisons test. p values <0.05 were considered statistically significant.
3. Results
STING deficiency protects against short-term toxic liver injury induced by tunicamycin. Whole liver protein was analyzed in order to confirm the absence of STING in
Tmem173gt mice (
Figure 1A). The animals were injected once with TM or vehicle control (DMSO in saline) and were euthanized at either 48 or 72 h later. TM-induced liver damage was assessed by ALT levels (
Figure 1B). At 48 h post-TM, both the wild-type (WT) and
Tmem173gt mice demonstrated similar ALT levels. However, at 72 h post-TM,
Tmem173gt mice demonstrated significantly lower ALT levels than their WT counterparts. All DMSO-injected mice demonstrated normal ALT levels. The data demonstrate a significant role for STING in developing liver damage following short-term toxic injury. However, the data also demonstrate that the role of STING is time-dependent and only significant at 72 h post-TM delivery.
Absence of STING during short-term high fructose feeding does not affect liver injury outcome. Knowing that STING played a significant role in toxic-induced liver damage, we wanted to investigate whether STING was also involved in other models of liver injury, such as NAFLD and high fructose feeding. WT and
Tmem173gt mice were fed the high-fructose diet for 4 wk. Histologically, both groups demonstrated the same morphology, with limited steatosis and very limited damage noted (
Figure 2A). The main histological findings were a few inflammatory foci per whole liver section as well as a rare mitotic figure or dead cell. ALT levels confirmed the histological findings and showed only a slight elevation for both groups after 4 wk of fructose feeding. While
Tmem173gt mice tended to have higher ALT levels, the differences were not statistically significant (
Figure 2B). We also investigated downstream STING targets,
Tbk1,
Irf3, and
Ifnb by gene expression and found no difference between the two groups (
Figure 2C). Together, these data demonstrate that short-term high fructose feeding and the possible resultant ER stress [
15] may not be sufficient to trigger STING activation and its downstream signaling cascade. Furthermore, the absence of STING did not make a difference in overall liver outcome and cell health.
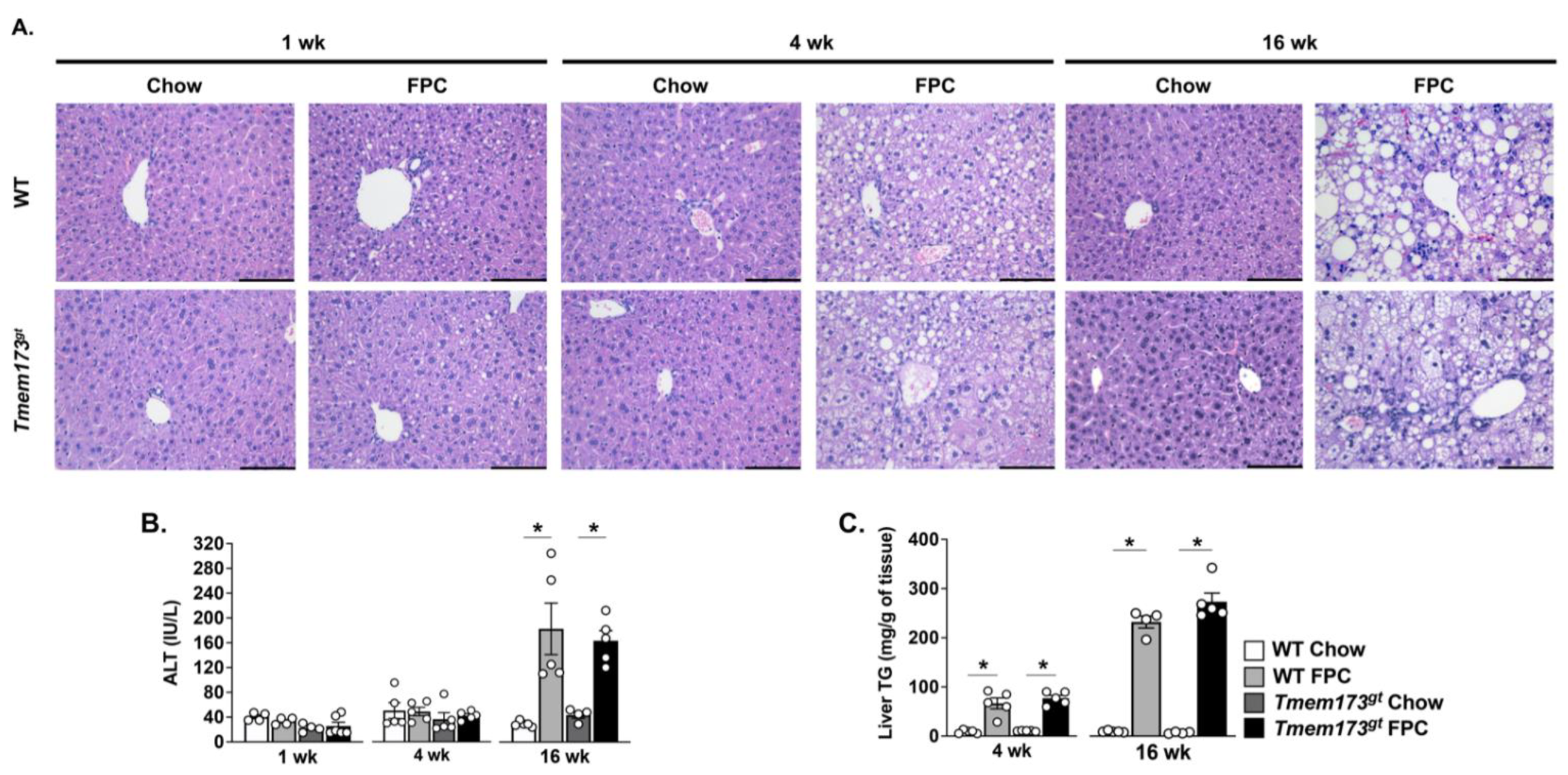
The absence of STING does not protect mice from steatosis or liver injury in response to FPC feeding. Given our mixed results regarding the contribution of STING to TM liver injury and high fructose feeding, we decided to investigate the role of STING in a more aggressive NASH-inducing model and thus fed mice a Fructose–Palmitate–Cholesterol (FPC) diet for varying time points. Both WT and
Tmem173gt mice were fed either control chow or FPC for 1, 4, or 16 wk. Chow-fed mice from both genotypes demonstrated no histological abnormalities throughout the experimental time-points (
Figure 3A). FPC-fed mice began to exhibit hepatic steatosis as early as 1 wk, but there was no apparent difference between WT and
Tmem173gt mice (
Figure 3A). By 4 wk of FPC feeding, significant steatosis could be seen in both WT and
Tmem173gt FPC-fed mice, but no significant liver injury was noted, as underscored by the lack of elevation in ALT levels (
Figure 3A,B). At 16 wk, both WT and
Tmem173gt FPC groups demonstrated significant liver steatosis and injury, clearly demonstrating a NASH phenotype, as underlined by their liver TG, ALT values, and histological features (
Figure 3A–C). Of note, the
Tmem173gt FPC-fed mice appeared to develop both micro- and macrosteatosis, compared to their WT counterparts, which displayed mostly macrosteatosis. However, throughout the varying time points, no significant differences in overall steatosis or injury were noted between the WT FPC-fed mice and their
Tmem173gt counterparts.
STING neither protects nor worsens liver fibrosis or inflammation in the dietary NASH model FPC. In order to fully understand the potential role of STING during NASH and long-term liver injury, we examined liver inflammation and fibrosis in our 16 wk animals. Liver sections were stained with Sirius Red and positive staining quantified for all groups (
Figure 4A,B). Both FPC-fed mice accumulated significantly more collagen deposition by 16 wk than their chow counterparts; however, no significant difference was seen between WT and
Tmem173gt mice. This was further confirmed by gene expression analysis for
Col1a1 (
Figure 4C). The same was true for liver inflammation. Both FPC-fed groups had significantly more CD68+ cells than their chow counterparts, but no significant difference was noted between WT and
Tmem173gt mice. These findings were confirmed by gene expression levels for
Ccl2 and
Tnfa, which also demonstrated significant increases in the FPC-fed mice compared to their chow counterparts but no differences between genotypes. Interestingly, the chow-fed
Tmem173gt mice did have significantly more CD68+ cells than their WT counterparts.
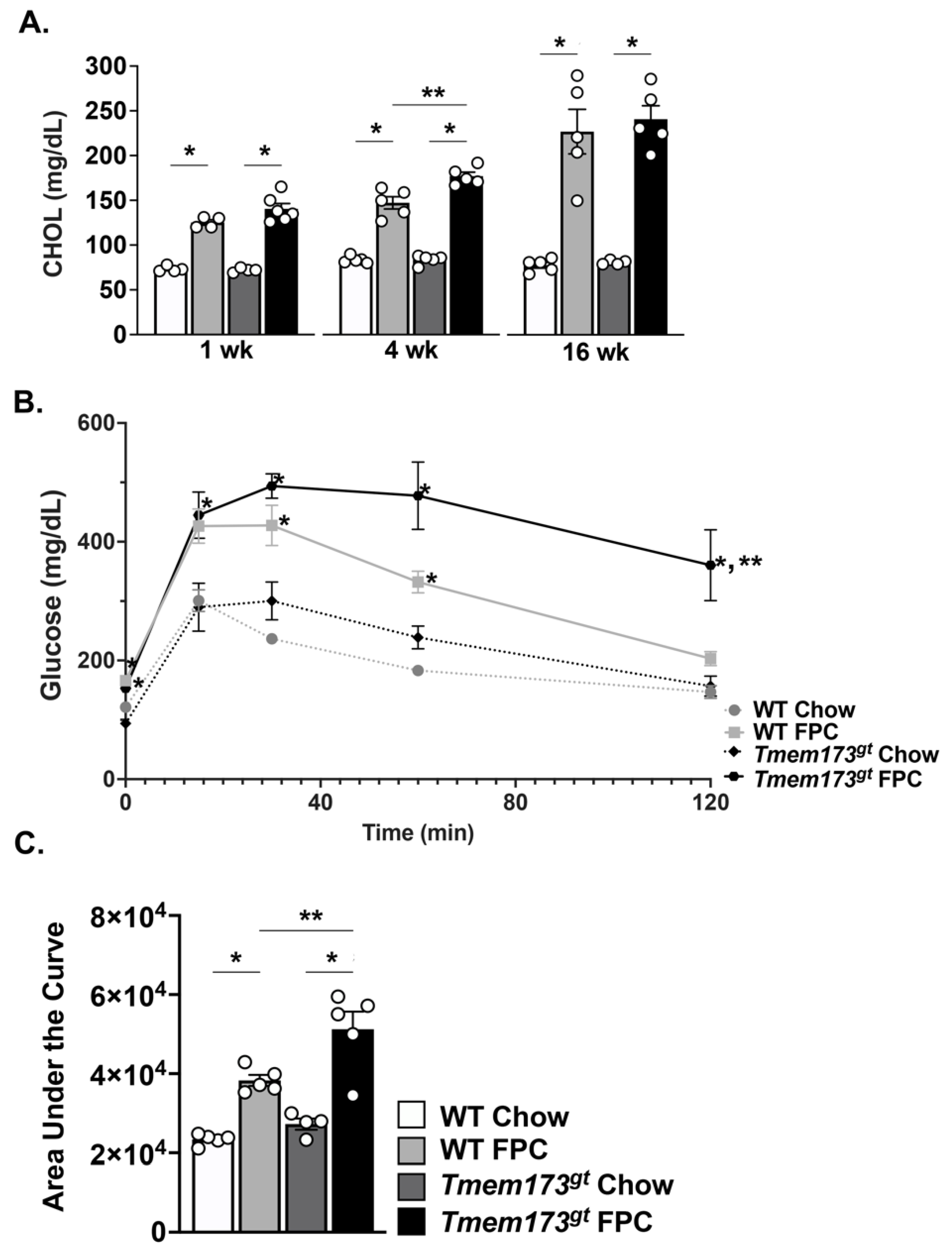
Insulin resistance is worse in the absence of STING during long-term FPC feeding. We further investigated the WT and
Tmem173gt mice for abnormalities in their metabolic function following FPC feeding (
Figure 5). FPC-fed WT and
Tmem173gt mice showed significant increases in their serum cholesterol compared to their chow counterparts at all timepoints; however, no significant differences between the two genotypes were seen (
Figure 5A). A glucose tolerance test was administered and showed that while both FPC-fed groups had significantly higher blood glucose levels in the beginning, only the
Tmem173gt FPC mice remained high for the duration of the test (
Figure 5B,C). Taken together, these data point towards a heightened state of insulin resistance for the
Tmem173gt FPC mice compared to all other groups at 16 wk.
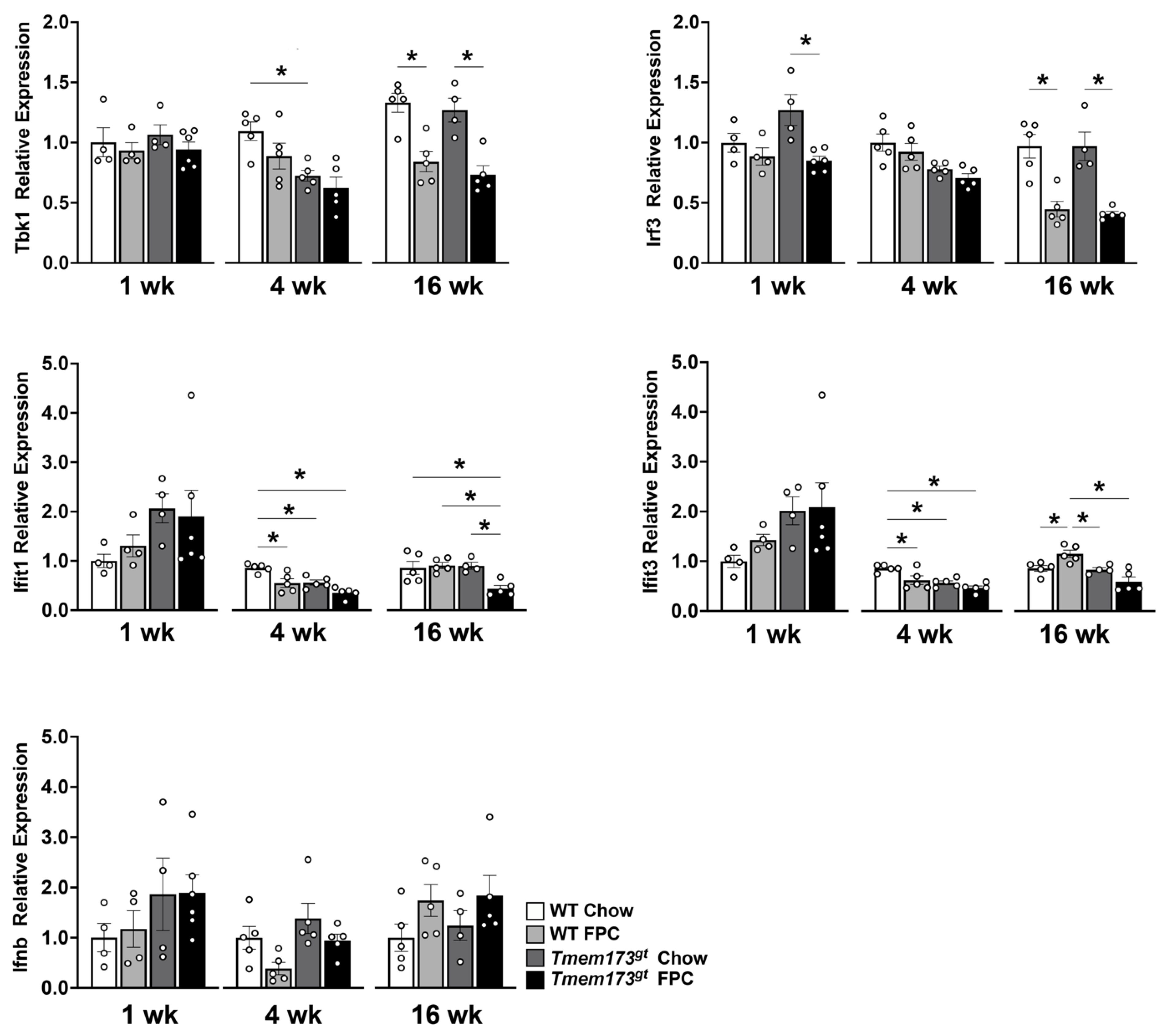
Whole body STING deficiency in a dietary NASH model has conflicting effects on the type I IFN pathway. Finally, we investigated whether FPC feeding activates STING signaling in the liver and whether these signals were suppressed in STING-deficient mice by qtPCR. Little to no changes were noted prior to 16 wk in both
Tbk1 and
Irf3 expression levels, but by 16 wk, a paradoxical significant decrease in expression levels was noted in both FPC-fed groups (
Figure 6). Again no significant difference was noted between genotypes. The expression patterns for both
Ifit1 and
Ifit3 were unique. Both genes demonstrated a significant decrease in
Tmem173gt FPC-fed mice at the 4 wk timepoint compared to WT chow mice, with non-significant trends of being lower than their WT counterparts. At 16 wk, the
Ifit1 expression levels for
Tmem173gt FPC-fed mice were significantly lower than all other groups. While at 16 wk, the
Ifit3 levels for
Tmem173gt FPC-fed mice were significantly lower than their wild-type counterparts.
Ifnb, the final molecule in the STING pathway demonstrated no significant differences whatsoever, neither by timepoint or by diet, nor by genotype group. Taken together, this points to a limited role for STING in NASH pathogenesis.
4. Discussion
In the present study, we demonstrated a stimulus-dependent role for STING in liver injury. In TM-treated mice, the absence of whole-body STING appeared to be beneficial to liver health as indicated by reduced ALT levels 3 d post-injection. However, in contrast, STING appeared to play no role in mice-fed high fructose diet for 4 wk, although admittedly the overall injury due to high fructose feeding was very limited. These data thus indicate that the role of STING in toxic liver injury may in fact be stimulus-, time-, and intensity-dependent. Furthermore, when mice were fed a FPC NASH-inducing diet for up to 16 wk, the absence of whole-body STING appeared to play a very limited role in overall health, affecting only insulin resistance but not liver health, for example, having no effect on levels of steatosis, inflammation, or fibrosis. Based on previously published reports using HFD and MCD models, these results were unexpected [
9,
10,
11,
16]. We propose a role for lipotoxcity in the activation of STING, possibly through ER stress activation during HFD, that may not be as prominent in a diet higher in carbohydrate.
Other groups have shown an important role for STING in both HFD and MCD models. One group, for example, using a HFD feeding model for 12 wk demonstrated that
Tmem173gt mice developed significantly lower ALT, liver triglyceride content, and inflammation [
11]. Another group using a MCD feeding model established that
Tmem173gt mice had significantly less steatosis, ballooning, inflammation, and fibrosis at 8 wk [
10]. However, in contrast, another group also using a HFD model demonstrated a beneficial role for IRF3, a downstream molecule of STING [
9]. This study established that mice, in the absence of IRF3, when fed HFD developed significantly more liver injury and fibrosis, as demonstrated by a significant increase in ALT and Sirius red staining, respectively [
9]. This study thus indicates a potential beneficial role for STING. In our present study, however, we demonstrated no difference in steatosis, injury, or fibrosis in the absence of STING. One significant difference between our current study and those mentioned above is the type of diet given to the animals. All of these other studies used high fat diets, whereas we used a diet high in both fat and carbohydrate; it could be that in the presence of high carbohydrates the role of STING is less important. It could also be that our diet is overall less inflammatory than HFD and thus does not interact with the STING pathway as aggressively as in these other studies. Additionally, the FPC diet was originally used in describing the role of TAZ (Transcriptional Co-Activator With PDZ-Binding Motif) in hepatic fibrogenesis, and it could be that this pathway outweighs the potential role of STING during this dietary insult [
17].
An interesting finding was a significant increase in CD68% cells in
Tmem173gt chow-fed mice compared to WT chow-fed mice. It has been previously documented that STING is highly expressed in macrophages [
10,
11] within the liver. Recently, a group demonstrated that HFD fed
Tmem173gt mice with wild-type bone marrow had a significant increase in their hepatic macrophage populations, as shown by increased F4/80 staining [
11] compared to
Tmem173gt mice with
Tmem173gt bone marrow. This group also demonstrated a significant decrease in overall hepatic macrophages when
Tmem173gt mice were fed MCD diet. While we appear to show the opposite findings in our STING mutant mice, both findings beg the question whether STING may be involved in macrophage migration into the liver.
One unexpected finding was the decrease in
Irf3 gene expression levels in our
Tmem173gt mice compared to WT mice. However, this has in fact already been described by two other groups [
9,
18]. Furthermore
Ifnb gene levels were not affected in either of the models employed in this paper, but no other papers report on
Ifnb gene expression levels during a NAFLD stimulus, while one group does demonstrate a significant increase in IFNβ protein levels with HFD [
19]. It may be that these dietary models are not inflammatory enough to induce
Ifnb with STING recruitment.
Overall, while our study demonstrates a detrimental role for STING during chemically induced toxic liver injury, it did not demonstrate a role for STING in a dietary models of NAFLD. Furthermore, we demonstrated no important role for STING in liver steatosis, inflammation, and fibrogenesis during short- and long-term feeding of a NASH-inducing diet, although we did note a significant reduction in whole-body insulin resistance in the presence of STING.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}