
Exposure to the Amino Acids Histidine, Lysine, and Threonine Reduces mTOR Activity and Affects Neurodevelopment in a Human Cerebral Organoid Model


 , , , ,
, , , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. iPSC line Generation and Maintenance
2.2. Ethical Approval
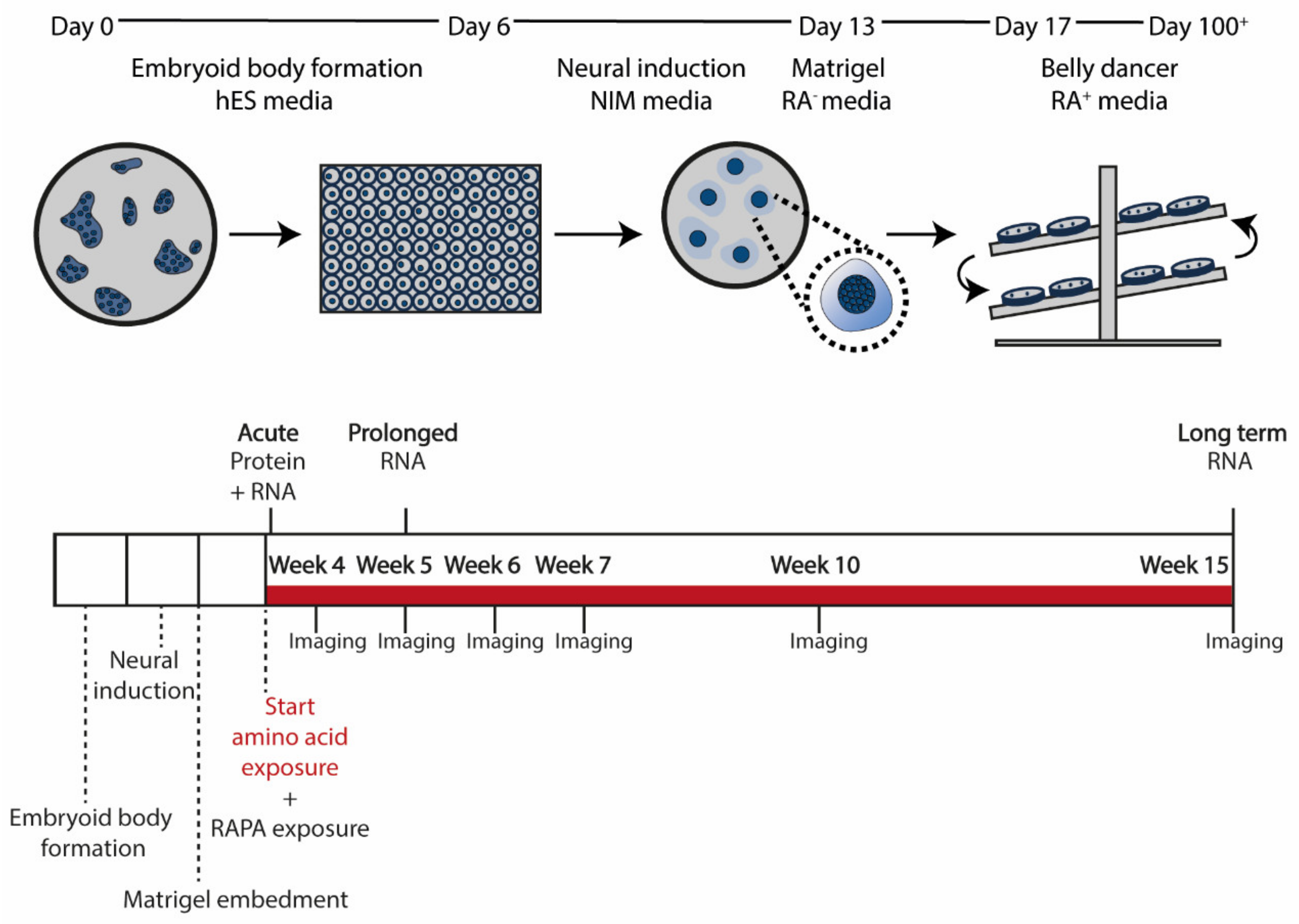
2.3. Organoid Differentiation
2.4. Exposure
2.5. Size Measurements
2.6. Western Blot
2.7. RNA Isolation
2.8. RNA Sequencing
2.8.1. Bulk RNAseq Analysis
2.8.2. Quality Control
2.8.3. Pre-Processing and Confounders
2.8.4. Differential Gene Expression Analysis
2.8.5. Ingenuity Pathway Analysis
2.8.6. Comparison with Previously Published Datasets
2.8.7. Data Availability
2.9. Weighted Gene Co-Expression Network Analysis
2.10. Statistics
3. Results
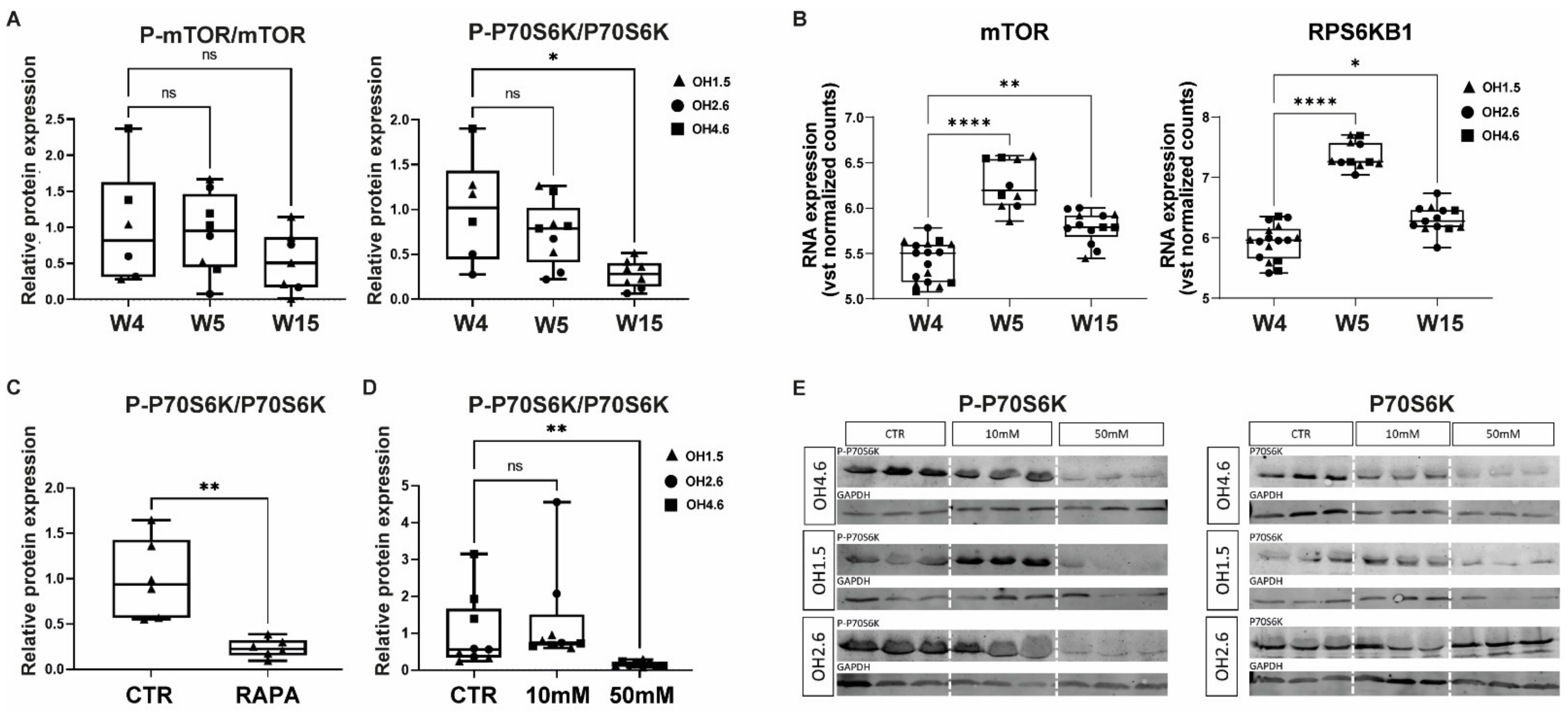
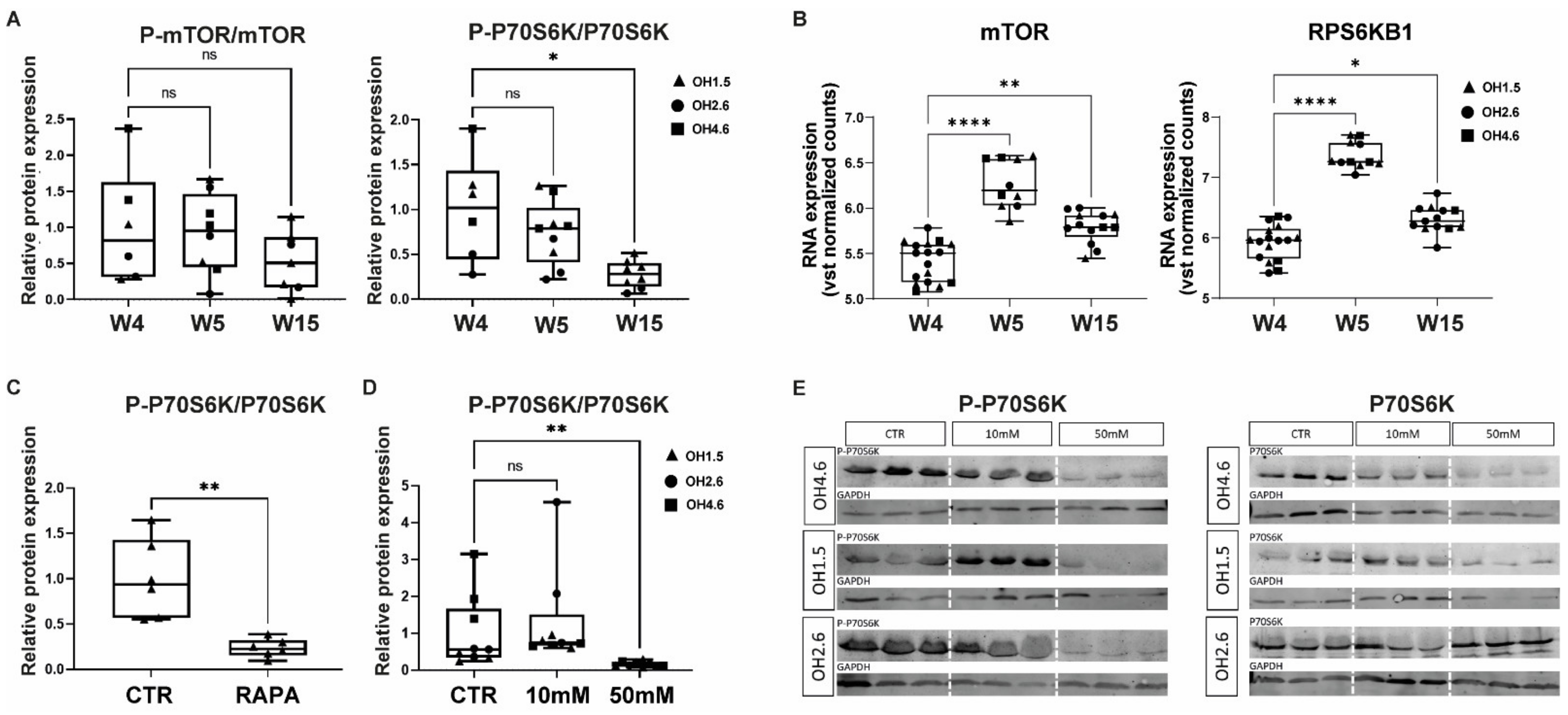
3.1. mTOR and P70S6K in Human Cerebral Organoids
3.2. Acute Effects of AA Exposure on mTOR Activity in Human Cerebral Organoids
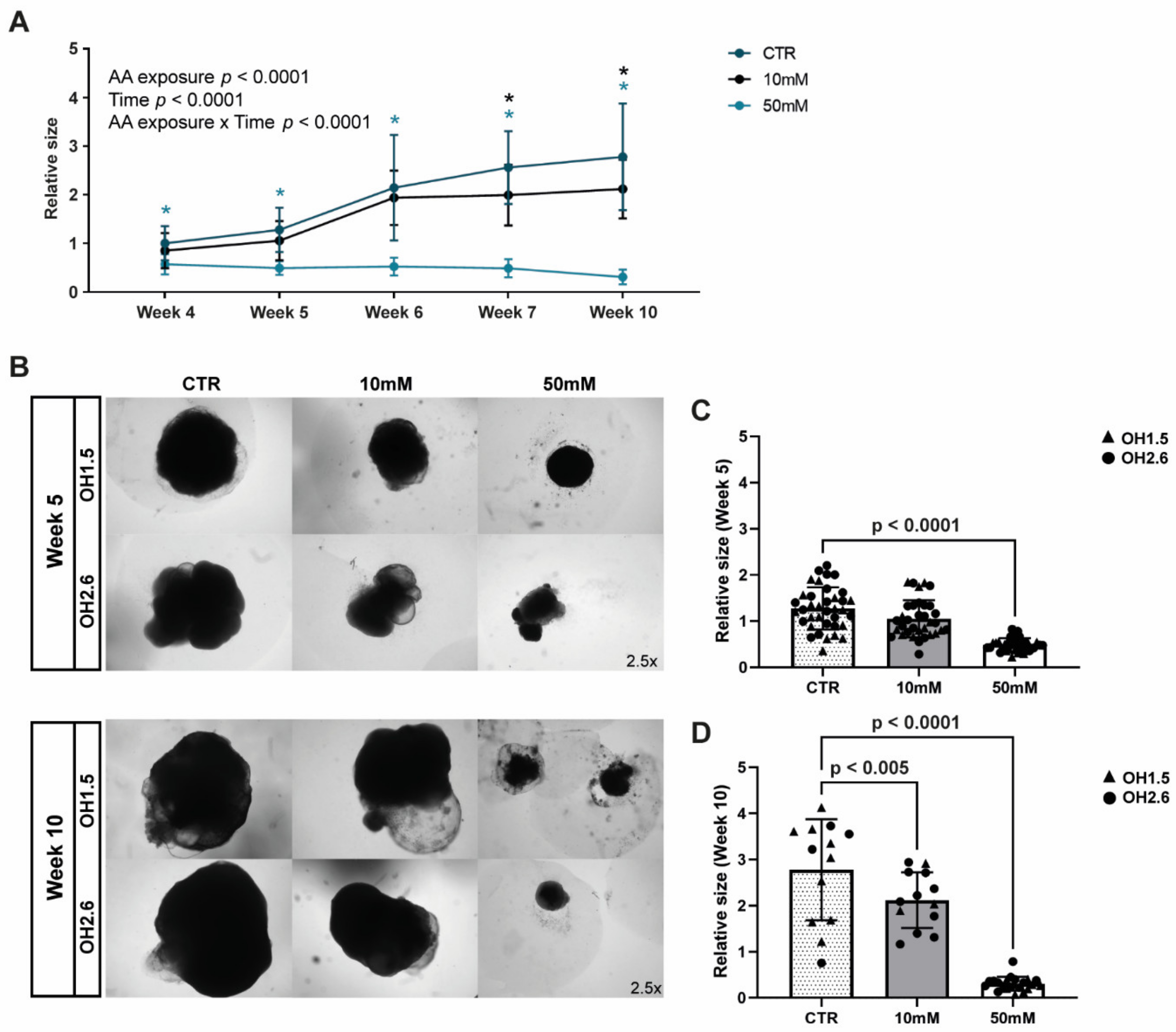
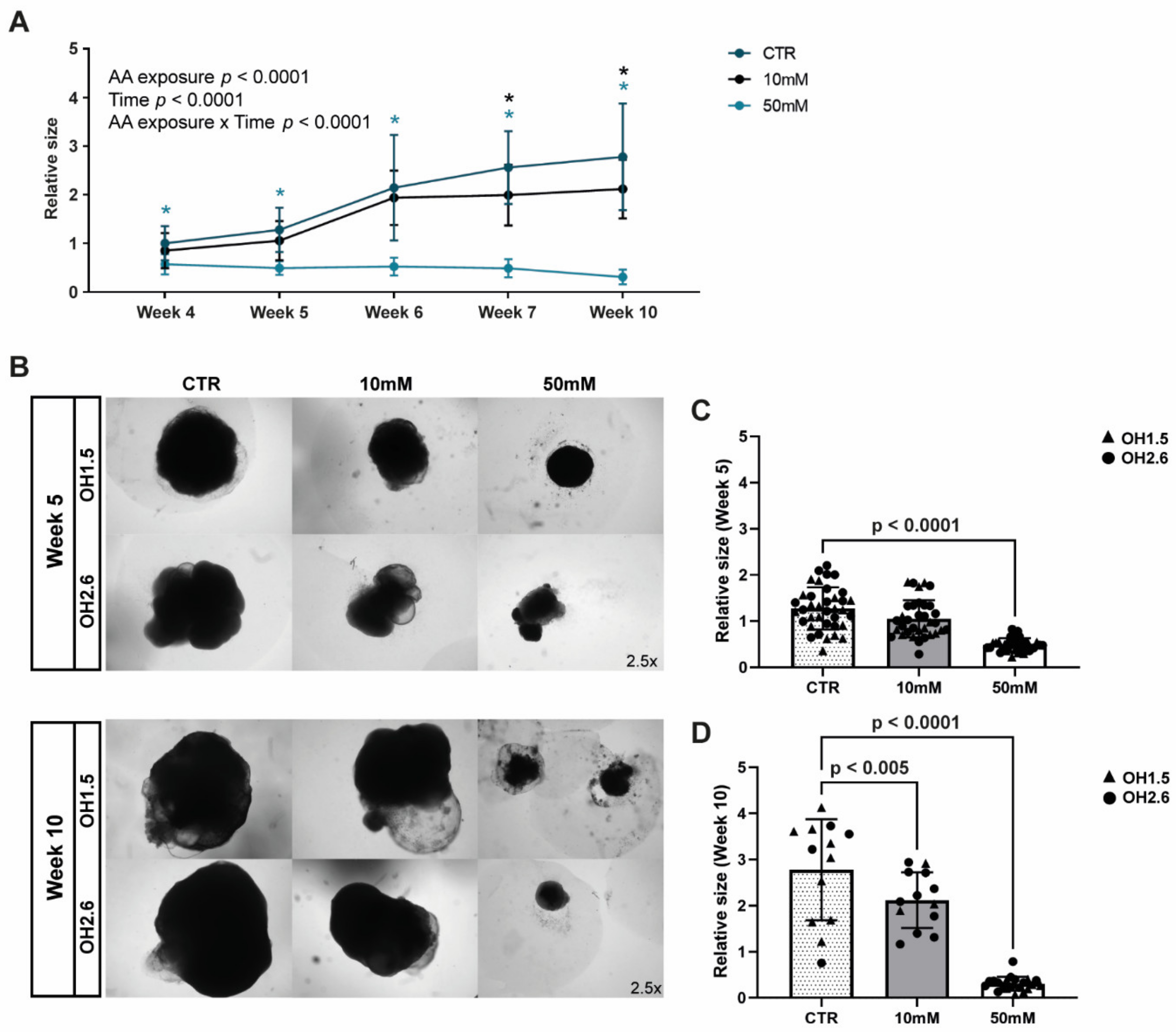
3.3. Chronic Amino Acid Exposure Causes Size Deficits in Cerebral Organoids
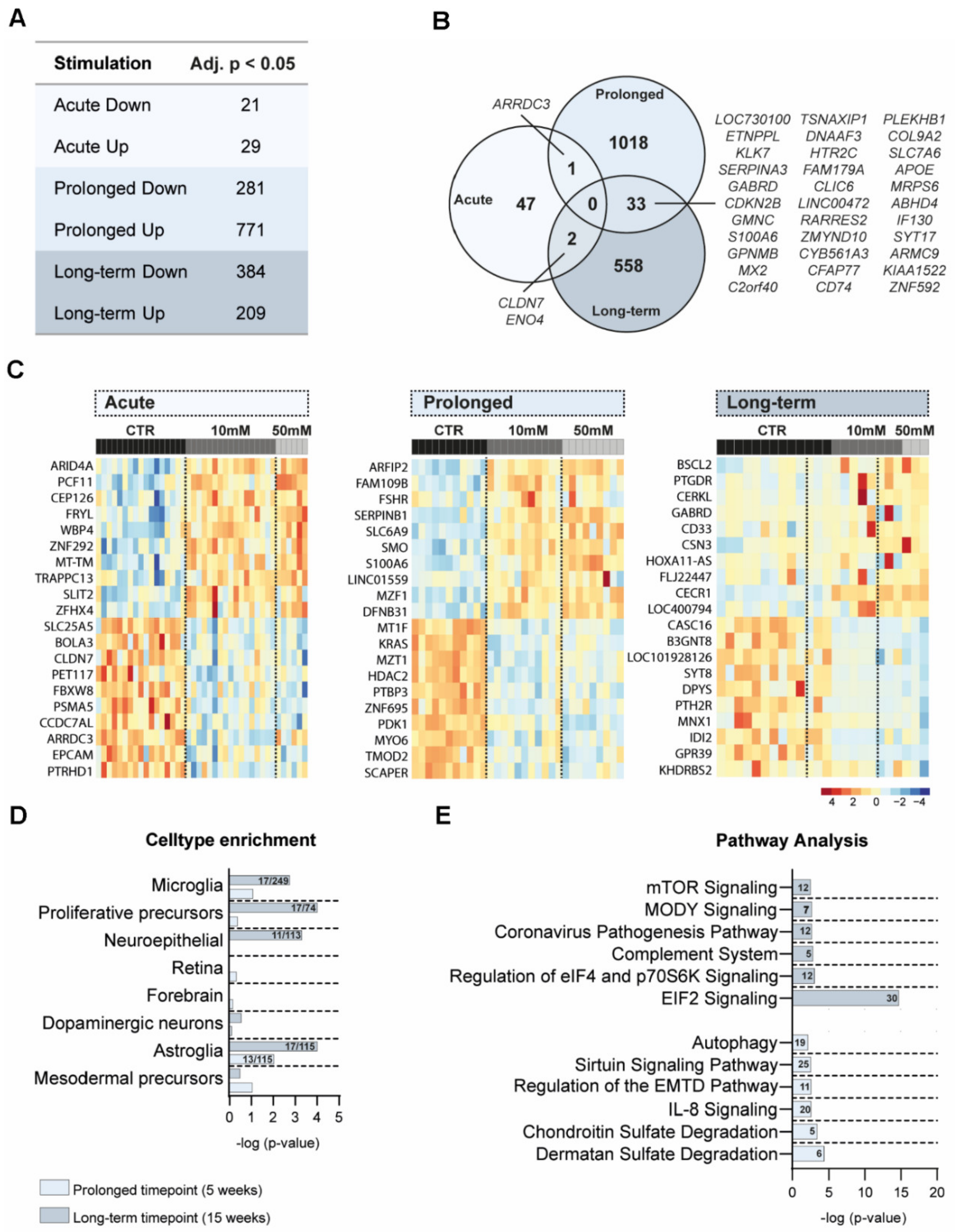
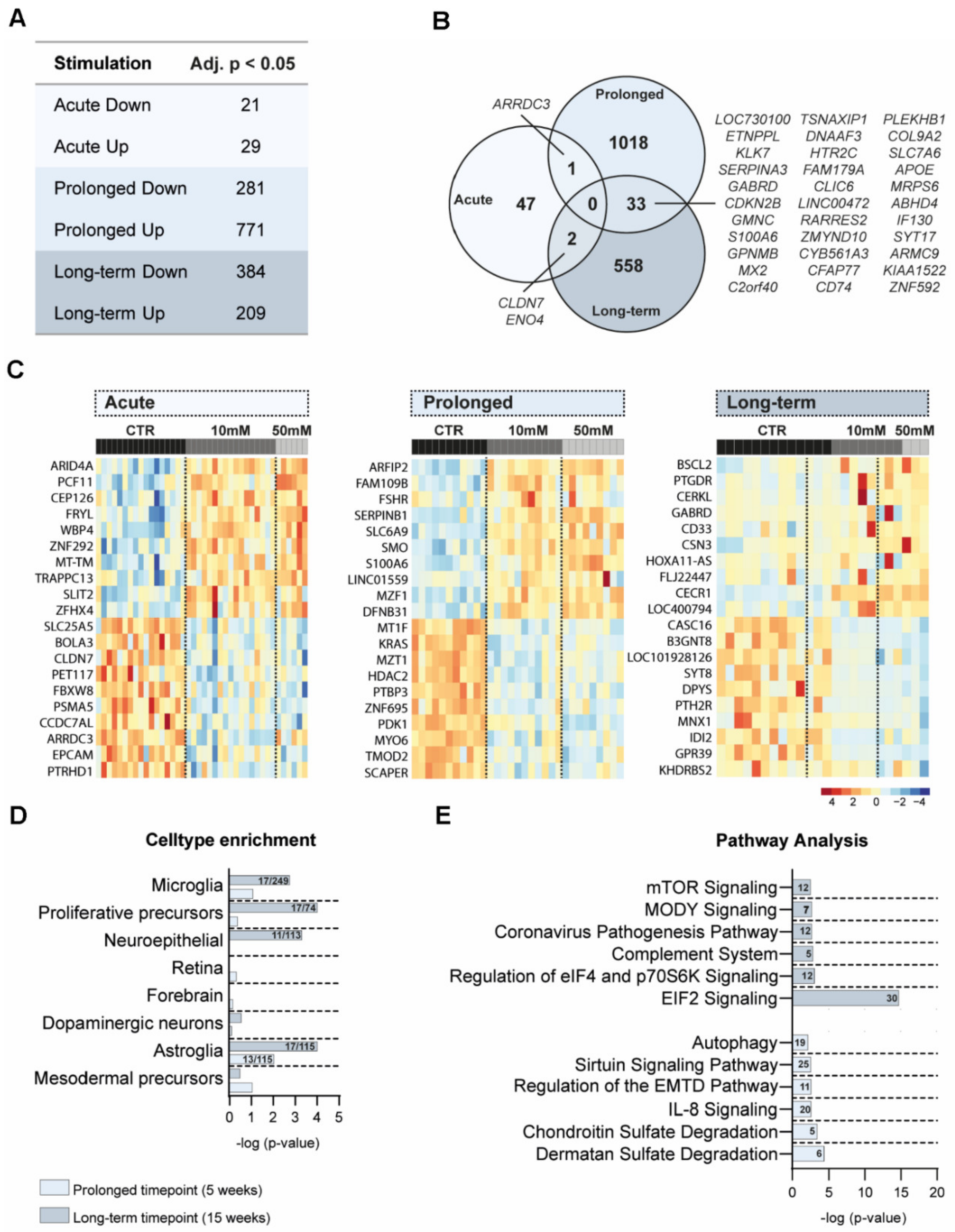
3.4. Transcriptomic Differences after Amino Acid Exposure in Cerebral Organoids
3.5. Cell Type and Pathway Analyses of DEG
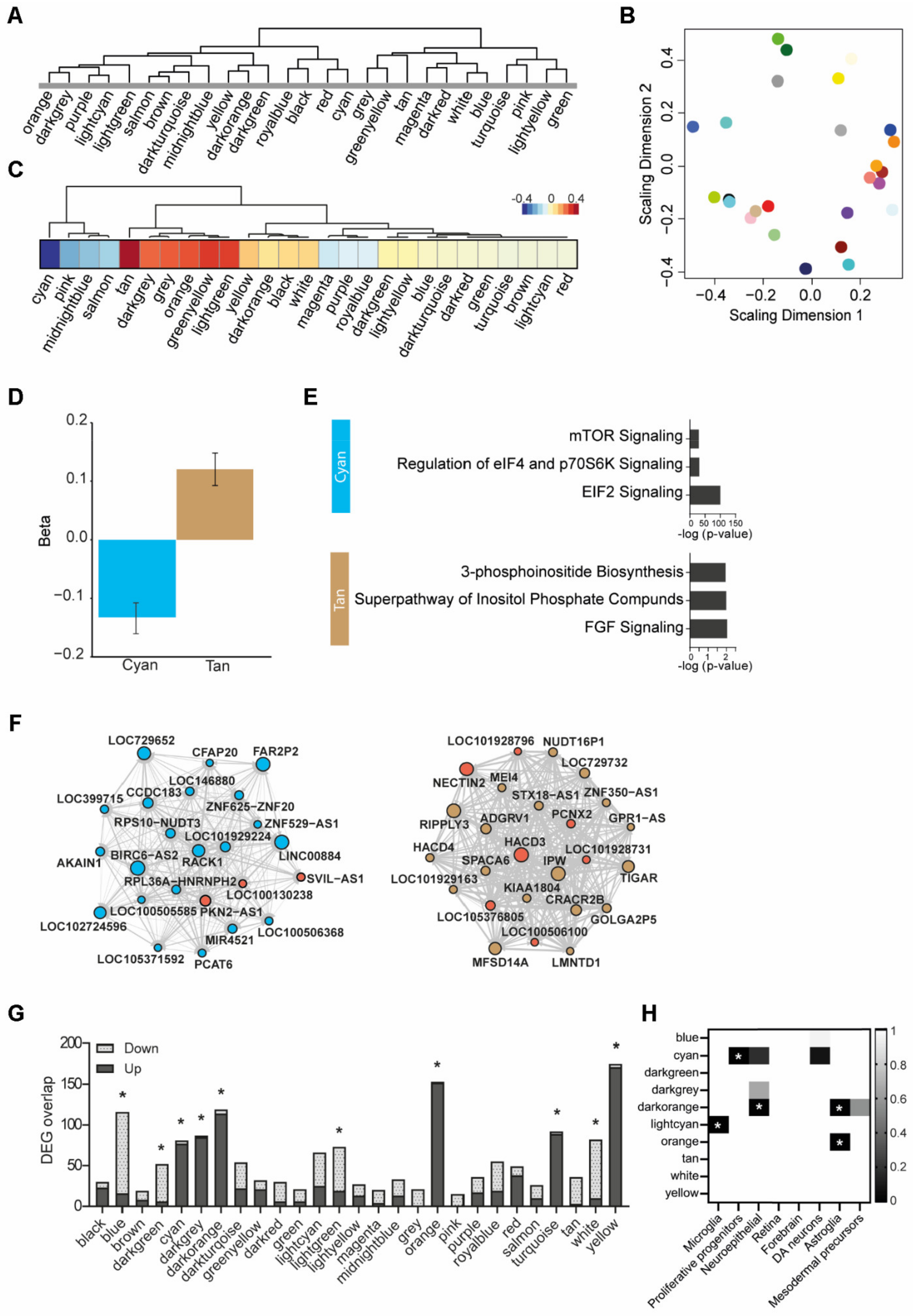
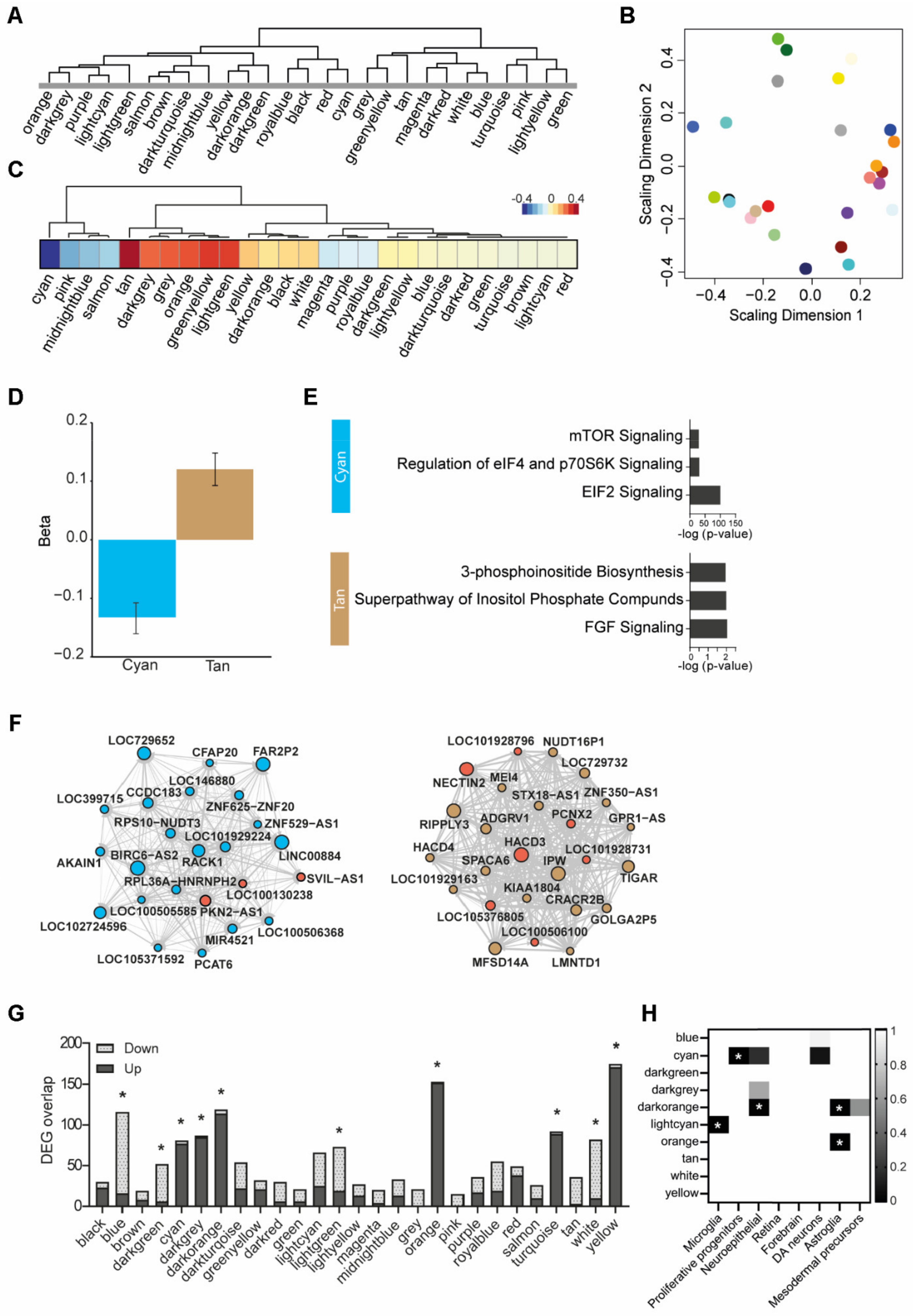
3.6. Weighted Gene Co-Expression Network Analysis
4. Discussion
4.1. Future Perspectives
4.2. Limitations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Rooij, S.R.; Wouters, H.; Yonker, J.E.; Painter, R.C.; Roseboom, T.J. Prenatal undernutrition and cognitive function in late adulthood. Proc. Natl. Acad. Sci. USA 2010, 107, 16881–16886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, Y.; Zhang, Y.; Feng, Z.; Liu, M.; Li, Y.; Yang, H.; Wang, D.; Zheng, L.; Lou, D.; Cheng, L.; et al. Nutritional Deficiency in Early Life Facilitates Aging-Associated Cognitive Decline. Curr. Alzheimer Res. 2017, 14, 841–849. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Francis, E.; Hinkle, S.N.; Ajjarapu, A.S.; Zhang, C. Preconception and Prenatal Nutrition and Neurodevelopmental Disorders: A Systematic Review and Meta-Analysis. Nutrients 2019, 11, 1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peretti, S.; Mariano, M.; Mazzocchetti, C.; Mazza, M.; Pino, M.C.; Verrotti Di Pianella, A.; Valenti, M. Diet: The Keystone of Autism Spectrum Disorder? Nutr. Neurosci. 2019, 22, 825–839. [Google Scholar] [CrossRef]
- Clair, D.S. Rates of Adult Schizophrenia Following Prenatal Exposure to the Chinese Famine of 1959–1961. JAMA 2005, 294, 557–562. [Google Scholar] [CrossRef] [Green Version]
- Hoek, H.W.; Brown, A.S.; Susser, E. The Dutch Famine and schizophrenia spectrum disorders. Soc. Psychiatry 1998, 33, 373–379. [Google Scholar] [CrossRef]
- Abbink, M.R.; van Deijk, A.-L.F.; Heine, V.M.; Verheijen, M.H.; Korosi, A. The involvement of astrocytes in early-life adversity induced programming of the brain. Glia 2019, 67, 1637–1653. [Google Scholar] [CrossRef]
- Marques, A.H.; Bjørke-Monsen, A.-L.; Teixeira, A.L.; Silverman, M.N. Maternal stress, nutrition and physical activity: Impact on immune function, CNS development and psychopathology. Brain Res. 2015, 1617, 28–46. [Google Scholar] [CrossRef]
- Salas, M.; Torrero, C.; Rubio, L.; Regalado, M. Effects of perinatal undernutrition on the development of neurons in the rat insular cortex. Nutr. Neurosci. 2012, 15, 20–25. [Google Scholar] [CrossRef]
- Watanabe, K.; Furumizo, Y.; Usui, T.; Hattori, Y.; Uemura, T. Nutrient-dependent increased dendritic arborization of somatosensory neurons. Genes Cells 2016, 22, 105–114. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.A.; Rinaldi, S.; Scalbert, A.; Ferrari, P.; Achaintre, D.; Gunter, M.J.; Appleby, P.N.; Key, T.J.; Travis, R.C. Plasma concentrations and intakes of amino acids in male meat-eaters, fish-eaters, vegetarians and vegans: A cross-sectional analysis in the EPIC-Oxford cohort. Eur. J. Clin. Nutr. 2016, 70, 306–312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G. Amino acids: Metabolism, functions, and nutrition. Amino Acids 2009, 37, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Bar-Peled, L.; Sabatini, D.M. Regulation of mTORC1 by amino acids. Trends Cell Biol. 2014, 24, 400–406. [Google Scholar] [CrossRef]
- Efeyan, A.; Zoncu, R.; Sabatini, D.M. Amino acids and mTORC1: From lysosomes to disease. Trends Mol. Med. 2012, 18, 524–533. [Google Scholar] [CrossRef] [PubMed]
- Sengupta, S.; Peterson, T.R.; Sabatini, D.M. Regulation of the mTOR Complex 1 Pathway by Nutrients, Growth Factors, and Stress. Mol. Cell 2010, 40, 310–322. [Google Scholar] [CrossRef] [Green Version]
- Andrews, M.G.; Subramanian, L.; Kriegstein, A.R. mTOR signaling regulates the morphology and migration of outer radial glia in developing human cortex. eLife 2020, 9, e58737. [Google Scholar] [CrossRef]
- Blair, J.D.; Hockemeyer, D.; Bateup, H.S. Genetically engineered human cortical spheroid models of tuberous sclerosis. Nat. Med. 2018, 24, 1568–1578. [Google Scholar] [CrossRef]
- Bockaert, J.; Marin, P. mTOR in Brain Physiology and Pathologies. Physiol. Rev. 2015, 95, 1157–1187. [Google Scholar] [CrossRef]
- Cloëtta, D.; Thomanetz, V.; Baranek, C.; Lustenberger, R.M.; Lin, S.; Oliveri, F.; Atanasoski, S.; Rüegg, M.A. Inactivation of mTORC1 in the Developing Brain Causes Microcephaly and Affects Gliogenesis. J. Neurosci. 2013, 33, 7799–7810. [Google Scholar] [CrossRef] [Green Version]
- Swiech, L.; Perycz, M.; Malik, A.; Jaworski, J. Role of mTOR in physiology and pathology of the nervous system. Biochim. Biophys. Acta (BBA) Proteins Proteom. 2008, 1784, 116–132. [Google Scholar] [CrossRef]
- Curatolo, P.; Moavero, R.; de Vries, P.J. Neurological and neuropsychiatric aspects of tuberous sclerosis complex. Lancet Neurol. 2015, 14, 733–745. [Google Scholar] [CrossRef]
- Howell, K.R.; Law, A.J. Neurodevelopmental concepts of schizophrenia in the genome-wide association era: AKT/mTOR signaling as a pathological mediator of genetic and environmental programming during development. Schizophr. Res. 2019, 217, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Winden, K.D.; Ebrahimi-Fakhari, D.; Sahin, M. Abnormal mTOR Activation in Autism. Annu. Rev. Neurosci. 2018, 41, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Zimmer, T.S.; Broekaart, D.W.M.; Gruber, V.-E.; Van Vliet, E.A.; Mühlebner, A.; Aronica, E. Tuberous Sclerosis Complex as Disease Model for Investigating mTOR-Related Gliopathy During Epileptogenesis. Front. Neurol. 2020, 11, 1028. [Google Scholar] [CrossRef]
- Prizant, R.L.; Barash, I. Negative effects of the amino acids Lys, His, and Thr on S6K1 phosphorylation in mammary epithelial cells. J. Cell. Biochem. 2008, 105, 1038–1047. [Google Scholar] [CrossRef]
- Wu, J.; de Theije, C.; da Silva, S.L.; Abbring, S.; van der Horst, H.; Broersen, L.M.; Willemsen, L.; Kas, M.; Garssen, J.; Kraneveld, A.D. Dietary interventions that reduce mTOR activity rescue autistic-like behavioral deficits in mice. Brain, Behav. Immun. 2017, 59, 273–287. [Google Scholar] [CrossRef]
- Lui, J.H.; Hansen, D.V.; Kriegstein, A.R. Development and Evolution of the Human Neocortex. Cell 2011, 146, 18–36. [Google Scholar] [CrossRef] [Green Version]
- Oberheim, N.A.; Takano, T.; Han, X.; He, W.; Lin, J.H.C.; Wang, F.; Xu, Q.; Wyatt, J.D.; Pilcher, W.; Ojemann, J.; et al. Uniquely Hominid Features of Adult Human Astrocytes. J. Neurosci. 2009, 29, 3276–3287. [Google Scholar] [CrossRef]
- Patir, A.; Shih, B.; McColl, B.W.; Freeman, T.C. A core transcriptional signature of human microglia: Derivation and utility in describing region-dependent alterations associated with Alzheimer’s disease. Glia 2019, 67, 1240–1253. [Google Scholar] [CrossRef]
- Di Lullo, E.; Kriegstein, A.R. The use of brain organoids to investigate neural development and disease. Nat. Rev. Neurosci. 2017, 18, 573–584. [Google Scholar] [CrossRef] [Green Version]
- Lancaster, M.A.; Renner, M.; Martin, C.-A.; Wenzel, D.; Bicknell, L.S.; Hurles, M.E.; Homfray, T.; Penninger, J.M.; Jackson, A.P.; Knoblich, J.A. Cerebral organoids model human brain development and microcephaly. Nature 2013, 501, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Pollen, A.A.; Nowakowski, T.J.; Chen, J.; Retallack, H.; Sandoval-Espinosa, C.; Nicholas, C.R.; Shuga, J.; Liu, S.J.; Oldham, M.C.; Diaz, A.; et al. Molecular Identity of Human Outer Radial Glia during Cortical Development. Cell 2015, 163, 55–67. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ormel, P.R.; Vieira de Sá, R.; Van Bodegraven, E.J.; Karst, H.; Harschnitz, O.; Sneeboer, M.A.M.; Johansen, L.E.; Van Dijk, R.E.; Scheefhals, N.; Berdenis van Berlekom, A.; et al. Microglia innately develop within cerebral organoids. Nat. Commun. 2018, 9, 4167. [Google Scholar] [CrossRef] [PubMed]
- Renner, M.; A Lancaster, M.; Bian, S.; Choi, H.; Ku, T.; Peer, A.; Chung, K.; A Knoblich, J. Self-organized developmental patterning and differentiation in cerebral organoids. EMBO J. 2017, 36, 1316–1329. [Google Scholar] [CrossRef]
- Kelava, I.; Lancaster, M.A. Dishing out mini-brains: Current progress and future prospects in brain organoid research. Dev. Biol. 2016, 420, 199–209. [Google Scholar] [CrossRef] [Green Version]
- Luo, C.; Lancaster, M.; Castanon, R.; Nery, J.R.; Knoblich, J.A.; Ecker, J.R. Cerebral Organoids Recapitulate Epigenomic Signatures of the Human Fetal Brain. Cell Rep. 2016, 17, 3369–3384. [Google Scholar] [CrossRef] [Green Version]
- Harschnitz, O.; Berg, L.H.V.D.; Johansen, L.E.; DI, M.D.J.; Kling, S.; Msc, R.V.d.S.; Vlam, L.; van Rheenen, W.; Karst, H.; Wierenga, C.J.; et al. Autoantibody pathogenicity in a multifocal motor neuropathy induced pluripotent stem cell-derived model. Ann. Neurol. 2016, 80, 71–88. [Google Scholar] [CrossRef]
- Quadrato, G.; Nguyen, T.; Macosko, E.Z.; Sherwood, J.L.; Min Yang, S.; Berger, D.R.; Maria, N.; Scholvin, J.; Goldman, M.; Kinney, J.P.; et al. Cell diversity and network dynamics in photosensitive human brain organoids. Nature 2017, 545, 48–53. [Google Scholar] [CrossRef] [Green Version]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]
- Muraro, M.J.; Dharmadhikari, G.; Grün, D.; Groen, N.; Dielen, T.; Jansen, E.; van Gurp, L.; Engelse, M.A.; Carlotti, F.; De Koning, E.J.; et al. A Single-Cell Transcriptome Atlas of the Human Pancreas. Cell Syst. 2016, 3, 385–394.e3. [Google Scholar] [CrossRef] [Green Version]
- Hashimshony, T.; Wagner, F.; Sher, N.; Yanai, I. CEL-Seq: Single-Cell RNA-Seq by Multiplexed Linear Amplification. Cell Rep. 2012, 2, 666–673. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 2010, 26, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sha, Y.; Phan, J.H.; Wang, M.D. Effect of low-expression gene filtering on detection of differentially expressed genes in RNA-seq data. In Proceedings of the 2015 37th Annual International Conference of the IEEE Engineering in Medicine and Biology Society (EMBC), Milano, Italy, 25–29 August 2015; pp. 6461–6464. [Google Scholar] [CrossRef] [Green Version]
- Hoffman, G.E.; Roussos, P. Dream: Powerful Differential Expression Analysis for Repeated Measures Designs. Bioinformatics 2021, 37, 192–201. [Google Scholar] [CrossRef] [PubMed]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Blighe, K.; Rana, S.; Lewis, M. EnhancedVolcano: Publication-Ready Volcano Plots with Enhanced Colouring and Labeling. R Package Version 1.12.0. Available online: https://github.com/kevinblighe/EnhancedVolcano (accessed on 1 January 2022).
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- Botía, J.A.; the United Kingdom Brain Expression Consortium; Vandrovcova, J.; Forabosco, P.; Guelfi, S.; D’Sa, K.; Hardy, J.; Lewis, C.M.; Ryten, M.; Weale, M.E. An additional k-means clustering step improves the biological features of WGCNA gene co-expression networks. BMC Syst. Biol. 2017, 11, 47. [Google Scholar] [CrossRef]
- Langfelder, P.; Horvath, S. WGCNA: An R package for weighted correlation network analysis. BMC Bioinform. 2008, 9, 559. [Google Scholar] [CrossRef] [Green Version]
- Ikeda, M.; Kanao, Y.; Yamanaka, M.; Sakuraba, H.; Mizutani, Y.; Igarashi, Y.; Kihara, A. Characterization of four mammalian 3-hydroxyacyl-CoA dehydratases involved in very long-chain fatty acid synthesis. FEBS Lett. 2008, 582, 2435–2440. [Google Scholar] [CrossRef] [Green Version]
- Erbil, S.; Oral, O.; Mitou, G.; Kig, C.; Timucin, E.; Maiorov, E.G.; Gulacti, F.; Gokce, G.; Dengjel, J.; Sezerman, O.U.; et al. RACK1 Is an Interaction Partner of ATG5 and a Novel Regulator of Autophagy. J. Biol. Chem. 2016, 291, 16753–16765. [Google Scholar] [CrossRef] [Green Version]
- Morita, M.; Gravel, S.-P.; Hulea, L.; Larsson, O.; Pollak, M.; St-Pierre, J.; Topisirovic, I. mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell Cycle 2015, 14, 473–480. [Google Scholar] [CrossRef] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976, Erratum in Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [Green Version]
- Magnuson, B.; Ekim, B.; Fingar, D.C. Regulation and function of ribosomal protein S6 kinase (S6K) within mTOR signalling networks. Biochem. J. 2011, 441, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tavares, M.R.; Pavan, I.C.; Amaral, C.L.; Meneguello, L.; Luchessi, A.D.; Simabuco, F.M. The S6K protein family in health and disease. Life Sci. 2015, 131, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, S.F.; Maag, D.; Maxwell, M.J.; Resnick, A.C.; Juluri, K.R.; Chakraborty, A.; Koldobskiy, M.A.; Cha, S.H.; Barrow, R.; et al. Amino Acid Signaling to mTOR Mediated by Inositol Polyphosphate Multikinase. Cell Metab. 2011, 13, 215–221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghu, P.; Joseph, A.; Krishnan, H.; Singh, P.; Saha, S. Phosphoinositides: Regulators of Nervous System Function in Health and Disease. Front. Mol. Neurosci. 2019, 12, 208. [Google Scholar] [CrossRef] [Green Version]
- Wengrod, J.; Wang, D.; Weiss, S.; Zhong, H.; Osman, I.; Gardner, L.B. Phosphorylation of eIF2α triggered by mTORC1 inhibition and PP6C activation is required for autophagy and is aberrant in PP6C-mutated melanoma. Sci. Signal. 2015, 8, ra27. [Google Scholar] [CrossRef] [Green Version]
- Misra, J.; Holmes, M.J.; Mirek, E.T.; Langevin, M.; Kim, H.-G.; Carlson, K.R.; Watford, M.; Dong, X.C.; Anthony, T.G.; Wek, R.C. Discordant regulation of eIF2 kinase GCN2 and mTORC1 during nutrient stress. Nucleic Acids Res. 2021, 49, 5726–5742. [Google Scholar] [CrossRef]
- Klann, K.; Tascher, G.; Münch, C. Functional Translatome Proteomics Reveal Converging and Dose-Dependent Regulation by mTORC1 and eIF2α. Mol. Cell 2019, 77, 913–925.e4. [Google Scholar] [CrossRef] [Green Version]
- Parveen, S.; Parthasarathy, H.; Vedagiri, D.; Gupta, D.; Nair, H.G.; Harshan, K.H. Regulation of EIF2α Phosphorylation by MAPKs Influences Polysome Stability and Protein Translation. bioRxiv 2021. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.-K.; Noda, M.; Verkhratsky, A. Physiology of Microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Parpura, V.; Vardjan, N.; Zorec, R. Physiology of Astroglia. Adv. Exp. Med. Biol. 2019, 1175, 45–91. [Google Scholar] [CrossRef]
- Mills, J.D.; Iyer, A.M.; Van Scheppingen, J.; Bongaarts, A.; Anink, J.J.; Janssen, B.; Zimmer, T.S.; Spliet, W.G.; Van Rijen, P.C.; Jansen, F.E.; et al. Coding and small non-coding transcriptional landscape of tuberous sclerosis complex cortical tubers: Implications for pathophysiology and treatment. Sci. Rep. 2017, 7, 8089. [Google Scholar] [CrossRef] [PubMed]
- Levine, B.; Mizushima, N.; Virgin, H.W. Autophagy in immunity and inflammation. Nature 2011, 469, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Stephenson, E.L.; Yong, V.W. Pro-inflammatory roles of chondroitin sulfate proteoglycans in disorders of the central nervous system. Matrix Biol. 2018, 71, 432–442. [Google Scholar] [CrossRef] [PubMed]
- Chadha, R.; Alganem, K.; Mccullumsmith, R.E.; Meador-Woodruff, J.H. mTOR kinase activity disrupts a phosphorylation signaling network in schizophrenia brain. Mol. Psychiatry 2021, 26, 6868–6879. [Google Scholar] [CrossRef] [PubMed]
- Henske, E.P.; Jozwiak, S.; Kingswood, J.C.; Sampson, J.R.; Thiele, E.A. Tuberous sclerosis complex. Nat. Rev. Dis. Primers 2016, 2, 16035. [Google Scholar] [CrossRef]
- Bhaduri, A.; Andrews, M.G.; Leon, W.M.; Jung, D.; Shin, D.; Allen, D.; Jung, D.; Schmunk, G.; Haeussler, M.; Salma, J.; et al. Cell stress in cortical organoids impairs molecular subtype specification. Nature 2020, 578, 142–148. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Module | Annotation | Increased Expression | Decreased Expression | ||
|---|---|---|---|---|---|
| Enrichment | q-Value | Enrichment | q-Value | ||
| Blue | DNA replication | 100/883 | 1.03 × 10−21 | ||
| Cyan | EIF2 and mTOR signaling + proliferative precursors | 46/382 | 6.44 × 10−11 | ||
| Dark green | Interleukin signaling | 54/689 | 1.62 × 10−10 | ||
| Dark grey | Neuroinflammation | 77/620 | 5.04 × 10−16 | ||
| Dark orange | Undetermined | 85/571 | 1.09 × 10−18 | ||
| Light cyan | Microglia | 114/827 | 2.88 × 10−6 | ||
| Orange | Astrocyte | 152/801 | 3.5 × 10−41 | ||
| Tan | Inositol signaling | 89/545 | 3.29 × 10−19 | ||
| White | Undetermined | 72/890 | 1.04 × 10−8 | ||
| Yellow | Mitochondrial (dys)function | 171/853 | 7.2 × 10−50 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Berdenis van Berlekom, A.; Kübler, R.; Hoogeboom, J.W.; Vonk, D.; Sluijs, J.A.; Pasterkamp, R.J.; Middeldorp, J.; Kraneveld, A.D.; Garssen, J.; Kahn, R.S.; et al. Exposure to the Amino Acids Histidine, Lysine, and Threonine Reduces mTOR Activity and Affects Neurodevelopment in a Human Cerebral Organoid Model. Nutrients 2022, 14, 2175. https://doi.org/10.3390/nu14102175
Berdenis van Berlekom A, Kübler R, Hoogeboom JW, Vonk D, Sluijs JA, Pasterkamp RJ, Middeldorp J, Kraneveld AD, Garssen J, Kahn RS, et al. Exposure to the Amino Acids Histidine, Lysine, and Threonine Reduces mTOR Activity and Affects Neurodevelopment in a Human Cerebral Organoid Model. Nutrients. 2022; 14(10):2175. https://doi.org/10.3390/nu14102175
Chicago/Turabian StyleBerdenis van Berlekom, Amber, Raphael Kübler, Jeske W. Hoogeboom, Daniëlle Vonk, Jacqueline A. Sluijs, R. Jeroen Pasterkamp, Jinte Middeldorp, Aletta D. Kraneveld, Johan Garssen, René S. Kahn, and et al. 2022. "Exposure to the Amino Acids Histidine, Lysine, and Threonine Reduces mTOR Activity and Affects Neurodevelopment in a Human Cerebral Organoid Model" Nutrients 14, no. 10: 2175. https://doi.org/10.3390/nu14102175
APA StyleBerdenis van Berlekom, A., Kübler, R., Hoogeboom, J. W., Vonk, D., Sluijs, J. A., Pasterkamp, R. J., Middeldorp, J., Kraneveld, A. D., Garssen, J., Kahn, R. S., Hol, E. M., de Witte, L. D., & Boks, M. P. (2022). Exposure to the Amino Acids Histidine, Lysine, and Threonine Reduces mTOR Activity and Affects Neurodevelopment in a Human Cerebral Organoid Model. Nutrients, 14(10), 2175. https://doi.org/10.3390/nu14102175