
Beneficial Effects of Exogenous Ketogenic Supplements on Aging Processes and Age-Related Neurodegenerative Diseases




Abstract
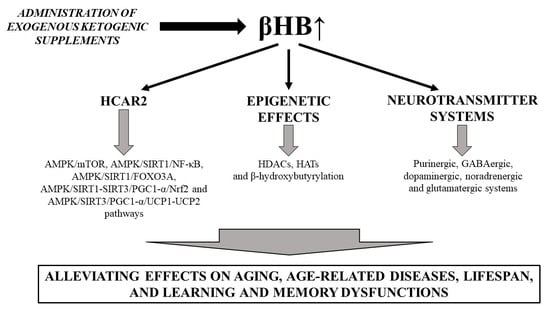
1. Introduction
2. Main Features of Aging Processes
2.1. Nutrient Sensing Pathways
2.2. Telomere Shortening and Genome Instability
2.3. Epigenetic Alterations
2.4. Mitochondrial Dysfunction
2.5. Altered Intercellular Communication: Increased Inflammatory Processes
2.6. Cellular Senescence
2.7. Loss of Proteostasis and Stem Cell Exhaustion
2.8. Effects of Senotherapeutic Drugs on Aging Hallmarks and Neurodegenerative Diseases: Main Signaling Pathways
3. Alleviating Effects of Ketosis on Lifespan, Aging and Age-Related Neurodegenerative Diseases
3.1. Ketosis-Evoked Neuroprotective Effects and Downstream Signaling Pathways
3.2. Beneficial Effects of EKSs-Evoked Ketosis (βHB) on Lifespan, Aging, Age-Related Diseases, as Well as Learning and Memory Dysfunctions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Campisi, J.; Kapahi, P.; Lithgow, G.J.; Melov, S.; Newman, J.C.; Verdin, E. From discoveries in ageing research to therapeutics for healthy ageing. Nature 2019, 571, 183–192. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Zhang, Z.; Ren, Y.; Wang, Y.; Fang, J.; Yue, H.; Ma, S.; Guan, F. Aging and age-related diseases: From mechanisms to therapeutic strategies. Biogerontology 2021, 22, 165–187. [Google Scholar] [CrossRef] [PubMed]
- Sen, A.; Capelli, V.; Husain, M. Cognition and dementia in older patients with epilepsy. Brain 2018, 141, 1592–1608. [Google Scholar] [CrossRef]
- United Nations, Department of Economic and Social Affairs, Population Division. World Population Ageing 2019: Highlights; United Nations: New York, NY, USA, 2019; ISBN 978-92-1-148325-3. [Google Scholar]
- Drygalski, K.; Fereniec, E.; Koryciński, K.; Chomentowski, A.; Kiełczewska, A.; Odrzygóźdź, C.; Modzelewska, B. Resveratrol and Alzheimer’s disease. From molecular pathophysiology to clinical trials. Exp. Gerontol. 2018, 113, 36–47. [Google Scholar] [CrossRef]
- Yang, C.; Zhang, W.; Dong, X.; Fu, C.; Yuan, J.; Xu, M.; Liang, Z.; Qiu, C.; Xu, C. A natural product solution to aging and aging-associated diseases. Pharmacol. Ther. 2020, 216, 107673. [Google Scholar] [CrossRef] [PubMed]
- De Magalhães, J.P.; Stevens, M.; Thornton, D. The Business of Anti-Aging Science. Trends Biotechnol. 2017, 35, 1062–1073. [Google Scholar] [CrossRef] [PubMed]
- Anisimov, V.N.; Zabezhinski, M.A.; Popovich, I.G.; Piskunova, T.S.; Semenchenko, A.V.; Tyndyk, M.L.; Yurova, M.N.; Rosenfeld, S.V.; Blagosklonny, M.V. Rapamycin increases lifespan and inhibits spontaneous tumorigenesis in inbred female mice. Cell Cycle 2011, 10, 4230–4236. [Google Scholar] [CrossRef] [PubMed]
- Bitto, A.; Ito, T.K.; Pineda, V.V.; LeTexier, N.J.; Huang, H.Z.; Sutlief, E.; Tung, H.; Vizzini, N.; Chen, B.; Smith, K.; et al. Transient rapamycin treatment can increase lifespan and healthspan in middle-aged mice. Elife 2016, 5, e16351. [Google Scholar] [CrossRef]
- Mannick, J.B.; Morris, M.; Hockey, H.P.; Roma, G.; Beibel, M.; Kulmatycki, K.; Watkins, M.; Shavlakadze, T.; Zhou, W.; Quinn, D.; et al. TORC1 inhibition enhances immune function and reduces infections in the elderly. Sci. Transl. Med. 2018, 10, eaaq1564. [Google Scholar] [CrossRef] [PubMed]
- Bannister, C.A.; Holden, S.E.; Jenkins-Jones, S.; Morgan, C.L.; Halcox, J.P.; Schernthaner, G.; Mukherjee, J.; Currie, C.J. Can people with type 2 diabetes live longer than those without? A comparison of mortality in people initiated with metformin or sulphonylurea monotherapy and matched, non-diabetic controls. Diabetes Obes. Metab. 2014, 16, 1165–1173. [Google Scholar] [CrossRef]
- Barzilai, N.; Crandall, J.P.; Kritchevsky, S.B.; Espeland, M.A. Metformin as a Tool to Target Aging. Cell Metab. 2016, 23, 1060–1065. [Google Scholar] [CrossRef] [PubMed]
- Bonkowski, M.S.; Sinclair, D.A. Slowing ageing by design: The rise of NAD+ and sirtuin-activating compounds. Nat. Rev. Mol. Cell Biol. 2016, 17, 679–690. [Google Scholar] [CrossRef]
- Kane, A.E.; Sinclair, D.A. Sirtuins and NAD+ in the Development and Treatment of Metabolic and Cardiovascular Diseases. Circ. Res. 2018, 123, 868–885. [Google Scholar] [CrossRef] [PubMed]
- Kirkland, J.L.; Tchkonia, T.; Zhu, Y.; Niedernhofer, L.J.; Robbins, P.D. The Clinical Potential of Senolytic Drugs. J. Am. Geriatr. Soc. 2017, 65, 2297–2301. [Google Scholar] [CrossRef] [PubMed]
- Brownlow, M.L.; Jung, S.H.; Moore, R.J.; Bechmann, N.; Jankord, R. Nutritional Ketosis Affects Metabolism and Behavior in Sprague-Dawley Rats in Both Control and Chronic Stress Environments. Front. Mol. Neurosci. 2017, 10, 129. [Google Scholar] [CrossRef]
- Brunengraber, H. Potential of ketone body esters for parenteral and oral nutrition. Nutrition 1997, 13, 233–235. [Google Scholar] [CrossRef]
- Clarke, K.; Tchabanenko, K.; Pawlosky, R.; Carter, E.; Knight, N.S.; Murray, A.J.; Cochlin, L.E.; King, M.T.; Wong, A.W.; Roberts, A.; et al. Oral 28-day and developmental toxicity studies of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate. Regul. Toxicol. Pharmacol. 2012, 63, 196–208. [Google Scholar] [CrossRef]
- Clarke, K.; Tchabanenko, K.; Pawlosky, R.; Carter, E.; Todd King, M.; Musa-Veloso, K.; Ho, M.; Roberts, A.; Robertson, J.; Vanitallie, T.B.; et al. Kinetics, safety and tolerability of (R)-3-hydroxybutyl (R)-3-hydroxybutyrate in healthy adult subjects. Regul. Toxicol. Pharmacol. 2012, 63, 401–408. [Google Scholar] [CrossRef]
- Schönfeld, P.; Wojtczak, L. Short- and medium-chain fatty acids in energy metabolism: The cellular perspective. J. Lipid Res. 2016, 57, 943–954. [Google Scholar] [CrossRef]
- Kovács, Z.; Brunner, B.; D’Agostino, D.P.; Ari, C. Age- and Sex-Dependent Modulation of Exogenous Ketone Supplement-Evoked Effects on Blood Glucose and Ketone Body Levels in Wistar Albino Glaxo Rijswijk Rats. Front. Neurosci. 2021, 14, 618422. [Google Scholar] [CrossRef]
- Achanta, L.B.; Rae, C.D. β-Hydroxybutyrate in the Brain: One Molecule, Multiple Mechanisms. Neurochem. Res. 2017, 42, 35–49. [Google Scholar] [CrossRef]
- Koppel, S.J.; Swerdlow, R.H. Neuroketotherapeutics: A modern review of a century-old therapy. Neurochem. Int. 2018, 117, 114–125. [Google Scholar] [CrossRef] [PubMed]
- Newman, J.C.; Verdin, E. Ketone bodies as signaling metabolites. Trends Endocrinol. Metab. 2014, 25, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Soto-Mota, A.; Norwitz, N.G.; Clarke, K. Why a d-β-hydroxybutyrate monoester? Biochem. Soc. Trans. 2020, 48, 51–59. [Google Scholar] [CrossRef]
- Ari, C.; Kovács, Z.; Juhasz, G.; Murdun, C.; Goldhagen, C.R.; Koutnik, A.P.; Poff, A.M.; Kesl, S.L.; D’Agostino, D.P. Exogenous Ketone Supplements Reduce Anxiety-Related Behavior in Sprague-Dawley and Wistar Albino Glaxo/Rijswijk Rats. Front. Mol. Neurosci. 2016, 9, 137. [Google Scholar] [CrossRef]
- Kesl, S.L.; Poff, A.M.; Ward, N.P.; Fiorelli, T.N.; Ari, C.; Van Putten, A.J.; Sherwood, J.W.; Arnold, P.; D’Agostino, D.P. Effects of exogenous ketone supplementation on blood ketone, glucose, triglyceride, and lipoprotein levels in Sprague-Dawley rats. Nutr. Metab. 2016, 13, 9. [Google Scholar] [CrossRef]
- Myette-Côté, É.; Neudorf, H.; Rafiei, H.; Clarke, K.; Little, J.P. Prior ingestion of exogenous ketone monoester attenuates the glycaemic response to an oral glucose tolerance test in healthy young individuals. J. Physiol. 2018, 596, 1385–1395. [Google Scholar] [CrossRef] [PubMed]
- Stubbs, B.J.; Cox, P.J.; Evans, R.D.; Santer, P.; Miller, J.J.; Faull, O.K.; Magor-Elliott, S.; Hiyama, S.; Stirling, M.; Clarke, K. On the Metabolism of Exogenous Ketones in Humans. Front. Physiol. 2017, 8, 848. [Google Scholar] [CrossRef]
- Hashim, S.A.; VanItallie, T.B. Ketone body therapy: From the ketogenic diet to the oral administration of ketone ester. J. Lipid Res. 2014, 55, 1818–1826. [Google Scholar] [CrossRef]
- McDonald, T.J.; Cervenka, M.C. Lessons learned from recent clinical trials of ketogenic diet therapies in adults. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 418–424. [Google Scholar] [CrossRef]
- Fortier, M.; Castellano, C.A.; St-Pierre, V.; Myette-Côté, É.; Langlois, F.; Roy, M.; Morin, M.C.; Bocti, C.; Fulop, T.; Godin, J.P.; et al. A ketogenic drink improves cognition in mild cognitive impairment: Results of a 6-month RCT. Alzheimers Dement. 2021, 17, 543–552. [Google Scholar] [CrossRef]
- Soto-Mota, A.; Norwitz, N.G.; Evans, R.; Clarke, K.; Barber, T.M. Exogenous ketosis in patients with type 2 diabetes: Safety, tolerability and effect on glycaemic control. Endocrinol. Diabetes Metab. 2021, e00264. [Google Scholar] [CrossRef]
- Camberos-Luna, L.; Massieu, L. Therapeutic strategies for ketosis induction and their potential efficacy for the treatment of acute brain injury and neurodegenerative diseases. Neurochem. Int. 2020, 133, 104614. [Google Scholar] [CrossRef]
- Han, Y.M.; Ramprasath, T.; Zou, M.H. β-hydroxybutyrate and its metabolic effects on age-associated pathology. Exp. Mol. Med. 2020, 52, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Kovács, Z.; D’Agostino, D.P.; Dobolyi, A.; Ari, C. Adenosine A1 Receptor Antagonism Abolished the Anti-seizure Effects of Exogenous Ketone Supplementation in Wistar Albino Glaxo Rijswijk Rats. Front. Mol. Neurosci. 2017, 10, 235. [Google Scholar] [CrossRef]
- Branco, A.F.; Ferreira, A.; Simões, R.F.; Magalhães-Novais, S.; Zehowski, C.; Cope, E.; Silva, A.M.; Pereira, D.; Sardão, V.A.; Cunha-Oliveira, T. Ketogenic diets: From cancer to mitochondrial diseases and beyond. Eur. J. Clin. Investig. 2016, 46, 285–298. [Google Scholar] [CrossRef]
- Kim, D.Y.; Simeone, K.A.; Simeone, T.A.; Pandya, J.D.; Wilke, J.C.; Ahn, Y.; Geddes, J.W.; Sullivan, P.G.; Rho, J.M. Ketone bodies mediate antiseizure effects through mitochondrial permeability transition. Ann. Neurol. 2015, 78, 77–87. [Google Scholar] [CrossRef] [PubMed]
- Kovács, Z.; D’Agostino, D.P.; Diamond, D.; Kindy, M.S.; Rogers, C.; Ari, C. Therapeutic Potential of Exogenous Ketone Supplement Induced Ketosis in the Treatment of Psychiatric Disorders: Review of Current Literature. Front. Psychiatry 2019, 10, 363. [Google Scholar] [CrossRef]
- Berk, B.A.; Law, T.H.; Packer, R.M.A.; Wessmann, A.; Bathen-Nöthen, A.; Jokinen, T.S.; Knebel, A.; Tipold, A.; Pelligand, L.; Meads, Z.; et al. A multicenter randomized controlled trial of medium-chain triglyceride dietary supplementation on epilepsy in dogs. J. Vet. Intern. Med. 2020, 34, 1248–1259. [Google Scholar] [CrossRef]
- Ciarlone, S.L.; Grieco, J.C.; D’Agostino, D.P.; Weeber, E.J. Ketone ester supplementation attenuates seizure activity, and improves behavior and hippocampal synaptic plasticity in an Angelman syndrome mouse model. Neurobiol. Dis. 2016, 96, 38–46. [Google Scholar] [CrossRef]
- D’Agostino, D.P.; Pilla, R.; Held, H.E.; Landon, C.S.; Puchowicz, M.; Brunengraber, H.; Ari, C.; Arnold, P.; Dean, J.B. Therapeutic ketosis with ketone ester delays central nervous system oxygen toxicity seizures in rats. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2013, 304, 829–836. [Google Scholar] [CrossRef]
- Kashiwaya, Y.; Bergman, C.; Lee, J.H.; Wan, R.; King, M.T.; Mughal, M.R.; Okun, E.; Clarke, K.; Mattson, M.P.; Veech, R.L. A ketone ester diet exhibits anxiolytic and cognition-sparing properties, and lessens amyloid and tau pathologies in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1530–1539. [Google Scholar] [CrossRef]
- Kovács, Z.; D’Agostino, D.P.; Ari, C. Anxiolytic Effect of Exogenous Ketone Supplementation Is Abolished by Adenosine A1 Receptor Inhibition in Wistar Albino Glaxo/Rijswijk Rats. Front. Behav. Neurosci. 2018, 12, 29. [Google Scholar] [CrossRef]
- Ari, C.; Zippert, M.; D’Agostino, D.P. Neuroregeneration improved by ketones. FASEB J. 2018, 32, 545–549. [Google Scholar] [CrossRef]
- Ari, C.; Pilla, R.; D’Agostino, D. Nutritional/metabolic therapies in animal models of amyotrophic lateral sclerosis, Alzheimer’s disease, and seizures. In Bioactive Nutraceuticals and Dietary Supplements in Neurological and Brain Disease; Ross Watson, R., Preedy, V.R., Eds.; Academic Press: New York, NY, USA, 2015; pp. 449–459. ISBN 978-0-12-411462-3. [Google Scholar]
- Newport, M.T.; VanItallie, T.B.; Kashiwaya, Y.; King, M.T.; Veech, R.L. A new way to produce hyperketonemia: Use of ketone ester in a case of Alzheimer’s disease. Alzheimers Dement. 2015, 11, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Tefera, T.W.; Wong, Y.; Barkl-Luke, M.E.; Ngo, S.T.; Thomas, N.K.; McDonald, T.S.; Borges, K. Triheptanoin Protects Motor Neurons and Delays the Onset of Motor Symptoms in a Mouse Model of Amyotrophic Lateral Sclerosis. PLoS ONE 2016, 11, e0161816. [Google Scholar] [CrossRef]
- Maalouf, M.; Rho, J.M.; Mattson, M.P. The neuroprotective properties of calorie restriction, the ketogenic diet, and ketone bodies. Brain Res. Rev. 2009, 59, 293–315. [Google Scholar] [CrossRef]
- Wu, Y.; Gong, Y.; Luan, Y.; Li, Y.; Liu, J.; Yue, Z.; Yuan, B.; Sun, J.; Xie, C.; Li, L.; et al. BHBA treatment improves cognitive function by targeting pleiotropic mechanisms in transgenic mouse model of Alzheimer’s disease. FASEB J. 2020, 34, 1412–1429. [Google Scholar] [CrossRef]
- Roberts, M.N.; Wallace, M.A.; Tomilov, A.A.; Zhou, Z.; Marcotte, G.R.; Tran, D.; Perez, G.; Gutierrez-Casado, E.; Koike, S.; Knotts, T.A.; et al. A Ketogenic Diet Extends Longevity and Healthspan in Adult Mice. Cell Metab. 2017, 26, 539–546. [Google Scholar] [CrossRef]
- Veech, R.L.; Bradshaw, P.C.; Clarke, K.; Curtis, W.; Pawlosky, R.; King, M.T. Ketone bodies mimic the life span extending properties of caloric restriction. IUBMB Life 2017, 69, 305–314. [Google Scholar] [CrossRef]
- Newman, J.C.; Verdin, E. β-hydroxybutyrate: Much more than a metabolite. Diabetes Res. Clin. Pract. 2014, 106, 173–181. [Google Scholar] [CrossRef]
- Rahman, M.; Muhammad, S.; Khan, M.A.; Chen, H.; Ridder, D.A.; Müller-Fielitz, H.; Pokorná, B.; Vollbrandt, T.; Stölting, I.; Nadrowitz, R.; et al. The β-hydroxybutyrate receptor HCA2 activates a neuroprotective subset of macrophages. Nat. Commun. 2014, 5, 3944. [Google Scholar] [CrossRef]
- Fu, S.P.; Wang, J.F.; Xue, W.J.; Liu, H.M.; Liu, B.R.; Zeng, Y.L.; Li, S.N.; Huang, B.X.; Lv, Q.K.; Wang, W.; et al. Anti-inflammatory effects of BHBA in both in vivo and in vitro Parkinson’s disease models are mediated by GPR109A-dependent mechanisms. J. Neuroinflamm. 2015, 12, 9. [Google Scholar] [CrossRef]
- Rezq, S.; Abdel-Rahman, A.A. Central GPR109A Activation Mediates Glutamate-Dependent Pressor Response in Conscious Rats. J. Pharmacol. Exp. Ther. 2016, 356, 456–465. [Google Scholar] [CrossRef]
- Graff, E.C.; Fang, H.; Wanders, D.; Judd, R.L. Anti-inflammatory effects of the hydroxycarboxylic acid receptor 2. Metabolism 2016, 65, 102–113. [Google Scholar] [CrossRef]
- Wakade, C.; Chong, R.; Bradley, E.; Thomas, B.; Morgan, J. Upregulation of GPR109A in Parkinson’s disease. PLoS ONE 2014, 9, e109818. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.M.; Bedarida, T.; Ding, Y.; Somba, B.K.; Lu, Q.; Wang, Q.; Song, P.; Zou, M.H. β-Hydroxybutyrate Prevents Vascular Senescence through hnRNP A1-Mediated Upregulation of Oct4. Mol. Cell 2018, 71, 1064–1078. [Google Scholar] [CrossRef] [PubMed]
- Edwards, C.; Canfield, J.; Copes, N.; Rehan, M.; Lipps, D.; Bradshaw, P.C. D-beta-hydroxybutyrate extends lifespan in C. elegans. Aging 2014, 6, 621–644. [Google Scholar] [CrossRef]
- Trevisan, K.; Cristina-Pereira, R.; Silva-Amaral, D.; Aversi-Ferreira, T.A. Theories of Aging and the Prevalence of Alzheimer’s Disease. Biomed. Res. Int. 2019, 2019, 9171424. [Google Scholar] [CrossRef] [PubMed]
- Ascherio, A.; Schwarzschild, M.A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 2016, 15, 1257–1272. [Google Scholar] [CrossRef]
- Hou, Y.; Dan, X.; Babbar, M.; Wei, Y.; Hasselbalch, S.G.; Croteau, D.L.; Bohr, V.A. Ageing as a risk factor for neurodegenerative disease. Nat. Rev. Neurol. 2019, 15, 565–581. [Google Scholar] [CrossRef] [PubMed]
- Pandya, V.A.; Patani, R. Decoding the relationship between ageing and amyotrophic lateral sclerosis: A cellular perspective. Brain 2020, 143, 1057–1072. [Google Scholar] [CrossRef]
- Broughton, S.; Partridge, L. Insulin/IGF-like signalling, the central nervous system and aging. Biochem. J. 2009, 418, 1–12. [Google Scholar] [CrossRef]
- Klement, R.J.; Fink, M.K. Dietary and pharmacological modification of the insulin/IGF-1 system: Exploiting the full repertoire against cancer. Oncogenesis 2016, 5, e193. [Google Scholar] [CrossRef]
- Martin, B.; Mattson, M.P.; Maudsley, S. Caloric restriction and intermittent fasting: Two potential diets for successful brain aging. Ageing Res. Rev. 2006, 5, 332–353. [Google Scholar] [CrossRef]
- Santos, J.; Leitão-Correia, F.; Sousa, M.J.; Leão, C. Dietary Restriction and Nutrient Balance in Aging. Oxid. Med. Cell Longev. 2016, 2016, 4010357. [Google Scholar] [CrossRef] [PubMed]
- López-Otín, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef] [PubMed]
- Nugent, S.; Tremblay, S.; Chen, K.W.; Ayutyanont, N.; Roontiva, A.; Castellano, C.A.; Fortier, M.; Roy, M.; Courchesne-Loyer, A.; Bocti, C.; et al. Brain glucose and acetoacetate metabolism: A comparison of young and older adults. Neurobiol. Aging 2014, 35, 1386–1395. [Google Scholar] [CrossRef] [PubMed]
- Schultz, M.B.; Sinclair, D.A. When stem cells grow old: Phenotypes and mechanisms of stem cell aging. Development 2016, 143, 3–14. [Google Scholar] [CrossRef]
- Mathew, R.; Pal Bhadra, M.; Bhadra, U. Insulin/insulin-like growth factor-1 signalling (IIS) based regulation of lifespan across species. Biogerontology 2017, 18, 35–53. [Google Scholar] [CrossRef]
- Cohen, E.; Bieschke, J.; Perciavalle, R.M.; Kelly, J.W.; Dillin, A. Opposing activities protect against age-onset proteotoxicity. Science 2006, 313, 1604–1610. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kim, S.G.; Blenis, J. Rapamycin: One drug, many effects. Cell Metab. 2014, 19, 373–379. [Google Scholar] [CrossRef] [PubMed]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 168, 960–976. [Google Scholar] [CrossRef] [PubMed]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Menzies, F.M.; Fleming, A.; Caricasole, A.; Bento, C.F.; Andrews, S.P.; Ashkenazi, A.; Füllgrabe, J.; Jackson, A.; Jimenez Sanchez, M.; Karabiyik, C.; et al. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron 2017, 93, 1015–1034. [Google Scholar] [CrossRef] [PubMed]
- Sen, P.; Shah, P.P.; Nativio, R.; Berger, S.L. Epigenetic Mechanisms of Longevity and Aging. Cell 2016, 166, 822–839. [Google Scholar] [CrossRef]
- Carling, D. The AMP-activated protein kinase cascade—A unifying system for energy control. Trends Biochem. Sci. 2004, 29, 18–24. [Google Scholar] [CrossRef]
- Steinberg, G.R.; Kemp, B.E. AMPK in Health and Disease. Physiol. Rev. 2009, 89, 1025–1078. [Google Scholar] [CrossRef]
- Chung, M.M.; Nicol, C.J.; Cheng, Y.C.; Lin, K.H.; Chen, Y.L.; Pei, D.; Lin, C.H.; Shih, Y.N.; Yen, C.H.; Chen, S.J.; et al. Metformin activation of AMPK suppresses AGE-induced inflammatory response in hNSCs. Exp. Cell Res. 2017, 352, 75–83. [Google Scholar] [CrossRef]
- Salminen, A.; Hyttinen, J.M.; Kaarniranta, K. AMP-activated protein kinase inhibits NF-κB signaling and inflammation: Impact on healthspan and lifespan. J. Mol. Med. 2011, 89, 667–676. [Google Scholar] [CrossRef]
- Cantó, C.; Gerhart-Hines, Z.; Feige, J.N.; Lagouge, M.; Noriega, L.; Milne, J.C.; Elliott, P.J.; Puigserver, P.; Auwerx, J. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature 2009, 458, 1056–1060. [Google Scholar] [CrossRef]
- Kapahi, P.; Kaeberlein, M.; Hansen, M. Dietary restriction and lifespan: Lessons from invertebrate models. Ageing Res. Rev. 2017, 39, 3–14. [Google Scholar] [CrossRef]
- He, W.; Newman, J.C.; Wang, M.Z.; Ho, L.; Verdin, E. Mitochondrial sirtuins: Regulators of protein acylation and metabolism. Trends Endocrinol. Metab. 2012, 23, 467–476. [Google Scholar] [CrossRef]
- Gillum, M.P.; Kotas, M.E.; Erion, D.M.; Kursawe, R.; Chatterjee, P.; Nead, K.T.; Muise, E.S.; Hsiao, J.J.; Frederick, D.W.; Yonemitsu, S.; et al. SirT1 regulates adipose tissue inflammation. Diabetes 2011, 60, 3235–3245. [Google Scholar] [CrossRef]
- Rodgers, J.T.; Lerin, C.; Haas, W.; Gygi, S.P.; Spiegelman, B.M.; Puigserver, P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature 2005, 434, 113–118. [Google Scholar] [CrossRef]
- Imai, S.; Guarente, L. NAD+ and sirtuins in aging and disease. Trends Cell Biol. 2014, 24, 464–471. [Google Scholar] [CrossRef] [PubMed]
- Morigi, M.; Perico, L.; Benigni, A. Sirtuins in Renal Health and Disease. J. Am. Soc. Nephrol. 2018, 29, 1799–1809. [Google Scholar] [CrossRef]
- Wątroba, M.; Dudek, I.; Skoda, M.; Stangret, A.; Rzodkiewicz, P.; Szukiewicz, D. Sirtuins, epigenetics and longevity. Ageing Res. Rev. 2017, 40, 11–19. [Google Scholar] [CrossRef]
- Cho, S.H.; Chen, J.A.; Sayed, F.; Ward, M.E.; Gao, F.; Nguyen, T.A.; Krabbe, G.; Sohn, P.D.; Lo, I.; Minami, S.; et al. SIRT1 deficiency in microglia contributes to cognitive decline in aging and neurodegeneration via epigenetic regulation of IL-1β. J. Neurosci. 2015, 35, 807–818. [Google Scholar] [CrossRef] [PubMed]
- Patel, N.V.; Gordon, M.N.; Connor, K.E.; Good, R.A.; Engelman, R.W.; Mason, J.; Morgan, D.G.; Morgan, T.E.; Finch, C.E. Caloric restriction attenuates Abeta-deposition in Alzheimer transgenic models. Neurobiol. Aging 2005, 26, 995–1000. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Fivecoat, H.; Ho, L.; Pan, Y.; Ling, E.; Pasinetti, G.M. The role of Sirt1: At the crossroad between promotion of longevity and protection against Alzheimer’s disease neuropathology. Biochim. Biophys. Acta 2010, 1804, 1690–1694. [Google Scholar] [CrossRef]
- Kim, D.; Nguyen, M.D.; Dobbin, M.M.; Fischer, A.; Sananbenesi, F.; Rodgers, J.T.; Delalle, I.; Baur, J.A.; Sui, G.; Armour, S.M.; et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007, 26, 3169–3179. [Google Scholar] [CrossRef] [PubMed]
- Marambaud, P.; Zhao, H.; Davies, P. Resveratrol promotes clearance of Alzheimer’s disease amyloid-beta peptides. J. Biol. Chem. 2005, 280, 37377–37382. [Google Scholar] [CrossRef]
- Luchsinger, J.A.; Tang, M.X.; Shea, S.; Mayeux, R. Caloric intake and the risk of Alzheimer disease. Arch. Neurol. 2002, 59, 1258–1263. [Google Scholar] [CrossRef] [PubMed]
- Julien, C.; Tremblay, C.; Emond, V.; Lebbadi, M.; Salem, N., Jr.; Bennett, D.A.; Calon, F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Oosterhof, N.; Dekens, D.W.; Lawerman, T.F.; van Dijk, M. Yet another role for SIRT1: Reduction of α-synuclein aggregation in stressed neurons. J. Neurosci. 2012, 32, 6413–6414. [Google Scholar] [CrossRef] [PubMed]
- Araki, T.; Sasaki, Y.; Milbrandt, J. Increased nuclear NAD biosynthesis and SIRT1 activation prevent axonal degeneration. Science 2004, 305, 1010–1013. [Google Scholar] [CrossRef]
- Virgili, M.; Contestabile, A. Partial neuroprotection of in vivo excitotoxic brain damage by chronic administration of the red wine antioxidant agent, trans-resveratrol in rats. Neurosci. Lett. 2000, 281, 123–126. [Google Scholar] [CrossRef]
- Brunet, A.; Sweeney, L.B.; Sturgill, J.F.; Chua, K.F.; Greer, P.L.; Lin, Y.; Tran, H.; Ross, S.E.; Mostoslavsky, R.; Cohen, H.Y.; et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004, 303, 2011–2015. [Google Scholar] [CrossRef]
- Zhao, L.; Cao, J.; Hu, K.; He, X.; Yun, D.; Tong, T.; Han, L. Sirtuins and their Biological Relevance in Aging and Age-Related Diseases. Aging Dis. 2020, 11, 927–945. [Google Scholar] [CrossRef]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef]
- Prozorovski, T.; Schulze-Topphoff, U.; Glumm, R.; Baumgart, J.; Schröter, F.; Ninnemann, O.; Siegert, E.; Bendix, I.; Brüstle, O.; Nitsch, R.; et al. Sirt1 contributes critically to the redox-dependent fate of neural progenitors. Nat. Cell Biol. 2008, 10, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Chang, H.C.; Guarente, L. SIRT1 mediates central circadian control in the SCN by a mechanism that decays with aging. Cell 2013, 153, 1448–1460. [Google Scholar] [CrossRef] [PubMed]
- Yaku, K.; Okabe, K.; Nakagawa, T. NAD metabolism: Implications in aging and longevity. Ageing Res. Rev. 2018, 47, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Blüher, M.; Kahn, B.B.; Kahn, C.R. Extended longevity in mice lacking the insulin receptor in adipose tissue. Science 2003, 299, 572–574. [Google Scholar] [CrossRef]
- Kenyon, C.J. The genetics of ageing. Nature 2010, 464, 504–512. [Google Scholar] [CrossRef] [PubMed]
- Greer, E.L.; Brunet, A. Different dietary restriction regimens extend lifespan by both independent and overlapping genetic pathways in C. elegans. Aging Cell 2009, 8, 113–127. [Google Scholar] [CrossRef]
- Zhu, Y.; Liu, X.; Ding, X.; Wang, F.; Geng, X. Telomere and its role in the aging pathways: Telomere shortening, cell senescence and mitochondria dysfunction. Biogerontology 2019, 20, 1–16. [Google Scholar] [CrossRef]
- Herrmann, M.; Pusceddu, I.; März, W.; Herrmann, W. Telomere biology and age-related diseases. Clin. Chem. Lab. Med. 2018, 56, 1210–1222. [Google Scholar] [CrossRef]
- Sahin, E.; DePinho, R.A. Axis of ageing: Telomeres, p53 and mitochondria. Nat. Rev. Mol. Cell Biol. 2012, 13, 397–404. [Google Scholar] [CrossRef]
- Bernardes de Jesus, B.; Vera, E.; Schneeberger, K.; Tejera, A.M.; Ayuso, E.; Bosch, F.; Blasco, M.A. Telomerase gene therapy in adult and old mice delays aging and increases longevity without increasing cancer. EMBO Mol. Med. 2012, 4, 691–704. [Google Scholar] [CrossRef] [PubMed]
- Shay, J.W. Role of Telomeres and Telomerase in Aging and Cancer. Cancer Discov. 2016, 6, 584–593. [Google Scholar] [CrossRef]
- Palacios, J.A.; Herranz, D.; De Bonis, M.L.; Velasco, S.; Serrano, M.; Blasco, M.A. SIRT1 contributes to telomere maintenance and augments global homologous recombination. J. Cell Biol. 2010, 191, 1299–1313. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Wilson, D.M., 3rd. DNA Damage and Associated DNA Repair Defects in Disease and Premature Aging. Am. J. Hum. Genet. 2019, 105, 237–257. [Google Scholar] [CrossRef]
- Foo, M.X.R.; Ong, P.F.; Dreesen, O. Premature aging syndromes: From patients to mechanism. J. Dermatol. Sci. 2019, 96, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Hoeijmakers, J.H. DNA damage, aging, and cancer. N. Engl. J. Med. 2009, 361, 1475–1485. [Google Scholar] [CrossRef] [PubMed]
- Kauppila, T.E.S.; Bratic, A.; Jensen, M.B.; Baggio, F.; Partridge, L.; Jasper, H.; Grönke, S.; Larsson, N.G. Mutations of mitochondrial DNA are not major contributors to aging of fruit flies. Proc. Natl. Acad. Sci. USA 2018, 115, 9620–9629. [Google Scholar] [CrossRef]
- Thanan, R.; Oikawa, S.; Hiraku, Y.; Ohnishi, S.; Ma, N.; Pinlaor, S.; Yongvanit, P.; Kawanishi, S.; Murata, M. Oxidative stress and its significant roles in neurodegenerative diseases and cancer. Int. J. Mol. Sci. 2014, 16, 193–217. [Google Scholar] [CrossRef]
- Feinberg, A.P.; Tycko, B. The history of cancer epigenetics. Nat. Rev. Cancer 2004, 4, 143–153. [Google Scholar] [CrossRef]
- Kane, A.E.; Sinclair, D.A. Epigenetic changes during aging and their reprogramming potential. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 61–83. [Google Scholar] [CrossRef]
- Burzynski, S.R. Aging: Gene silencing or gene activation? Med. Hypotheses 2005, 64, 201–208. [Google Scholar] [CrossRef]
- Ben-Avraham, D.; Muzumdar, R.H.; Atzmon, G. Epigenetic genome-wide association methylation in aging and longevity. Epigenomics 2012, 4, 503–509. [Google Scholar] [CrossRef]
- Benayoun, B.A.; Pollina, E.A.; Brunet, A. Epigenetic regulation of ageing: Linking environmental inputs to genomic stability. Nat. Rev. Mol. Cell Biol. 2015, 16, 593–610. [Google Scholar] [CrossRef]
- Hernandez, D.G.; Nalls, M.A.; Gibbs, J.R.; Arepalli, S.; van der Brug, M.; Chong, S.; Moore, M.; Longo, D.L.; Cookson, M.R.; Traynor, B.J.; et al. Distinct DNA methylation changes highly correlated with chronological age in the human brain. Hum. Mol. Genet. 2011, 20, 1164–1172. [Google Scholar] [CrossRef]
- Waki, T.; Tamura, G.; Sato, M.; Motoyama, T. Age-related methylation of tumor suppressor and tumor-related genes: An analysis of autopsy samples. Oncogene 2003, 22, 4128–4133. [Google Scholar] [CrossRef]
- Casillas, M.A., Jr.; Lopatina, N.; Andrews, L.G.; Tollefsbol, T.O. Transcriptional control of the DNA methyltransferases is altered in aging and neoplastically-transformed human fibroblasts. Mol. Cell. Biochem. 2003, 252, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Liu, L.; Tollefsbol, T.O. Glucose restriction can extend normal cell lifespan and impair precancerous cell growth through epigenetic control of hTERT and p16 expression. FASEB J. 2010, 24, 1442–1453. [Google Scholar] [CrossRef] [PubMed]
- Wakeling, L.A.; Ions, L.J.; Ford, D. Could Sirt1-mediated epigenetic effects contribute to the longevity response to dietary restriction and be mimicked by other dietary interventions? Age 2009, 31, 327–341. [Google Scholar] [CrossRef]
- Alageel, A.; Tomasi, J.; Tersigni, C.; Brietzke, E.; Zuckerman, H.; Subramaniapillai, M.; Lee, Y.; Iacobucci, M.; Rosenblat, J.D.; Mansur, R.B.; et al. Evidence supporting a mechanistic role of sirtuins in mood and metabolic disorders. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 86, 95–101. [Google Scholar] [CrossRef]
- De Ruijter, A.J.; van Gennip, A.H.; Caron, H.N.; Kemp, S.; van Kuilenburg, A.B. Histone deacetylases (HDACs): Characterization of the classical HDAC family. Biochem. J. 2003, 370, 737–749. [Google Scholar] [CrossRef] [PubMed]
- Satoh, A.; Brace, C.S.; Rensing, N.; Cliften, P.; Wozniak, D.F.; Herzog, E.D.; Yamada, K.A.; Imai, S. Sirt1 extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab. 2013, 18, 416–430. [Google Scholar] [CrossRef]
- Siebold, A.P.; Banerjee, R.; Tie, F.; Kiss, D.L.; Moskowitz, J.; Harte, P.J. Polycomb Repressive Complex 2 and Trithorax modulate Drosophila longevity and stress resistance. Proc. Natl. Acad. Sci. USA 2010, 107, 169–174. [Google Scholar] [CrossRef]
- Pasyukova, E.G.; Vaiserman, A.M. HDAC inhibitors: A new promising drug class in anti-aging research. Mech. Ageing Dev. 2017, 166, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Taliyan, R. Targeting histone deacetylases: A novel approach in Parkinson’s disease. Parkinsons Dis. 2015, 2015, 303294. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.E.; Ko, C.P. Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2011, 231, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Ricobaraza, A.; Cuadrado-Tejedor, M.; Marco, S.; Pérez-Otaño, I.; García-Osta, A. Phenylbutyrate rescues dendritic spine loss associated with memory deficits in a mouse model of Alzheimer disease. Hippocampus 2012, 22, 1040–1050. [Google Scholar] [CrossRef]
- Wiley, J.C.; Pettan-Brewer, C.; Ladiges, W.C. Phenylbutyric acid reduces amyloid plaques and rescues cognitive behavior in AD transgenic mice. Aging Cell 2011, 10, 418–428. [Google Scholar] [CrossRef]
- Harrison, I.F.; Crum, W.R.; Vernon, A.C.; Dexter, D.T. Neurorestoration induced by the HDAC inhibitor sodium valproate in the lactacystin model of Parkinson’s is associated with histone acetylation and up-regulation of neurotrophic factors. Br. J. Pharmacol. 2015, 172, 4200–4215. [Google Scholar] [CrossRef]
- Grillari, J.; Grillari-Voglauer, R. Novel modulators of senescence, aging, and longevity: Small non-coding RNAs enter the stage. Exp. Gerontol. 2010, 45, 302–311. [Google Scholar] [CrossRef]
- Nelson, P.T.; Wang, W.X.; Rajeev, B.W. MicroRNAs (miRNAs) in neurodegenerative diseases. Brain Pathol. 2008, 18, 130–138. [Google Scholar] [CrossRef]
- Rodriguez-Ortiz, C.J.; Baglietto-Vargas, D.; Martinez-Coria, H.; LaFerla, F.M.; Kitazawa, M. Upregulation of miR-181 decreases c-Fos and SIRT-1 in the hippocampus of 3xTg-AD mice. J. Alzheimers Dis. 2014, 42, 1229–1238. [Google Scholar] [CrossRef]
- Mao, K.; Zhang, G. The role of PARP1 in neurodegenerative diseases and aging. FEBS J 2021. [Google Scholar] [CrossRef]
- Martire, S.; Mosca, L.; d’Erme, M. PARP-1 involvement in neurodegeneration: A focus on Alzheimer’s and Parkinson’s diseases. Mech. Ageing Dev. 2015, 146-148, 53–64. [Google Scholar] [CrossRef] [PubMed]
- Narne, P.; Pandey, V.; Simhadri, P.K.; Phanithi, P.B. Poly(ADP-ribose)polymerase-1 hyperactivation in neurodegenerative diseases: The death knell tolls for neurons. Semin. Cell Dev. Biol. 2017, 63, 154–166. [Google Scholar] [CrossRef] [PubMed]
- Beneke, S.; Cohausz, O.; Malanga, M.; Boukamp, P.; Althaus, F.; Bürkle, A. Rapid regulation of telomere length is mediated by poly(ADP-ribose) polymerase-1. Nucleic Acids Res. 2008, 36, 6309–6317. [Google Scholar] [CrossRef] [PubMed]
- Ye, T.J.; Lu, Y.L.; Yan, X.F.; Hu, X.D.; Wang, X.L. High mobility group box-1 release from H2O2-injured hepatocytes due to sirt1 functional inhibition. World J. Gastroenterol. 2019, 25, 5434–5450. [Google Scholar] [CrossRef]
- Kam, T.I.; Mao, X.; Park, H.; Chou, S.C.; Karuppagounder, S.S.; Umanah, G.E.; Yun, S.P.; Brahmachari, S.; Panicker, N.; Chen, R.; et al. Poly(ADP-ribose) drives pathologic α-synuclein neurodegeneration in Parkinson’s disease. Science 2018, 362, eaat8407. [Google Scholar] [CrossRef] [PubMed]
- Rulten, S.L.; Rotheray, A.; Green, R.L.; Grundy, G.J.; Moore, D.A.; Gómez-Herreros, F.; Hafezparast, M.; Caldecott, K.W. PARP-1 dependent recruitment of the amyotrophic lateral sclerosis-associated protein FUS/TLS to sites of oxidative DNA damage. Nucleic Acids Res. 2014, 42, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Chini, C.C.S.; Tarragó, M.G.; Chini, E.N. NAD and the aging process: Role in life, death and everything in between. Mol. Cell. Endocrinol. 2017, 455, 62–74. [Google Scholar] [CrossRef]
- Xie, Z.; Zhang, D.; Chung, D.; Tang, Z.; Huang, H.; Dai, L.; Qi, S.; Li, J.; Colak, G.; Chen, Y.; et al. Metabolic Regulation of Gene Expression by Histone Lysine β-Hydroxybutyrylation. Mol. Cell 2016, 62, 194–206. [Google Scholar] [CrossRef]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Moehle, E.A.; Shen, K.; Dillin, A. Mitochondrial proteostasis in the context of cellular and organismal health and aging. J. Biol. Chem. 2019, 294, 5396–5407. [Google Scholar] [CrossRef]
- Hekimi, S.; Lapointe, J.; Wen, Y. Taking a “good” look at free radicals in the aging process. Trends Cell Biol. 2011, 21, 569–576. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, M.M.; Zheng, B.; Lu, T.; Yan, Z.; Py, B.F.; Ng, A.; Xavier, R.J.; Li, C.; Yankner, B.A.; Scherzer, C.R.; et al. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2010, 107, 14164–14169. [Google Scholar] [CrossRef]
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. α-Synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037. [Google Scholar] [CrossRef] [PubMed]
- Youle, R.J.; Narendra, D.P. Mechanisms of mitophagy. Nat. Rev. Mol. Cell. Biol. 2011, 12, 9–14. [Google Scholar] [CrossRef]
- Gottlieb, R.A.; Mentzer, R.M. Autophagy during cardiac stress: Joys and frustrations of autophagy. Annu. Rev. Physiol. 2010, 72, 45–59. [Google Scholar] [CrossRef]
- Madeo, F.; Tavernarakis, N.; Kroemer, G. Can autophagy promote longevity? Nat. Cell Biol. 2010, 12, 842–846. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Herzig, S.; Shaw, R.J. AMPK: Guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018, 19, 121–135. [Google Scholar] [CrossRef]
- Lee, I.H.; Cao, L.; Mostoslavsky, R.; Lombard, D.B.; Liu, J.; Bruns, N.E.; Tsokos, M.; Alt, F.W.; Finkel, T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc. Natl. Acad. Sci. USA 2008, 105, 3374–3379. [Google Scholar] [CrossRef]
- McCarty, M.F.; DiNicolantonio, J.J.; O’Keefe, J.H. Ketosis may promote brain macroautophagy by activating Sirt1 and hypoxia-inducible factor-1. Med. Hypotheses 2015, 85, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Brown, K.; Hirschey, M.D.; Verdin, E.; Chen, D. Calorie restriction reduces oxidative stress by SIRT3-mediated SOD2 activation. Cell Metab. 2010, 12, 662–667. [Google Scholar] [CrossRef]
- Hafner, A.V.; Dai, J.; Gomes, A.P.; Xiao, C.Y.; Palmeira, C.M.; Rosenzweig, A.; Sinclair, D.A. Regulation of the mPTP by SIRT3-mediated deacetylation of CypD at lysine 166 suppresses age-related cardiac hypertrophy. Aging 2010, 2, 914–923. [Google Scholar] [CrossRef]
- Halling, J.F.; Pilegaard, H. PGC-1α-mediated regulation of mitochondrial function and physiological implications. Appl. Physiol. Nutr. Metab. 2020, 45, 927–936. [Google Scholar] [CrossRef] [PubMed]
- Austin, S.; St-Pierre, J. PGC1α and mitochondrial metabolism--emerging concepts and relevance in ageing and neurodegenerative disorders. J. Cell Sci. 2012, 125, 4963–4971. [Google Scholar] [CrossRef] [PubMed]
- Cantó, C.; Auwerx, J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 2009, 20, 98–105. [Google Scholar] [CrossRef]
- Puigserver, P.; Spiegelman, B.M. Peroxisome proliferator-activated receptor-gamma coactivator 1 alpha (PGC-1α): Transcriptional coactivator and metabolic regulator. Endocr. Rev. 2003, 24, 78–90. [Google Scholar] [CrossRef]
- Scirpo, R.; Fiorotto, R.; Villani, A.; Amenduni, M.; Spirli, C.; Strazzabosco, M. Stimulation of nuclear receptor peroxisome proliferator-activated receptor-γ limits NF-κB-dependent inflammation in mouse cystic fibrosis biliary epithelium. Hepatology 2015, 62, 1551–1562. [Google Scholar] [CrossRef]
- Tyagi, S.; Gupta, P.; Saini, A.S.; Kaushal, C.; Sharma, S. The peroxisome proliferator-activated receptor: A family of nuclear receptors role in various diseases. J. Adv. Pharm. Technol. Res. 2011, 2, 236–240. [Google Scholar] [CrossRef]
- Qin, W.; Yang, T.; Ho, L.; Zhao, Z.; Wang, J.; Chen, L.; Zhao, W.; Thiyagarajan, M.; MacGrogan, D.; Rodgers, J.T.; et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J. Biol. Chem. 2006, 281, 21745–21754. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.D.; Pasinetti, G.M. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar] [CrossRef] [PubMed]
- Eisele, P.S.; Salatino, S.; Sobek, J.; Hottiger, M.O.; Handschin, C. The peroxisome proliferator-activated receptor γ coactivator 1α/β (PGC-1) coactivators repress the transcriptional activity of NF-κB in skeletal muscle cells. J. Biol. Chem. 2013, 288, 2246–2260. [Google Scholar] [CrossRef]
- Mookerjee, S.A.; Divakaruni, A.S.; Jastroch, M.; Brand, M.D. Mitochondrial uncoupling and lifespan. Mech. Ageing Dev. 2010, 131, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Garagnani, P.; Parini, P.; Giuliani, C.; Santoro, A. Inflammaging: A new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol. 2018, 14, 576–590. [Google Scholar] [CrossRef]
- Josephson, A.M.; Bradaschia-Correa, V.; Lee, S.; Leclerc, K.; Patel, K.S.; Muinos Lopez, E.; Litwa, H.P.; Neibart, S.S.; Kadiyala, M.; Wong, M.Z.; et al. Age-related inflammation triggers skeletal stem/progenitor cell dysfunction. Proc. Natl. Acad. Sci. USA 2019, 116, 6995–7004. [Google Scholar] [CrossRef]
- Bauernfeind, F.; Ablasser, A.; Bartok, E.; Kim, S.; Schmid-Burgk, J.; Cavlar, T.; Hornung, V. Inflammasomes: Current understanding and open questions. Cell. Mol. Life Sci. 2011, 68, 765–783. [Google Scholar] [CrossRef]
- Salminen, A.; Kaarniranta, K.; Kauppinen, A. Inflammaging: Disturbed interplay between autophagy and inflammasomes. Aging 2012, 4, 166–175. [Google Scholar] [CrossRef]
- Martinez-Vicente, M.; Cuervo, A.M. Autophagy and neurodegeneration: When the cleaning crew goes on strike. Lancet Neurol. 2007, 6, 352–361. [Google Scholar] [CrossRef]
- Nixon, R.A.; Yang, D.S. Autophagy failure in Alzheimer’s disease--locating the primary defect. Neurobiol. Dis. 2011, 43, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Masters, S.L.; O’Neill, L.A. Disease-associated amyloid and misfolded protein aggregates activate the inflammasome. Trends Mol. Med. 2011, 17, 276–282. [Google Scholar] [CrossRef]
- Salminen, A.; Huuskonen, J.; Ojala, J.; Kauppinen, A.; Kaarniranta, K.; Suuronen, T. Activation of innate immunity system during aging: NF-kB signaling is the molecular culprit of inflamm-aging. Ageing Res. Rev. 2008, 7, 83–105. [Google Scholar] [CrossRef]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef] [PubMed]
- Levy, M.; Thaiss, C.A.; Elinav, E. Taming the inflammasome. Nat. Med. 2015, 21, 213–215. [Google Scholar] [CrossRef]
- Patel, M.N.; Carroll, R.G.; Galván-Peña, S.; Mills, E.L.; Olden, R.; Triantafilou, M.; Wolf, A.I.; Bryant, C.E.; Triantafilou, K.; Masters, S.L. Inflammasome Priming in Sterile Inflammatory Disease. Trends Mol. Med. 2017, 23, 165–180. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Abdullah, A.; Mohd Murshid, N.; Makpol, S. Antioxidant Modulation of mTOR and Sirtuin Pathways in Age-Related Neurodegenerative Diseases. Mol. Neurobiol. 2020, 57, 5193–5207. [Google Scholar] [CrossRef]
- Osorio, F.G.; Bárcena, C.; Soria-Valles, C.; Ramsay, A.J.; de Carlos, F.; Cobo, J.; Fueyo, A.; Freije, J.M.; López-Otín, C. Nuclear lamina defects cause ATM-dependent NF-κB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 2012, 26, 2311–2324. [Google Scholar] [CrossRef]
- Amaya-Montoya, M.; Pérez-Londoño, A.; Guatibonza-García, V.; Vargas-Villanueva, A.; Mendivil, C.O. Cellular Senescence as a Therapeutic Target for Age-Related Diseases: A Review. Adv. Ther. 2020, 37, 1407–1424. [Google Scholar] [CrossRef]
- Borghesan, M.; Hoogaars, W.M.H.; Varela-Eirin, M.; Talma, N.; Demaria, M. A Senescence-Centric View of Aging: Implications for Longevity and Disease. Trends Cell Biol. 2020, 30, 777–791. [Google Scholar] [CrossRef]
- Campisi, J. Aging, cellular senescence, and cancer. Annu. Rev. Physiol. 2013, 75, 685–705. [Google Scholar] [CrossRef]
- Di Micco, R.; Krizhanovsky, V.; Baker, D.; d’Adda di Fagagna, F. Cellular senescence in ageing: From mechanisms to therapeutic opportunities. Nat. Rev. Mol. Cell Biol. 2021, 22, 75–95. [Google Scholar] [CrossRef] [PubMed]
- He, S.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- Tran, D.; Bergholz, J.; Zhang, H.; He, H.; Wang, Y.; Zhang, Y.; Li, Q.; Kirkland, J.L.; Xiao, Z.X. Insulin-like growth factor-1 regulates the SIRT1-p53 pathway in cellular senescence. Aging Cell 2014, 13, 669–678. [Google Scholar] [CrossRef]
- Young, A.R.; Narita, M.; Ferreira, M.; Kirschner, K.; Sadaie, M.; Darot, J.F.; Tavaré, S.; Arakawa, S.; Shimizu, S.; Watt, F.M.; et al. Autophagy mediates the mitotic senescence transition. Genes Dev. 2009, 23, 798–803. [Google Scholar] [CrossRef]
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and aging related signaling pathways. Mech. Ageing Dev. 2020, 187, 111215. [Google Scholar] [CrossRef]
- Salmenperä, P.; Karhemo, P.R.; Räsänen, K.; Laakkonen, P.; Vaheri, A. Fibroblast spheroids as a model to study sustained fibroblast quiescence and their crosstalk with tumor cells. Exp. Cell Res. 2016, 345, 17–24. [Google Scholar] [CrossRef][Green Version]
- Cai, J.; Weiss, M.L.; Rao, M.S. In search of “stemness”. Exp. Hematol. 2004, 32, 585–598. [Google Scholar] [CrossRef]
- Kim, K.H.; Chen, C.C.; Monzon, R.I.; Lau, L.F. Matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol. Cell Biol. 2013, 33, 2078–2090. [Google Scholar] [CrossRef]
- Collado, M.; Serrano, M. Senescence in tumours: Evidence from mice and humans. Nat. Rev. Cancer 2010, 10, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Woods, G.; Demaria, M.; Rane, A.; Zou, Y.; McQuade, A.; Rajagopalan, S.; Limbad, C.; Madden, D.T.; Campisi, J.; et al. Cellular Senescence Is Induced by the Environmental Neurotoxin Paraquat and Contributes to Neuropathology Linked to Parkinson’s Disease. Cell Rep. 2018, 22, 930–940. [Google Scholar] [CrossRef]
- Finkel, T.; Serrano, M.; Blasco, M.A. The common biology of cancer and ageing. Nature 2007, 448, 767–774. [Google Scholar] [CrossRef]
- Wang, J.C.; Bennett, M. Aging and atherosclerosis: Mechanisms, functional consequences, and potential therapeutics for cellular senescence. Circ. Res. 2012, 111, 245–259. [Google Scholar] [CrossRef]
- Cao, L.; Li, W.; Kim, S.; Brodie, S.G.; Deng, C.X. Senescence, aging, and malignant transformation mediated by p53 in mice lacking the Brca1 full-length isoform. Genes Dev. 2003, 17, 201–213. [Google Scholar] [CrossRef]
- Jones, R.G.; Plas, D.R.; Kubek, S.; Buzzai, M.; Mu, J.; Xu, Y.; Birnbaum, M.J.; Thompson, C.B. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell 2005, 18, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Balch, W.E.; Morimoto, R.I.; Dillin, A.; Kelly, J.W. Adapting proteostasis for disease intervention. Science 2008, 319, 916–919. [Google Scholar] [CrossRef]
- Klaips, C.L.; Jayaraj, G.G.; Hartl, F.U. Pathways of cellular proteostasis in aging and disease. J. Cell Biol. 2018, 217, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Lapierre, L.R.; De Magalhaes Filho, C.D.; McQuary, P.R.; Chu, C.C.; Visvikis, O.; Chang, J.T.; Gelino, S.; Ong, B.; Davis, A.E.; Irazoqui, J.E.; et al. The TFEB orthologue HLH-30 regulates autophagy and modulates longevity in Caenorhabditis elegans. Nat. Commun. 2013, 4, 2267. [Google Scholar] [CrossRef]
- Basisty, N.; Meyer, J.G.; Schilling, B. Protein Turnover in Aging and Longevity. Proteomics 2018, 18, e1700108. [Google Scholar] [CrossRef]
- Wong, S.Q.; Kumar, A.V.; Mills, J.; Lapierre, L.R. Autophagy in aging and longevity. Hum. Genet. 2020, 139, 277–290. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef] [PubMed]
- Ryu, D.; Mouchiroud, L.; Andreux, P.A.; Katsyuba, E.; Moullan, N.; Nicolet-Dit-Félix, A.A.; Williams, E.G.; Jha, P.; Lo Sasso, G.; Huzard, D.; et al. Urolithin A induces mitophagy and prolongs lifespan in C. elegans and increases muscle function in rodents. Nat. Med. 2016, 22, 879–888. [Google Scholar] [CrossRef]
- Ou, X.; Lee, M.R.; Huang, X.; Messina-Graham, S.; Broxmeyer, H.E. SIRT1 positively regulates autophagy and mitochondria function in embryonic stem cells under oxidative stress. Stem Cells 2014, 32, 1183–1194. [Google Scholar] [CrossRef] [PubMed]
- Goodell, M.A.; Rando, T.A. Stem cells and healthy aging. Science 2015, 350, 1199–1204. [Google Scholar] [CrossRef] [PubMed]
- Keyes, B.E.; Fuchs, E. Stem cells: Aging and transcriptional fingerprints. J. Cell. Biol. 2018, 217, 79–92. [Google Scholar] [CrossRef]
- Zhang, P.; Kishimoto, Y.; Grammatikakis, I.; Gottimukkala, K.; Cutler, R.G.; Zhang, S.; Abdelmohsen, K.; Bohr, V.A.; Misra Sen, J.; Gorospe, M.; et al. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci. 2019, 22, 719–728. [Google Scholar] [CrossRef]
- Ehninger, D.; Neff, F.; Xie, K. Longevity, aging and rapamycin. Cell. Mol. Life Sci. 2014, 71, 4325–4346. [Google Scholar] [CrossRef]
- Kaeberlein, M.; Galvan, V. Rapamycin and Alzheimer’s disease: Time for a clinical trial? Sci. Transl. Med. 2019, 11, eaar4289. [Google Scholar] [CrossRef] [PubMed]
- Sharma, M.; Gupta, Y.K. Chronic treatment with trans resveratrol prevents intracerebroventricular streptozotocin induced cognitive impairment and oxidative stress in rats. Life Sci. 2002, 71, 2489–2498. [Google Scholar] [CrossRef]
- Lu, M.; Su, C.; Qiao, C.; Bian, Y.; Ding, J.; Hu, G. Metformin Prevents Dopaminergic Neuron Death in MPTP/P-Induced Mouse Model of Parkinson’s Disease via Autophagy and Mitochondrial ROS Clearance. Int. J. Neuropsychopharmacol. 2016, 19, pyw047. [Google Scholar] [CrossRef] [PubMed]
- Foretz, M.; Guigas, B.; Bertrand, L.; Pollak, M.; Viollet, B. Metformin: From mechanisms of action to therapies. Cell Metab. 2014, 20, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, A.S.; Gubbi, S.; Barzilai, N. Benefits of Metformin in Attenuating the Hallmarks of Aging. Cell Metab. 2020, 32, 15–30. [Google Scholar] [CrossRef]
- Najafi, M.; Cheki, M.; Rezapoor, S.; Geraily, G.; Motevaseli, E.; Carnovale, C.; Clementi, E.; Shirazi, A. Metformin: Prevention of genomic instability and cancer: A review. Mutat. Res. Genet. Toxicol. Environ. Mutagen. 2018, 827, 1–8. [Google Scholar] [CrossRef]
- Aatsinki, S.M.; Buler, M.; Salomäki, H.; Koulu, M.; Pavek, P.; Hakkola, J. Metformin induces PGC-1α expression and selectively affects hepatic PGC-1α functions. Br. J. Pharmacol. 2014, 171, 2351–2363. [Google Scholar] [CrossRef]
- Prasad, S.; Sajja, R.K.; Kaisar, M.A.; Park, J.H.; Villalba, H.; Liles, T.; Abbruscato, T.; Cucullo, L. Role of Nrf2 and protective effects of Metformin against tobacco smoke-induced cerebrovascular toxicity. Redox Biol. 2017, 12, 58–69. [Google Scholar] [CrossRef]
- Cuyàs, E.; Verdura, S.; Llorach-Parés, L.; Fernández-Arroyo, S.; Joven, J.; Martin-Castillo, B.; Bosch-Barrera, J.; Brunet, J.; Nonell-Canals, A.; Sanchez-Martinez, M.; et al. Metformin Is a Direct SIRT1-Activating Compound: Computational Modeling and Experimental Validation. Front. Endocrinol. 2018, 9, 657. [Google Scholar] [CrossRef]
- Moiseeva, O.; Deschênes-Simard, X.; St-Germain, E.; Igelmann, S.; Huot, G.; Cadar, A.E.; Bourdeau, V.; Pollak, M.N.; Ferbeyre, G. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-κB activation. Aging Cell 2013, 12, 489–498. [Google Scholar] [CrossRef]
- Tizazu, A.M.; Nyunt, M.S.Z.; Cexus, O.; Suku, K.; Mok, E.; Xian, C.H.; Chong, J.; Tan, C.; How, W.; Hubert, S.; et al. Metformin Monotherapy Downregulates Diabetes-Associated Inflammatory Status and Impacts on Mortality. Front. Physiol. 2019, 10, 572. [Google Scholar] [CrossRef]
- Wang, C.; Liu, C.; Gao, K.; Zhao, H.; Zhou, Z.; Shen, Z.; Guo, Y.; Li, Z.; Yao, T.; Mei, X. Metformin preconditioning provide neuroprotection through enhancement of autophagy and suppression of inflammation and apoptosis after spinal cord injury. Biochem. Biophys. Res. Commun. 2016, 477, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Markowicz-Piasecka, M.; Sikora, J.; Szydłowska, A.; Skupień, A.; Mikiciuk-Olasik, E.; Huttunen, K.M. Metformin—A Future Therapy for Neurodegenerative Diseases: Theme: Drug Discovery, Development and Delivery in Alzheimer’s Disease Guest Editor: Davide Brambilla. Pharm. Res. 2017, 34, 2614–2627. [Google Scholar] [CrossRef]
- Fang, J.; Yang, J.; Wu, X.; Zhang, G.; Li, T.; Wang, X.; Zhang, H.; Wang, C.C.; Liu, G.H.; Wang, L. Metformin alleviates human cellular aging by upregulating the endoplasmic reticulum glutathione peroxidase 7. Aging Cell 2018, 17, e12765. [Google Scholar] [CrossRef]
- Bridgeman, S.C.; Ellison, G.C.; Melton, P.E.; Newsholme, P.; Mamotte, C.D.S. Epigenetic effects of metformin: From molecular mechanisms to clinical implications. Diabetes Obes. Metab. 2018, 20, 1553–1562. [Google Scholar] [CrossRef]
- Noren Hooten, N.; Martin-Montalvo, A.; Dluzen, D.F.; Zhang, Y.; Bernier, M.; Zonderman, A.B.; Becker, K.G.; Gorospe, M.; de Cabo, R.; Evans, M.K. Metformin-mediated increase in DICER1 regulates microRNA expression and cellular senescence. Aging Cell 2016, 15, 572–581. [Google Scholar] [CrossRef] [PubMed]
- De Zegher, F.; Díaz, M.; Ibáñez, L. Association between Long Telomere Length and Insulin Sensitization in Adolescent Girls with Hyperinsulinemic Androgen Excess. JAMA Pediatr. 2015, 169, 787–788. [Google Scholar] [CrossRef]
- Diman, A.; Boros, J.; Poulain, F.; Rodriguez, J.; Purnelle, M.; Episkopou, H.; Bertrand, L.; Francaux, M.; Deldicque, L.; Decottignies, A. Nuclear respiratory factor 1 and endurance exercise promote human telomere transcription. Sci. Adv. 2016, 2, e1600031. [Google Scholar] [CrossRef] [PubMed]
- Bhullar, K.S.; Hubbard, B.P. Lifespan and healthspan extension by resveratrol. Biochim. Biophys. Acta 2015, 1852, 1209–1218. [Google Scholar] [CrossRef] [PubMed]
- Porquet, D.; Casadesús, G.; Bayod, S.; Vicente, A.; Canudas, A.M.; Vilaplana, J.; Pelegrí, C.; Sanfeliu, C.; Camins, A.; Pallàs, M.; et al. Dietary resveratrol prevents Alzheimer’s markers and increases life span in SAMP8. Age 2013, 35, 1851–1865. [Google Scholar] [CrossRef] [PubMed]
- Mancuso, R.; del Valle, J.; Modol, L.; Martinez, A.; Granado-Serrano, A.B.; Ramirez-Núñez, O.; Pallás, M.; Portero-Otin, M.; Osta, R.; Navarro, X. Resveratrol improves motoneuron function and extends survival in SOD1(G93A) ALS mice. Neurotherapeutics 2014, 11, 419–432. [Google Scholar] [CrossRef]
- Price, N.L.; Gomes, A.P.; Ling, A.J.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef]
- Morris, B.J. Seven sirtuins for seven deadly diseases of aging. Free Radic. Biol. Med. 2013, 56, 133–171. [Google Scholar] [CrossRef]
- Choi, H.I.; Kim, H.J.; Park, J.S.; Kim, I.J.; Bae, E.H.; Ma, S.K.; Kim, S.W. PGC-1α attenuates hydrogen peroxide-induced apoptotic cell death by upregulating Nrf-2 via GSK3β inactivation mediated by activated p38 in HK-2 Cells. Sci. Rep. 2017, 7, 4319. [Google Scholar] [CrossRef]
- Huang, K.; Gao, X.; Wei, W. The crosstalk between Sirt1 and Keap1/Nrf2/ARE anti-oxidative pathway forms a positive feedback loop to inhibit FN and TGF-β1 expressions in rat glomerular mesangial cells. Exp. Cell Res. 2017, 361, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Joo, M.S.; Kim, W.D.; Lee, K.Y.; Kim, J.H.; Koo, J.H.; Kim, S.G. AMPK Facilitates Nuclear Accumulation of Nrf2 by Phosphorylating at Serine 550. Mol. Cell Biol. 2016, 36, 1931–1942. [Google Scholar] [CrossRef]
- Mercken, E.M.; Mitchell, S.J.; Martin-Montalvo, A.; Minor, R.K.; Almeida, M.; Gomes, A.P.; Scheibye-Knudsen, M.; Palacios, H.H.; Licata, J.J.; Zhang, Y.; et al. SRT2104 extends survival of male mice on a standard diet and preserves bone and muscle mass. Aging Cell 2014, 13, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Carullo, G.; Cappello, A.R.; Frattaruolo, L.; Badolato, M.; Armentano, B.; Aiello, F. Quercetin and derivatives: Useful tools in inflammation and pain management. Future Med. Chem. 2017, 9, 79–93. [Google Scholar] [CrossRef]
- Feng, X.; Sureda, A.; Jafari, S.; Memariani, Z.; Tewari, D.; Annunziata, G.; Barrea, L.; Hassan, S.T.S.; Šmejkal, K.; Malaník, M.; et al. Berberine in Cardiovascular and Metabolic Diseases: From Mechanisms to Therapeutics. Theranostics 2019, 9, 1923–1951. [Google Scholar] [CrossRef] [PubMed]
- Sarker, M.R.; Franks, S.F. Efficacy of curcumin for age-associated cognitive decline: A narrative review of preclinical and clinical studies. Geroscience 2018, 40, 73–95. [Google Scholar] [CrossRef]
- Sato, K.; Kashiwaya, Y.; Keon, C.A.; Tsuchiya, N.; King, M.T.; Radda, G.K.; Chance, B.; Clarke, K.; Veech, R.L. Insulin, ketone bodies, and mitochondrial energy transduction. FASEB J. 1995, 9, 651–658. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.K.; Rani, E.; Waheed, A.; Rajput, S.K. Pharmacoresistant Epilepsy: A Current Update on Non-Conventional Pharmacological and Non-Pharmacological Interventions. J. Epilepsy Res. 2015, 5, 1–8. [Google Scholar] [CrossRef]
- VanItallie, T.B.; Nufert, T.H. Ketones: Metabolism’s ugly duckling. Nutr. Rev. 2003, 61, 327–341. [Google Scholar] [CrossRef] [PubMed]
- Almeida, C.G.; de Mendonça, A.; Cunha, R.A.; Ribeiro, J.A. Adenosine promotes neuronal recovery from reactive oxygen species induced lesion in rat hippocampal slices. Neurosci. Lett. 2003, 339, 127–130. [Google Scholar] [CrossRef]
- Choudhury, H.; Chellappan, D.K.; Sengupta, P.; Pandey, M.; Gorain, B. Adenosine Receptors in Modulation of Central Nervous System Disorders. Curr. Pharm. Des. 2019, 25, 2808–2827. [Google Scholar] [CrossRef] [PubMed]
- McNally, M.A.; Hartman, A.L. Ketone bodies in epilepsy. J. Neurochem. 2012, 121, 28–35. [Google Scholar] [CrossRef]
- Juge, N.; Gray, J.A.; Omote, H.; Miyaji, T.; Inoue, T.; Hara, C.; Uneyama, H.; Edwards, R.H.; Nicoll, R.A.; Moriyama, Y. Metabolic control of vesicular glutamate transport and release. Neuron 2010, 68, 99–112. [Google Scholar] [CrossRef]
- Tieu, K.; Perier, C.; Caspersen, C.; Teismann, P.; Wu, D.C.; Yan, S.D.; Naini, A.; Vila, M.; Jackson-Lewis, V.; Ramasamy, R.; et al. D-beta-hydroxybutyrate rescues mitochondrial respiration and mitigates features of Parkinson disease. J. Clin. Investig. 2003, 112, 892–901. [Google Scholar] [CrossRef]
- Veech, R.L.; Todd King, M.; Pawlosky, R.; Kashiwaya, Y.; Bradshaw, P.C.; Curtis, W. The “great” controlling nucleotide coenzymes. IUBMB Life 2019, 71, 565–579. [Google Scholar] [CrossRef]
- Shimazu, T.; Hirschey, M.D.; Newman, J.; He, W.; Shirakawa, K.; Le Moan, N.; Grueter, C.A.; Lim, H.; Saunders, L.R.; Stevens, R.D.; et al. Suppression of oxidative stress by β-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013, 339, 211–214. [Google Scholar] [CrossRef]
- Tseng, A.H.; Shieh, S.S.; Wang, D.L. SIRT3 deacetylates FOXO3 to protect mitochondria against oxidative damage. Free Radic. Biol. Med. 2013, 63, 222–234. [Google Scholar] [CrossRef]
- Zhao, L.; Ackerman, S.L. Endoplasmic reticulum stress in health and disease. Curr. Opin. Cell Biol. 2006, 18, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Sleiman, S.F.; Henry, J.; Al-Haddad, R.; El Hayek, L.; Abou Haidar, E.; Stringer, T.; Ulja, D.; Karuppagounder, S.S.; Holson, E.B.; Ratan, R.R.; et al. Exercise promotes the expression of brain derived neurotrophic factor (BDNF) through the action of the ketone body β-hydroxybutyrate. Elife 2016, 5, e15092. [Google Scholar] [CrossRef]
- Manning, B.D.; Cantley, L.C. AKT/PKB signaling: Navigating downstream. Cell 2007, 129, 1261–1274. [Google Scholar] [CrossRef]
- Xu, D.; Lian, D.; Wu, J.; Liu, Y.; Zhu, M.; Sun, J.; He, D.; Li, L. Brain-derived neurotrophic factor reduces inflammation and hippocampal apoptosis in experimental Streptococcus pneumoniae meningitis. J. Neuroinflamm. 2017, 14, 156. [Google Scholar] [CrossRef]
- Marosi, K.; Kim, S.W.; Moehl, K.; Scheibye-Knudsen, M.; Cheng, A.; Cutler, R.; Camandola, S.; Mattson, M.P. 3-Hydroxybutyrate regulates energy metabolism and induces BDNF expression in cerebral cortical neurons. J. Neurochem. 2016, 139, 769–781. [Google Scholar] [CrossRef] [PubMed]
- Lau, D.; Bengtson, C.P.; Buchthal, B.; Bading, H. BDNF Reduces Toxic Extrasynaptic NMDA Receptor Signaling via Synaptic NMDA Receptors and Nuclear-Calcium-Induced Transcription of inhba/Activin A. Cell Rep. 2015, 12, 1353–1366. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P.; Lovell, M.A.; Furukawa, K.; Markesbery, W.R. Neurotrophic factors attenuate glutamate-induced accumulation of peroxides, elevation of intracellular Ca2+ concentration, and neurotoxicity and increase antioxidant enzyme activities in hippocampal neurons. J. Neurochem. 1995, 65, 1740–1751. [Google Scholar] [CrossRef]
- Menzies, K.J.; Zhang, H.; Katsyuba, E.; Auwerx, J. Protein acetylation in metabolism—Metabolites and cofactors. Nat. Rev. Endocrinol. 2016, 12, 43–60. [Google Scholar] [CrossRef] [PubMed]
- Guil, S.; Long, J.C.; Cáceres, J.F. hnRNP A1 relocalization to the stress granules reflects a role in the stress response. Mol. Cell. Biol. 2006, 26, 5744–5758. [Google Scholar] [CrossRef] [PubMed]
- Jean-Philippe, J.; Paz, S.; Caputi, M. hnRNP A1: The Swiss army knife of gene expression. Int. J. Mol. Sci. 2013, 14, 18999–19024. [Google Scholar] [CrossRef]
- Parodi, B.; Rossi, S.; Morando, S.; Cordano, C.; Bragoni, A.; Motta, C.; Usai, C.; Wipke, B.T.; Scannevin, R.H.; Mancardi, G.L.; et al. Fumarates modulate microglia activation through a novel HCAR2 signaling pathway and rescue synaptic dysregulation in inflamed CNS. Acta Neuropathol. 2015, 130, 279–295. [Google Scholar] [CrossRef]
- Maalouf, M.; Sullivan, P.G.; Davis, L.; Kim, D.Y.; Rho, J.M. Ketones inhibit mitochondrial production of reactive oxygen species production following glutamate excitotoxicity by increasing NADH oxidation. Neuroscience 2007, 145, 256–264. [Google Scholar] [CrossRef] [PubMed]
- Pawlosky, R.J.; Kemper, M.F.; Kashiwaya, Y.; King, M.T.; Mattson, M.P.; Veech, R.L. Effects of a dietary ketone ester on hippocampal glycolytic and tricarboxylic acid cycle intermediates and amino acids in a 3xTgAD mouse model of Alzheimer’s disease. J. Neurochem. 2017, 141, 195–207. [Google Scholar] [CrossRef]
- Offermanns, S.; Schwaninger, M. Nutritional or pharmacological activation of HCA(2) ameliorates neuroinflammation. Trends Mol. Med. 2015, 21, 245–255. [Google Scholar] [CrossRef]
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2017, 127, 624–633. [Google Scholar] [CrossRef]
- Norwitz, N.G.; Hu, M.T.; Clarke, K. The Mechanisms by Which the Ketone Body D-β-Hydroxybutyrate May Improve the Multiple Cellular Pathologies of Parkinson’s disease. Front. Nutr. 2019, 6, 63. [Google Scholar] [CrossRef]
- Hasan-Olive, M.M.; Lauritzen, K.H.; Ali, M.; Rasmussen, L.J.; Storm-Mathisen, J.; Bergersen, L.H. A Ketogenic Diet Improves Mitochondrial Biogenesis and Bioenergetics via the PGC1α-SIRT3-UCP2 Axis. Neurochem. Res. 2019, 44, 22–37. [Google Scholar] [CrossRef] [PubMed]
- Zou, X.H.; Li, H.M.; Wang, S.; Leski, M.; Yao, Y.C.; Yang, X.D.; Huang, Q.J.; Chen, G.Q. The effect of 3-hydroxybutyrate methyl ester on learning and memory in mice. Biomaterials 2009, 30, 1532–1541. [Google Scholar] [CrossRef]
- Scheibye-Knudsen, M.; Mitchell, S.J.; Fang, E.F.; Iyama, T.; Ward, T.; Wang, J.; Dunn, C.A.; Singh, N.; Veith, S.; Hasan-Olive, M.M.; et al. A high-fat diet and NAD+ activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014, 20, 840–855. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Auwerx, J. Exploring the therapeutic space around NAD+. J. Cell Biol. 2012, 199, 205–209. [Google Scholar] [CrossRef]
- Kong, X.; Wang, R.; Xue, Y.; Liu, X.; Zhang, H.; Chen, Y.; Fang, F.; Chang, Y. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS ONE 2010, 5, e11707. [Google Scholar] [CrossRef]
- Shi, T.; Wang, F.; Stieren, E.; Tong, Q. SIRT3, a mitochondrial sirtuin deacetylase, regulates mitochondrial function and thermogenesis in brown adipocytes. J. Biol. Chem. 2005, 280, 13560–13567. [Google Scholar] [CrossRef] [PubMed]
- Cheng, A.; Wan, R.; Yang, J.L.; Kamimura, N.; Son, T.G.; Ouyang, X.; Luo, Y.; Okun, E.; Mattson, M.P. Involvement of PGC-1α in the formation and maintenance of neuronal dendritic spines. Nat. Commun. 2012, 3, 1250. [Google Scholar] [CrossRef] [PubMed]
- Jeong, E.A.; Jeon, B.T.; Shin, H.J.; Kim, N.; Lee, D.H.; Kim, H.J.; Kang, S.S.; Cho, G.J.; Choi, W.S.; Roh, G.S. Ketogenic diet-induced peroxisome proliferator-activated receptor-γ activation decreases neuroinflammation in the mouse hippocampus after kainic acid-induced seizures. Exp. Neurol. 2011, 232, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Morris, G.; Puri, B.K.; Carvalho, A.; Maes, M.; Berk, M.; Ruusunen, A.; Olive, L. Induced Ketosis as a Treatment for Neuroprogressive Disorders: Food for Thought? Int. J. Neuropsychopharmacol. 2020, 23, 366–384. [Google Scholar] [CrossRef]
- Simeone, T.A.; Matthews, S.A.; Samson, K.K.; Simeone, K.A. Regulation of brain PPARgamma2 contributes to ketogenic diet anti-seizure efficacy. Exp. Neurol. 2017, 287, 54–64. [Google Scholar] [CrossRef]
- Kashiwaya, Y.; Pawlosky, R.; Markis, W.; King, M.T.; Bergman, C.; Srivastava, S.; Murray, A.; Clarke, K.; Veech, R.L. A ketone ester diet increases brain malonyl-CoA and Uncoupling proteins 4 and 5 while decreasing food intake in the normal Wistar Rat. J. Biol. Chem. 2010, 285, 25950–25956. [Google Scholar] [CrossRef]
- Srivastava, S.; Kashiwaya, Y.; King, M.T.; Baxa, U.; Tam, J.; Niu, G.; Chen, X.; Clarke, K.; Veech, R.L. Mitochondrial biogenesis and increased uncoupling protein 1 in brown adipose tissue of mice fed a ketone ester diet. FASEB J. 2012, 26, 2351–2362. [Google Scholar] [CrossRef]
- Bae, H.R.; Kim, D.H.; Park, M.H.; Lee, B.; Kim, M.J.; Lee, E.K.; Chung, K.W.; Kim, S.M.; Im, D.S.; Chung, H.Y. β-Hydroxybutyrate suppresses inflammasome formation by ameliorating endoplasmic reticulum stress via AMPK activation. Oncotarget 2016, 7, 66444–66454. [Google Scholar] [CrossRef]
- Yamanashi, T.; Iwata, M.; Kamiya, N.; Tsunetomi, K.; Kajitani, N.; Wada, N.; Iitsuka, T.; Yamauchi, T.; Miura, A.; Pu, S.; et al. Beta-hydroxybutyrate, an endogenic NLRP3 inflammasome inhibitor, attenuates stress-induced behavioral and inflammatory responses. Sci. Rep. 2017, 7, 7677. [Google Scholar] [CrossRef]
- Youm, Y.H.; Nguyen, K.Y.; Grant, R.W.; Goldberg, E.L.; Bodogai, M.; Kim, D.; D’Agostino, D.; Planavsky, N.; Lupfer, C.; Kanneganti, T.D.; et al. The ketone metabolite β-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat. Med. 2015, 21, 263–269. [Google Scholar] [CrossRef]
- Ari, C.; Murdun, C.; Koutnik, A.P.; Goldhagen, C.R.; Rogers, C.; Park, C.; Bharwani, S.; Diamond, D.M.; Kindy, M.S.; D’Agostino, D.P.; et al. Exogenous Ketones Lower Blood Glucose Level in Rested and Exercised Rodent Models. Nutrients 2019, 11, 2330. [Google Scholar] [CrossRef]
- Edwards, C.; Copes, N.; Bradshaw, P.C. D-ß-hydroxybutyrate: An anti-aging ketone body. Oncotarget 2015, 6, 3477–3478. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C. The first long-lived mutants: Discovery of the insulin/IGF-1 pathway for ageing. Philos. Trans. R. Soc. Lond. B. Biol. Sci. 2011, 366, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Willcox, B.J.; Donlon, T.A.; He, Q.; Chen, R.; Grove, J.S.; Yano, K.; Masaki, K.H.; Willcox, D.C.; Rodriguez, B.; Curb, J.D. FOXO3A genotype is strongly associated with human longevity. Proc. Natl. Acad. Sci. USA 2008, 105, 13987–13992. [Google Scholar] [CrossRef] [PubMed]
- Hansen, M.; Chandra, A.; Mitic, L.L.; Onken, B.; Driscoll, M.; Kenyon, C. A role for autophagy in the extension of lifespan by dietary restriction in C. elegans. PLoS Genet. 2008, 4, e24. [Google Scholar] [CrossRef]
- Yamada, T.; Zhang, S.J.; Westerblad, H.; Katz, A. β-Hydroxybutyrate inhibits insulin-mediated glucose transport in mouse oxidative muscle. Am. J. Physiol. Endocrinol. Metab. 2010, 299, 364–373. [Google Scholar] [CrossRef]
- Barja, G. Free radicals and aging. Trends Neurosci. 2004, 27, 595–600. [Google Scholar] [CrossRef]
- Newman, J.C.; Covarrubias, A.J.; Zhao, M.; Yu, X.; Gut, P.; Ng, C.P.; Huang, Y.; Haldar, S.; Verdin, E. Ketogenic Diet Reduces Midlife Mortality and Improves Memory in Aging Mice. Cell Metab. 2017, 26, 547–557.e8. [Google Scholar] [CrossRef]
- Simeone, K.A.; Matthews, S.A.; Rho, J.M.; Simeone, T.A. Ketogenic diet treatment increases longevity in Kcna1-null mice, a model of sudden unexpected death in epilepsy. Epilepsia 2016, 57, 178–182. [Google Scholar] [CrossRef]
- Liu, K.; Li, F.; Sun, Q.; Lin, N.; Han, H.; You, K.; Tian, F.; Mao, Z.; Li, T.; Tong, T.; et al. p53 β-hydroxybutyrylation attenuates p53 activity. Cell Death Dis. 2019, 10, 243. [Google Scholar] [CrossRef]
- Habieb, M.E.; Mohamed, M.A.; El Gamal, D.M.; Hawas, A.M.; Mohamed, T.M. Anti-aging effect of DL-β-hydroxybutyrate against hepatic cellular senescence induced by D-galactose or γ-irradiation via autophagic flux stimulation in male rats. Arch. Gerontol. Geriatr. 2021, 92, 104288. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Han, L.; Zhao, G.; Shen, H.; Wang, P.; Sun, Z.; Xu, C.; Su, Y.; Li, G.; Tong, T.; et al. hnRNP A1 antagonizes cellular senescence and senescence-associated secretory phenotype via regulation of SIRT1 mRNA stability. Aging Cell 2016, 15, 1063–1073. [Google Scholar] [CrossRef] [PubMed]
- Armada-Moreira, A.; Gomes, J.I.; Pina, C.C.; Savchak, O.K.; Gonçalves-Ribeiro, J.; Rei, N.; Pinto, S.; Morais, T.P.; Martins, R.S.; Ribeiro, F.F.; et al. Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14, 90. [Google Scholar] [CrossRef]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Holper, L.; Ben-Shachar, D.; Mann, J.J. Multivariate meta-analyses of mitochondrial complex I and IV in major depressive disorder, bipolar disorder, schizophrenia, Alzheimer disease, and Parkinson disease. Neuropsychopharmacology 2019, 44, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Mosconi, L.; de Leon, M.; Murray, J.; Lu, J.; Javier, E.; McHugh, P.; Swerdlow, R.H. Reduced mitochondria cytochrome oxidase activity in adult children of mothers with Alzheimer’s disease. J. Alzheimers Dis. 2011, 27, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Lugrin, J.; Rosenblatt-Velin, N.; Parapanov, R.; Liaudet, L. The role of oxidative stress during inflammatory processes. Biol. Chem. 2014, 395, 203–230. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Yu, J.; Zhu, A.; Nakanishi, H. Nutrients, Microglia Aging, and Brain Aging. Oxid. Med. Cell Longev. 2016, 2016, 7498528. [Google Scholar] [CrossRef]
- Hatanpää, K.; Isaacs, K.R.; Shirao, T.; Brady, D.R.; Rapoport, S.I. Loss of proteins regulating synaptic plasticity in normal aging of the human brain and in Alzheimer disease. J. Neuropathol. Exp. Neurol. 1999, 58, 637–643. [Google Scholar] [CrossRef]
- Rao, J.S.; Kellom, M.; Kim, H.W.; Rapoport, S.I.; Reese, E.A. Neuroinflammation and synaptic loss. Neurochem. Res. 2012, 37, 903–910. [Google Scholar] [CrossRef]
- Scheff, S.W.; Price, D.A.; Schmitt, F.A.; Mufson, E.J. Hippocampal synaptic loss in early Alzheimer’s disease and mild cognitive impairment. Neurobiol. Aging 2006, 27, 1372–1384. [Google Scholar] [CrossRef]
- Appelberg, K.S.; Hovda, D.A.; Prins, M.L. The effects of a ketogenic diet on behavioral outcome after controlled cortical impact injury in the juvenile and adult rat. J. Neurotrauma 2009, 26, 497–506. [Google Scholar] [CrossRef]
- Krikorian, R.; Shidler, M.D.; Dangelo, K.; Couch, S.C.; Benoit, S.C.; Clegg, D.J. Dietary ketosis enhances memory in mild cognitive impairment. Neurobiol. Aging 2012, 33, e19–e27. [Google Scholar] [CrossRef]
- Maalouf, M.; Rho, J.M. Oxidative impairment of hippocampal long-term potentiation involves activation of protein phosphatase 2A and is prevented by ketone bodies. J. Neurosci. Res. 2008, 86, 3322–3330. [Google Scholar] [CrossRef]
- Rusek, M.; Pluta, R.; Ułamek-Kozioł, M.; Czuczwar, S.J. Ketogenic Diet in Alzheimer’s disease. Int. J. Mol. Sci. 2019, 20, 3892. [Google Scholar] [CrossRef] [PubMed]
- Chatterjee, P.; Fernando, M.; Fernando, B.; Dias, C.B.; Shah, T.; Silva, R.; Williams, S.; Pedrini, S.; Hillebrandt, H.; Goozee, K.; et al. Potential of coconut oil and medium chain triglycerides in the prevention and treatment of Alzheimer’s disease. Mech. Ageing Dev. 2020, 186, 111209. [Google Scholar] [CrossRef]
- Fernando, W.M.; Martins, I.J.; Goozee, K.G.; Brennan, C.S.; Jayasena, V.; Martins, R.N. The role of dietary coconut for the prevention and treatment of Alzheimer’s disease: Potential mechanisms of action. Br. J. Nutr. 2015, 114, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Rebello, C.J.; Keller, J.N.; Liu, A.G.; Johnson, W.D.; Greenway, F.L. Pilot feasibility and safety study examining the effect of medium chain triglyceride supplementation in subjects with mild cognitive impairment: A randomized controlled trial. BBA Clin. 2015, 3, 123–125. [Google Scholar] [CrossRef]
- Ota, M.; Matsuo, J.; Ishida, I.; Hattori, K.; Teraishi, T.; Tonouchi, H.; Ashida, K.; Takahashi, T.; Kunugi, H. Effect of a ketogenic meal on cognitive function in elderly adults: Potential for cognitive enhancement. Psychopharmacology 2016, 233, 3797–3802. [Google Scholar] [CrossRef]
- Pan, Y.; Larson, B.; Araujo, J.A.; Lau, W.; de Rivera, C.; Santana, R.; Gore, A.; Milgram, N.W. Dietary supplementation with medium-chain TAG has long-lasting cognition-enhancing effects in aged dogs. Br. J. Nutr. 2010, 103, 1746–1754. [Google Scholar] [CrossRef] [PubMed]
- Pawlosky, R.J.; Kashiwaya, Y.; King, M.T.; Veech, R.L. A Dietary Ketone Ester Normalizes Abnormal Behavior in a Mouse Model of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1044. [Google Scholar] [CrossRef] [PubMed]
- Alexander, G.E.; Chen, K.; Pietrini, P.; Rapoport, S.I.; Reiman, E.M. Longitudinal PET Evaluation of Cerebral Metabolic Decline in Dementia: A Potential Outcome Measure in Alzheimer’s Disease Treatment Studies. Am. J. Psychiatry 2002, 159, 738–745. [Google Scholar] [CrossRef]
- Reiman, E.M.; Chen, K.; Alexander, G.E.; Caselli, R.J.; Bandy, D.; Osborne, D.; Saunders, A.M.; Hardy, J. Functional brain abnormalities in young adults at genetic risk for late-onset Alzheimer’s dementia. Proc. Natl. Acad. Sci. USA 2004, 101, 284–289. [Google Scholar] [CrossRef] [PubMed]
- Henderson, S.T.; Vogel, J.L.; Barr, L.J.; Garvin, F.; Jones, J.J.; Costantini, L.C. Study of the ketogenic agent AC-1202 in mild to moderate Alzheimer’s disease: A randomized, double-blind, placebo-controlled, multicenter trial. Nutr. Metab. 2009, 6, 31. [Google Scholar] [CrossRef]
- Reger, M.A.; Henderson, S.T.; Hale, C.; Cholerton, B.; Baker, L.D.; Watson, G.S.; Hyde, K.; Chapman, D.; Craft, S. Effects of beta-hydroxybutyrate on cognition in memory-impaired adults. Neurobiol. Aging 2004, 25, 311–314. [Google Scholar] [CrossRef]
- Kashiwaya, Y.; Takeshima, T.; Mori, N.; Nakashima, K.; Clarke, K.; Veech, R.L. D-beta-hydroxybutyrate protects neurons in models of Alzheimer’s and Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2000, 97, 5440–5444. [Google Scholar] [CrossRef]
- Yin, J.X.; Maalouf, M.; Han, P.; Zhao, M.; Gao, M.; Dharshaun, T.; Ryan, C.; Whitelegge, J.; Wu, J.; Eisenberg, D.; et al. Ketones block amyloid entry and improve cognition in an Alzheimer’s model. Neurobiol. Aging 2016, 39, 25–37. [Google Scholar] [CrossRef]
- Singer, T.P.; Ramsay, R.R.; McKeown, K.; Trevor, A.; Castagnoli, N.E., Jr. Mechanism of the neurotoxicity of 1-methyl-4-phenylpyridinium (MPP+), the toxic bioactivation product of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Toxicology 1988, 49, 17–23. [Google Scholar] [CrossRef]
- Vanitallie, T.B.; Nonas, C.; Di Rocco, A.; Boyar, K.; Hyams, K.; Heymsfield, S.B. Treatment of Parkinson disease with diet-induced hyperketonemia: A feasibility study. Neurology 2005, 64, 728–730. [Google Scholar] [CrossRef]
- Ari, C.; Poff, A.M.; Held, H.E.; Landon, C.S.; Goldhagen, C.R.; Mavromates, N.; D’Agostino, D.P. Metabolic therapy with Deanna Protocol supplementation delays disease progression and extends survival in amyotrophic lateral sclerosis (ALS) mouse model. PLoS ONE 2014, 9, e103526. [Google Scholar] [CrossRef]
- Ari, C.; Murdun, C.; Goldhagen, C.; Koutnik, A.P.; Bharwani, S.R.; Diamond, D.M.; Kindy, M.; D’Agostino, D.P.; Kovacs, Z. Exogenous Ketone Supplements Improved Motor Performance in Preclinical Rodent Models. Nutrients 2020, 12, 2459. [Google Scholar] [CrossRef]
- Netzahualcoyotzi, C.; Tapia, R. Degeneration of spinal motor neurons by chronic AMPA-induced excitotoxicity in vivo and protection by energy substrates. Acta Neuropathol. Commun. 2015, 3, 27. [Google Scholar] [CrossRef]
- Zhao, Z.; Lange, D.J.; Voustianiouk, A.; MacGrogan, D.; Ho, L.; Suh, J.; Humala, N.; Thiyagarajan, M.; Wang, J.; Pasinetti, G.M. A ketogenic diet as a potential novel therapeutic intervention in amyotrophic lateral sclerosis. BMC Neurosci. 2006, 7, 29. [Google Scholar] [CrossRef]
- Zhao, W.; Varghese, M.; Vempati, P.; Dzhun, A.; Cheng, A.; Wang, J.; Lange, D.; Bilski, A.; Faravelli, I.; Pasinetti, G.M. Caprylic triglyceride as a novel therapeutic approach to effectively improve the performance and attenuate the symptoms due to the motor neuron loss in ALS disease. PLoS ONE 2012, 7, e49191. [Google Scholar] [CrossRef] [PubMed]
- Abg Abd Wahab, D.Y.; Gau, C.H.; Zakaria, R.; Muthu Karuppan, M.K.; A-Rahbi, B.S.; Abdullah, Z.; Alrafiah, A.; Abdullah, J.M.; Muthuraju, S. Review on Cross Talk between Neurotransmitters and Neuroinflammation in Striatum and Cerebellum in the Mediation of Motor Behaviour. Biomed. Res. Int. 2019, 14, 1767203. [Google Scholar] [CrossRef] [PubMed]
- Blasco, H.; Mavel, S.; Corcia, P.; Gordon, P.H. The glutamate hypothesis in ALS: Pathophysiology and drug development. Curr. Med. Chem. 2014, 21, 3551–3575. [Google Scholar] [CrossRef]
- Brichta, L.; Greengard, P.; Flajolet, M. Advances in the pharmacological treatment of Parkinson’s disease: Targeting neurotransmitter systems. Trends Neurosci. 2013, 36, 543–554. [Google Scholar] [CrossRef]
- Tisch, S.; Silberstein, P.; Limousin-Dowsey, P.; Jahanshahi, M. The basal ganglia: Anatomy, physiology, and pharmacology. Psychiatr. Clin. 2004, 27, 757–799. [Google Scholar] [CrossRef]
- D’Amelio, M.; Puglisi-Allegra, S.; Mercuri, N. The role of dopaminergic midbrain in Alzheimer’s disease: Translating basic science into clinical practice. Pharmacol. Res. 2018, 130, 414–419. [Google Scholar] [CrossRef]
- Huang, D.; Liu, D.; Yin, J.; Qian, T.; Shrestha, S.; Ni, H. Glutamate-glutamine and GABA in brain of normal aged and patients with cognitive impairment. Eur. Radiol. 2017, 27, 2698–2705. [Google Scholar] [CrossRef]
- Stanciu, G.D.; Luca, A.; Rusu, R.N.; Bild, V.; Beschea Chiriac, S.I.; Solcan, C.; Bild, W.; Ababei, D.C. Alzheimer’s Disease Pharmacotherapy in Relation to Cholinergic System Involvement. Biomolecules 2019, 10, 40. [Google Scholar] [CrossRef]
- Ma, S.; Hangya, B.; Leonard, C.S.; Wisden, W.; Gundlach, A.L. Dual-transmitter systems regulating arousal, attention, learning and memory. Neurosci. Biobehav. Rev. 2018, 85, 21–33. [Google Scholar] [CrossRef]
- Choong, C.J.; Sasaki, T.; Hayakawa, H.; Yasuda, T.; Baba, K.; Hirata, Y.; Uesato, S.; Mochizuki, H. A novel histone deacetylase 1 and 2 isoform-specific inhibitor alleviates experimental Parkinson’s disease. Neurobiol. Aging 2016, 37, 103–116. [Google Scholar] [CrossRef]
- D’Mello, S.R. Histone deacetylases as targets for the treatment of human neurodegenerative diseases. Drug News Perspect. 2009, 22, 513–524. [Google Scholar] [CrossRef]
- Feng, H.L.; Leng, Y.; Ma, C.H.; Zhang, J.; Ren, M.; Chuang, D.M. Combined lithium and valproate treatment delays disease onset, reduces neurological deficits and prolongs survival in an amyotrophic lateral sclerosis mouse model. Neuroscience 2008, 155, 567–572. [Google Scholar] [CrossRef]
- Lu, X.; Wang, L.; Yu, C.; Yu, D.; Yu, G. Histone Acetylation Modifiers in the Pathogenesis of Alzheimer’s Disease. Front. Cell. Neurosci. 2015, 9, 226. [Google Scholar] [CrossRef] [PubMed]
- Chuang, D.M.; Leng, Y.; Marinova, Z.; Kim, H.J.; Chiu, C.T. Multiple roles of HDAC inhibition in neurodegenerative conditions. Trends Neurosci. 2009, 32, 591–601. [Google Scholar] [CrossRef]
- Kazantsev, A.G.; Thompson, L.M. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat. Rev. Drug Discov. 2008, 7, 854–868. [Google Scholar] [CrossRef] [PubMed]
- Peleg, S.; Sananbenesi, F.; Zovoilis, A.; Burkhardt, S.; Bahari-Javan, S.; Agis-Balboa, R.C.; Cota, P.; Wittnam, J.L.; Gogol-Doering, A.; Opitz, L.; et al. Altered histone acetylation is associated with age-dependent memory impairment in mice. Science 2010, 328, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Taliyan, R.; Ramagiri, S. Histone deacetylase inhibitor, trichostatin A, improves learning and memory in high-fat diet-induced cognitive deficits in mice. J. Mol. Neurosci. 2015, 56, 1–11. [Google Scholar] [CrossRef]
- Peng, S.; Wuu, J.; Mufson, E.J.; Fahnestock, M. Precursor form of brain-derived neurotrophic factor and mature brain-derived neurotrophic factor are decreased in the pre-clinical stages of Alzheimer’s disease. J. Neurochem. 2005, 93, 1412–1421. [Google Scholar] [CrossRef] [PubMed]
- Qin, X.Y.; Cao, C.; Cawley, N.X.; Liu, T.T.; Yuan, J.; Loh, Y.P.; Cheng, Y. Decreased peripheral brain-derived neurotrophic factor levels in Alzheimer’s disease: A meta-analysis study (N = 7277). Mol. Psychiatry 2017, 22, 312–320. [Google Scholar] [CrossRef]
- Valenzuela, P.L.; Castillo-García, A.; Morales, J.S.; de la Villa, P.; Hampel, H.; Emanuele, E.; Lista, S.; Lucia, A. Exercise benefits on Alzheimer’s disease: State-of-the-science. Ageing Res. Rev. 2020, 62, 101108. [Google Scholar] [CrossRef]
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934. [Google Scholar] [CrossRef]
- Swanton, T.; Cook, J.; Beswick, J.A.; Freeman, S.; Lawrence, C.B.; Brough, D. Is Targeting the Inflammasome a Way Forward for Neuroscience Drug Discovery? SLAS Discov. 2018, 23, 991–1017. [Google Scholar] [CrossRef]
- Hyun, D.H.; Lee, M.; Halliwell, B.; Jenner, P. Proteasomal inhibition causes the formation of protein aggregates containing a wide range of proteins, including nitrated proteins. J. Neurochem. 2003, 86, 363–373. [Google Scholar] [CrossRef]
- Lim, J.E.; Song, M.; Jin, J.; Kou, J.; Pattanayak, A.; Lalonde, R.; Fukuchi, K. The effects of MyD88 deficiency on exploratory activity, anxiety, motor coordination, and spatial learning in C57BL/6 and APPswe/PS1dE9 mice. Behav. Brain Res. 2012, 227, 36–42. [Google Scholar] [CrossRef][Green Version]
- Liu, X.; Wu, Z.; Hayashi, Y.; Nakanishi, H. Age-dependent neuroinflammatory responses and deficits in long-term potentiation in the hippocampus during systemic inflammation. Neuroscience 2012, 216, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Hao, J.; Liu, R.; Turner, G.; Shi, F.D.; Rho, J.M. Inflammation-mediated memory dysfunction and effects of a ketogenic diet in a murine model of multiple sclerosis. PLoS ONE 2012, 7, e35476. [Google Scholar] [CrossRef] [PubMed]
- Pallàs, M.; Pizarro, J.G.; Gutierrez-Cuesta, J.; Crespo-Biel, N.; Alvira, D.; Tajes, M.; Yeste-Velasco, M.; Folch, J.; Canudas, A.M.; Sureda, F.X.; et al. Modulation of SIRT1 expression in different neurodegenerative models and human pathologies. Neuroscience 2008, 154, 1388–1397. [Google Scholar] [CrossRef]
- Herskovits, A.Z.; Guarente, L. SIRT1 in neurodevelopment and brain senescence. Neuron 2014, 81, 471–483. [Google Scholar] [CrossRef]
- Corpas, R.; Revilla, S.; Ursulet, S.; Castro-Freire, M.; Kaliman, P.; Petegnief, V.; Giménez-Llort, L.; Sarkis, C.; Pallàs, M.; Sanfeliu, C. SIRT1 Overexpression in Mouse Hippocampus Induces Cognitive Enhancement through Proteostatic and Neurotrophic Mechanisms. Mol. Neurobiol. 2017, 54, 5604–5619. [Google Scholar] [CrossRef]
- Wang, R.; Zhang, Y.; Li, J.; Zhang, C. Resveratrol ameliorates spatial learning memory impairment induced by Aβ1-42 in rats. Neuroscience 2017, 344, 39–47. [Google Scholar] [CrossRef]
- Gao, J.; Wang, W.Y.; Mao, Y.W.; Gräff, J.; Guan, J.S.; Pan, L.; Mak, G.; Kim, D.; Su, S.C.; Tsai, L.H. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 2010, 466, 1105–1109. [Google Scholar] [CrossRef]
- Michán, S.; Li, Y.; Chou, M.M.; Parrella, E.; Ge, H.; Long, J.M.; Allard, J.S.; Lewis, K.; Miller, M.; Xu, W.; et al. SIRT1 is essential for normal cognitive function and synaptic plasticity. J. Neurosci. 2010, 30, 9695–9707. [Google Scholar] [CrossRef] [PubMed]
- Han, S.; Choi, J.R.; Soon Shin, K.; Kang, S.J. Resveratrol upregulated heat shock proteins and extended the survival of G93A-SOD1 mice. Brain Res. 2012, 1483, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, Y.; Tang, L.; Zhang, N.; Fan, D. Protective effects of resveratrol through the up-regulation of SIRT1 expression in the mutant hSOD1-G93A-bearing motor neuron-like cell culture model of amyotrophic lateral sclerosis. Neurosci. Lett. 2011, 503, 250–255. [Google Scholar] [CrossRef] [PubMed]
- Mudò, G.; Mäkelä, J.; Di Liberto, V.; Tselykh, T.V.; Olivieri, M.; Piepponen, P.; Eriksson, O.; Mälkiä, A.; Bonomo, A.; Kairisalo, M.; et al. Transgenic expression and activation of PGC-1α protect dopaminergic neurons in the MPTP mouse model of Parkinson’s disease. Cell. Mol. Life Sci. 2012, 69, 1153–1165. [Google Scholar] [CrossRef]
- Karuppagounder, S.S.; Pinto, J.T.; Xu, H.; Chen, H.L.; Beal, M.F.; Gibson, G.E. Dietary supplementation with resveratrol reduces plaque pathology in a transgenic model of Alzheimer’s disease. Neurochem. Int. 2009, 54, 111–118. [Google Scholar] [CrossRef]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.; Wu, P.H.; Tarr, P.T.; Lindenberg, K.S.; St-Pierre, J.; Zhang, C.Y.; Mootha, V.K.; Jäger, S.; Vianna, C.R.; Reznick, R.M.; et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell 2004, 119, 121–135. [Google Scholar] [CrossRef]
- Wareski, P.; Vaarmann, A.; Choubey, V.; Safiulina, D.; Liiv, J.; Kuum, M.; Kaasik, A. PGC-1{alpha} and PGC-1{beta} regulate mitochondrial density in neurons. J. Biol. Chem. 2009, 284, 21379–21385. [Google Scholar] [CrossRef]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef]
- Gong, B.; Chen, F.; Pan, Y.; Arrieta-Cruz, I.; Yoshida, Y.; Haroutunian, V.; Pasinetti, G.M. SCFFbx2-E3-ligase-mediated degradation of BACE1 attenuates Alzheimer’s disease amyloidosis and improves synaptic function. Aging Cell 2010, 9, 1018–1031. [Google Scholar] [CrossRef] [PubMed]
- Kiaei, M.; Kipiani, K.; Chen, J.; Calingasan, N.Y.; Beal, M.F. Peroxisome proliferator-activated receptor-gamma agonist extends survival in transgenic mouse model of amyotrophic lateral sclerosis. Exp. Neurol. 2005, 191, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Varghese, M.; Yemul, S.; Pan, Y.; Cheng, A.; Marano, P.; Hassan, S.; Vempati, P.; Chen, F.; Qian, X.; et al. Peroxisome proliferator activator receptor gamma coactivator-1alpha (PGC-1α) improves motor performance and survival in a mouse model of amyotrophic lateral sclerosis. Mol. Neurodegener. 2011, 6, 51. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.; Yadav, A.; Chaturvedi, R.K. Peroxisome proliferator-activated receptors (PPARs) as therapeutic target in neurodegenerative disorders. Biochem. Biophys. Res. Commun. 2017, 483, 1166–1177. [Google Scholar] [CrossRef] [PubMed]
- D’ANGELO, M.; Castelli, V.; Catanesi, M.; Antonosante, A.; Dominguez-Benot, R.; Ippoliti, R.; Benedetti, E.; Cimini, A. PPARγ and Cognitive Performance. Int. J. Mol. Sci. 2019, 20, 5068. [Google Scholar] [CrossRef] [PubMed]
- Im, J.Y.; Lee, K.W.; Woo, J.M.; Junn, E.; Mouradian, M.M. DJ-1 induces thioredoxin 1 expression through the Nrf2 pathway. Hum. Mol. Genet. 2012, 21, 3013–3024. [Google Scholar] [CrossRef] [PubMed]
- Kensler, T.W.; Wakabayashi, N.; Biswal, S. Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 89–116. [Google Scholar] [CrossRef]
- Kincaid, B.; Bossy-Wetzel, E. Forever young: SIRT3 a shield against mitochondrial meltdown, aging, and neurodegeneration. Front. Aging Neurosci. 2013, 5, 48. [Google Scholar] [CrossRef] [PubMed]
- Ramesh, S.; Govindarajulu, M.; Lynd, T.; Briggs, G.; Adamek, D.; Jones, E.; Heiner, J.; Majrashi, M.; Moore, T.; Amin, R.; et al. SIRT3 activator Honokiol attenuates β-Amyloid by modulating amyloidogenic pathway. PLoS ONE 2018, 13, e0190350. [Google Scholar] [CrossRef]
- Song, W.; Song, Y.; Kincaid, B.; Bossy, B.; Bossy-Wetzel, E. Mutant SOD1G93A triggers mitochondrial fragmentation in spinal cord motor neurons: Neuroprotection by SIRT3 and PGC-1α. Neurobiol. Dis. 2013, 51, 72–81. [Google Scholar] [CrossRef] [PubMed]



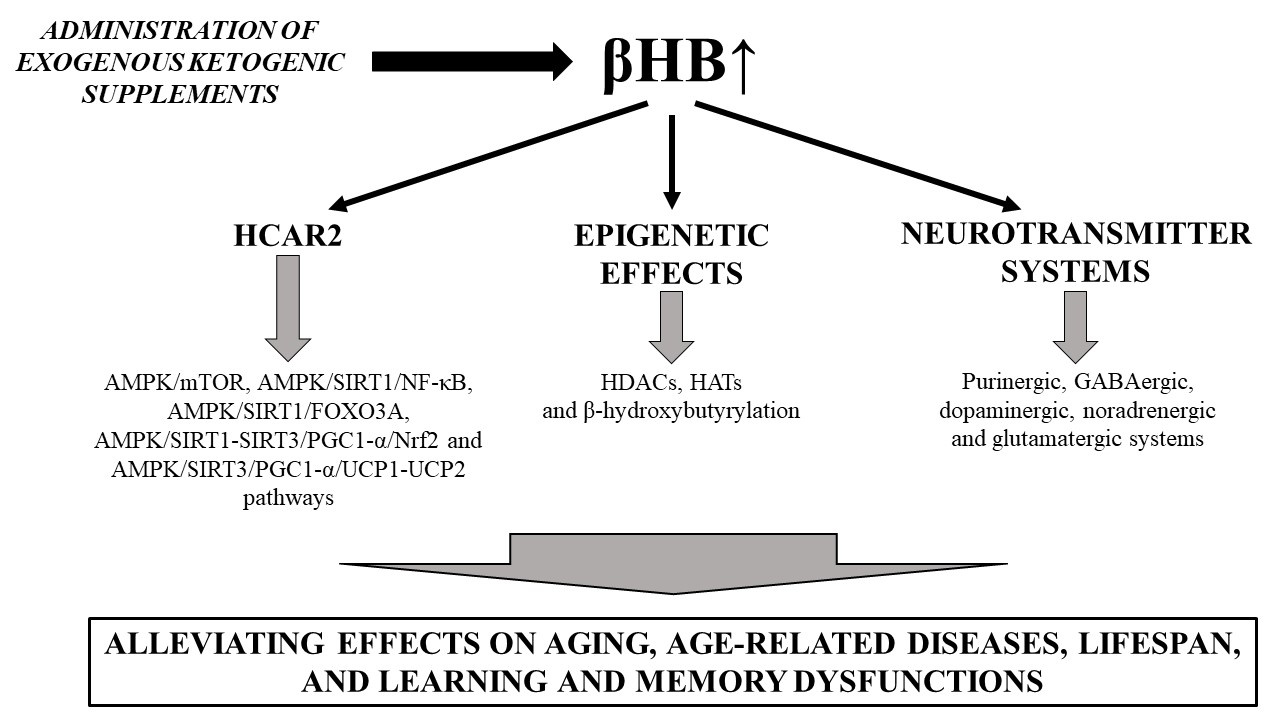{kind=link}
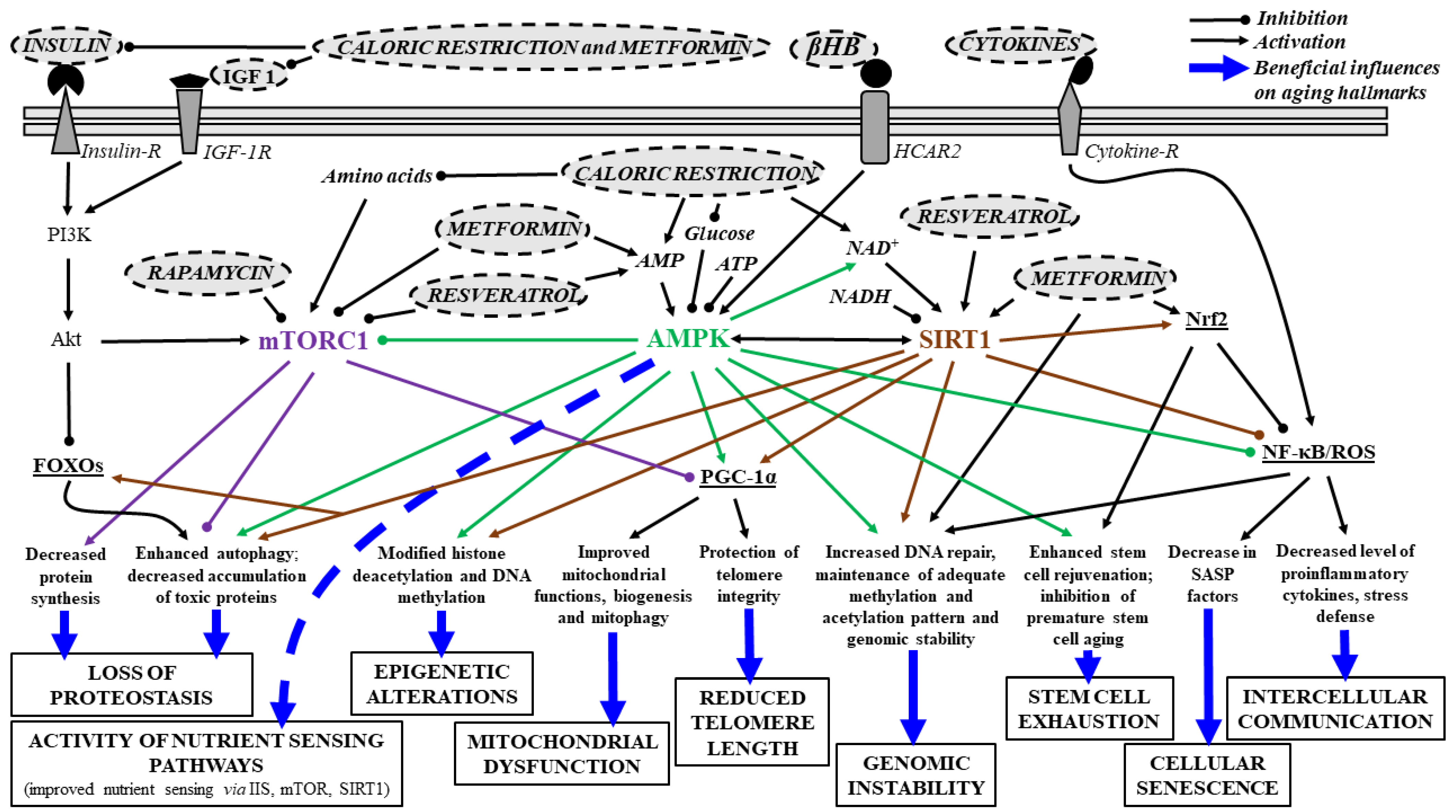{kind=link}
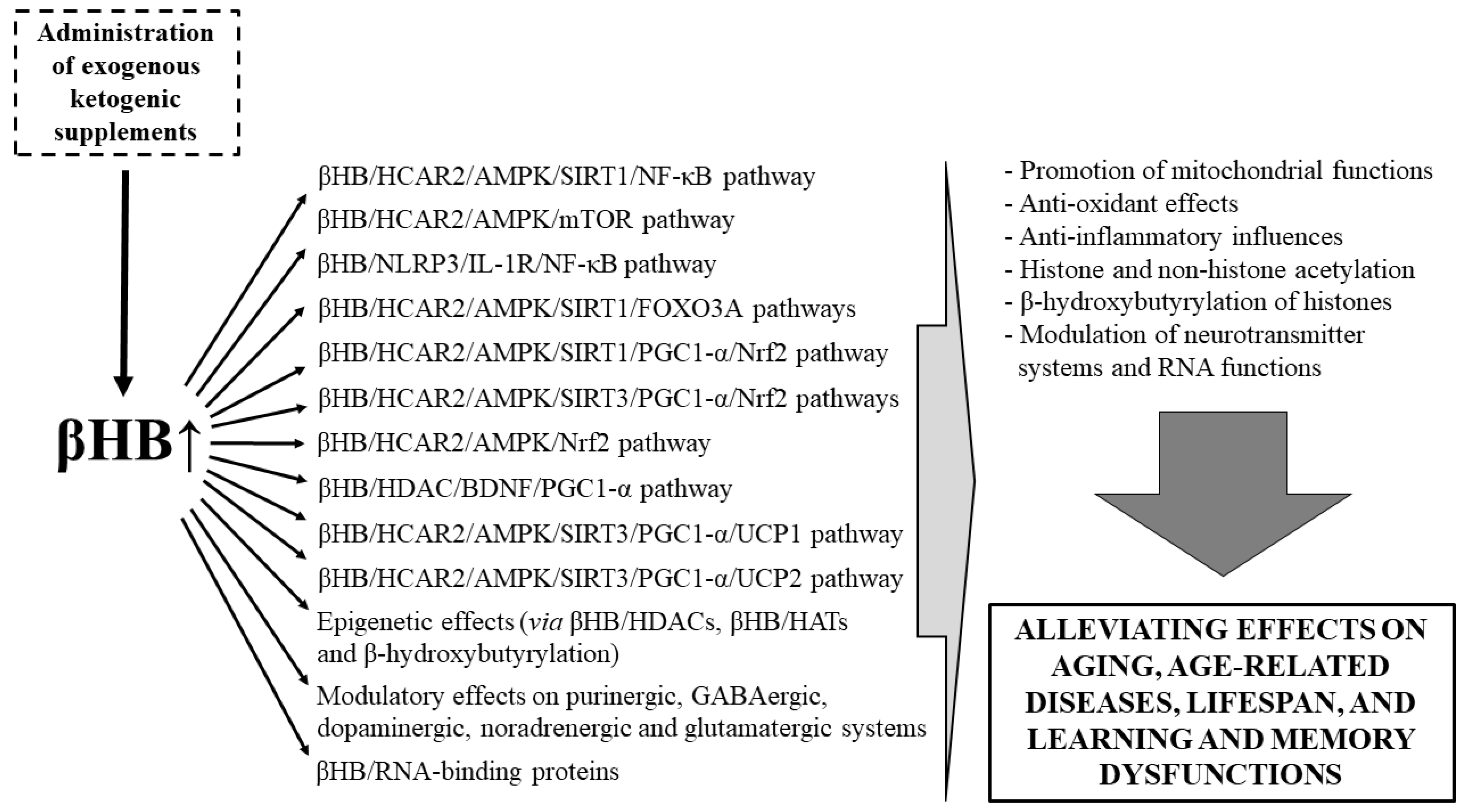{kind=link}
| Name (Components) | Dose and Route of Administration | Treatment Duration | Model Organism (Species) | Significant Increase in Blood βHB Level | Main Findings | Ref. |
|---|---|---|---|---|---|---|
| Beta-hydroxybutyrate (βHB) | ||||||
| βHB (DL-β-Hydroxybutyric acid sodium salt) | 1.5 mmol/kg/day (subcutaneous administration, 0.25 μL/h) | 4 weeks | A mouse model of Alzheimer’s disease (5XFAD) | No data | Improved learning and memory; attenuated Aβ accumulation | [50] |
| βHB + acetoacetate | 600 mg βHB/kg/day + 150 mg acetoacetate/kg/day (subcutaneous injection) | 2 months | A mouse model of Alzheimer’s disease (APPSwInd) | Yes | Improved cognitive performance; reduced Aβ accumulation | [329] |
| βHB | 0.4, 0.8, or 1.6 mmol/kg/day (subcutaneous administration, 1 μL/h) | 28 days | LPS-induced Parkinson’s disease rat model | No data | Beneficial effects on motor dysfunction; protection of dopaminergic neurons | [55] |
| βHB (D-βHB) | 0.4, 0.8, or 1.6 mmol/kg/day (subcutaneous administration, 1 μL/h) | 1 week | MPTP-induced Parkinson’s disease mouse model | Yes | Improved motor performance; decrease in MPTP-induced dopaminergic neurodegeneration | [258] |
| Ketone esters (KEs) | ||||||
| KE (R,S-1,3-butanediol acetoacetate diester: BD-AcAc2; standard rodent chow mixed at 10% BD-AcAc2 by volume and 1% saccharin) | Ad libitum (oral intake) | 8 weeks | A mouse model of Angelman syndrome (UBE3Atm1Alb/J null mutation mice) | Yes | Improved motor coordination, learning and memory | [41] |
| KE (comprised of D-β-hydroxybutyrate and (R)-1,3-butanediol; 125 g KE/1000 g diet) | Animals were fed a 4 to 5 g pellet/animal at approximately 06:00 hours each day (oral intake) | 8 months | A mouse model of Alzheimer’s disease (3xTgAD) | Yes | Improvements in performance on learning and memory tests; decreased Aβ and hyperphosphorylated tau deposition | [43] |
| KE [ketone monoester, (R)-3-hydroxybutyl (R)-3-hydroxybutyrate] + MCT and coconut oil (CO) mixture (4:3) | Normal diet + 28.7 g of the KE thrice daily + 165 mL/day of the MCT/CO mixture (oral intake) | 20 months | A patient with Alzheimer’s disease dementia | Yes | Improving behavior as well as cognitive and daily-activity performance | [47] |
| Medium chain triglycerides (MCTs) | ||||||
| MCT (97% caprylic acid + 3% capric acid; a normal diet supplemented with 5.5% MCT) | Dogs were fed once/day for about one hour; about 200 g supplemented diet/day/animal (oral intake) | 8 months | Aged dogs | Yes | Improvements in learning ability and attention | [322] |
| MCT (the diet was mixed with Deanna protocol/DP at 22% by weight; DP contained 10% MCT high in caprylic acid) | Ad libitum (oral intake) | 6–10 weeks | A mouse model of Amyotrophic lateral sclerosis (SOD1-G93A) | No | Better motor performance, improved (lower) neurological scores and extended survival time | [332] |
| MCT (a diet in which 35% of the calories was derived from triheptanoin) | Ad libitum (oral intake) | 24 weeks | A mouse model of Amyotrophic lateral sclerosis (SOD1-G93A) | Yes | Protection against motor neuron loss; improved motor function | [48] |
| MCT [a diet containing 10% (w/w) caprylic acid] | Ad libitum; about 3 g diet/day was consumed/animal (oral intake) | About 12 weeks | A mouse model of Amyotrophic lateral sclerosis (SOD1-G93A) | Yes | Protection against motor neuron loss; improved motor function | [336] |
| MCT (NeoBee 895, >95% of the fatty acids are caprylic acid; the remainder consists of caproic and capric acids) | 40 mL MCT (oral intake) | Single administration | Adult subjects with Alzheimer’s disease or mild cognitive impairment | Yes | Improvement in cognitive functions (in patients without APOE ε4 allele) | [327] |
| MCT (AC-1202, an MCT composed of glycerin and, almost entirely, caprylic acid, NeoBee 895) | Normal diet + 20 g MCT/day/patient (oral intake) | 3 months | Humans with mild to moderate Alzheimer’s disease | Yes | Improvement in cognitive performance (in patients without APOE ε4 allele) | [326] |
| MCT (50 g Ketogenic meal, Ketonformula containing 20 g of MCTs: 15 g caprylic acid + 5 g capric acid) | 50 g ketogenic meal (oral intake) | Single administration | Humans; elderly, non-demented | Yes | Positive effects on working memory, visual attention, and task switching | [321] |
| MCT (MCT drink: a 12% emulsion of Captex 355, containing 60% caprylic acid and 40% capric acid) | Normal diet + 15 g MCT twice/day/patient in a ketogenic drink (oral intake) | 6 months | Humans; aged participants with mild cognitive impairment | Yes | Improved executive function, memory, and language | [32] |
| MCT (MCT oil, Nestle™) | Normal diet + 56 g MCT/day/patient (oral intake) | 24 weeks | Humans; adults with mild cognitive impairment | Yes | Improved memory | [320] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kovács, Z.; Brunner, B.; Ari, C. Beneficial Effects of Exogenous Ketogenic Supplements on Aging Processes and Age-Related Neurodegenerative Diseases. Nutrients 2021, 13, 2197. https://doi.org/10.3390/nu13072197
Kovács Z, Brunner B, Ari C. Beneficial Effects of Exogenous Ketogenic Supplements on Aging Processes and Age-Related Neurodegenerative Diseases. Nutrients. 2021; 13(7):2197. https://doi.org/10.3390/nu13072197
Chicago/Turabian StyleKovács, Zsolt, Brigitta Brunner, and Csilla Ari. 2021. "Beneficial Effects of Exogenous Ketogenic Supplements on Aging Processes and Age-Related Neurodegenerative Diseases" Nutrients 13, no. 7: 2197. https://doi.org/10.3390/nu13072197
APA StyleKovács, Z., Brunner, B., & Ari, C. (2021). Beneficial Effects of Exogenous Ketogenic Supplements on Aging Processes and Age-Related Neurodegenerative Diseases. Nutrients, 13(7), 2197. https://doi.org/10.3390/nu13072197