
Vitamin C Deficiency in the Young Brain—Findings from Experimental Animal Models †


Abstract
1. Introduction
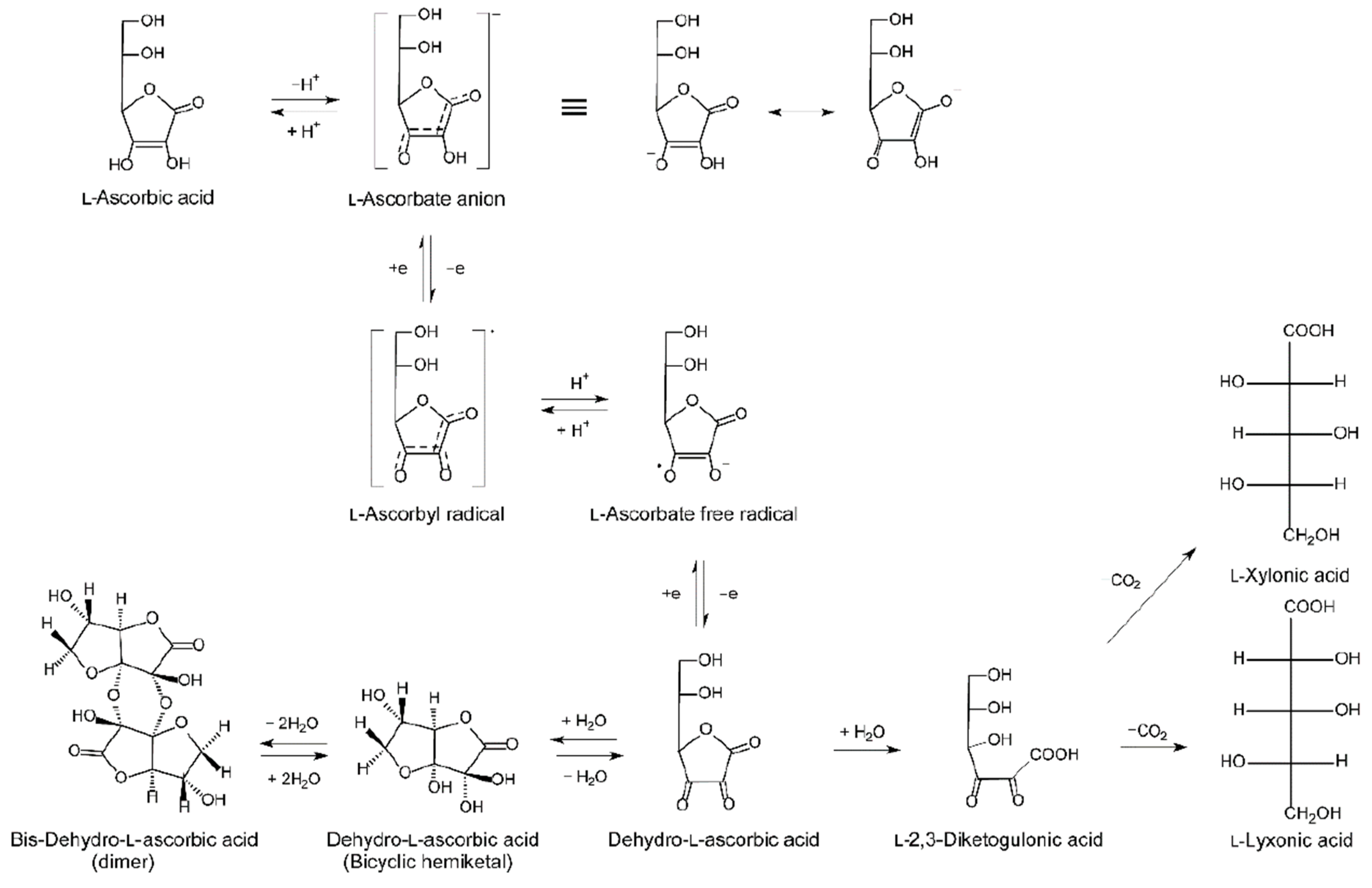
2. Vitamin C Regulation In Vivo
2.1. Cellular Vitamin C Uptake
2.1.1. Ascorbic Acid Transport
2.1.2. Dehydroascorbic Acid Transport
2.2. Cellular Vitamin C Efflux
2.3. Vitamin C Pharmacokinetics
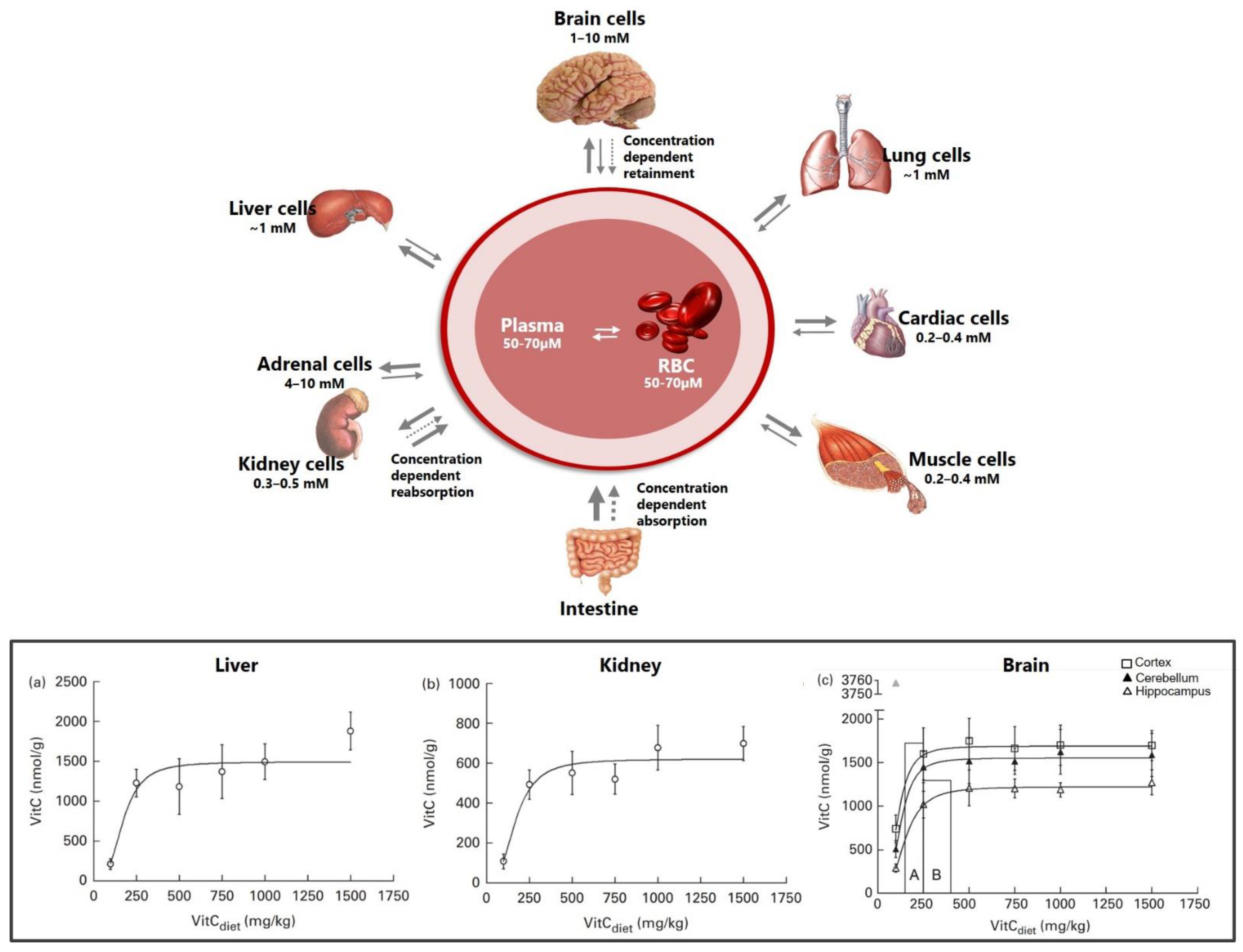
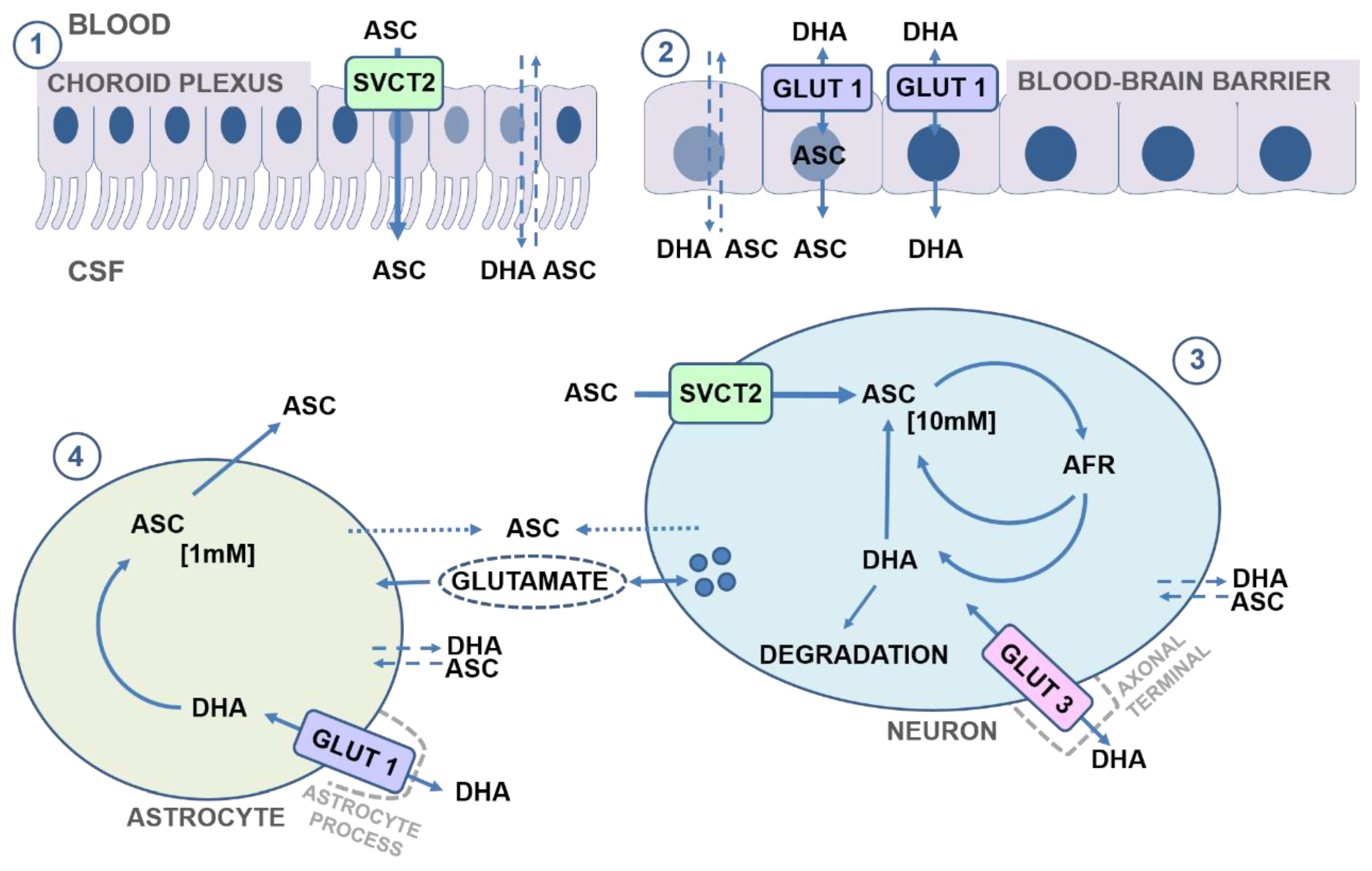
3. Vitamin C Transport to the Brain
3.1. Crossing the Blood–Brain Barrier
3.1.1. ASC Transport
3.1.2. DHA Transport
3.2. Inside the Brain
3.2.1. Vitamin C Transport to Neurons
3.2.2. Vitamin C Transport to Neuroglia
4. Vitamin C Functions in the Brain
4.1. Preventing Oxidation of Poly-Unsaturated Fatty Acids
4.2. Co-Factor for Fe2+-2-Oxogluterate-Dependent Dioxygenases
4.2.1. Collagen Synthesis
4.2.2. Hypoxia-Inducible Transcription Factors
4.2.3. Epigentic Regulation
4.2.4. Carnitine Availability
4.3. Signal Transduction
4.3.1. Monoaminergic Neurotransmitters
4.3.2. Glutamate Signaling
5. Effects of Vitamin C Deficiency on Brain Development
5.1. Prenatal Effects of Vitamin C Deficiency
5.1.1. Fetal Vitamin C Levels
5.1.2. Neuronal Consequences
5.1.3. Effect of Prenatal vitC Deficiency on Offspring Growth
5.1.4. Clinical Studies
5.2. Postnatal Effects of Vitamin C Deficiency
5.2.1. Perinatal Period and Early Life
5.2.2. Lipid Peroxidation
5.2.3. Changes in Brain Structure and Function
5.2.4. Infants Born Preterm
5.3. Vitamin C Deficiency in Young Life
5.3.1. Redox Homeostasis
5.3.2. Changes in Brain Structure and Function
5.3.3. Vitamin C Status in Children
6. Potential Challenges When Evaluating Clinical Studies of Vitamin C
7. Concluding Remarks
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Burns, J. Missing step in man, monkey and guinea pig required for the biosynthesis of L-ascorbic acid. Nature 1957, 180, 553. [Google Scholar] [CrossRef] [PubMed]
- Pauling, L. Evolution and the need for ascorbic acid. Proc. Natl. Acad. Sci. USA 1970, 67, 1643–1648. [Google Scholar] [CrossRef]
- Chatterjee, I.B. Evolution and the biosynthesis of ascorbic acid. Science 1973, 182, 1271–1272. [Google Scholar] [CrossRef] [PubMed]
- Chaudhuri, C.R.; Chatterjee, I. L–ascorbic acid synthesis in birds: Phylogenetic trend. Science 1969, 164, 435–436. [Google Scholar] [CrossRef]
- Birney, E.C.; Jennes, R.; Ayaz, K.M. Inability of bats to synthesise L-ascorbic acid. Nature 1976, 260, 626–628. [Google Scholar] [CrossRef]
- Lachapelle, M.Y.; Drouin, G. Inactivation dates of the human and guinea pig vitamin C genes. Genetica 2011, 139, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Jenness, R.; Birney, E.C.; Ayaz, K.L.; Buzzell, D.M. Ontogenetic development of L-gulonolactone oxidase activity in several vertebrates. Comp. Biochem. Physiol. B Comp. Biochem. 1984, 78, 167–173. [Google Scholar] [CrossRef]
- Nishikimi, M.; Fukuyama, R.; Minoshima, S.; Shimizu, N.; Yagi, K. Cloning and chromosomal mapping of the human nonfunctional gene for L-gulono-gamma-lactone oxidase, the enzyme for L–ascorbic acid biosynthesis missing in man. J. Biol. Chem. 1994, 269, 13685–13688. [Google Scholar] [CrossRef]
- Nishikimi, M.; Kawai, T.; Yagi, K. Guinea pigs possess a highly mutated gene for L-gulono-gamma-lactone oxidase, the key enzyme for L-ascorbic acid biosynthesis missing in this species. J. Biol. Chem. 1992, 267, 21967–21972. [Google Scholar] [CrossRef]
- Carr, A.C.; Lykkesfeldt, J. Discrepancies in global vitamin C recommendations: A review of RDA criteria and underlying health perspectives. Crit. Rev. Food. Sci. Nutr. 2021, 61, 742–755. [Google Scholar] [CrossRef]
- Levine, M.; Conry-Cantilena, C.; Wang, Y.; Welch, R.W.; Washko, P.W.; Dhariwal, K.R.; Park, J.B.; Lazarev, A.; Graumlich, J.F.; King, J.; et al. Vitamin C pharmacokinetics in healthy volunteers: Evidence for a recommended dietary allowance. Proc. Natl. Acad. Sci. USA 1996, 93, 3704–3709. [Google Scholar] [CrossRef] [PubMed]
- Frei, B.; Birlouez-Aragon, I.; Lykkesfeldt, J. Authors’ perspective: What is the optimum intake of vitamin C in humans? Crit. Rev. Food. Sci. Nutr. 2012, 52, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.L.; Hodges, R.E. Serum Levels of Vitamin-C in Relation to Dietary and Supplemental Intake of Vitamin-C in Smokers and Nonsmokers. Ann. N.Y. Acad. Sci. 1987, 498, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Lykkesfeldt, J. Smoking depletes vitamin C: Should smokers be recommended to take supplements? In Cigarette Smoke and Oxidative Stress; Halliwell, B., Poulsen, H.E., Eds.; Springer: Berlin/Heidelberg, Germany, 2006; pp. 237–260. [Google Scholar]
- Schleicher, R.L.; Carroll, M.D.; Ford, E.S.; Lacher, D.A. Serum vitamin C and the prevalence of vitamin C deficiency in the United States: 2003–2004 National Health and Nutrition Examination Survey (NHANES). Am. J. Clin. Nutr. 2009, 90, 1252–1263. [Google Scholar] [CrossRef] [PubMed]
- Madruga de Oliveira, A.; Rondo, P.H.; Barros, S.B. Concentrations of ascorbic acid in the plasma of pregnant smokers and nonsmokers and their newborns. Int. J. Vitam. Nutr. Res. 2004, 74, 193–198. [Google Scholar] [CrossRef]
- Madruga de Oliveira, A.; Rondo, P.H.; Mastroeni, S.S.; Oliveira, J.M. Plasma concentrations of ascorbic acid in parturients from a hospital in Southeast Brazil. Clin. Nutr. 2008, 27, 228–232. [Google Scholar] [CrossRef]
- Villalpando, S.; Montalvo-Velarde, I.; Zambrano, N.; Garcia-Guerra, A.; Ramirez-Silva, C.I.; Shamah-Levy, T.; Rivera, J.A. Vitamins A, and C and folate status in Mexican children under 12 years and women 12–49 years: A probabilistic national survey. Salud Publica Mex. 2003, 45, S508–S519. [Google Scholar] [CrossRef]
- Ma, A.-G.M.D.; Schouten, E.G.M.D.; Wang, Y.M.D.; Xu, R.-X.M.D.; Zheng, M.-C.M.D.; Li, Y.; Wang, Q.M.M.; Sun, Y.M.M. Micronutrient status in anemic and non–anemic Chinese women in the third trimester of pregnancy. Asia Pac. J. Clin. Nutr. 2009, 18, 41–47. [Google Scholar]
- Rowe, S.; Carr, A.C. Global Vitamin C Status and Prevalence of Deficiency: A Cause for Concern? Nutrients 2020, 12, 2008. [Google Scholar] [CrossRef]
- Carr, A.C.; Rowe, S. Factors Affecting Vitamin C Status and Prevalence of Deficiency: A Global Health Perspective. Nutrients 2020, 12, 1963. [Google Scholar] [CrossRef]
- Lykkesfeldt, J. On the effect of vitamin C intake on human health: How to (mis) interprete the clinical evidence. Redox Biol. 2020, 23, 101532. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.E.; Forman, R.E.; Chen, B.T.; Avshalumov, M.V.; Cragg, S.J.; Drew, K.L. Brain antioxidant regulation in mammals and anoxia–tolerant reptiles: Balanced for neuroprotection and neuromodulation. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2002, 133, 515–525. [Google Scholar] [CrossRef]
- Lykkesfeldt, J.; Trueba, G.P.; Poulsen, H.E.; Christen, S. Vitamin C deficiency in weanling guinea pigs: Differential expression of oxidative stress and DNA repair in liver and brain. Br. J. Nutr. 2007, 98, 1116–1119. [Google Scholar] [CrossRef] [PubMed]
- Institute of Medicine (US). Dietary Reference Intakes for Vitamin C, Vitamin E, Selenium, and Carotenoids: A Report of the Panel on Dietary Antioxidants and Related Compounds, Subcommittees on Upper Reference Levels of Nutrients and of Interpretation and Use of Dietary Reference Intakes, and the Standing Committee on the Scientific Evaluation of Dietary Reference Intakes, Food and Nutrition Board, Institute of Medicine; National Academy Press: Washington, DC, USA, 2000; p. 506. [Google Scholar]
- Sotiriou, S.; Gispert, S.; Cheng, J.; Wang, Y.H.; Chen, A.; Hoogstraten-Miller, S.; Miller, G.F.; Kwon, O.; Levine, M.; Guttentag, S.H.; et al. Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nat. Med. 2002, 8, 514–517. [Google Scholar] [CrossRef]
- Harrison, F.E.; Dawes, S.M.; Meredith, M.E.; Babaev, V.R.; Li, L.; May, J.M. Low vitamin C and increased oxidative stress and cell death in mice that lack the sodium–dependent vitamin C transporter SVCT2. Free Radic. Biol. Med. 2010, 49, 821–829. [Google Scholar] [CrossRef]
- Lykkesfeldt, J.; Tveden-Nyborg, P. The Pharmacokinetics of Vitamin C. Nutrients 2019, 11, 2412. [Google Scholar] [CrossRef] [PubMed]
- Buettner, G.R. The pecking order of free radicals and antioxidants: Lipid peroxidation, a–tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef]
- Lindblad, M.; Tveden-Nyborg, P.; Lykkesfeldt, J. Regulation of vitamin C homeostasis during deficiency. Nutrients 2013, 5, 2860–2879. [Google Scholar] [CrossRef]
- Wilson, J.X. Regulation of vitamin C transport. Annu. Rev. Nutr. 2005, 25, 105–125. [Google Scholar] [CrossRef]
- Tsukaguchi, H.; Tokui, T.; Mackenzie, B.; Berger, U.V.; Chen, X.Z.; Wang, Y.; Brubaker, R.F.; Hediger, M.A. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature 1999, 399, 70–75. [Google Scholar] [CrossRef]
- Hediger, M.A. New view at C. Nat. Med. 2002, 8, 445–446. [Google Scholar] [CrossRef]
- Padayatty, S.; Levine, M. Vitamin C: The known and the unknown and Goldilocks. Oral Dis. 2016, 22, 463–493. [Google Scholar] [CrossRef] [PubMed]
- Savini, I.; Rossi, A.; Pierro, C.; Avigliano, L.; Catani, M.V. SVCT1 and SVCT2: Key proteins for vitamin C uptake. Amino. Acids 2008, 34, 347–355. [Google Scholar] [CrossRef]
- May, J.M. The SLC23 family of ascorbate transporters: Ensuring that you get and keep your daily dose of vitamin C. Br. J. Pharmacol. 2011, 164, 1793–1801. [Google Scholar] [CrossRef]
- Daruwala, R.; Song, J.; Koh, W.S.; Rumsey, S.C.; Levine, M. Cloning and functional characterization of the human sodium-dependent vitamin C transporters hSVCT1 and hSVCT2. FEBS Lett. 1999, 460, 480–484. [Google Scholar] [CrossRef]
- Wang, Y.; Mackenzie, B.; Tsukaguchi, H.; Weremowicz, S.; Morton, C.C.; Hediger, M.A. Human vitamin C (L-ascorbic acid) transporter SVCT1. Biochem. Biophys. Res. Commun. 2000, 267, 488–494. [Google Scholar] [CrossRef]
- Boyer, J.C.; Campbell, C.E.; Sigurdson, W.J.; Kuo, S.M. Polarized localization of vitamin C transporters, SVCT1 and SVCT2, in epithelial cells. Biochem. Biophys. Res. Commun. 2005, 334, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Godoy, A.; Ormazabal, V.; Moraga-Cid, G.; Zuniga, F.A.; Sotomayor, P.; Barra, V.; Vasquez, O.; Montecinos, V.; Mardones, L.; Guzman, C.; et al. Mechanistic insights and functional determinants of the transport cycle of the ascorbic acid transporter SVCT2. Activation by sodium and absolute dependence on bivalent cations. J. Biol. Chem. 2007, 282, 615–624. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, S.; Inoue, K.; Murata, T.; Kamigaso, S.; Yasujima, T.; Maeda, J.Y.; Yoshida, Y.; Ohta, K.Y.; Yuasa, H. Identification and Functional Characterization of the First Nucleobase Transporter in Mammals Implication in the Species Difference in the Intestinal Absorption Mechanism of Nucleobases and their Analogs Between Higher Primates and Other Mammals. J. Biol. Chem. 2010, 285, 6522–6531. [Google Scholar] [CrossRef]
- Bürzle, M.; Suzuki, Y.; Ackermann, D.; Miyazaki, H.; Maeda, N.; Clémençon, B.; Burrier, R.; Hediger, M.A. The sodium-dependent ascorbic acid transporter family SLC23. Mol. Asp. Med. 2013, 34, 436–454. [Google Scholar] [CrossRef]
- Michels, A.J.; Hagen, T.M.; Frei, B. Human genetic variation influences vitamin C homeostasis by altering vitamin C transport and antioxidant enzyme function. Annu. Rev. Nutr. 2013, 33, 45–70. [Google Scholar] [CrossRef] [PubMed]
- Corpe, C.P.; Tu, H.; Eck, P.; Wang, J.; Faulhaber–Walter, R.; Schnermann, J.; Margolis, S.; Padayatty, S.; Sun, H.; Wang, Y.; et al. Vitamin C transporter Slc23a1 links renal reabsorption, vitamin C tissue accumulation, and perinatal survival in mice. J. Clin. Investig. 2010, 120, 1069–1083. [Google Scholar] [CrossRef] [PubMed]
- Lykkesfeldt, J. Increased oxidative damage in vitamin C deficiency is accompanied by induction of ascorbic acid recycling capacity in young but not mature guinea pigs. Free Radic. Res. 2002, 36, 567–574. [Google Scholar] [CrossRef] [PubMed]
- May, J.M.; Qu, Z.-C.; Whitesell, R.R.; Cobb, C.E. Ascorbate recycling in human erythrocytes: Role of GSH in reducing dehydroascorbate. Free Radic. Biol. Med. 1996, 20, 543–551. [Google Scholar] [CrossRef]
- Rumsey, S.C.; Daruwala, R.; Al-Hasani, H.; Zarnowski, M.J.; Simpson, I.A.; Levine, M. Dehydroascorbic Acid Transport by GLUT4 in XenopusOocytes and Isolated Rat Adipocytes. J. Biol. Chem. 2000, 275, 28246–28253. [Google Scholar] [CrossRef] [PubMed]
- Rumsey, S.C.; Kwon, O.; Xu, G.W.; Burant, C.F.; Simpson, I.; Levine, M. Glucose transporter isoforms GLUT1 and GLUT3 transport dehydroascorbic acid. J. Biol. Chem. 1997, 272, 18982–18989. [Google Scholar] [CrossRef]
- Corpe, C.P.; Eck, P.; Wang, J.; Al-Hasani, H.; Levine, M. Intestinal dehydroascorbic acid (DHA) transport mediated by the facilitative sugar transporters, GLUT2 and GLUT8. J. Biol. Chem. 2013, 288, 9092–9101. [Google Scholar] [CrossRef]
- Korcok, J.; Dixon, S.J.; Lo, T.C.; Wilson, J.X. Differential effects of glucose on dehydroascorbic acid transport and intracellular ascorbate accumulation in astrocytes and skeletal myocytes. Brain Res. 2003, 993, 201–207. [Google Scholar] [CrossRef]
- Malo, C.; Wilson, J. Glucose modulates vitamin C transport in adult human small intestinal brush border membrane vesicles. J. Nutr. 2000, 130, 63–69. [Google Scholar] [CrossRef]
- Padayatty, S.J.; Levine, M. New insights into the physiology and pharmacology of vitamin C. Can. Med. Assoc. J. 2001, 164, 353–355. [Google Scholar]
- Linster, C.L.; Van, S.E. Vitamin C. Biosynthesis, recycling and degradation in mammals. FEBS J. 2007, 274, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Eck, P.; Kwon, O.; Chen, S.; Mian, O.; Levine, M. The human sodium-dependent ascorbic acid transporters SLC23A1 and SLC23A2 do not mediate ascorbic acid release in the proximal renal epithelial cell. Physiol. Rep. 2013, 1. [Google Scholar] [CrossRef] [PubMed]
- Mendiratta, S.; Qu, Z.-C.; May, J.M. Erythrocyte ascorbate recycling: Antioxidant effects in blood. Free Radic. Biol. Med. 1998, 24, 789–797. [Google Scholar] [CrossRef]
- Upston, J.M.; Karjalainen, A.; Bygrave, F.L.; Stocker, R. Efflux of hepatic ascorbate: A potential contributor to the maintenance of plasma vitamin C. Biochem. J. 1999, 342, 49–56. [Google Scholar] [CrossRef]
- May, J.M.; Qu, Z.-C. Ascorbic acid efflux and re-uptake in endothelial cells: Maintenance of intracellular ascorbate. Mol. Cell. Biochem. 2009, 325, 79–88. [Google Scholar] [CrossRef]
- Viscovich, M.; Lykkesfeldt, J.; Poulsen, H.E. Vitamin C pharmacokinetics of plain and slow release formulations in smokers. Clin. Nutr. 2004, 23, 1043–1050. [Google Scholar] [CrossRef]
- Fürst, J.; Gschwentner, M.; Ritter, M.; Botta, G.; Jakab, M.; Mayer, M.; Garavaglia, L.; Bazzini, C.; Rodighiero, S.; Meyer, G. Molecular and functional aspects of anionic channels activated during regulatory volume decrease in mammalian cells. Pflügers. Archiv. 2002, 444, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Siushansian, R.; Dixon, S.J.; Wilson, J.X. Osmotic swelling stimulates ascorbate efflux from cerebral astrocytes. J. Neurochem. 1996, 66, 1227–1233. [Google Scholar] [CrossRef]
- May, J.M.; Li, L.; Hayslett, K.; Qu, Z.-C. Ascorbate transport and recycling by SH-SY5Y neuroblastoma cells: Response to glutamate toxicity. Neurochem. Res. 2006, 31, 785–794. [Google Scholar] [CrossRef]
- Padayatty, S.J.; Doppman, J.L.; Chang, R.; Wang, Y.; Gill, J.; Papanicolaou, D.A.; Levine, M. Human adrenal glands secrete vitamin C in response to adrenocorticotrophic hormone. Am. J. Clin. Nutr. 2007, 86, 145–149. [Google Scholar] [CrossRef]
- Wilson, J.X.; Peters, C.E.; Sitar, S.M.; Daoust, P.; Gelb, A.W. Glutamate stimulates ascorbate transport by astrocytes. Brain Res. 2000, 858, 61–66. [Google Scholar] [CrossRef]
- Yusa, T. Increased extracellular ascorbate release reflects glutamate re-uptake during the early stage of reperfusion after forebrain ischemia in rats. Brain Res. 2001, 897, 104–113. [Google Scholar] [CrossRef]
- Levine, M.; Wang, Y.; Padayatty, S.J.; Morrow, J. A new recommended dietary allowance of vitamin C for healthy young women. Proc. Natl. Acad. Sci. USA 2001, 98, 9842–9846. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.E. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000, 23, 209–216. [Google Scholar] [CrossRef]
- Sogaard, D.; Lindblad, M.M.; Paidi, M.D.; Hasselholt, S.; Lykkesfeldt, J.; Tveden–Nyborg, P. In vivo vitamin C deficiency in guinea pigs increases ascorbate transporters in liver but not kidney and brain. Nutr. Res. 2014, 34, 639–645. [Google Scholar] [CrossRef]
- Harrison, F.E.; Green, R.J.; Dawes, S.M.; May, J.M. Vitamin C distribution and retention in the mouse brain. Brain Res. 2010, 1348, 181–186. [Google Scholar] [CrossRef] [PubMed]
- Maeda, N.; Hagihara, H.; Nakata, Y.; Hiller, S.; Wilder, J.; Reddick, R. Aortic wall damage in mice unable to synthesize ascorbic acid. Proc. Natl. Acad. Sci. USA 2000, 97, 841–846. [Google Scholar] [CrossRef]
- Takahashi, M.; Miyata, S.; Fujii, J.; Inai, Y.; Ueyama, S.; Araki, M.; Soga, T.; Fujinawa, R.; Nishitani, C.; Ariki, S. In vivo role of aldehyde reductase. Biochim. Biophys. Acta (BBA) Gen. Subj. 2012, 1820, 1787–1796. [Google Scholar] [CrossRef] [PubMed]
- Furusawa, H.; Sato, Y.; Tanaka, Y.; Inai, Y.; Amano, A.; Iwama, M.; Kondo, Y.; Handa, S.; Murata, A.; Nishikimi, M. Vitamin C is not essential for carnitine biosynthesis in vivo: Verification in vitamin C–depleted senescence marker protein–30/gluconolactonase knockout mice. Biol. Pharmaceut. Bull. 2008, 31, 1673–1679. [Google Scholar] [CrossRef]
- Kondo, Y.; Inai, Y.; Sato, Y.; Handa, S.; Kubo, S.; Shimokado, K.; Goto, S.; Nishikimi, M.; Maruyama, N.; Ishigami, A. Senescence marker protein 30 functions as gluconolactonase in L–ascorbic acid biosynthesis, and its knockout mice are prone to scurvy. Proc. Natl. Acad. Sci. USA 2006, 103, 5723–5728. [Google Scholar] [CrossRef]
- Hasselholt, S.; Tveden-Nyborg, P.; Lykkesfeldt, J. Distribution of vitamin C is tissue specific with early saturation of the brain and adrenal glands following differential oral dose regimens in guinea pigs. Br. J. Nutr. 2015, 113, 1539–1549. [Google Scholar] [CrossRef]
- Tveden-Nyborg, P.; Vogt, L.; Schjoldager, J.G.; Jeannet, N.; Hasselholt, S.; Paidi, M.D.; Christen, S.; Lykkesfeldt, J. Maternal vitamin C deficiency during pregnancy persistently impairs hippocampal neurogenesis in offspring of guinea pigs. PLoS ONE 2012, 7, e48488. [Google Scholar] [CrossRef] [PubMed]
- Rice, M.E.; Russo-Menna, I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience 1998, 82, 1213–1223. [Google Scholar] [CrossRef]
- Rice, M.E.; Lee, E.J.; Choy, Y. High levels of ascorbic acid, not glutathione, in the CNS of anoxia-tolerant reptiles contrasted with levels in anoxia-intolerant species. J. Neurochem. 1995, 64, 1790–1799. [Google Scholar] [CrossRef] [PubMed]
- Tveden-Nyborg, P.; Hasselholt, S.; Miyashita, N.; Moos, T.; Poulsen, H.E.; Lykkesfeldt, J. Chronic Vitamin C Deficiency does not Accelerate Oxidative Stress in Ageing Brains of Guinea Pigs. Basic Clin. Pharmacol. Toxicol. 2012, 110, 524–529. [Google Scholar] [CrossRef] [PubMed]
- Lykkesfeldt, J.; Moos, T. Age-dependent change in Vitamin C status: A phenomenon of maturation rather than of ageing. Mech. Ageing Dev. 2005, 126, 892–898. [Google Scholar] [CrossRef] [PubMed]
- Paidi, M.D.; Schjoldager, J.G.; Lykkesfeldt, J.; Tveden-Nyborg, P. Chronic Vitamin C Deficiency Promotes Redox Imbalance in the Brain but Does Not Alter Sodium-Dependent Vitamin C Transporter 2 Expression. Nutrients 2014, 6, 1809–1822. [Google Scholar] [CrossRef] [PubMed]
- Spector, R. Micronutrient homeostasis in mammalian brain and cerebrospinal fluid. J. Neurochem. 1989, 53, 1667–1674. [Google Scholar] [CrossRef] [PubMed]
- Berger, U.V.; Lu, X.C.; Liu, W.; Tang, Z.; Slusher, B.S.; Hediger, M.A. Effect of middle cerebral artery occlusion on mRNA expression for the sodium-coupled vitamin C transporter SVCT2 in rat brain. J. Neurochem. 2003, 86, 896–906. [Google Scholar] [CrossRef]
- Garcia, M.D.L.; Salazar, K.; Millan, C.; Rodriguez, F.; Montecinos, H.; Caprile, T.; Silva, C.; Cortes, C.; Reinicke, K.; Vera, J.C.; et al. Sodium vitamin C cotransporter SVCT2 is expressed in hypothalamic glial cells. Glia 2005, 50, 32–47. [Google Scholar] [CrossRef]
- Mun, G.H.; Kim, M.J.; Lee, J.H.; Kim, H.J.; Chung, Y.H.; Chung, Y.B.; Kang, J.S.; Hwang, Y.I.; Oh, S.H.; Kim, J.G.; et al. Immunohistochemical study of the distribution of sodium–dependent vitamin C transporters in adult rat brain. J. Neurosci. Res. 2006, 83, 919–928. [Google Scholar] [CrossRef]
- Hammarström, L. Autoradiographic studies on the distribution of C14-labelled ascorbic acid and dehydroascorbic acid. Acta Physiol. Scand. 1966, 70, 1–83. [Google Scholar] [CrossRef]
- Angelow, S.; Haselbach, M.; Galla, H.-J. Functional characterisation of the active ascorbic acid transport into cerebrospinal fluid using primary cultured choroid plexus cells. Brain Res. 2003, 988, 105–113. [Google Scholar] [CrossRef]
- Mack, W.J.; Mocco, J.; Ducruet, A.F.; Laufer, I.; King, R.G.; Zhang, Y.; Guo, W.; Pinsky, D.J.; Connolly, E.S., Jr. A cerebroprotective dose of intravenous citrate/sorbitol-stabilized dehydroascorbic acid is correlated with increased cerebral ascorbic acid and inhibited lipid peroxidation after murine reperfused stroke. Neurosurgery 2006, 59, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Nualart, F.; Mack, L.; García, A.; Cisternas, P.; Bongarzone, E.R.; Heitzer, M.; Jara, N.; Martínez, F.; Ferrada, L.; Espinoza, F. Vitamin C transporters, recycling and the bystander effect in the nervous system: SVCT2 versus gluts. J. Stem Cell. Res. Therapy 2014, 4, 209. [Google Scholar] [CrossRef] [PubMed]
- Leino, R.L.; Gerhart, D.Z.; van Bueren, A.M.; McCall, A.L.; Drewes, L.R. Ultrastructural localization of GLUT 1 and GLUT 3 glucose transporters in rat brain. J. Neurosci. Res. 1997, 49, 617–626. [Google Scholar] [CrossRef]
- Lykkesfeldt, J. Ascorbate and dehydroascorbic acid as reliable biomarkers of oxidative stress: Analytical reproducibility and long-term stability of plasma samples subjected to acidic deproteinization. Cancer Epidemiol. Biomark. Prev. 2007, 16, 2513–2516. [Google Scholar] [CrossRef] [PubMed]
- Lykkesfeldt, J.; Loft, S.; Nielsen, J.B.; Poulsen, H.E. Ascorbic acid and dehydroascorbic acid as biomarkers of oxidative stress caused by smoking. Am. J. Clin. Nutr. 1997, 65, 959–963. [Google Scholar] [CrossRef]
- Castro, M.; Caprile, T.; Astuya, A.; Millan, C.; Reinicke, K.; Vera, J.C.; Vasquez, O.; Aguayo, L.G.; Nualart, F. High-affinity sodium-vitamin C co-transporters (SVCT) expression in embryonic mouse neurons. J. Neurochem. 2001, 78, 815–823. [Google Scholar] [CrossRef]
- Caprile, T.; Salazar, K.; Astuya, A.; Cisternas, P.; Silva-Alvarez, C.; Montecinos, H.; Millán, C.; de los Angeles García, M.; Nualart, F. The Na+-dependent L-ascorbic acid transporter SVCT2 expressed in brainstem cells, neurons, and neuroblastoma cells is inhibited by flavonoids. J. Neurochem. 2009, 108, 563–577. [Google Scholar] [CrossRef] [PubMed]
- Qiu, S.; Li, L.; Weeber, E.J.; May, J.M. Ascorbate transport by primary cultured neurons and its role in neuronal function and protection against excitotoxicity. J. Neurosci. Res. 2007, 85, 1046–1056. [Google Scholar] [CrossRef]
- Kratzing, C.; Kelly, J.; Oelrichs, B. Ascorbic acid in neural tissues. J. Neurochem. 1982, 39, 625–627. [Google Scholar] [CrossRef] [PubMed]
- Milby, K.; Oke, A.; Adams, R. Detailed mapping of ascorbate distribution in rat brain. Neurosciletters 1982, 28, 15–20. [Google Scholar]
- Mefford, I.N.; Oke, A.F.; Adams, R.N. Regional distribution of ascorbate in human brain. Brain Res. 1981, 212, 223–226. [Google Scholar] [CrossRef]
- Meredith, M.E.; Harrison, F.E.; May, J.M. Differential regulation of the ascorbic acid transporter SVCT2 during development and in response to ascorbic acid depletion. Biochem. Biophys. Res. Commun. 2011, 414, 737–742. [Google Scholar] [CrossRef]
- Vera, J.C.; Rivas, C.I.; Fischbarg, J.; Golde, D.W. Mammalian facilitative hexose transporters mediate the transport of dehydroascorbic acid. Nature 1993, 364, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Mantych, G.J.; James, D.E.; Chung, H.D.; Devaskar, S.U. Cellular localization and characterization of Glut 3 glucose transporter isoform in human brain. Endocrinology 1992, 131, 1270–1278. [Google Scholar] [CrossRef]
- Morgello, S.; Uson, R.R.; Schwartz, E.J.; Haber, R.S. The human blood-brain barrier glucose transporter (GLUT1) is a glucose transporter of gray matter astrocytes. Glia 1995, 14, 43–54. [Google Scholar] [CrossRef]
- García-Krauss, A.; Ferrada, L.; Astuya, A.; Salazar, K.; Cisternas, P.; Martínez, F.; Ramírez, E.; Nualart, F. Dehydroascorbic acid promotes cell death in neurons under oxidative stress: A protective role for astrocytes. Mol. Neurobiol. 2016, 53, 5847–5863. [Google Scholar] [CrossRef]
- Song, J.H.; Shin, S.H.; Ross, G.M. Oxidative stress induced by ascorbate causes neuronal damage in an in vitro system. Brain Res. 2001, 895, 66–72. [Google Scholar] [CrossRef]
- Silva–Alvarez, C.; Salazar, K.; Cisternas, P.; Martinez, F.; Liour, S.; Jara, N.; Bertinat, R.; Nualart, F. Apical polarization of SVCT2 in apical radial glial cells and progenitors during brain development. Mol. Neurobiol. 2017, 54, 5449–5467. [Google Scholar] [CrossRef] [PubMed]
- Salazar, K.; Cerda, G.; Martínez, F.; Sarmiento, J.M.; González, C.; Rodríguez, F.; García-Robles, M.; Tapia, J.C.; Cifuentes, M.; Nualart, F. SVCT2 transporter expression is post-natally induced in cortical neurons and its function is regulated by its short isoform. J. Neurochem. 2014, 130, 693–706. [Google Scholar] [CrossRef] [PubMed]
- Oyarce, K.; Silva-Alvarez, C.; Ferrada, L.; Martínez, F.; Salazar, K.; Nualart, F. SVCT2 is expressed by cerebellar precursor cells, which differentiate into neurons in response to ascorbic acid. Mol. Neurobiol. 2018, 55, 1136–1149. [Google Scholar] [CrossRef]
- Berger, U.V.; Hediger, M.A. The vitamin C transporter SVCT2 is expressed by astrocytes in culture but not in situ. Neuroreport 2000, 11, 1395–1399. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.A.; Pozo, M.; Cortés, C.; de los Angeles García, M.; Concha, I.I.; Nualart, F. Intracellular ascorbic acid inhibits transport of glucose by neurons, but not by astrocytes. J. Neurochem. 2007, 102, 773–782. [Google Scholar] [CrossRef] [PubMed]
- Astuya, A.; Caprile, T.; Castro, M.; Salazar, K.; de los Angeles García, M.; Reinicke, K.; Rodríguez, F.; Vera, J.C.; Millán, C.; Ulloa, V. Vitamin C uptake and recycling among normal and tumor cells from the central nervous system. J. Neurosci. Res 2005, 79, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Nualart, F.J.; Rivas, C.I.; Montecinos, V.P.; Godoy, A.S.; Guaiquil, V.H.; Golde, D.W.; Vera, J.C. Recycling of vitamin C by a bystander effect. J. Biol. Chem. 2003, 278, 10128–10133. [Google Scholar] [CrossRef]
- Siushansian, R.; Tao, L.; Dixon, S.J.; Wilson, J.X. Cerebral astrocytes transport ascorbic acid and dehydroascorbic acid through distinct mechanisms regulated by cyclic AMP. J. Neurochem. 1997, 68, 2378–2385. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.X.; Dragan, M. Sepsis inhibits recycling and glutamate-stimulated export of ascorbate by astrocytes. Free Radic. Biol. Med. 2005, 39, 990–998. [Google Scholar] [CrossRef]
- Iwata, N.; Okazaki, M.; Xuan, M.; Kamiuchi, S.; Matsuzaki, H.; Hibino, Y. Orally administrated ascorbic acid suppresses neuronal damage and modifies expression of SVCT2 and GLUT1 in the brain of diabetic rats with cerebral ischemia-reperfusion. Nutrients 2014, 6, 1554–1577. [Google Scholar] [CrossRef]
- Gess, B.; Sevimli, S.; Strecker, J.K.; Young, P.; Schabitz, W.R. Sodium-dependent vitamin C transporter 2 (SVCT2) expression and activity in brain capillary endothelial cells after transient ischemia in mice. PLoS ONE 2011, 6, e17139. [Google Scholar] [CrossRef]
- Salazar, K.; Martínez, F.; Pérez-Martín, M.; Cifuentes, M.; Trigueros, L.; Ferrada, L.; Espinoza, F.; Saldivia, N.; Bertinat, R.; Forman, K. SVCT2 expression and function in reactive astrocytes is a common event in different brain pathologies. Mol. Neurobiol. 2018, 55, 5439–5452. [Google Scholar] [CrossRef]
- Frei, B.; England, L.; Ames, B.N. Ascorbate is an outstanding antioxidant in human blood plasma. Proc. Natl. Acad. Sci. USA 1989, 86, 6377–6381. [Google Scholar] [CrossRef] [PubMed]
- Frei, B.; Stocker, R.; Ames, B.N. Antioxidant defenses and lipid peroxidation in human blood plasma. Proc. Natl. Acad. Sci. USA 1988, 85, 9748–9752. [Google Scholar] [CrossRef]
- Packer, J.; Slater, T.; Willson, R.L. Direct observation of a free radical interaction between vitamin E and vitamin C. Nature 1979, 278, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Nandi, A.; Mukhopadhyay, M.; Mukhopadhyay, C.K.; Chatterjee, I.B. Ascorbate protects guinea pig tissues against lipid peroxidation. Free Radic. Biol. Med. 1994, 16, 417–426. [Google Scholar] [CrossRef]
- Nandi, A.; Mukhopadhyay, C.K.; Ghosh, M.K.; Chattopadhyay, D.J.; Chatterjee, I.B. Evolutionary significance of vitamin C biosynthesis in terrestrial vertebrates. Free Radic. Biol. Med. 1997, 22, 1047–1054. [Google Scholar] [CrossRef]
- Halliwell, B. Reactive oxygen species and the central nervous system. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef]
- Driver, A.S.; Kodavanti, P.R.S.; Mundy, W.R. Age-related changes in reactive oxygen species production in rat brain homogenates. Neurotoxicol. Teratol. 2000, 22, 175–181. [Google Scholar] [CrossRef]
- Bazinet, R.P.; Layé, S. Polyunsaturated fatty acids and their metabolites in brain function and disease. Nature Rev. Neurosci. 2014, 15, 771–785. [Google Scholar] [CrossRef]
- Tveden-Nyborg, P.; Lykkesfeldt, J. Does vitamin C deficiency result in impaired brain development in infants? Redox Rep. 2009, 14, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Di Miceli, M.; Bosch-Bouju, C.; Layé, S. PUFA and their derivatives in neurotransmission and synapses: A new hallmark of synaptopathies. Proc. Nutr. Soc. 2020, 79, 1–16. [Google Scholar] [CrossRef]
- Sastry, P.S. Lipids of nervous tissue: Composition and metabolism. Prog. Lipid Res. 1985, 24, 69–176. [Google Scholar] [CrossRef]
- Postila, P.A.; Róg, T. A Perspective: Active Role of Lipids in Neurotransmitter Dynamics. Mol. Neurobiol. 2020, 57, 910–925. [Google Scholar] [CrossRef] [PubMed]
- Araque, A.; Castillo, P.E.; Manzoni, O.J.; Tonini, R. Synaptic functions of endocannabinoid signaling in health and disease. Neuropharmacology 2017, 124, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Haj–Dahmane, S.; Shen, R.-Y.; Elmes, M.W.; Studholme, K.; Kanjiya, M.P.; Bogdan, D.; Thanos, P.K.; Miyauchi, J.T.; Tsirka, S.E.; Deutsch, D.G. Fatty-acid-binding protein 5 controls retrograde endocannabinoid signaling at central glutamate synapses. Proc. Natl. Acad. Sci. USA 2018, 115, 3482–3487. [Google Scholar] [CrossRef]
- Stella, N. Endocannabinoid signaling in microglial cells. Neuropharmacology 2009, 56, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Montine, K.S.; Olson, S.J.; Amarnath, V.; Whetsell, W.O., Jr.; Graham, D.G.; Montine, T.J. Immunohistochemical detection of 4–hydroxy-2-nonenal adducts in Alzheimer’s disease is associated with inheritance of APOE4. Am. J. Path. 1997, 150, 437. [Google Scholar]
- Pamplona, R.; Naudí, A.; Gavín, R.; Pastrana, M.A.; Sajnani, G.; Ilieva, E.V.; del Río, J.A.; Portero-Otín, M.; Ferrer, I.; Requena, J.R. Increased oxidation, glycoxidation, and lipoxidation of brain proteins in prion disease. Free Radic. Biol. Med. 2008, 45, 1159–1166. [Google Scholar] [CrossRef]
- Schippling, S.; Kontush, A.; Arlt, S.; Buhmann, C.; Stürenburg, H.-J.; Mann, U.; Müller-Thomsen, T.; Beisiegel, U. Increased lipoprotein oxidation in Alzheimer’s disease. Free Radic. Biol. Med. 2000, 28, 351–360. [Google Scholar] [CrossRef]
- Long, J.; Liu, C.; Sun, L.; Gao, H.; Liu, J. Neuronal mitochondrial toxicity of malondialdehyde: Inhibitory effects on respiratory function and enzyme activities in rat brain mitochondria. Neurochem. Res. 2009, 34, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Aguado, T.; Palazuelos, J.; Monory, K.; Stella, N.; Cravatt, B.; Lutz, B.; Marsicano, G.; Kokaia, Z.; Guzmán, M.; Galve-Roperh, I. The endocannabinoid system promotes astroglial differentiation by acting on neural progenitor cells. J. Neurosci. 2006, 26, 1551–1561. [Google Scholar] [CrossRef] [PubMed]
- Harkany, T.; Guzmán, M.; Galve–Roperh, I.; Berghuis, P.; Devi, L.A.; Mackie, K. The emerging functions of endocannabinoid signaling during CNS development. Trends Pharm. Sci. 2007, 28, 83–92. [Google Scholar] [CrossRef]
- Rashid, M.A.; Katakura, M.; Kharebava, G.; Kevala, K.; Kim, H.Y. N-docosahexaenoylethanolamine is a potent neurogenic factor for neural stem cell differentiation. J. Neurochem. 2013, 125, 869–884. [Google Scholar] [CrossRef]
- Harrison, F.E.; Meredith, M.E.; Dawes, S.M.; Saskowski, J.L.; May, J.M. Low ascorbic acid and increased oxidative stress in gulo(-/-) mice during development. Brain Res. 2010, 1349, 143–152. [Google Scholar] [CrossRef]
- Hasselholt, S.; Tveden–Nyborg, P.; Lykkesfeldt, J. Vitamin C: It’s role in brain development and cognition. In Nutrition and Cognitive Performance: A Developmental Perspective; Riby, L., Smith, M.A., Foster, J.K., Eds.; Palgrave-MacMillan: London, UK, 2012; pp. 29–52. [Google Scholar]
- Walmsley, A.R.; Batten, M.R.; Lad, U.; Bulleid, N.J. Intracellular retention of procollagen within the endoplasmic reticulum is mediated by prolyl 4-hydroxylase. J. Biol. Chem. 1999, 274, 14884–14892. [Google Scholar] [CrossRef]
- Yoshikawa, K.; Takahashi, S.; Imamura, Y.; Sado, Y.; Hayashi, T. Secretion of Non-helical collagenous pblypeptides of α1 (IV) and α2 (IV) chains upon depletion of ascorbate by cultured human cells. J. Biochem. 2001, 129, 929–936. [Google Scholar] [CrossRef]
- Hara, K.; Akiyama, Y. Collagen-related abnormalities, reduction in bone quality, and effects of menatetrenone in rats with a congenital ascorbic acid deficiency. J. Bone Miner. Metab. 2009, 27, 324–332. [Google Scholar] [CrossRef]
- Hodges, R.E.; Baker, E.M.; Hood, J.; SAUBERLICH, H.E.; March, S.C. Experimental scurvy in man. Am. J. Clin. Nutr. 1969, 22, 535–548. [Google Scholar] [CrossRef]
- Gone, I.; Wadu, M.; Goodman, M. Capillary hemorrhage in ascorbic-acid-deficient guinea pigs. Ultrastructural basis. Arch. Pathol. 1968, 85, 493–502. [Google Scholar]
- Weinstein, M.; Babyn, P.; Zlotkin, S. An orange a day keeps the doctor away: Scurvy in the year 2000. Pediatrics 2001, 108, E55. [Google Scholar] [CrossRef] [PubMed]
- Flashman, E.; Davies, S.L.; Yeoh, K.K.; Schofield, C.J. Investigating the dependence of the hypoxia-inducible factor hydroxylases (factor inhibiting HIF and prolyl hydroxylase domain 2) on ascorbate and other reducing agents. Biochem. J. 2010, 427, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Osipyants, A.I.; Poloznikov, A.A.; Smirnova, N.A.; Hushpulian, D.M.; Khristichenko, A.Y.; Chubar, T.A.; Zakhariants, A.A.; Ahuja, M.; Gaisina, I.N.; Thomas, B. L–ascorbic acid: A true substrate for HIF prolyl hydroxylase? Biochimie 2018, 147, 46–54. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Lai, U.H.; Zhu, L.; Singh, A.; Ahmed, M.; Forsyth, N.R. Reactive oxygen species formation in the brain at different oxygen levels: The role of hypoxia inducible factors. Front. Cell Develop. Biol. 2018, 6, 132. [Google Scholar] [CrossRef]
- Schönenberger, M.J.; Kovacs, W.J. Hypoxia signaling pathways: Modulators of oxygen-related organelles. Front. Cell Dev. Biol. 2015, 3, 42. [Google Scholar] [CrossRef]
- Chandel, N.; Maltepe, E.; Goldwasser, E.; Mathieu, C.; Simon, M.; Schumacker, P. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc. Natl. Acad. Sci. USA 1998, 95, 11715–11720. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Qu, Y.; Li, J.; Xiong, Y.; Mao, M.; Mu, D. Relationship between HIF-1α expression and neuronal apoptosis in neonatal rats with hypoxia–ischemia brain injury. Brain Res. 2007, 1180, 133–139. [Google Scholar] [CrossRef]
- Chen, W.; Jadhav, V.; Tang, J.; Zhang, J.H. HIF–1α inhibition ameliorates neonatal brain injury in a rat pup hypoxic-ischemic model. Neurobiol. Dis. 2008, 31, 433–441. [Google Scholar] [CrossRef]
- Bae, Y.-H.; Joo, H.; Bae, J.; Hyeon, S.J.; Her, S.; Ko, E.; Choi, H.G.; Ryu, H.; Hur, E.-M.; Bu, Y. Brain injury induces HIF-1α-dependent transcriptional activation of LRRK2 that exacerbates brain damage. Cell Death Disease 2018, 9, 1–19. [Google Scholar] [CrossRef]
- Fan, X.; Heijnen, C.J.; van der Kooij, M.A.; Groenendaal, F.; van Bel, F. The role and regulation of hypoxia–inducible factor-1α expression in brain development and neonatal hypoxic–ischemic brain injury. Brain Res. Rev. 2009, 62, 99–108. [Google Scholar] [CrossRef]
- Sharp, F.R.; Bernaudin, M. HIF1 and oxygen sensing in the brain. Nature Rev. Neurosci. 2004, 5, 437–448. [Google Scholar] [CrossRef]
- Lee, Y.M.; Jeong, C.H.; Koo, S.Y.; Son, M.J.; Song, H.S.; Bae, S.K.; Raleigh, J.A.; Chung, H.Y.; Yoo, M.A.; Kim, K.W. Determination of hypoxic region by hypoxia marker in developing mouse embryos in vivo: A possible signal for vessel development. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2001, 220, 175–186. [Google Scholar] [CrossRef]
- Tomita, S.; Ueno, M.; Sakamoto, M.; Kitahama, Y.; Ueki, M.; Maekawa, N.; Sakamoto, H.; Gassmann, M.; Kageyama, R.; Ueda, N. Defective brain development in mice lacking the Hif-1α gene in neural cells. Mol. Cell. Biol. 2003, 23, 6739–6749. [Google Scholar] [CrossRef]
- Chen, J.; Guo, L.; Zhang, L.; Wu, H.; Yang, J.; Liu, H.; Wang, X.; Hu, X.; Gu, T.; Zhou, Z. Vitamin C modulates TET1 function during somatic cell reprogramming. Nat. Genet. 2013, 45, 1504. [Google Scholar] [CrossRef]
- Wang, T.; Chen, K.; Zeng, X.; Yang, J.; Wu, Y.; Shi, X.; Qin, B.; Zeng, L.; Esteban, M.A.; Pan, G. The histone demethylases Jhdm1a/1b enhance somatic cell reprogramming in a vitamin-C dependent manner. Cell Stem Cell 2011, 9, 575–587. [Google Scholar] [CrossRef] [PubMed]
- Yin, R.; Mao, S.-Q.; Zhao, B.; Chong, Z.; Yang, Y.; Zhao, C.; Zhang, D.; Huang, H.; Gao, J.; Li, Z.; et al. Ascorbic Acid Enhances Tet–Mediated 5–Methylcytosine Oxidation and Promotes DNA Demethylation in Mammals. J. Am. Chem. Soc. 2013, 135, 10396–10403. [Google Scholar] [CrossRef]
- Blaschke, K.; Ebata, K.T.; Karimi, M.M.; Zepeda-Martínez, J.A.; Goyal, P.; Mahapatra, S.; Tam, A.; Laird, D.J.; Hirst, M.; Rao, A. Vitamin C induces Tet-dependent DNA demethylation and a blastocyst–like state in ES cells. Nature 2013, 500, 222–226. [Google Scholar] [CrossRef]
- DiTroia, S.P.; Percharde, M.; Guerquin, M.-J.; Wall, E.; Collignon, E.; Ebata, K.T.; Mesh, K.; Mahesula, S.; Agathocleous, M.; Laird, D.J. Maternal vitamin C regulates reprogramming of DNA methylation and germline development. Nature 2019, 573, 271–275. [Google Scholar] [CrossRef]
- Kawahori, K.; Kondo, Y.; Yuan, X.; Kawasaki, Y.; Hanzawa, N.; Tsujimoto, K.; Wada, F.; Kohda, T.; Ishigami, A.; Yamada, T. Ascorbic acid during the suckling period is required for proper DNA demethylation in the liver. Sci. Rep. 2020, 10, 1–13. [Google Scholar] [CrossRef] [PubMed]
- He, X.B.; Kim, M.; Kim, S.Y.; Yi, S.H.; Rhee, Y.H.; Kim, T.; Lee, E.H.; Park, C.H.; Dixit, S.; Harrison, F.E. Vitamin C Facilitates Dopamine Neuron Differentiation in Fetal Midbrain Through TET 1-and JMJD 3-Dependent Epigenetic Control Manner. Stem Cells 2015, 33, 1320–1332. [Google Scholar] [CrossRef]
- Hahn, M.A.; Qiu, R.; Wu, X.; Li, A.X.; Zhang, H.; Wang, J.; Jui, J.; Jin, S.-G.; Jiang, Y.; Pfeifer, G.P. Dynamics of 5-hydroxymethylcytosine and chromatin marks in Mammalian neurogenesis. Cell Rep. 2013, 3, 291–300. [Google Scholar] [CrossRef]
- Vaz, F.M.; Wanders, R.J. Carnitine biosynthesis in mammals. Biochem. J. 2002, 361, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Rebouche, C.J. Ascorbic-Acid and Carnitine Biosynthesis. Am. J. Clin. Nutr. 1991, 54, S1147–S1152. [Google Scholar] [CrossRef]
- Nelson, P.J.; Pruitt, R.E.; Henderson, L.L.; Jenness, R.; Henderson, L.M. Effect of ascorbic acid deficiency on the in vivo synthesis of carnitine. Biochim. Biophys. Acta (BBA) Gen. Subj. 1981, 672, 123–127. [Google Scholar] [CrossRef]
- Rebouche, C.J. The ability of guinea pigs to synthesize carnitine at a normal rate from ϵ-N-trimethyllysine or γ-butyrobetaine in vivo is not compromised by experimental vitamin C deficiency. Metabolism 1995, 44, 624–629. [Google Scholar] [CrossRef]
- Alkonyi, I.; Cseko, J.; Sandor, A. Role of the liver in carnitine metabolism: The mechanism of development of carnitine-deficient status in guinea-pigs. J. Clin. Chem. Clin. Biochem. 1990, 28, 319–321. [Google Scholar]
- Ferreira, G.C.; McKenna, M.C. L-Carnitine and acetyl-L-carnitine roles and neuroprotection in developing brain. Neurochem. Res. 2017, 42, 1661–1675. [Google Scholar] [CrossRef]
- Celestino–Soper, P.B.; Violante, S.; Crawford, E.L.; Luo, R.; Lionel, A.C.; Delaby, E.; Cai, G.; Sadikovic, B.; Lee, K.; Lo, C. A common X–linked inborn error of carnitine biosynthesis may be a risk factor for nondysmorphic autism. Proc. Natl. Acad. Sci. USA 2012, 109, 7974–7981. [Google Scholar] [CrossRef]
- Ueno, Y.; Koike, M.; Shimada, Y.; Shimura, H.; Hira, K.; Tanaka, R.; Uchiyama, Y.; Hattori, N.; Urabe, T. L-carnitine enhances axonal plasticity and improves white–matter lesions after chronic hypoperfusion in rat brain. J. Cerebral Blood Flow Metab. 2015, 35, 382–391. [Google Scholar] [CrossRef] [PubMed]
- Wainwright, M.S.; Mannix, M.K.; Brown, J.; Stumpf, D.A. L-carnitine reduces brain injury after hypoxia-ischemia in newborn rats. Pediatric Res. 2003, 54, 688–695. [Google Scholar] [CrossRef]
- Rau, T.F.; Lu, Q.; Sharma, S.; Sun, X.; Leary, G.; Beckman, M.L.; Hou, Y.; Wainwright, M.S.; Kavanaugh, M.; Poulsen, D.J. Oxygen glucose deprivation in rat hippocampal slice cultures results in alterations in carnitine homeostasis and mitochondrial dysfunction. PLoS ONE 2012, 7, e40881. [Google Scholar] [CrossRef] [PubMed]
- Xu, S.; Waddell, J.; Zhu, W.; Shi, D.; Marshall, A.D.; McKenna, M.C.; Gullapalli, R.P. In vivo longitudinal proton magnetic resonance spectroscopy on neonatal hypoxic-ischemic rat brain injury: Neuroprotective effects of acetyl-L-carnitine. Magn. Reson. Med. 2015, 74, 1530–1542. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Xu, S.; Lu, X.; Gullapalli, R.P.; McKenna, M.C.; Waddell, J. Neuroprotective effects of acetyl-l-carnitine on neonatal hypoxia ischemia-induced brain injury in rats. Dev. Neurosci. 2016, 38, 384–396. [Google Scholar] [CrossRef] [PubMed]
- Scafidi, S.; Racz, J.; Hazelton, J.; McKenna, M.C.; Fiskum, G. Neuroprotection by acetyl-L-carnitine after traumatic injury to the immature rat brain. Dev. Neurosci. 2010, 32, 480–487. [Google Scholar] [CrossRef]
- López–Suárez, O.; Concheiro-Guisán, A.; Sánchez–Pintos, P.; Cocho, J.A.; Lorenzo, J.R.F.; Couce, M.L. Acylcarnitine profile in neonatal hypoxic-ischemic encephalopathy: The value of butyrylcarnitine as a prognostic marker. Medicine 2019, 98, e15221. [Google Scholar] [CrossRef] [PubMed]
- Johnston, C.S.; Corte, C.; Swan, P.D. Marginal vitamin C status is associated with reduced fat oxidation during submaximal exercise in young adults. Nutr. Metab. 2006, 3, 1–5. [Google Scholar] [CrossRef]
- Ward, M.S.; Lamb, J.; May, J.M.; Harrison, F.E. Behavioral and monoamine changes following severe vitamin C deficiency. J. Neurochem. 2013, 124, 363–375. [Google Scholar] [CrossRef]
- Meredith, M.E.; May, J.M. Regulation of embryonic neurotransmitter and tyrosine hydroxylase protein levels by ascorbic acid. Brain Res. 2013, 1539, 7–14. [Google Scholar] [CrossRef]
- Levine, M. Ascorbic acid specifically enhances dopamine beta–monooxygenase activity in resting and stimulated chromaffin cells. J. Biol. Chem. 1986, 261, 7347–7356. [Google Scholar] [CrossRef]
- Levine, M.; Morita, K.; Heldman, E.; Pollard, H.B. Ascorbic acid regulation of norepinephrine biosynthesis in isolated chromaffin granules from bovine adrenal medulla. J. Biol. Chem. 1985, 260, 15598–15603. [Google Scholar] [CrossRef]
- Hansen, S.N.; Jørgensen, J.M.B.; Nyengaard, J.R.; Lykkesfeldt, J.; Tveden–Nyborg, P. Early Life Vitamin C Deficiency Does Not Alter Morphology of Hippocampal CA1 Pyramidal Neurons or Markers of Synaptic Plasticity in a Guinea Pig Model. Nutrients 2018, 10, 749. [Google Scholar] [CrossRef] [PubMed]
- Tveden-Nyborg, P.; Johansen, L.K.; Raida, Z.; Villumsen, C.K.; Larsen, J.O.; Lykkesfeldt, J. Vitamin C deficiency in early postnatal life impairs spatial memory and reduces the number of hippocampal neurons in guinea pigs. Am. J. Clin. Nutr. 2009, 90, 540–546. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, N.R.; Ledo, A.; Laranjinha, J.; Gerhardt, G.A.; Barbosa, R.M. Simultaneous measurements of ascorbate and glutamate in vivo in the rat brain using carbon fiber nanocomposite sensors and microbiosensor arrays. Bioelectrochemistry 2018, 121, 142–150. [Google Scholar] [CrossRef] [PubMed]
- Sandstrom, M.I.; Rebec, G.V. Extracellular ascorbate modulates glutamate dynamics: Role of behavioral activation. BMC Neurosci. 2007, 8, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Rebec, G.V.; Witowski, S.R.; Sandstrom, M.I.; Rostand, R.D.; Kennedy, R.T. Extracellular ascorbate modulates cortically evoked glutamate dynamics in rat striatum. Neurosci. Lett. 2005, 378, 166–170. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; May, J.M. Vitamin C function in the brain: Vital role of the ascorbate transporter SVCT2. Free Radic. Biol. Med. 2009, 46, 719–730. [Google Scholar] [CrossRef]
- Mi, D.J.; Dixit, S.; Warner, T.A.; Kennard, J.A.; Scharf, D.A.; Kessler, E.S.; Moore, L.M.; Consoli, D.C.; Bown, C.W.; Eugene, A.J.; et al. Altered glutamate clearance in ascorbate deficient mice increases seizure susceptibility and contributes to cognitive impairment in APP/PSEN1 mice. Neurobiol. Aging 2018, 71, 241–254. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.; Day, B.J.; Crapo, J.D.; Fridovich, I.; McNamara, J.O. Requirement for superoxide in excitotoxic cell death. Neuron 1996, 16, 345–355. [Google Scholar] [CrossRef]
- Johnston, M.V. Excitotoxicity in neonatal hypoxia. Mental Retard. Dev. Disabil. Res. Rev. 2001, 7, 229–234. [Google Scholar] [CrossRef]
- Johnston, M.V. Excitotoxicity in perinatal brain injury. Brain Pathol. 2005, 15, 234–240. [Google Scholar] [CrossRef]
- Warner, T.A.; Kang, J.-Q.; Kennard, J.A.; Harrison, F.E. Low brain ascorbic acid increases susceptibility to seizures in mouse models of decreased brain ascorbic acid transport and Alzheimer’s disease. Epilepsy Res. 2015, 110, 20–25. [Google Scholar] [CrossRef]
- Dyer, A.H.; Vahdatpour, C.; Sanfeliu, A.; Tropea, D. The role of Insulin-Like Growth Factor 1 (IGF–1) in brain development, maturation and neuroplasticity. Neuroscience 2016, 325, 89–99. [Google Scholar] [CrossRef] [PubMed]
- Mattson, M.P. Glutamate and Neurotrophic Factors in Neuronal Plasticity and Disease. Ann. N.Y. Acad. Sci. 2008, 1144, 97–112. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Larsen, R.S.; Philpot, B.D.; Paulsen, O. Roles of Presynaptic NMDA Receptors in Neurotransmission and Plasticity. Trends Neurosci. 2016, 39, 26–39. [Google Scholar] [CrossRef] [PubMed]
- Calero, C.I.; Vickers, E.; Cid, G.M.; Aguayo, L.G.; von Gersdorff, H.; Calvo, D.J. Allosteric modulation of retinal GABA receptors by ascorbic acid. J. Neurosci. 2011, 31, 9672–9682. [Google Scholar] [CrossRef]
- Rosa, P.B.; Neis, V.B.; Ribeiro, C.M.; Moretti, M.; Rodrigues, A.L.S. Antidepressant-like effects of ascorbic acid and ketamine involve modulation of GABAA and GABAB receptors. Pharm. Rep. 2016, 68, 996–1001. [Google Scholar] [CrossRef]
- Moretti, M.; Werle, I.; da Rosa, P.B.; Neis, V.B.; Platt, N.; Souza, S.V.S.; Rodrigues, A.L.S. A single coadministration of subeffective doses of ascorbic acid and ketamine reverses the depressive–like behavior induced by chronic unpredictable stress in mice. Pharmacol. Biochem. Behav. 2019, 187, 172800. [Google Scholar] [CrossRef]
- Moretti, M.; Fraga, D.B.; Rodrigues, A.L.S. Ascorbic Acid to Manage Psychiatric Disorders. CNS Drugs 2017, 31, 571–583. [Google Scholar] [CrossRef]
- Plevin, D.; Galletly, C. The neuropsychiatric effects of vitamin C deficiency: A systematic review. BMC Psychiatry 2020, 20, 1–9. [Google Scholar] [CrossRef]
- Isaacs, E.B.; Gadian, D.G.; Sabatini, S.; Chong, W.K.; Quinn, B.T.; Fischl, B.R.; Lucas, A. The effect of early human diet on caudate volumes and IQ. Pediatric Res. 2008, 63, 308–314. [Google Scholar] [CrossRef]
- Rosales, F.J.; Reznick, J.S.; Zeisel, S.H. Understanding the role of nutrition in the brain and behavioral development of toddlers and preschool children: Identifying and addressing methodological barriers. Nutr. Neurosci. 2009, 12, 190–202. [Google Scholar] [CrossRef]
- Georgieff, M.K.; Ramel, S.E.; Cusick, S.E. Nutritional influences on brain development. Acta Paediatrica 2018, 107, 1310–1321. [Google Scholar] [CrossRef] [PubMed]
- Prado, E.L.; Dewey, K.G. Nutrition and brain development in early life. Nutr. Rev. 2014, 72, 267–284. [Google Scholar] [CrossRef]
- Dobbing, J.; Sands, J. Growth and Development of Brain and Spinal Cord of Guinea Pig. Brain Res. 1970, 17, 115–123. [Google Scholar] [CrossRef]
- Carter, A.M. Animal models of human placentation––a review. Placenta 2007, 28, S41–S47. [Google Scholar] [CrossRef] [PubMed]
- Enders, A.C. A comparative study of the fine structure of the trophoblast in several hemochorial placentas. Am. J. Anat. 1965, 116, 29–67. [Google Scholar] [CrossRef]
- Nitsos, I.; Rees, S. The effects of intrauterine growth retardation on the development of neuroglia in fetal guinea pigs. An immunohistochemical and an ultrastructural study. Int. J. Dev. Neurosci. 1990, 8, 233–244. [Google Scholar] [CrossRef]
- Back, S.A.; Luo, N.L.; Borenstein, N.S.; Levine, J.M.; Volpe, J.J.; Kinney, H.C. Late oligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J. Neurosci. 2001, 21, 1302–1312. [Google Scholar] [CrossRef] [PubMed]
- Workman, A.D.; Charvet, C.J.; Clancy, B.; Darlington, R.B.; Finlay, B.L. Modeling Transformations of Neurodevelopmental Sequences across Mammalian Species. J. Neurosci. 2013, 33, 7368–7383. [Google Scholar] [CrossRef]
- Clancy, B.; Finlay, B.L.; Darlington, R.B.; Anand, K. Extrapolating brain development from experimental species to humans. Neurotoxicology 2007, 28, 931–937. [Google Scholar] [CrossRef]
- Gladen, B.C.; Tabacova, S.; Baird, D.D.; Little, R.E.; Balabaeva, L. Variability of lipid hydroperoxides in pregnant and nonpregnant women. Reprod. Toxicol. 1999, 13, 41–44. [Google Scholar] [CrossRef]
- Morris, J.M.; Gopaul, N.K.; Endresen, M.J.; Knight, M.; Linton, E.A.; Dhir, S.; Ängård, E.E.; Redman, C.W. Circulating markers of oxidative stress are raised in normal pregnancy and pre-eclampsia. BJOG Int. J. Obstet. Gynaecol. 1998, 105, 1195–1199. [Google Scholar] [CrossRef]
- Toescu, V.; Nuttall, S.L.; Martin, U.; Kendall, M.J.; Dunne, F. Oxidative stress and normal pregnancy. Clin. Endocrinol. 2002, 57, 609–613. [Google Scholar] [CrossRef]
- Domínguez-Perles, R.; Gil–Izquierdo, A.; Ferreres, F.; Medina, S. Update on oxidative stress and inflammation in pregnant women, unborn children (nasciturus), and newborns-Nutritional and dietary effects. Free Radic. Biol. Med. 2019, 142, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Rajan, D.P.; Huang, W.; Dutta, B.; Devoe, L.D.; Leibach, F.H.; Ganapathy, V.; Prasad, P.D. Human placental sodium–dependent vitamin C transporter (SVCT2): Molecular cloning and transport function. Biochem. Biophys. Res. Commun. 1999, 262, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Scaife, A.R.; McNeill, G.; Campbell, D.M.; Martindale, S.; Devereux, G.; Seaton, A. Maternal intake of antioxidant vitamins in pregnancy in relation to maternal and fetal plasma levels at delivery. Br. J. Nutr. 2006, 95, 771–778. [Google Scholar] [CrossRef] [PubMed]
- Baydas, G.; Karatas, F.; Gursu, M.F.; Bozkurt, H.A.; Ilhan, N.; Yasar, A.; Canatan, H. Antioxidant vitamin levels in term and preterm infants and their relation to maternal vitamin status. Arch. Med. Res. 2002, 33, 276–280. [Google Scholar] [CrossRef]
- Schjoldager, J.G.; Tveden–Nyborg, P.; Lykkesfeldt, J. Prolonged maternal vitamin C deficiency overrides preferential fetal ascorbate transport but does not influence perinatal survival in guinea pigs. Br. J. Nutr. 2013, 110, 1573–1579. [Google Scholar] [CrossRef]
- Schjoldager, J.G.; Paidi, M.D.; Lindblad, M.M.; Birck, M.M.; Kjærgaard, A.B.; Dantzer, V.; Lykkesfeldt, J.; Tveden-Nyborg, P. Maternal vitamin C deficiency during pregnancy results in transient fetal and placental growth retardation in guinea pigs. Eur. J. Nutr. 2015, 54, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Paidi, M.D.; Schjoldager, J.G.; Lykkesfeldt, J.; Tveden–Nyborg, P. Prenatal vitamin C deficiency results in differential levels of oxidative stress during late gestation in foetal guinea pig brains. Redox. Biol. 2014, 2, 361–367. [Google Scholar] [CrossRef]
- Dobbing, J.; Sands, J. Comparative aspects of the brain growth spurt. Early Hum. Dev. 1979, 3, 79–83. [Google Scholar] [CrossRef]
- Ikonomidou, C.; Kaindl, A.M. Neuronal death and oxidative stress in the developing brain. Antioxid. Redox Sign 2011, 14, 1535–1550. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, Y.; Bae, S.; Lim, S.H.; Jang, M.; Choi, J.; Jeon, J.; Hwang, Y.-I.; Kang, J.S.; Lee, W.J. Vitamin C deficiency causes severe defects in the development of the neonatal cerebellum and in the motor behaviors of Gulo -/- mice. Antioxid. Redox Sign 2015, 23, 1270–1283. [Google Scholar] [CrossRef]
- Hansen, S.N.; Schjoldager, J.G.; Paidi, M.D.; Lykkesfeldt, J.; Tveden–Nyborg, P. Maternal vitamin C deficiency does not reduce hippocampal volume and β–tubulin III intensity in prenatal Guinea pigs. Nutr. Res. 2016, 36, 696–702. [Google Scholar] [CrossRef]
- Nam, S.M.; Seo, J.S.; Go, T.-H.; Nahm, S.-S.; Chang, B.-J. Ascorbic acid supplementation prevents the detrimental effects of prenatal and postnatal lead exposure on the Purkinje cell and related proteins in the cerebellum of developing rats. Biol. Trace Elem. Res. 2019, 190, 446–456. [Google Scholar] [CrossRef] [PubMed]
- Kishimoto, Y.; Kanai, T.; Sato, K.; Lee, J.; Jeong, K.-S.; Shimokado, K.; Maruyama, N.; Ishigami, A. Insufficient ascorbic acid intake during gestation induces abnormal cardiac dilation in fetal and neonatal SMP30/GNL knockout mice. Pediatric Res. 2013, 73, 578–584. [Google Scholar] [CrossRef]
- Mallard, C.; Loeliger, M.; Copolov, D.; Rees, S. Reduced number of neurons in the hippocampus and the cerebellum in the postnatal guinea-pig following intrauterine growth-restriction. Neuroscience 2000, 100, 327–333. [Google Scholar] [CrossRef]
- Mallard, E.C.; Rehn, A.; Rees, S.; Tolcos, M.; Copolov, D. Ventriculomegaly and reduced hippocampal volume following intrauterine growth-restriction: Implications for the aetiology of schizophrenia. Schizophr. Res. 1999, 40, 11–21. [Google Scholar] [CrossRef]
- Dieni, S.; Rees, S. Dendritic morphology is altered in hippocampal neurons following prenatal compromise. J. Neurobiol. 2003, 55, 41–52. [Google Scholar] [CrossRef]
- Tolcos, M.; McDougall, A.; Shields, A.; Chung, Y.; O’Dowd, R.; Turnley, A.; Wallace, M.; Rees, S. Intrauterine Growth Restriction Affects Cerebellar Granule Cells in the Developing Guinea Pig Brain. Dev. Neurosci. 2018, 40, 162–174. [Google Scholar] [CrossRef]
- Tolcos, M.; Bateman, E.; O’Dowd, R.; Markwick, R.; Vrijsen, K.; Rehn, A.; Rees, S. Intrauterine growth restriction affects the maturation of myelin. Exp. Neurol. 2011, 232, 53–65. [Google Scholar] [CrossRef]
- Brender, J.D.; Werler, M.M.; Kelley, K.E.; Vuong, A.M.; Shinde, M.U.; Zheng, Q.; Huber Jr, J.C.; Sharkey, J.R.; Griesenbeck, J.S.; Romitti, P.A. Nitrosatable drug exposure during early pregnancy and neural tube defects in offspring: National Birth Defects Prevention Study. Am. J. Epidemiol. 2011, 174, 1286–1295. [Google Scholar] [CrossRef]
- Camarena, V.; Wang, G. The epigenetic role of vitamin C in health and disease. Cell. Mol. Life Sci. 2016, 73, 1645–1658. [Google Scholar] [CrossRef] [PubMed]
- Madruga de Oliveira, A.; Rondó, P.H.C.; Oliveira, J.M. Maternal alcohol consumption may influence cord blood ascorbic acid concentration: Findings from a study of Brazilian mothers and their newborns. Br. J. Nutr. 2009, 102, 895–898. [Google Scholar] [CrossRef] [PubMed]
- Mathews, F.; Neil, H. Nutrient intakes during pregnancy in a cohort of nulliparous women. J. Hum. Nutr. Diet. 1998, 11, 151–161. [Google Scholar] [CrossRef]
- Mathews, F.; Yudkin, P.; Neil, A. Influence of maternal nutrition on outcome of pregnancy: Prospective cohort study. BMJ 1999, 319, 339–343. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Hong, Y.-C.; Lee, K.; Kim, Y.; Kim, W.; Chang, N.; Park, E.; Park, H.; Hann, H. Influence of maternal serum levels of vitamins C and E during the second trimester on birth weight and length. Eur. J. Clin. Nutr. 2004, 58, 1365–1371. [Google Scholar] [CrossRef]
- Jang, W.; Kim, H.; Lee, B.-E.; Chang, N. Maternal fruit and vegetable or vitamin C consumption during pregnancy is associated with fetal growth and infant growth up to 6 months: Results from the Korean Mothers and Children’s Environmental Health (MOCEH) cohort study. Nutr. J. 2018, 17, 105. [Google Scholar] [CrossRef]
- Leitner, Y.; Fattal-Valevski, A.; Geva, R.; Eshel, R.; Toledano-Alhadef, H.; Rotstein, M.; Bassan, H.; Radianu, B.; Bitchonsky, O.; Jaffa, A.J. Neurodevelopmental outcome of children with intrauterine growth retardation: A longitudinal, 10-year prospective study. J. Child Neurol. 2007, 22, 580–587. [Google Scholar] [CrossRef]
- Bellido–González, M.; Díaz-López, M.Á.; López–Criado, S.; Maldonado–Lozano, J. Cognitive functioning and academic achievement in children aged 6–8 years, born at term after intrauterine growth restriction and fetal cerebral redistribution. J. Pediatric Psychol. 2017, 42, 345–354. [Google Scholar] [CrossRef]
- Wang, Y.; Fu, W.; Liu, J. Neurodevelopment in children with intrauterine growth restriction: Adverse effects and interventions. J. Matern.-Fetal Neonatal Med. 2016, 29, 660–668. [Google Scholar] [CrossRef]
- Chen, J.; Chen, P.; Bo, T.; Luo, K. Cognitive and behavioral outcomes of intrauterine growth restriction school-age children. Pediatrics 2016, 137, e20153868. [Google Scholar] [CrossRef] [PubMed]
- Dede, H.; Takmaz, O.; Ozbasli, E.; Dede, S.; Gungor, M. Higher level of oxidative stress markers in small for gestational age newborns delivered by cesarean section at term. Fetal Pediatr. Pathol. 2017, 36, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Wixey, J.A.; Chand, K.K.; Colditz, P.B.; Bjorkman, S.T. Neuroinflammation in intrauterine growth restriction. Placenta 2017, 54, 117–124. [Google Scholar] [CrossRef] [PubMed]
- Chappell, L.C.; Seed, P.T.; Kelly, F.J.; Briley, A.; Hunt, B.J.; Charnock-Jones, D.S.; Mallet, A.; Poston, L. Vitamin C and E supplementation in women at risk of preeclampsia is associated with changes in indices of oxidative stress and placental function. Am. J. Obstet. Gynecol. 2002, 187, 777–784. [Google Scholar] [CrossRef] [PubMed]
- Rumbold, A.R.; Crowther, C.A.; Haslam, R.R.; Dekker, G.A.; Robinson, J.S. Vitamins C and E and the risks of preeclampsia and perinatal complications. NEJ Med. 2006, 354, 1796–1806. [Google Scholar] [CrossRef] [PubMed]
- Chappell, L.C.; Seed, P.T.; Briley, A.; Kelly, F.J.; Hunt, B.J.; Charnock–Jones, D.S.; Mallet, A.I.; Poston, L. A longitudinal study of biochemical variables in women at risk of preeclampsia. Am. J. Obstet. Gynecol. 2002, 187, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Spinnato, J.A.; Freire, S.; e Silva, J.L.P.; Rudge, M.V.C.; Martins-Costa, S.; Koch, M.A.; Goco, N.; de Barros Santos, C.; Cecatti, J.G.; Costa, R. Antioxidant therapy to prevent preeclampsia: A randomized controlled trial. Obstet. Gynecol. 2007, 110, 1311–1318. [Google Scholar] [CrossRef]
- Ahn, Y.; Kim, Y.; Park, H.; Park, B.; Lee, H. Prenatal vitamin C status is associated with placental apoptosis in normal–term human pregnancies. Placenta 2007, 28, 31–38. [Google Scholar] [CrossRef] [PubMed]
- Crowther, C.A.; Rumbold, A.; Robinson, J. The authors reply. NEJ Med. 2006, 355, 1066. [Google Scholar] [CrossRef]
- Lavoie, J.-C.; Chessex, P. Parenteral nutrition and oxidant stress in the newborn: A narrative review. Free Radic. Biol. Med. 2019, 142, 155–167. [Google Scholar] [CrossRef] [PubMed]
- Perrone, S.; Laschi, E.; Buonocore, G. Biomarkers of oxidative stress in the fetus and in the newborn. Free Radic. Biol. Med. 2019, 142, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Ortega, R.M.; Quintas, M.E.; Andrés, P.; Martínez, R.M.; López-Sobaler, A.M. Ascorbic acid levels in maternal milk: Differences with respect to ascorbic acid status during the third trimester of pregnancy. Br. J. Nutr. 1998, 79, 431–437. [Google Scholar] [CrossRef]
- Picciano, M.F. Nutrient Composition of Human Milk. Pediatric Clin. N. Am. 2001, 48, 53–67. [Google Scholar] [CrossRef]
- Salmenperä, L. Vitamin C nutrition during prolonged lactation: Optimal in infants while marginal in some mothers. Am. J. Clin. Nutr. 1984, 40, 1050–1056. [Google Scholar] [CrossRef] [PubMed]
- Johnston, C.S.; Thompson, L.L. Vitamin C status of an outpatient population. J. Am. Coll. Nutr. 1998, 17, 366–370. [Google Scholar] [CrossRef]
- Hong, J.; Lee, H.; Park, E.; Kim, Y.-J.; Lee, H.; Park, B.-H.; Ha, E.-H.; Kong, K.; Chang, N.; Park, H. Association of mid-pregnancy antioxidative vitamin and oxidative stress levels with infant growth during the first 3 years of life. Food Nutr. Res. 2014, 58. [Google Scholar] [CrossRef] [PubMed]
- Roberts, L.J., II; Fessel, J.P. The biochemistry of the isoprostane, neuroprostane, and isofuran pathways of lipid peroxidation. Chem. Phys. Lipids 2004, 128, 173–186. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Curran, C.P.; Nebert, D.W.; Patel, K.V.; Williams, M.T.; Vorhees, C.V. Effect of vitamin C deficiency during postnatal development on adult behavior: Functional phenotype of Gulo (-/-) knockout mice. Genes Brain Behav. 2012, 11, 269–277. [Google Scholar] [CrossRef] [PubMed]
- Harrison, F.E.; Yu, S.S.; Van Den Bossche, K.L.; Li, L.; May, J.M.; McDonald, M.P. Elevated oxidative stress and sensorimotor deficits but normal cognition in mice that cannot synthesize ascorbic acid. J. Neurochem. 2008, 106, 1198–1208. [Google Scholar] [CrossRef]
- Kurihara, K.; Homma, T.; Kobayashi, S.; Shichiri, M.; Fujiwara, H.; Fujii, S.; Yamada, K.-I.; Nakane, M.; Kawamae, K.; Fujii, J. Ascorbic acid insufficiency impairs spatial memory formation in juvenile AKR1A-knockout mice. J. Clin. Biochem. Nutr. 2019, 65, 209–216. [Google Scholar] [CrossRef]
- Kondo, Y.; Sasaki, T.; Sato, Y.; Amano, A.; Aizawa, S.; Iwama, M.; Handa, S.; Shimada, N.; Fukuda, M.; Akita, M. Vitamin C depletion increases superoxide generation in brains of SMP30/GNL knockout mice. Biochem. Biophys. Res. Commun. 2008, 377, 291–296. [Google Scholar] [CrossRef]
- Hansen, S.N.; Schou-Pedersen, A.M.V.; Lykkesfeldt, J.; Tveden-Nyborg, P. Spatial Memory Dysfunction Induced by Vitamin C Deficiency Is Associated with Changes in Monoaminergic Neurotransmitters and Aberrant Synapse Formation. Antioxidants 2018, 7, 82. [Google Scholar] [CrossRef]
- Falsaperla, R.; Lombardo, F.; Filosco, F.; Romano, C.; Saporito, M.A.N.; Puglisi, F.; Piro, E.; Ruggieri, M.; Pavone, P. Oxidative Stress in Preterm Infants: Overview of Current Evidence and Future Prospects. Pharmaceuticals 2020, 13, 145. [Google Scholar] [CrossRef]
- Burd, I.; Welling, J.; Kannan, G.; Johnston, M.V. Excitotoxicity as a common mechanism for fetal neuronal injury with hypoxia and intrauterine inflammation. In Advances in Pharmacology; Elsevier: Amsterdam, The Netherlands, 2016; Volume 76, pp. 85–101. [Google Scholar]
- Pregnolato, S.; Chakkarapani, E.; Isles, A.R.; Luyt, K. Glutamate Transport and Preterm Brain Injury. Front. Physiol. 2019, 10. [Google Scholar] [CrossRef]
- Parikh, P.; Juul, S.E. Neuroprotective Strategies in Neonatal Brain Injury. J. Pediatrics 2018, 192, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Friel, J.K.; Martin, S.M.; Langdon, M.; Herzberg, G.R.; Buettner, G.R. Milk from mothers of both premature and full–term infants provides better antioxidant protection than does infant formula. Pediatric Res. 2002, 51, 612–618. [Google Scholar] [CrossRef] [PubMed]
- Minić, S.; Ješić, M.; Đurović, D.; Miletić, S.; Lugonja, N.; Marinković, V.; Nikolić-Kokić, A.; Spasić, S.; Vrvić, M.M. Redox properties of transitional milk from mothers of preterm infants. J. Paediatr. Child Health 2018, 54, 160–164. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, I.; Elremaly, W.; Rouleau, T.; Lavoie, J.C. Ascorbylperoxide contaminating parenteral nutrition is associated with bronchopulmonary dysplasia or death in extremely preterm infants. J. Parenter. Enteral Nutr. 2017, 41, 1023–1029. [Google Scholar] [CrossRef] [PubMed]
- Morin, G.; Guiraut, C.; Perez Marcogliese, M.; Mohamed, I.; Lavoie, J.-C. Glutathione Supplementation of Parenteral Nutrition Prevents Oxidative Stress and Sustains Protein Synthesis in Guinea Pig Model. Nutrients 2019, 11, 2063. [Google Scholar] [CrossRef]
- Chessex, P.; Harrison, A.; Khashu, M.; Lavoie, J.-C. In preterm neonates, is the risk of developing bronchopulmonary dysplasia influenced by the failure to protect total parenteral nutrition from exposure to ambient light? J. Pediatrics 2007, 151, 213–214. [Google Scholar] [CrossRef]
- Nualart, F.; Castro, T.; Low, M.; Henríquez, J.P.; Oyarce, K.; Cisternas, P.; García, A.; Yáñez, A.J.; Bertinat, R.; Montecinos, V.P. Dynamic expression of the sodium-vitamin C co-transporters, SVCT1 and SVCT2, during perinatal kidney development. Histochem. Cell Biol. 2013, 139, 233–247. [Google Scholar] [CrossRef] [PubMed]
- Strahle, J.M.; Triplett, R.L.; Alexopoulos, D.; Smyser, T.A.; Rogers, C.E.; Limbrick, D.D.; Smyser, C.D. Impaired hippocampal development and outcomes in very preterm infants with perinatal brain injury. NeuroImage Clin. 2019, 22, 101787. [Google Scholar] [CrossRef] [PubMed]
- Twilhaar, E.S.; Wade, R.M.; De Kieviet, J.F.; Van Goudoever, J.B.; Van Elburg, R.M.; Oosterlaan, J. Cognitive outcomes of children born extremely or very preterm since the 1990s and associated risk factors: A meta-analysis and meta-regression. JAMA Pediatrics 2018, 172, 361–367. [Google Scholar] [CrossRef]
- Popovich, D.; McAlhany, A.; Adewumi, A.O.; Barnes, M.M. Scurvy: Forgotten But Definitely Not Gone. J. Pediatric Health Care 2009, 23, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, A.; Shaharyar, A.; Kumar, A.; Bhat, M.S.; Mishra, M. Scurvy in pediatric age group—A disease often forgotten? J. Clin. Orthop. Trauma 2015, 6, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Levitt, P. Structural and functional maturation of the developing primate brain. J. Pediatrics 2003, 143, 35–45. [Google Scholar] [CrossRef]
- Fox, S.E.; Levitt, P.; Nelson, C.A., III. How the timing and quality of early experiences influence the development of brain architecture. Child Dev. 2010, 81, 28–40. [Google Scholar] [CrossRef]
- Cusick, S.E.; Georgieff, M.K. The role of nutrition in brain development: The golden opportunity of the “first 1000 days”. J. Pediatrics 2016, 175, 16–21. [Google Scholar] [CrossRef]
- Guidi, S.; Ciani, E.; Severi, S.; Contestabile, A.; Bartesaghi, R. Postnatal neurogenesis in the dentate gyrus of the guinea pig. Hippocampus 2005, 15, 285–301. [Google Scholar] [CrossRef]
- Frikke-Schmidt, H.; Tveden-Nyborg, P.; Lykkesfeldt, J. L-dehydroascorbic acid can substitute l-ascorbic acid as dietary vitamin C source in guinea pigs. Redox. Biol. 2016, 7, 8–13. [Google Scholar] [CrossRef] [PubMed]
- Padayatty, S.J.; Katz, A.; Wang, Y.; Eck, P.; Kwon, O.; Lee, J.H.; Chen, S.; Corpe, C.; Dutta, A.; Dutta, S.K.; et al. Vitamin C as an antioxidant: Evaluation of its role in disease prevention. J. Am. Coll. Nutr. 2003, 22, 18–35. [Google Scholar] [CrossRef] [PubMed]
- Shohaimi, S.; Bingham, S.; Welch, A.; Luben, R.; Day, N.; Wareham, N.; Khaw, K. Occupational social class, educational level and area deprivation independently predict plasma ascorbic acid concentration: A cross-sectional population based study in the Norfolk cohort of the European Prospective Investigation into Cancer (EPIC–Norfolk). Eur. J. Clin. Nutr. 2004, 58, 1432–1435. [Google Scholar] [CrossRef] [PubMed]
- Dror, D.K.; Allen, L.H. Interventions with Vitamins B6, B12 and C in Pregnancy. Paediatr. Perinat. Epidemiol. 2012, 26, 55–74. [Google Scholar] [CrossRef]
- Lykkesfeldt, J.; Poulsen, H.E. Is vitamin C supplementation beneficial? Lessons learned from randomised controlled trials. Br. J. Nutr. 2010, 103, 1251–1259. [Google Scholar] [CrossRef]
- Kobylecki, C.J.; Afzal, S.; Nordestgaard, B.G. Genetically high plasma vitamin C and urate: A Mendelian randomization study in 106 147 individuals from the general population. Rheumatology 2018, 57, 1769–1776. [Google Scholar] [CrossRef]
- Boeing, H.; Bohlscheid–Thomas, S.; Voss, S.; Schneeweiss, S.; Wahrendorf, J. The relative validity of vitamin intakes derived from a food frequency questionnaire compared to 24–hour recalls and biological measurements: Results from the EPIC pilot study in Germany. European Prospective Investigation into Cancer and Nutrition. Int. J. Epidemiol. 1997, 26, S82. [Google Scholar] [CrossRef]
- Sichieri, R.; Everhart, J. Validity of a Brazilian food frequency questionnaire against dietary recalls and estimated energy intake. Nutr. Res. 1998, 18, 1649–1659. [Google Scholar] [CrossRef]
- Ocke, M.C.; Bueno-de-Mesquita, H.B.; Pols, M.A.; Smit, H.A.; van Staveren, W.A.; Kromhout, D. The Dutch EPIC food frequency questionnaire. II. Relative validity and reproducibility for nutrients. Int. J. Epidemiol. 1997, 26, S49. [Google Scholar] [CrossRef]
- Pullar, J.M.; Bayer, S.; Carr, A.C. Appropriate handling, processing and analysis of blood samples is essential to avoid oxidation of vitamin C to dehydroascorbic acid. Antioxidants 2018, 7, 29. [Google Scholar] [CrossRef]
- Wang, S.; Schram, I.M.; Sund, R.B. Determination of plasma ascorbic acid by HPLC: Method and stability studies. Eur. J. Pharmaceut. Sci. 1995, 3, 231–239. [Google Scholar] [CrossRef]
- Washko, P.W.; Welch, R.W.; Dhariwal, K.R.; Wang, Y.; Levine, M. Ascorbic acid and dehydroascorbic acid analyses in biological samples. Anal. Biochem. 1992, 204, 1–14. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| VitC | Species/Strain | Time-Point | Principal Findings | Ref |
|---|---|---|---|---|
| Depletion (to the brain) | Mice/svct2−/− | E18.5–19.5 Term | Neonatal deaths. Petechial bleedings on brain surface an in parenchyma, reflecting weakened capillary walls. Increased lipid peroxidation (isoketals). Neuronal apoptosis in cerebral cortex and brain stem. Altered regulation of norepinephrine and dopamine and reduced dopaminergic neurons (decreased tyrosine kinase positive neurons). Aberrant DNA and histone methylation status. | [26,27,181] |
| Depletion | Mice/gulo−/− | Term | Neonatal deaths. Petechial bleedings in brain parenchyma. Increased lipid peroxidation (MDA, 8-isoprostane), redox imbalance (increased GSH:GSSG and NO). Deviated structural development in cerebral cortex, hippocampus and cerebellum. Reduced BDNF and GDNF. | [226] |
| Deficiency | Mice/gulo−/− | Term (E20) | Increased lipid peroxidation (MDA) in cerebellum but not cortex. | [137] |
| Deficiency | Guinea pig/Dunkin Hartley | GD45 and GD 56 | Increased lipid peroxidation (MDA) at GD 56 not 45. Redox imbalance marker (SOD) was increased in both GD45 and 56. No effect on hippocampal volume or β-tubulin III in hippocampal stratum lucidum. Transitional growth reduction reported for GD45. | [74,227] |
| VitC | Strain | Time-Point | Principal Findings | Ref |
|---|---|---|---|---|
| Deficiency | gulo−/− | PD1 | No reported change in lipid peroxidation. | [137] |
| PD10 PD18 | Increased lipid peroxidation (MDA) in cerebellum, not cortex. | |||
| Increased lipid peroxidation (F2-isoprostanes) in cortex not cerebellum. Increased redox imbalance (GSH) in cortex. Possible increase in GFAP stained cells (astrocytes) albeit not quantified. No functional effects on locomotion, agility or strength were detected. | ||||
| Depleted | gulo−/− | PD21 | Increased redox imbalance (GSH). | [262] |
| Deficient | PD60–100 | No redox imbalance. Reduced locomotion but no effect on spatial learning (MWM). Spatial memory was not assessed. Enhanced response to dopaminergic agonist indicating deviated regulation of dopaminergic signaling. | ||
| Depletion | gulo−/− | Young adults (20 gr) | Increased lipid peroxidation (MDA) and increased protein carbonyls in cortex. Decreased dopamine and serotonin metabolites in cortex and striatum. Locomotor deficits and reduced social dominance. | [180] |
| Depletion | gulo−/− | 4 wks–8 wks | Increased lipid peroxidation (MDA) in cortex, not cerebellum. | [97] |
| Deficiency | 4 wks–8 wks | Increased lipid peroxidation (MDA) in cortex, not cerebellum. | ||
| Deficient | gulo−/− | 6–18 wks old | Increased F4-neuroprostanes (also in vitC supplemented gulo−/− counterparts). Reduced sensimotory competence, most significant in deficient gulo−/−. Memory and cognition was not affected. | [263] |
| Depletion (acute) | akr1−/− | Juvenile (5 wks old–1 wk deplet.) | No apparent redox imbalance. No recorded changes in hippocampal histology (n = 2). Reduced spatial memory competence. No effect on neurotransmitters (dopamine, norepinephrine, glutamic acid, GABA, acetylcholine and selected metabolites). | [264] |
| Deficiency (long term) | Adult (12–13 wks) | No effect on spatial memory competence. | ||
| Depletion | SMP30/ GNL−/− | PD30- 2,4,8 wks depletion | 4- and 8-wk depletion increased superoxide production ex vivo; reduced cells in cerebellar cortex after 8-wk depletion (though data not shown). No effect on SOD expression or activity. | [265] |
| VitC | Strain | Time-Point | Principal Findings | Ref |
|---|---|---|---|---|
| Depletion | Dunkin Hartley | PD2–3 wks | Increased lipid peroxidation (MDA), increased protein carbonyls, induced DNA-based excision. | [24] |
| Severe deficiency | Dunkin Hartley | PD7–11 wks | No effects on the investigated hippocampal structures or synaptic plasticity markers and BDNF in cortex, hippocampus or striatum. | [184] |
| Deficiency | No additional apparent differences compared to severe deficiency. | |||
| Pre- and postnatal deficiency | Dunkin Hartley | PD2–7 | No effect on lipid peroxidation (MDA, 8-F2-isoprostane); GSH not different. | [221] |
| PD10 | Reduced hippocampal volume and reduced proliferation in hippocampal granular layer. | [74] | ||
| PD27 | Reduced hippocampal volume and increased proliferation in granular layer and subgranluar zones. | |||
| PD70 | Increased lipid peroxidation (MDA). Hippocampal volume reduction. Persistent decrease in hippocampal volume despite vitC repletion after birth. | [74,79] | ||
| Deficiency | Dunkin Hartley | PD7–9 wks | No effect on lipid peroxidation (MDA) or redox markers (SOD, GSH). Reduced neuron numbers in hippocampus. Deviated serotonin metabolites and reduced synaptophysin. Reduced spatial memory competence. | [185,266] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tveden-Nyborg, P. Vitamin C Deficiency in the Young Brain—Findings from Experimental Animal Models. Nutrients 2021, 13, 1685. https://doi.org/10.3390/nu13051685
Tveden-Nyborg P. Vitamin C Deficiency in the Young Brain—Findings from Experimental Animal Models. Nutrients. 2021; 13(5):1685. https://doi.org/10.3390/nu13051685
Chicago/Turabian StyleTveden-Nyborg, Pernille. 2021. "Vitamin C Deficiency in the Young Brain—Findings from Experimental Animal Models" Nutrients 13, no. 5: 1685. https://doi.org/10.3390/nu13051685
APA StyleTveden-Nyborg, P. (2021). Vitamin C Deficiency in the Young Brain—Findings from Experimental Animal Models. Nutrients, 13(5), 1685. https://doi.org/10.3390/nu13051685