
Small and Large Intestine (I): Malabsorption of Nutrients



Abstract
1. Introduction
2. Causes of Nutrient Malabsorption
2.1. Luminal Phase
- the adequate hydrolysis of the substrate, dependent on a sufficient concentration and mixture of pancreatic enzymes;
- the correct solubilization of fats in water (emulsion), which depends, in turn, on the adequate synthesis, transport, and concentration of bile salts in the intestinal lumen;
- the adequate availability of specific nutrients, especially cobalamin (vitamin B12), also dependent on a sufficient secretion of intrinsic factor in the stomach.
- adequate intragastric conversion of dietary proteins into amino acids, which play a role in releasing cholecystokinin (CCK) from duodenal and jejunal endocrine epithelial cells and releasing pancreatic enzymes to the duodenojejunal lumen;
- an adequate concentration and mixture of pancreatic ferments with the ingested nutrients;
- an adequate concentration of bile salts in the intestinal lumen (responsible for the release of enterokinase).
2.1.1. Substrate Hydrolysis
Digestive Enzyme Deficiency
Digestive Enzyme Inactivation
Dyssynchrony of Enzyme Release, Inadequate Mixing
2.1.2. Fat Solubilization
Decreased Synthesis and/or Secretion of Conjugated Bile Acids
Bile Salt Deconjugation
- Anatomic disorders, such as gastric bypass for the treatment of obesity, tumors of the small bowel, adhesions from previous surgery or strictures due to radiation or inflammatory bowel disease, blind intestinal loops, and small intestinal diverticulosis. In all these circumstances, intestinal stasis occurs, favoring the growth of bacteria such as Escherichia coli, Klebsiella spp, and Aeromonas, among others.
- Immune disorders. Intestinal immunity is important for maintaining the correct microbial composition in the small intestine. Combined variable immunodeficiency, IgA deficiency, acquired immunodeficiency (e.g., HIV), and even immunosenescence (often aggravated by using antisecretory drugs) [27] are associated with an increased risk of SIBO.
- Metabolic and systemic disorders. Diabetes mellitus and Parkinson’s disease can cause SIBO due to intestinal neuropathy. Disorders such as pancreatic insufficiency and cirrhosis can predispose to SIBO by changing the quantity and composition of these digestive enzymes or bile, thereby allowing microbes to grow. SIBO has long been regarded as a potential complication of celiac disease and one of the causes of failure to respond to gluten withdrawal. Overall, SIBO is responsible for approximately 10‒20% of nonresponsive celiac disease [28,29,30], though some studies suggest that SIBO may coexist with other disorders associated with nonresponsive CD [30]. SIBO has been frequently documented in association with alcoholic or nonalcoholic liver disease [31,32], and is more likely in patients with decompensated cirrhosis of the liver with portal hypertension, ascites, and jaundice. Intestinal bacteria are clearly fundamental to overt hepatic encephalopathy and even minimal hepatic encephalopathy.
Increased Bile Salt Loss
2.1.3. Luminal Availability of Specific Nutrients
Diminished Gastric Acid
Diminished Intrinsic Factor
Bacterial Consumption of Nutrients
2.2. Mucosal (Absorptive) Phase
2.2.1. Brush Border Hydrolysis
Acquired Disaccharidase Defect (Lactase Deficiency)
- congenital lactase deficiency (CLD): an extremely rare autosomal recessive disease characterized by absent or reduced enzymatic activity from birth;
- primary lactose intolerance or adult-type lactase deficiency: a common autosomal recessive condition resulting from a developmentally regulated change of the lactase gene expression;
- secondary lactase deficiency: a transient condition deriving from intestinal damage secondary to several diseases such as infections, celiac disease, food allergy, small bowel bacterial overgrowth, Crohn’s disease, or radiation/chemotherapy-induced enteritis.
Congenital Disaccharidase Defect (Sucrase-Isomaltase Deficiency)
Trehalase Deficiency
Fructose Intolerance
2.3. Epithelial Transport
2.3.1. Nutrient-Specific Defects in Transport (Hartnup Disease and Cystinuria)
2.3.2. Global Defects in Transport
Celiac Disease
Other Diseases Causing Enteropathy
- Immunomediated Enteropathy
- anorexia from underlying disease,
- chronic nausea and/or vomiting,
- oropharyngeal ulceration,
- malabsorption,
- gastrointestinal tract obstruction,
- fistulizing disease,
- increased energy expenditure,
- hypercatabolism, associated with sepsis and systemic inflammatory responses syndrome,
- extensive surgical resection of the small bowel,
- small intestinal bacterial overgrowth,
- medication side effects, and
- NPO for diagnostic workup.
- Ipilimumab. The drug is well known for gastrointestinal side effects including enterocolitis, which is probably initiated by T-cell activation in the gut [90].
- Immune checkpoint inhibitors such as anti-programmed death-1 antibodies (anti-PD-1, pembroluzimab, and nevolimab) are associated with a similar risk of immune mediated entero-(colitis) [91].
- Methotrexate, capecitabine, and TNFα inhibitors, indicating that clinicians and pathologists should be aware of the expanding spectrum of drugs that can cause apoptotic enteropathy [96].
- Olmesartan-associated enteropathy. Up to now, more than 100 cases of olmesartan-associated enteropathy have been described [78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97]. The mechanism by which olmesartan exerts its effect is not yet fully understood, but a role for cell-mediated immunity has been suggested based on the long delay between the onset of olmesartan therapy and the development of diarrhea (and enteropathy). Patients usually present with nonbloody diarrhea and weight loss, but some develop severe dehydration with electrolyte imbalances and acute renal failure. Up to 64% of patients carry the HLA-DQ2 and/or HLA-DQ8 gene, in comparison to only 40% in the general population; this suggests a genetic susceptibility in its pathogenesis [74]. Histopathology can mimic AIE, especially with respect to intra-epithelial lymphocytosis within or close to normal limits, loss of Paneth and goblet cells, and crypt apoptosis.
- Infectious
- Infiltrative
- Neoplastic
- Diet-Related
- Ischemic
- Others
2.4. Postabsorptive, Processing Phase
2.4.1. Enterocyte Processing
Abetalipoproteinemia
2.4.2. Lymphatic
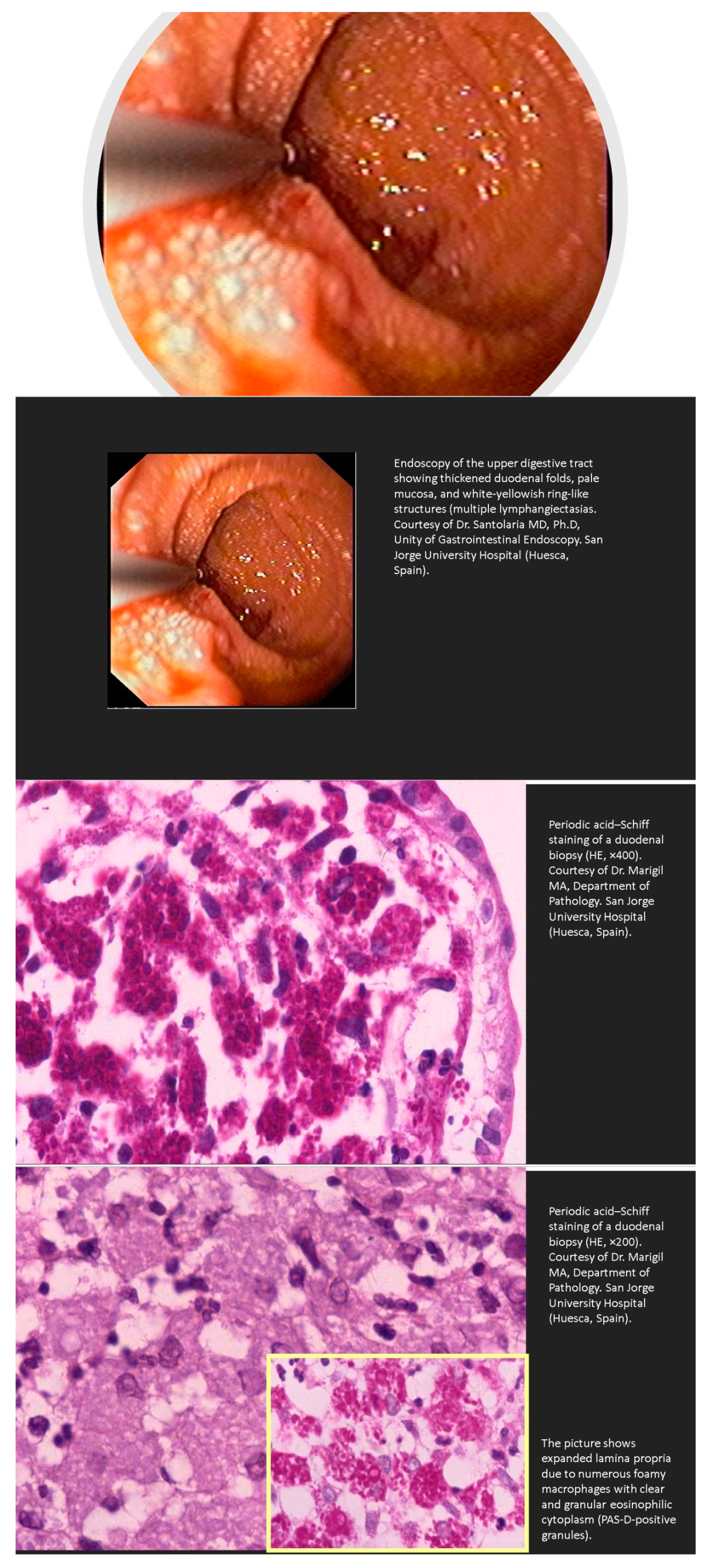
Intestinal Lymphangiectasia
Radiation
3. Consequences of Nutrient Malabsorption

{kind=link}
{kind=link}
{kind=link}
| Specific Nutritional Deficiency | Causes | Symptoms and Signs Due to Micronutrient Maldigestion or Malabsorption |
|---|---|---|
| Vitamin A 1 | Bariatric surgery (gastric bypass, biliopancreatic, or duodenal switch procedures) Disorders associated with fat malabsorption:
| [A]Serum retinol levels (levels less than 20 μg/dL (0.7 μmol/L) suggest deficiency. Xerophthalmia Night blindness (nyctalopia) and retinopathy Poor bone growth Hyperkeratosis, phrynoderma (follicular hyperkeratosis) Impairment of the humoral and cell-mediated immune system via direct and indirect effects on the phagocytes and T cells |
| Vitamin D 2,3,4 | [D] Deficiency (serum 25[OH]D <12 ng/mL (30 nmol/L) or insufficiency (12 to 20 ng/mL (30 to 50 nmol/L) Reduced intestinal absorption of calcium and phosphorus Secondary hyperparathyroidism and phosphaturia Bone pain and tenderness, muscle weakness Low bone mass on bone densitometry and fractures | |
| Vitamin K 5 | [K]Vitamin K status also can be determined indirectly by measuring vitamin K-dependent factors (i.e., prothrombin, factors VII, IX, X, or protein C) Easy bruisability Mucosal bleeding Splinter hemorrhages Epistaxis, melena, hematuria, and any other manifestations of impaired coagulation | |
| Vitamin E 6 | [E]Defined as below 0.5 mg/dL Neuropathy (spinocerebellar syndrome) Skeletal myopathy Pigmented retinopathy Brown bowel syndrome (intestinal lipofuscinosis) Hemolytic anemia |
| Specific Nutritional Deficiency | Causes | Symptoms and Signs Due to Micronutrient Maldigestion-Malabsorption. |
|---|---|---|
| Vitamin B1 (thiamine) | Anorexia nervosa Bariatric surgery Malabsorptive syndromes such as active celiac disease, malignancies, and short bowel syndrome | [B1] Measurement:the normal range for blood thiamine concentration varies somewhat between laboratories, 70 to 180 nmol/L (3.0 to 7.7 mcg/dL). Thiamine deficiency in the diet causes two clinical phenotypes:
|
| Vitamin B2 (Riboflavin) Ariboflavinosis | [B2] Measurement:Erythrocyte glutathione reductase assay is a better functional index of insufficient riboflavin intake (a coefficient >1.4 indicates riboflavin insufficiency). Symptoms of ariboflavonosis: Sore throat, hyperemia of pharyngeal mucous membranes, edema of mucous membranes, cheilitis, stomatitis, glossitis, normocytic-normochromic anemia, and seborrheic dermatitis | |
| Vitamin B3 (Niacin) 1 | [B3] Measurement:High levels of a metabolic product of a vitamin, such as N-methylnicotinamide, reflect adequate concentrations of intracellular niacin. Photosensitive pigmented dermatitis (typically located in sun-exposed areas), diarrhea, and dementia | |
| Vitamin B5 (Pantothenic acid) | [B5] Measurement:Urinary excretion below 1 mg/day generally indicates low dietary intake Paresthesias and dysesthesias, referred to as “burning feet syndrome” | |
| Vitamin B6 (Pyridoxine) 2 | [B6] Measurement:20 to 30 nmol/L (4.9 to 7.4 ng/mL) is generally considered marginal and >30 nmol/L (>7.4 ng/mL) is sufficient. Marginal deficiencies: nonspecific stomatitis, glossitis, cheilosis, irritability, confusion, depression, and possibly peripheral neuropathy. Severe deficiency is associated with seborrheic dermatitis, microcytic anemia, and seizures. | |
| Biotin | [Biotin] Measurement:Normal urine biotin excretion is around 75 to 195 μmol/L day Dermatitis around the eyes, nose and mouth, conjunctivitis, alopecia, and neurologic symptoms (changes in mental status, lethargy, hallucinations, and paresthesias). | |
| Vitamin C (Ascorbic acid)3 | [Vitamin C] Measurement:Symptoms of scurvy generally occur when the plasma concentration of ascorbic acid is less than 0.2 mg/dL (11 μmol L) Follicular hyperkeratosis and perifollicular hemorrhage, with petechiae and coiled hairs. ecchymoses, gingivitis (with bleeding and receding gums and dental caries). Sjogren’s syndrome, arthralgias, edema, anemia, and impaired wound healing. Musculoskeletal pain may be caused by hemorrhage into the muscles or periosteum. Generalized systemic symptoms are weakness, malaise, joint swelling, arthralgias, anorexia, depression, neuropathy, and vasomotor instability. | |
| Vitamin B12 and folate4 | Decreased intake (e.g., reduced intake of animal products, strict vegan diet, breastfeeding by a vitamin B12-deficient mother) Autoantibodies to intrinsic factor or gastric parietal cells (e.g., pernicious anemia) Decreased absorption (e.g., gastrectomy, bariatric surgery, pancreatic insufficiency, celiac disease, Crohn’s disease, bacterial overgrowth, fish tapeworm infection, gastric atrophy associated with aging, extensive ileal resection, or bypass) Medications and drugs that interfere with absorption or stability (e.g., metformin, histamine receptor antagonists, proton pump inhibitors, nitrous oxide) | [Cobalamin] Measurement:Below 200 pg/mL (below 148 pmol/L)—low; consistent with deficiency; 200 to 300 pg/mL (148 to 221 pmol/L)–borderline; deficiency is possible and additional testing is useful. [Folate] Measurement:From 2 to 4 ng/mL (from 4.5 to 9.1 nmol/L)—borderline: Below 2 ng/mL (below 4.5 nmol/L)—low, consistent with folate deficiency Macrocytic anemia, mild leukopenia and/or thrombocytopenia, low reticulocyte count, hypersegmented neutrophils Cognitive slowing and neuropathy Fatigue, irritability Cognitive decline, forgetfulness, dementia, psychosis Glossitis, oral ulcers (folate deficiency) Symmetric paresthesias or numbness and gait problems Restless legs syndrome Subacute combined degeneration of the dorsal (posterior) and lateral columns (white matter) of the spinal cord due to demyelination (weakness, ataxia, and paresthesias that may progress to spasticity and paraplegia) Abnormal deep tendon reflexes Extrapyramidal signs (i.e., dystonia, dysarthria, rigidity) Skin hyperpigmentation Increased risk of osteoporosis and fractures of the hip or spine Increased risk of gastric cancer in individuals with pernicious anemia (B12 deficiency) Risk of neural tube defects (folate deficiency) |
- Secretin and cholecystokinin release in response to a meal are impaired in CeD, thus diminishing the delivery of bile and pancreatic secretion into the duodenal lumen.
- The delivery of excessive dietary fat into the colon results in bacterial production of hydroxy fatty acids, which are potent cathartics.
- The stool volume and osmotic load delivered to the colon are increased by malabsorption.
- There is active electrolyte secretion in the lumen of the small intestine.
- If the disease extends to and involves the ileum, patients can experience the direct cathartic action of malabsorbed bile salts on the colon.
3.1. Iron
3.2. Folate and Cobalamin
3.3. Fat-Soluble Vitamins
3.4. Other Micronutrient Deficiencies
| Specific Nutritional Deficiency | Causes | Symptoms and Signs Due to Micronutrient Maldigestion-Malabsorption |
|---|---|---|
| Iron deficiency 1 | Disorders that affect the mucosal cells responsible for iron absorption, such as celiac disease, atrophic gastritis, Helicobacter pylori infection, and bariatric surgery, proton pump inhibitors1 | [Iron] Measurement:Transferrin saturation <19%; Ferritin 30 ng/mL (a higher ferritin level may be “falsely normal” and cannot be used to eliminate the possibility of iron deficiency in individuals with comorbidities). Fatigue, Pica (Pagophagia), restless legs syndrome, headache, exercise intolerance, exertional dyspnea, weakness. Pallor, dry or rough skin, atrophic glossitis with loss of tongue papillae, cheilosis (also called angular cheilitis), koilonychia (spoon nails), esophageal web, which may be accompanied by dysphagia (e.g., Plummer‒Vinson or Patterson‒Kelly syndrome; rare), alopecia (rare) in especially severe cases, chlorosis (pale, faintly green complexion; extremely rare). |
| Calcium deficiency | (See causes of steatorrhea and vitamin D malabsorption) Severe acute pancreatitis | [Calcium] Measurement:The normal range for serum calcium is approximately 8.6 to 10.2 mg/dL (2.15 to 2.54 mmol/L). Neuromuscular irritability (paresthesias), carpopedal spasm, tetany, seizures, Heart failure or prolonged QT interval. |
| Magnesium | Acute or chronic diarrhea, malabsorption and steatorrhea, and small bowel bypass surgery Severe acute pancreatitis Proton pump inhibitors (in patients taking diuretics). Calcineurin inhibitors Alcohol Uncontrolled diabetes mellitus Posttransplant patients Hypercalcemia | [Magnesium] Measurement:Hypomagnesemia is defined as a serum magnesium less than 1.6 mg/dL [0.66 mmol/L]) Neuromuscular manifestations, including neuromuscular hyperexcitability (e.g., tremor, tetany, convulsions), weakness, apathy, delirium, and coma Insulin resistance and the metabolic syndrome Migraine headaches and asthma Cardiovascular manifestations, including widening of the QRS and peaking of T waves with moderate magnesium depletion, and widening of the PR interval, diminution of T waves, and atrial and ventricular arrhythmias with severe depletion Abnormalities of calcium metabolism, including hypocalcemia, hypoparathyroidism, parathyroid hormone (PTH) resistance, and decreased synthesis of calcitriol Hypokalemia |
| Copper | Foregut surgery, including gastrectomy or gastric bypass Chronic diarrhea or other malabsorptive conditions including celiac disease Chronic peritoneal dialysis or hemodialysis Excessive zinc ingestion (copper and zinc are competitively absorbed from the jejunum) Total parenteral nutrition (TPN) with insufficient copper | [Copper] Measurement:The normal range for total copper in the blood is 85 to 180 μcg/dL. Fragile, abnormally formed hair, depigmentation of the skin, muscle weakness (myeloneuropathy), neurologic abnormalities, edema, and hepatosplenomegaly, and osteoporosis. The neurologic manifestations (ataxia, neuropathy, and cognitive deficits) can mimic vitamin B12 deficiency. Hematologic features: anemia (usually normocytic; sometimes macrocytic and occasionally with microcytic cells) and neutropenia. Thrombocytopenia may also occur but is relatively rare. |
| Zinc 2 | Gastric bypass for obesity Necrotizing enterocolitis Malabsorption syndromes (such as celiac disease and chronic inflammatory bowel disease) Alcohol-related cirrhosis Medications that increase urinary losses of zinc, including thiazides, loop diuretics, and angiotensin receptor blockers Patients receiving chronic TPN solutions lacking adequate zinc supplementation. | [Zinc] Measurement:The normal range for zinc in the blood is 64.7–139.8 μcg/dL Alkaline phosphatase activity can serve as a supportive marker of zinc status. Delayed sexual maturation, impotence, hypogonadism, oligospermia Alopecia, dysgeusia (impaired taste) Immune dysfunction Night blindness Impaired wound healing Skin lesions (erythematous, vesiculobullous, and pustular lesions) Change in hair color and hair loss Impaired appetite Decubitus ulcers |
| Selenium | Gastric bypass for obesity Chronic diarrhea or other malabsorptive conditions including celiac disease Extensive intestinal resection TPN with insufficient copper | [Selenium] Measurement:The normal range for selenium in the blood is 70‒150 ng/mL (0.15 parts per million) Skeletal muscle dysfunction Cardiomyopathy Mood disorders Impaired immune function Macrocytosis Whitened nailbeds |
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hogenauer, C.; Hammer, H.F. Maldigestion and Malabsorption. In Sleisenger and Fordtran’s Gastrointestinal and Liver Disease, 10th ed.; Feldman, M., Friedman, L.S., Brandt, L.J., Eds.; Saunders: Philadelphia, PA, USA, 2016; pp. 1788–1825. [Google Scholar]
- Goodman, B.E. Insights into digestion and absorption of major nutrients in humans. Adv. Physiol. Educ. 2010, 34, 44–53. [Google Scholar] [CrossRef]
- Aronson, P.S.; Boron, W.F.; Boulpaep, E.L. Physiology of membranes. In Medical Physiology: A Cellular and Molecular Approach; Boron, W.F., Boulpaep, E.L., Eds.; Saunders: Philadelphia, PA, USA, 2003; pp. 66–67. [Google Scholar]
- Lowe, M.E. The triglyceride lipases of the pancreas. J. Lipid Res. 2002, 43, 2007–2016. [Google Scholar] [CrossRef]
- Kleeff, J.; Whitcomb, D.C.; Shimosegawa, T.; Esposito, I.; Lerch, M.M.; Gress, T.; Mayerle, J.; Drewes, A.M.; Rebours, V.; Akisik, F.; et al. Chronic pancreatitis. Nat. Rev. Dis. Primers 2017, 3, 17060. [Google Scholar] [CrossRef] [PubMed]
- Bobadilla, J.L.; Macek, M.; Fine, J.P.; Farrell, P.M. Cystic fibrosis: A worldwide analysis ofCFTR mutations—Correlation with incidence data and application to screening. Hum. Mutat. 2002, 19, 575–606. [Google Scholar] [CrossRef]
- Sikkens, E.C.; Cahen, D.L.; De Wit, J.; Looman, C.W.; Van Eijck, C.; Bruno, M.J. A Prospective Assessment of the Natural Course of the Exocrine Pancreatic Function in Patients with a Pancreatic Head Tumor. J. Clin. Gastroenterol. 2014, 48, e43–e46. [Google Scholar] [CrossRef] [PubMed]
- Ahn, D.H.; Bekaii-Saab, T. Ampullary Cancer: An Overview. Am. Soc. Clin. Oncol. Educ. Book 2014, 2014, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Sikkens, E.C.M.; Cahen, D.L.; de Wit, J.; Looman, C.W.N.; van Eijck, C.; Bruno, M.J. Prospective assessment of the influence of pancreatic cancer resection on exocrine pancreatic function. BJS 2013, 101, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Townes, P.L.; Byrson, M.F.; Miller, G. 50 Years Ago in The Journal of Pediatrics. J. Pediatr. 1967, 71, 220. [Google Scholar] [CrossRef]
- Zheng, X.L.; Kitamoto, Y.; Sadler, J.E. Enteropeptidase, a type II transmembrane serine protease. Front. Biosci. 2009, 1, 242–249. [Google Scholar]
- Mann, N.S.; Mann, S.K. Enterokinase. Exp. Biol. Med. 1994, 206, 114–118. [Google Scholar] [CrossRef] [PubMed]
- Lebenthal, E.; Antonowicz, I.; Shwachman, H. Enterokinase and trypsin activities in pancreatic insufficiency and diseases of the small intestine. Gastroenterology 1976, 70, 508–512. [Google Scholar] [CrossRef]
- Tan, H.; Su, D.; Zhuo, Z. Shwachman-diamond syndrome: A case report. Medicine 2021, 100, e24712. [Google Scholar] [CrossRef] [PubMed]
- Cho, M.S.; Kasi, A. Zollinger Ellison Syndrome. StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, 24 November 2020. [Google Scholar]
- Nuzzo, A.; Czernichow, S.; Hertig, A.; Ledoux, S.; Poghosyan, T.; Quilliot, D.; Le Gall, M.; Bado, A.; Joly, F. Prevention and treatment of nutritional complications after bariatric surgery. Lancet Gastroenterol. Hepatol. 2021, 6, 238–251. [Google Scholar] [CrossRef]
- Lupoli, R.; Lembo, E.; Saldalamacchia, G.; Avola, C.K.; Angrisani, L.; Capaldo, B. Bariatric surgery and long-term nutritional issues. World J. Diabetes 2017, 8, 464–474. [Google Scholar] [CrossRef]
- Binder, H.J.; Reuben, A. Nutrient digestion and absorption. In Medical Physiology: A Cellular and Molecular Approach; Boron, W.F., Boulpaep, E.L., Eds.; Saunders: Philadelphia, PA, USA, 2009; pp. 949–979. [Google Scholar]
- Kalaitzakis, E. Gastrointestinal dysfunction in liver cirrhosis. World J. Gastroenterol. 2014, 20, 14686–14695. [Google Scholar] [CrossRef] [PubMed]
- Romiti, A.; Merli, M.; Martorano, M.; Parrilli, G.; Martino, F.; Riggio, O.; Truscelli, A.; Capocaccia, L.; Budillon, G. Malabsorption and nutritional abnormalities in patients with liver cirrhosis. Ital. J. Gastroenterol. 1990, 22, 118–123. [Google Scholar] [PubMed]
- Brimo Alsaman, M.Z.; Mazketly, M.; Ziadeh, M.; Aleter, O.; Ghazal, A. Cholecystocutaneous fistula incidence, etiology, clinical manifestations, diagnosis and treatment. A literature review. Ann. Med. Surg. 2020, 59, 180–185. [Google Scholar] [CrossRef]
- Crespi, M.; Montecamozzo, G.; Foschi, D. Diagnosis and treatment of biliary fistulas in the laparoscopic era. Gastroenterol. Res. Pract. 2016, 2016, 6293538. [Google Scholar] [CrossRef]
- Sundaram, S.S.; Bove, K.E.; Lovell, M.A.; Sokol, R.J. Mechanisms of Disease: Inborn errors of bile acid synthesis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2008, 5, 456–468. [Google Scholar] [CrossRef]
- Wang, H.H.; Liu, M.; Li, X.; Portincasa, P.; Wang, D.Q.-H. Impaired intestinal cholecystokinin secretion, a fascinating but overlooked link between coeliac disease and cholesterol gallstone disease. Eur. J. Clin. Investig. 2017, 47, 328–333. [Google Scholar] [CrossRef]
- Takakura, W.; Pimentel, M. Small Intestinal Bacterial Overgrowth and Irritable Bowel Syndrome—An Update. Front. Psychiatry 2020, 11, 664. [Google Scholar] [CrossRef]
- Chen, B.; Kim, J.J.-W.; Zhang, Y.; Du, L.; Dai, N. Prevalence and predictors of small intestinal bacterial overgrowth in irritable bowel syndrome: A systematic review and meta-analysis. J. Gastroenterol. 2018, 53, 807–818. [Google Scholar] [CrossRef]
- Lo, W.; Chan, W.W. Proton Pump Inhibitor Use and the Risk of Small Intestinal Bacterial Overgrowth: A Meta-Analysis. Clin. Gastroenterol. Hepatol. 2013, 11, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Tursi, A.; Brandimarte, G.; Giorgetti, G. High prevalence of small intestinal bacterial overgrowth in celiac patients with persistence of gastrointestinal symptoms after gluten withdrawal. Am. J. Gastroenterol. 2003, 98, 839–843. [Google Scholar] [CrossRef]
- Losurdo, G.; Marra, A.M.; Shahini, E.; Girardi, B.; Giorgio, F.; Amoruso, A.; Pisani, A.; Piscitelli, D.; Barone, M.; Principi, M.; et al. Small intestinal bacterial overgrowth and celiac disease: A systematic review with pooled-data analysis. Neurogastroenterol. Motil. 2017, 29, e13028. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Tapia, A.; Barton, S.H.; Rosenblatt, J.E.; Murray, J.A. Prevalence of Small Intestine Bacterial Overgrowth Diagnosed by Quantitative Culture of Intestinal Aspirate in Celiac Disease. J. Clin. Gastroenterol. 2009, 43, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Kwong, E.K.; Puri, P. Gut microbiome changes in Nonalcoholic fatty liver disease & alcoholic liver disease. Transl. Gastroenterol. Hepatol. 2021, 6, 3. [Google Scholar] [PubMed]
- Quigley, E.M.; Stanton, C.; Murphy, E. The gut microbiota and the liver. Pathophysiological and clinical implications. J. Hepatol. 2013, 58, 1020–1027. [Google Scholar] [CrossRef]
- Hofmann, A.F.; Poley, J.R. Role of bile acid malabsorption in pathogenesis of diarrhea and steatorrhea in patients with ileal resection. I. Response to cholestyramine or replacement of dietary long chain triglyceride by medium chain triglyceride. Gastroenterology 1972, 62, 918. [Google Scholar] [CrossRef]
- Poley, J.R.; Hofmann, A.F. Role of fat maldigestion in pathogenesis of steatorrhea in ileal resection. Fat digestion after two sequential test meals with and without cholestyramine. Gastroenterology 1976, 71, 38–44. [Google Scholar] [CrossRef]
- Lapidus, A.; Einarsson, K. Effects of ileal resection on biliary lipids and bile acid composition in patients with Crohn’s disease. Gut 1991, 32, 1488–1491. [Google Scholar] [CrossRef]
- Camaschella, C. New insights into iron deficiency and iron deficiency anemia. Blood Rev. 2017, 31, 225–233. [Google Scholar] [CrossRef]
- Carabotti, M.; Annibale, B.; Lahner, E. Common Pitfalls in the Management of Patients with Micronutrient Deficiency: Keep in Mind the Stomach. Nutrients 2021, 13, 208. [Google Scholar] [CrossRef]
- Figlin, E.; Chetrit, A.; Shahar, A.; Shpilberg, O.; Zivelin, A.; Rosenberg, N.; Brok-Simoni, F.; Gadoth, N.; Sela, B.-A.; Seligsohn, U. High prevalences of vitamin B12 and folic acid deficiency in elderly subjects in Israel. Br. J. Haematol. 2003, 123, 696. [Google Scholar] [CrossRef]
- Quigley, E.M.M.; Murray, J.A.; Pimentel, M. AGA Clinical Practice Update on Small Intestinal Bacterial Overgrowth: Expert Review. Gastroenterology 2020, 159, 1526–1532. [Google Scholar] [CrossRef] [PubMed]
- Di Constanzo, M.; Berni, R. Lactose Intolerance: Common Misunderstandings. Ann. Nutr. Metab. 2018, 73 (Suppl. 4), 30–37. [Google Scholar] [CrossRef] [PubMed]
- Lomer, M.C. Review article: The aetiology, diagnosis, mechanisms, and clinical evidence for food intolerance. Aliment. Pharmacol. Ther. 2015, 41, 262–275. [Google Scholar] [CrossRef] [PubMed]
- Suchy, F.J.; Brannon, P.M.; Carpenter, T.O.; Fernandez, J.R.; Gilsanz, V.; Gould, J.B.; Hall, K.; Hui, S.L.; Lupton, J.; Mennella, J.; et al. NIH consensus development conference statement: Lactose intolerance and health. NIH Consens. State Sci. Statements 2010, 27, 1–27. [Google Scholar] [PubMed]
- Walsh, J.; Meyer, R.; Shah, N.; Quekett, J.; Fox, A.T. Differentiating milk allergy (IgE and non-IgE mediated) from lactose intolerance: Understanding the underlying mechanisms and presentations. Br. J. Gen. Pract. 2016, 66, e609–e611. [Google Scholar] [CrossRef]
- Gericke, B.; Amiri, M.; Naim, H.Y. The multiple roles of sucrase-isomaltase in the intestinal physiology. Mol. Cell Pediatr. 2016, 3, 2. [Google Scholar] [CrossRef]
- Treem, W.R. Clinical aspects and treatment of congenital sucrase-isomaltase deficiency. J. Pediatr. Gastroenterol. Nutr. 2012, 55, S7–S13. [Google Scholar] [CrossRef]
- Naim, H.Y.; Heine, M.; Zimmer, K.P. Congenital sucrase-isomaltase deficiency: Heterogeneity of inheritance, trafficking, and function of an intestinal enzyme complex. J. Pediatr. Gastroenterol. Nutr. 2012, 55 (Suppl. 2), S13–S20. [Google Scholar] [CrossRef] [PubMed]
- Murray, I.A.; Coupland, K.; Smith, J.A.; Ansell, I.D.; Long, R.G. Intestinal trehalase activity in a UK population: Establishing a normal range and the effect of disease. Br. J. Nutr. 2000, 83, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Skoog, S.M.; Bharucha, A.E. Dietary fructose, and gastrointestinal symptoms: A review. Am. J. Gastroenterol. 2004, 99, 2046–2050. [Google Scholar] [CrossRef]
- Rumessen, J.J.; Gudmand-Høyer, E. Absorption capacity of fructose in healthy adults. Comparison with sucrose and its constituent monosaccharides. Gut 1986, 27, 1161. [Google Scholar] [CrossRef]
- Hyams, J.S. Sorbitol intolerance: An unappreciated cause of functional gastrointestinal complaints. Gastroenterology 1983, 84, 30–33. [Google Scholar] [CrossRef]
- Daniel, H. Molecular and integrative physiology of intestinal peptide transport. Annu. Rev. Physiol. 2004, 66, 361–384. [Google Scholar] [CrossRef]
- Iqbal, J.; Hussain, M.M. Intestinal lipid absorption. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1183–E1194. [Google Scholar] [CrossRef]
- Mansbach, C.M.; Gorelick, F. Development, and physiological regulation of intestinal lipid absorption. II. Dietary lipid absorption, complex lipid synthesis, and the intracellular packaging and secretion of chylomicrons. Am. J. Physiol. Gastrointest. Liver Physiol. 2007, 293, G645–G650. [Google Scholar] [CrossRef] [PubMed]
- Johnson, L.R. Essential Medical Physiology; Lippincott-Raven: Philadelphia, PA, USA, 1998. [Google Scholar]
- Mansbach, C.M.; Siddiqi, S.A. The biogenesis of chylomicrons. Annu. Rev. Physiol. 2010, 72, 315–333. [Google Scholar] [CrossRef]
- Kleta, R.; Romeo, E.; Ristic, Z.; Ohura, T.; Stuart, C.; Arcos-Burgos, M.; Wagner, C.A.; Camargo, S.R.M.; Inoue, S.; Matsuura, N.; et al. Mutations in SLC6A19, encoding B0AT1, cause Hartnup disorder. Nat. Genet. 2004, 36, 999–1002. [Google Scholar] [CrossRef]
- Arboleda Bustán, J.E.; Bujons Tur, A.; de Knecht, E.L. WITHDRAWN: Clinical characteristics of pediatric patients with cystinuria at the Puigvert Foundation in the last 20 years. Actas Urol. Esp. 2020. [Google Scholar] [CrossRef]
- Lebwohl, B.; Sanders, D.S.; Green, P.H.R. Coeliac disease. Lancet 2018, 391, 70–81. [Google Scholar] [CrossRef]
- Chaudhry, N.A.; Jacobs, C.; Green, P.H.R.; Rampertab, A.D. All Things Gluten: A Review. Gastroenterol. Clin. N. Am. 2021, 50, 29–40. [Google Scholar] [CrossRef]
- Walker, M.M.; Ludvigsson, J.F.; Sanders, D.S. Coeliac disease: Review of diagnosis and management. Med. J. Aust. 2017, 207, 173–178. [Google Scholar] [CrossRef]
- Ciccocioppo, R.; Kruzliak, P.; Cangemi, G.C.; Pohanka, M.; Betti, E.; Lauret, E.; Rodrigo, L. The spectrum of differences between childhood and adulthood celiac disease. Nutrients 2015, 7, 8733–8751. [Google Scholar] [CrossRef]
- Abu Daya, H.; Lebwohl, B.; Lewis, S.K.; Green, P.H. Celiac disease patients presenting with anemia have more severe disease than those presenting with diarrhea. Clin. Gastroenterol. Hepatol. 2013, 11, 1472–1477. [Google Scholar] [CrossRef] [PubMed]
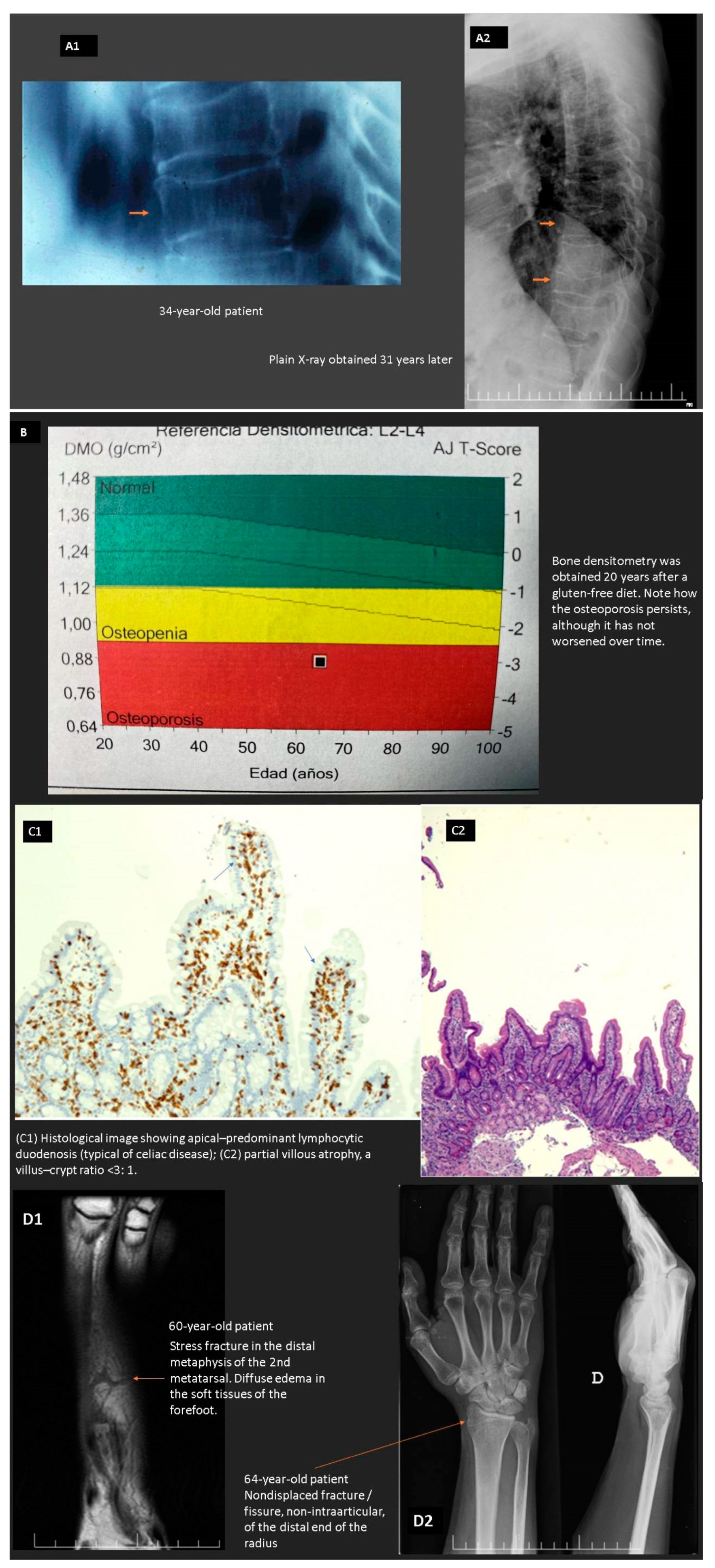
- Stuckey, B.G.A.; Mahoney, L.A.; Dragovic, S.; Brown, S.J. Celiac disease and bone health: Is there a gap in the management of postmenopausal osteoporosis? Climacteric 2020, 23, 559–565. [Google Scholar] [CrossRef] [PubMed]
- Duerksen, D.R.; Lix, L.M.; Johansson, H.; McCloskey, E.V.; Harvey, N.C.; Kanis, J.A.; Leslie, W.D. Fracture risk assessment in celiac disease: A registry-based cohort study. Osteoporos. Int. 2021, 32, 93–99. [Google Scholar] [CrossRef]
- Al-Toma, A.; Verbeek, W.H.; Hadithi, M.; von Blomberg, B.M.; Mulder, C.J. Survival in refractory coeliac disease and enteropathy-associated T-cell lymphoma: Retrospective evaluation of single-centre experience. Gut 2007, 56, 1373–1378. [Google Scholar] [CrossRef]
- Malamut, G.; Afchain, P.; Verkarre, V.; Lecomte, T.; Amiot, A.; Damotte, D.; Bouhnik, Y.; Colombel, J.F.; Delchier, J.C.; Allez, M.; et al. Presentation and long-term follow-up of refractory celiac disease: Comparison of type I with type II. Gastroenterology 2009, 136, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Rubio-Tapia, A.; Kelly, D.G.; Lahr, B.D.; Dogan, A.; Wu, T.T.; Murray, J.A. Clinical staging, and survival in refractory celiac disease: A single center experience. Gastroenterology 2009, 136, 99–107, quiz 352-3. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, B.; Vadala, M.; Laurino, C. Gluten-free diet in non-celiac patients: Beliefs, truths, advantages, and disadvantages. Minerva Gastroenterol. Dietol. 2019, 65, 153–162. [Google Scholar] [CrossRef]
- Vici, G.; Belli, L.; Biondi, M.; Polzonetti, V. Gluten free diet and nutrient deficiencies: A review. Clin. Nutr. 2016, 35, 1236–1241. [Google Scholar] [CrossRef] [PubMed]
- Saturni, L.; Ferretti, G.; Bacchetti, T. The gluten-free diet: Safety and nutritional quality. Nutrients 2010, 2, 16–34. [Google Scholar] [CrossRef]
- Pellegrini, N.; Agostoni, C. Nutritional aspects of gluten-free products. J. Sci. Food Agric. 2015, 95, 2380–2385. [Google Scholar] [CrossRef]
- Foschia, M.; Horstmann, S.; Arendt, E.K.; Zannini, E. Nutritional therapy—Facing the gap between coeliac disease and gluten-free food. Int. J. Food Microbiol. 2016, 239, 113–124. [Google Scholar] [CrossRef]
- Jansson-Knodell, C.L.; Muray, J.A.; Rubio-Tapia, A. Management of Small Bowel Villous Atrophy in Patients Seronegative for Celiac Disease. Am. J. Gastroenterol. 2020, 115, 492–497. [Google Scholar] [CrossRef]
- Mekhjian, H.S.; Switz, D.M.; Melnyk, C.S.; Rankin, G.B.; Brooks, R.K. Clinical features and natural history of Crohn’s disease. Gastroenterology 1979, 77, 898–906. [Google Scholar] [CrossRef]
- Gupta, N.; Bostrom, A.G.; Kirschner, B.S.; Cohen, S.A.; Abramson, O.; Ferry, G.D.; Gold, B.D.; Winter, H.S.; Baldassano, R.N.; Smith, T.; et al. Presentation and disease course in early- compared to later-onset pediatric Crohn’s disease. Am. J. Gastroenterol. 2008, 103, 2092–2098. [Google Scholar] [CrossRef]
- van Wanrooij, R.L.J.; Bontkes, H.J.; Neefjes-Borst, E.A.; Mulder, C.J.; Bouma, G. Immune-mediated enteropathies: From bench to bedside. J. Autoimmun. 2021, 118, 102609. [Google Scholar] [CrossRef] [PubMed]
- Akram, S.; Murray, J.A.; Pardi, D.S.; Alexander, G.L.; Schaffner, J.A.; Russo, P.A. Adult Autoimmune Enteropathy: Mayo Clinic Rochester Experience. Clin. Gastroenterol. Hepatol. 2007, 5, 1282–1290. [Google Scholar] [CrossRef]
- Cunningham-Rundles, C.; Bodian, C. Common variable immunodeficiency: Clinical and immunological features of 248 patients. Clin. Immunol. 1999, 92, 34–48. [Google Scholar] [CrossRef]
- Hermans, P.E.; Diaz-Buxo, J.A.; Stobo, J.D. Idiopathic late-onset immunoglobulin deficiency. Clinical observations in 50 patients. Am. J. Med. 1976, 61, 221–237. [Google Scholar] [CrossRef]
- Malamut, G.; Verkarre, V.; Suarez, F.; Viallard, J.F.; Lascaux, A.S.; Cosnes, J.; Bouhnik, Y.; Lambotte, O.; Béchade, D.; Ziol, M.; et al. The enteropathy associated with common variable immunodeficiency: The delineated frontiers with celiac disease. Am. J. Gastroenterol. 2020, 105, 2262–2275. [Google Scholar] [CrossRef]
- Nilssen, D.E.; Halstensen, T.S.; Froland, S.S.; Fausa, O.; Brandtzaeg, P. Intestinal intraepithelial gamma/delta T cells in B-cell deficiency. Immunodeficiency 1993, 4, 71–72. [Google Scholar]
- Biagi, F.; Bianchi, P.I.; Zilli, A.; Marchese, A.; Luinetti, O.; Lougaris, V.; Plebani, A.; Villanacci, V.; Corazza, G.R. The significance of duodenal mucosal atrophy in patients with common variable immunodeficiency: A clinical and histopathologic study. Am. J. Clin. Pathol. 2012, 138, 185–189. [Google Scholar] [CrossRef] [PubMed]
- Rothenberg, M.E. Eosinophilic gastrointestinal disorders (EGID). J. Allergy Clin. Immunol. 2004, 113, 11–28, quiz 9. [Google Scholar] [CrossRef]
- Talley, N.J.; Shorter, R.G.; Phillips, S.F.; Zinsmeister, A.R. Eosinophilic gastroenteritis: A clinicopathological study of patients with disease of the mucosa, muscle layer, and subserosal tissues. Gut 1990, 31, 54–58. [Google Scholar] [CrossRef] [PubMed]
- Pineton de Chambrun, G.; Dufour, G.; Tassy, B.; Rivière, B.; Bouta, N.; Bismuth, M.; Panaro, F.; Funakoshi, N.; Ramos, R.; Valats, J.C.; et al. Diagnosis, Natural History and Treatment of Eosinophilic Enteritis: A Review. Curr. Gastroenterol. Rep. 2018, 20, 37. [Google Scholar] [CrossRef]
- Naylor, A.R. Eosinophilic gastroenteritis. Scott. Med. J. 1990, 35, 163–165. [Google Scholar] [CrossRef]
- Sullivan, K.M.; Deeg, H.J.; Sanders, J.; Klosterman, A.; Amos, D.; Shulman, H.; Sale, G.; Martin, P.; Witherspoo, R.R.; Appelbaum, F.; et al. Hyperacute graft-v-host disease in patients not given immunosuppression after allogeneic marrow transplantation. Blood 1986, 67, 1172–1175. [Google Scholar] [CrossRef]
- Weisdorf, D.J.; Snover, D.C.; Haake, R.; Miller, W.J.; McGlave, P.B.; Blazar, B.; Ramsay, N.K.; Kersey, J.H.; Filipovich, A. Acute upper gastrointestinal graft-versus-host disease: Clinical significance and response to immunosuppressive therapy. Blood 1990, 76, 624–629. [Google Scholar] [CrossRef] [PubMed]
- Wakui, M.; Okamoto, S.; Ishida, A.; Kobayashi, H.; Watanabe, R.; Yajima, T.; Iwao, Y.; Hisamatsu, T.; Hibi, T.; Ikeda, Y. Prospective evaluation for upper gastrointestinal tract acute graft-versus-host disease after hematopoietic stem cell transplantation. Bone Marrow Transplant. 1999, 23, 573. [Google Scholar] [CrossRef][Green Version]
- Weber, J.S.; Kahler, K.C.; Hauschild, A. Management of immune-related adverse events and kinetics of response with ipilimumab. J. Clin. Oncol. 2012, 30, 2691–2697. [Google Scholar] [CrossRef]
- Weber, J.S.; Postow, M.; Lao, C.D.; Schadendorf, D. Management of adverse events following treatment with anti-programmed death-1 agents. Oncology 2016, 21, 1230–1240. [Google Scholar] [CrossRef] [PubMed]
- Gentile, N.M.; D’Souza, A.; Fujii, L.L.; Wu, T.T.; Murray, J.A. Association between ipilimumab and celiac disease. Mayo Clin. Proc. 2013, 88, 414–417. [Google Scholar] [CrossRef]
- Cammarota, G.; Cuoco, L.; Cianci, R.; Pandolfi, F.; Gasbarrini, G. Onset of coeliac disease during treatment with interferon for chronic hepatitis C. Lancet 2000, 356, 1494–1495. [Google Scholar] [CrossRef]
- Fernández-Salazar, L.; Álvarez-Quiñones, M.; Hernández, J.M.G.; Fraile, A.; Mayor, E.; Arranz, E.; Garrote, J.A. IFN beta 1b induced celiac disease. Scand. J. Gastroenterol. 2011, 46, 1525–1526. [Google Scholar] [CrossRef]
- Kamar, N.; Faure, P.; Dupuis, E.; Cointault, O.; Joseph-Hein, K.; Durand, D.; Moreau, J.; Rostaing, L. Villous atrophy induced by mycophenolate mofetil in renal-transplant patients. Transpl. Int. 2004, 17, 463–467. [Google Scholar] [CrossRef]
- Soldini, D.; Gaspert, A.; Montani, M.; Reineke, T.; Rogler, G.; Odze, R.; Weber, A. Apoptotic enteropathy caused by antimetabolites and TNF-alpha antagonists. J. Clin. Pathol. 2014, 67, 582–586. [Google Scholar] [CrossRef] [PubMed]
- Ianiro, G.; Bibbo, S.; Montalto, M.; Ricci, R.; Gasbarrini, A.; Cammarota, G. Systematic review: Sprue-like enteropathy associated with Olmesartan. Aliment. Pharmacol. Ther. 2014, 40, 16–23. [Google Scholar] [CrossRef] [PubMed]
- Marth, T. Whipple’s disease. Acta Clin. Belg. 2016, 71, 373–378. [Google Scholar] [CrossRef]
- Dolmans, R.A.; Boel, C.H.; Lacle, M.M.; Kusters, J.G. Clinical manifestations, treatment, and diagnosis of Tropheryma whipplei infections. Clin. Microbiol. Rev. 2017, 30, 529–555. [Google Scholar] [CrossRef]
- Durand, D.V.; Lecomte, C.; Cathébras, P.; Rousset, H.; Godeau, P. Whipple disease. Clinical review of 52 cases. The SNFMI Research Group on Whipple Disease. Société Nationale Française de Médecine Interne [Baltimore]. Medicine 1997, 76, 170–184. [Google Scholar] [CrossRef]
- Brar, H.S.; Aloysius, M.M.; Shah, N.J. Tropical Sprue 2021 Feb 9. In StatPearls [Internet]; StatPearls Publishing: Treasure Island, FL, USA, January 2021. [Google Scholar]
- Brown, I.; Bettington, M.; Rosty, C. The role of histopathology in the diagnosis and management of coeliac disease and other malabsorptive conditions. Histopathology 2021, 78, 88–105. [Google Scholar] [CrossRef]
- Sousa, D.; Neto Gonçalves, T.; Marto, N.; Horta, A.B. Malabsorption Due to Chronic Giardiasis as a Presenting Symptom of Common Variable Immunodeficiency. Cureus 2020, 12, e12201. [Google Scholar] [PubMed]
- Allain, T.; Amat, C.B.; Motta, J.P.; Anna Manko Buret, A.G. Interactions of Giardia sp. with the intestinal barrier: Epithelium, mucus, and microbiota. Tissue Barriers 2017, 5, e1274354. [Google Scholar] [CrossRef] [PubMed]
- Aziz, I.; Peerally, M.F.; Barnes, J.H.; Kandasamy, V.; Whiteley, J.C.; Partridge, D. The clinical and phenotypical assessment of seronegative villous atrophy; a prospective UK centre experience evaluating 200 adult cases over a 15-year period (2000–2015). Gut 2017, 66, 1563–1572. [Google Scholar] [CrossRef]
- Fung, W.P.; Tan, K.K.; Yu, S.F.; Sho, K.M. Malabsorption and subtotal villous atrophy secondary to pulmonary and intestinal tuberculosis. Gut 1970, 11, 212–216. [Google Scholar] [CrossRef][Green Version]
- Desai, H.G.; Zaveri, M.P.; Gupta, N.I.; Antia, F.P. Histological and functional studies of small intestinal mucosa in intestinal tuberculosis. Indian J. Med. Sci. 1976, 30, 1–4. [Google Scholar]
- Batman, P.A.; Kapembwa, M.S.; Griffin, G.E. Enteropathy associated with HIV. Gut 1990, 31, 960. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cummins, A.G.; LaBrooy, J.T.; Stanley, D.P.; Rowland, R.; Shearman, D.J.C. Quantitative histological study of enteropathy associated with HIV infection. Gut 1990, 31, 317–321. [Google Scholar] [CrossRef]
- Ullrich, R.; Zeitz, M.; Heise, W.; L’Age, M.; Hoffken, G.; Riecken, E.O. Small intestinal structure and function in patients infected with human immunodeficiency virus (HIV): Evidence for HIV induced enteropathy. Ann. Intern. Med. 1989, 111, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Batman, P.A.; Miller, A.R.O.; Forster, S.M.; Harris, J.R.W.; Pinching, A.J.; Griffin, G.E. Jejunal enteropathy associated with human immunodeficiency virus infection: Quantitative histology. J. Clin. Pathol. 1989, 42, 275–281. [Google Scholar] [CrossRef]
- Zafar, H.; Jain, A.G.; Khetpal, N.; Ali, S.; Rashid, M.U.; Ahmad, S. Small Intestinal Bacterial Overgrowth: A Critical Review of an Underrecognized but Disrupting Entity. Crit. Rev. Oncog. 2020, 25, 365–379. [Google Scholar] [CrossRef]
- Brown, I.S.; Bettington, A.; Bettington, M.; Rosty, C. Self-limited coeliac- like enteropathy: A series of 18 cases highlighting another coeliac disease mimic. Histopathology 2016, 68, 254–261. [Google Scholar] [CrossRef] [PubMed]
- Troeger, H.; Loddenkemper, C.; Schneider, T.; Fromm, M.; Schulzke, J.D. Structura and functional changes of the duodenum in human norovirus infection. Gut 2009, 58, 1070–1077. [Google Scholar] [CrossRef]
- Wolf, J.L.; Schreiber, D.S. Viral gastroenteritis. Med. Clin. N. Am. 1982, 66, 575–595. [Google Scholar] [CrossRef]
- Lerardi, E.; Losurdo, G.; Iannone, A.; Piscitelli, D.; Amoruso, A.; Barone, M.; Principi, M.B.; Pisani, A.; Di Leo, A. Lymphocytic duodenitis or microscopic enteritis and gluten-related conditions: What needs to be explored? Ann. Gastroenterol. 2017, 30, 380–392. [Google Scholar]
- Al-Bawardy, B.; Ramos, G.; Wu, T.; Wu, T.T.; Rubio-Tapia, A.; Murray, J.A. Sa1409 Collagenous Sprue: 15 Year Experience of a Large Tertiary Center. Gastroenterology 2016, 150, S307–S308. [Google Scholar] [CrossRef]
- Rejeski, J.; Conway, J.; Zhou, Y. Collagenous Sprue. Am. J. Med. Sci. 2020, 359, 310–311. [Google Scholar] [CrossRef]
- Rowe, K.; Pankow, J.; Nehme, F.; Salyers, W. Gastrointestinal Amyloidosis: Review of the Literature. Cureus 2017, 9, e1228. [Google Scholar] [CrossRef]
- Ebert, E.C.; Nagar, M. Gastrointestinal manifestations of amyloidosis. Am. J. Gastroenterol. 2008, 103, 776–787. [Google Scholar] [CrossRef]
- Fonnesu, C.; Giovinale, M.; Verrecchia, E.; De Socio, G.; Cerquaglia, C.; Curigliano, V.; Soriano, A.; Obici, L.; Grieco, A.; Lauriola, L.L.; et al. Gastrointestinal amyloidosis: A case of chronic diarrhoea. Eur. Rev. Med. Pharmacol. Sci. 2009, 13, 45–50. [Google Scholar]
- Lim, A.Y.; Lee, J.H.; Jung, K.S.; Gwag, H.B.; Kim, D.H.; Kim, S.J.; Lee, G.Y.; Kim, J.S.; Kim, H.J.; Lee, S.Y.; et al. Clinical features and outcomes of systemic amyloidosis with gastrointestinal involvement: A single-center experience. Korean J. Intern. Med. 2015, 30, 496–505. [Google Scholar] [CrossRef]
- Ewers, E.C.; Sheffler, R.L.; Wang, J.; Ngauy, V. Immunoproliferative Small Intestinal Disease Associated with Overwhelming Polymicrobial Gastrointestinal Infection with Transformation to Diffuse Large B-Cell Lymphoma. Am. J. Trop. Med. Hyg. 2016, 94, 1177–1181. [Google Scholar] [CrossRef] [PubMed]
- Lecuit, M.; Abachin, E.; Martin, A.; Poyart, C.; Pochart, P.; Suarez, F.; Bengoufa, D.; Feuillard, J.; Lavergne, A.; Gordon, J.I.; et al. Immunoproliferative small intestinal disease associated with Campylobacter jejuni. N. Engl. J. Med. 2004, 350, 239–248. [Google Scholar] [CrossRef]
- Fine, K.D.; Stone, M.J. Alpha-heavy chain disease, Mediterranean lymphoma, and immunoproliferative small intestinal disease: A review of clinicopathological features, pathogenesis, and differential diagnosis. Am. J. Gastroenterol. 1999, 94, 1139–1152. [Google Scholar] [PubMed]
- Cording, S.; Lhermitte, L.; Malamut, G.; Berrabah, S.; Trinquand, A.; Guegan, N.; Villarese, P.; Kaltenbach, S.; Meresse, M.; Khater, S.; et al. Oncogenetic landscape of lymphomagenesis in coeliac disease. CELAC network. Gut 2021. [Google Scholar] [CrossRef] [PubMed]
- Cellier, C.; Delabesse, E.; Helmer, C.; Patey, N.; Matuchansky, C.; Jabri, B.; Macintyre, E.; Cerf-Bensussan, N.; Brousse, N. Refractory sprue, coeliac disease, and enteropathy-associated T-cell lymphoma. French coeliac disease Study Group. Lancet 2000, 356, 203–208. [Google Scholar] [CrossRef]
- Ho-Yen, C.; Chang, F.; van der Walt, J.; Mitchell, T.; Ciclitira, P. Recent advances in refractory coeliac disease: A review. Histopathology 2009, 54, 783–795. [Google Scholar] [CrossRef] [PubMed]
- Chibbar, R.; Nostedt, J.; Mihalicz, D.; Deschenes, J.; McLean, R.; Dieleman, L.A. Refractory Celiac Disease Type II: A Case Report and Literature Review. Front. Med. (Lausanne) 2020, 7, 56487. [Google Scholar]
- Malamut, G.; Chandesris, O.; Verkarre, V.; Meresse, B.; Callens, C.; Macintyre, E.; Bouhnik, Y.; Gornet, J.M.; Allez, M.; Jian, R.; et al. Enteropathy associated T cell lymphoma in celiac disease: A large retrospective study. Dig. Liver Dis. 2013, 45, 377–384. [Google Scholar] [CrossRef]
- Sabbah, M.; Farhat, F.B.; Bellil, N.; Khanchel, F.; Ouakaa, A.; Gargouri, D. An unusual case of diffuse large B-cell lymphoma complicating a Crohn’s disease. Clin. Case Rep. 2020, 8, 3062–3065. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Broséus, J.; Feugier, P.; Thieblemont, C.; Laurent, B.; Danese, S.; Arnone, D.; Ndiaye, N.C.; Kokten, T.; Houlgatte, R.; et al. Characteristics of Lymphoma in Patients with Inflammatory Bowel Disease: A Systematic Review. J. Crohns Colitis 2020, jjaa193. [Google Scholar] [CrossRef]
- Severyns, T.; Kirchgesner, J.; Lambert, J.; Thieblemont, C.; Amiot, A.; Abitbol, V.; Treton, K.; Cazals-Hatem, D.; Malamut, G.; Coppo, P.; et al. Prognosis of Lymphoma in Patients with Known Inflammatory Bowel Disease: A French Multicentre Cohort Study. J. Crohns Colitis 2020, 14, 1222–1230. [Google Scholar] [CrossRef]
- Tickell, K.D.; Atlas, H.E.; Walson, J.L. Environmental enteric dysfunction: A review of potential mechanisms, consequences and management strategies. BMC Med. 2019, 17, 181–190. [Google Scholar] [CrossRef]
- Keusch, G.T.; Denno, D.M.; Black, R.E.; Duggan, C.; Guerrant, R.L.; Lavery, J.V.; Nataro, J.O.; Rosenberg, I.H.; Ryan, E.T.; Tarr, P.I.; et al. Environmental enteric dysfunction: Pathogenesis, diagnosis, and clinical consequences. Clin. Infect. Dis. 2014, 59 (Suppl. 4), S207–S212. [Google Scholar] [CrossRef]
- Harper, K.M.; Mutasa, M.; Prendergast, A.J.; Humphrey, J.; Manges, A.R. Environmental enteric dysfunction pathways and child stunting: A systematic review. PLoS Negl. Trop. Dis. 2018, 12, e0006205. [Google Scholar] [CrossRef]
- Syed, S.; Ali, A.; Duggan, C. Environmental enteric dysfunction in children. J. Pediatr. Gastroenterol. Nutr. 2016, 63, 6–14. [Google Scholar] [CrossRef]
- Sreenarasimhaiah, J. Chronic mesenteric ischemia. Best Pract. Res. Clin. Gastroenterol. 2005, 19, 283. [Google Scholar] [CrossRef] [PubMed]
- Moawad, J.; Gewertz, B.L. Chronic mesenteric ischemia. Clinical presentation and diagnosis. Surg. Clin. N. Am. 1997, 77, 357–369. [Google Scholar] [CrossRef]
- Galland, R.B.; Spencer, J. The natural history of clinically established radiation enteritis. Lancet 1985, 1, 1257–1258. [Google Scholar] [CrossRef]
- Scolapio, J.S.; Ukleja, A.; Burnes, J.U.; Kelly, D.G. Outcome of patients with radiation enteritis treated with home parenteral nutrition. Am. J. Gastroenterol. 2002, 97, 662–666. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Hegele, R.A. Abetalipoproteinemia and homozygous hypobetalipoproteinemia: A framework for diagnosis and management. J. Inherit. Metab. Dis. 2014, 37, 333–339. [Google Scholar] [CrossRef]
- Hussain, M.M.; Shi, J.; Dreizen, P. Microsomal triglyceride transfer protein and its role in apolipoprotein Blipoprotein assembly. J. Lipid Res. 2003, 44, 22–32. [Google Scholar] [CrossRef]
- Di Filippo, M.; Moulin, P.; Roy, P.; Samson-Bouma, M.E.; Collardeau-Frachon, S.; Chebel-Dumont, S.; Peretti, N.; Dumortier, J.; Zoulim, F.; Fontanges, T.; et al. Homozygous MTTP and APOB mutations may lead to hepatic steatosis and fibrosis despite metabolic differences in congenital hypocholesterolemia. J. Hepatol. 2014, 61, 891–902. [Google Scholar] [CrossRef]
- Walsh, M.T.; Di Leo, E.; Okur, I.; Tarugi, P.; Hussain, M.M. Structure-function analyses of microsomal triglyceride transfer protein missense mutations in abetalipoproteinemia and hypobetalipoproteinemia subjects. Biochim. Biophys. Acta 2016, 1861, 1623–1633. [Google Scholar] [CrossRef]
- Tarugi, P.; Averna, M. Hypobetalipoproteinemia: Genetics, biochemistry, and clinical spectrum. Adv. Clin. Chem. 2011, 54, 81–107. [Google Scholar] [PubMed]
- Peretti, N.; Sassolas, A.; Roy, C.C.; Deslandres, C.; Charcosset, M.; Castagnetti, J.; Pugnet-Chardon, L.; Moulin, P.; Labarge, S.; Bouthillier, L.; et al. Guidelines for the diagnosis and management of chylomicron retention disease based on a review of the literature and the experience of two centers. Orphanet J. Rare Dis. 2010, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Bredefeld, C.; Peretti, N.; Hussain, M.M. Medical Advisory Panel, New Classification and Management of Abetalipoproteinemia and Related Disorders. Gastroenterology 2021. [Google Scholar] [CrossRef]
- Huber, R.; Semmler, G.; Mayr, A.; Offner, F.; Datz, C. Primary intestinal lymphangiectasia in an adult patient: A case report and review of literature. World J. Gastroenterol. 2020, 26, 7707–7718. [Google Scholar] [CrossRef]
- Freeman, H.J.; Nimmo, M. Intestinal lymphangiectasia in adults. World J. Gastrointest. Oncol. 2011, 3, 19–23. [Google Scholar] [CrossRef]
- Lopez, R.N.; Day, A.S. Primary intestinal lymphangiectasia in children: A review. J. Paediatr. Child. Health 2020, 56, 1719–1723. [Google Scholar] [CrossRef]
- Crutzen, B.; Poncelet, P.A. Protein-Losing Enteropathy in Primary Lymphangiectasia. J. Belg. Soc. Radiol. 2020, 104, 34. [Google Scholar] [CrossRef]
- Iwamuro, M.; Tanaka, T.; Kanzaki, H.; Kawano, S.; Kawahara, Y.; Iwasaki, Y. Deterioration of duodenal lymphangiectasia after radiotherapy for gastric MALT lymphoma. Ecancermedicalscience 2017, 11, 752–757. [Google Scholar] [CrossRef] [PubMed]
- Rao, S.S.; Dundas, S.; Holdsworth, C.D. Intestinal lymphangiectasia secondary to radiotherapy and chemotherapy. Dig. Dis. Sci. 1987, 32, 939–942. [Google Scholar] [CrossRef]
- Greenberger, N.J.; Isselbacher, K.J. Malabsorption following radiation injury to the gastrointestinal tract. Am. J. Med. 1964, 36, 50–56. [Google Scholar] [CrossRef]
- Tankel, H.I.; Clark, D.H.; Lee, F.D. Radiation enteritis with malabsorption. Gut 1965, 6, 560–569. [Google Scholar] [CrossRef]
- Dominguez-Muñoz, J.E.; Phillips, M. Nutritional therapy in chronic pancreatitis. Gastroenterol. Clin. N. Am. 2018, 47, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Tan, B.H.; Birdsell, L.A.; Martin, L.; Baracos, V.E.; Fearon, K.C.H. Sarcopenia in an overweight or obese patient is an adverse prognostic factor in pancreatic cancer. Clin. Cancer Res. 2009, 15, 6973–6979. [Google Scholar] [CrossRef]
- Engelen, M.P.; Schroder, R.; Van der Hoorn, K.; Deutz, N.E.P.; Comet, G. Use of body mass index percentile to identify fat-free mass depletion in children with cystic fibrosis. Clin. Nutr. 2012, 31, 927–933. [Google Scholar] [CrossRef]
- Livingstone, C.; Davis, J.; Marvin, V.; Morton, K. Vitamin A deficiency presenting as night blindness during pregnancy. Ann. Clin. Biochem. 2003, 40, 292–294. [Google Scholar] [CrossRef] [PubMed]
- Duggan, S.N.; Smyth, N.D.; O’Sullivan, M.; Feehan, S.; Ridgway, P.F.; Conlon, K.C. The prevalence of malnutrition and fat-soluble vitamin deficiencies in chronic pancreatitis. Nutr. Clin. Pract. 2014, 29, 348–354. [Google Scholar] [CrossRef] [PubMed]
- Lindkvist, B.; Dominguez-Munoz, J.E.; Luaces-Regueira, M.; Castiñeiras-Alvariño, M.; Nieto-Garcia, L.; Iglesias-Garcia, J. Serum nutritional markers for prediction of pancreatic exocrine insufficiency in chronic pancreatitis. Pancreatology 2012, 12, 305–310. [Google Scholar] [CrossRef]
- Lindkvist, B.; Phillips, M.E.; Dominguez-Munoz, J.E. Clinical, anthropometric and laboratory nutritional markers of pancreatic exocrine insufficiency: Prevalence and diagnostic use. Pancreatology 2015, 15, 589–597. [Google Scholar] [CrossRef] [PubMed]
- Montalto, G.; Sores, M.; Carroccio, A.; Scafidi, E.; Barbagallo, C.M.; Ippolito, S.; Notarbartolo, A. Lipoproteins and Chronic Pancreatitis. Pancreas 1994, 9, 137–138. [Google Scholar] [CrossRef] [PubMed]
- De la Iglesia-Garcia, D.; Vallejo-Senra, N.; Iglesias-Garcia, J.; López-López, A.; Nieto, L.; Domínguez-Muñoz, J.E. Increased risk of mortality associated with pancreatic exocrine insufficiency in patients with chronic pancreatitis. J. Clin. Gastroenterol. 2018, 52, e63–e72. [Google Scholar] [CrossRef] [PubMed]
- Sikkens, E.C.; Cahen, D.L.; Koch, A.D.; Braat, H.; Poley, J.W.; Kuipers, E.J. The prevalence of fat-soluble vitamin deficiencies and a decreased bone mass in patients with chronic pancreatitis. Pancreatology 2013, 13, 238–242. [Google Scholar] [CrossRef] [PubMed]
- Barton, S.H.; Kelly, D.G.; Murray, J.A. Nutritional deficiencies in celiac disease. Gastroenterol. Clin. N. Am. 2007, 36, 93–108. [Google Scholar] [CrossRef] [PubMed]
- Food and Nutrition Board of the Institute of Medicine. Dietary Reference Intakes for Vitamin A, Vitamin K, Arsenic, Boron, Chromium, Copper, Iodine, Iron, Manganese, Molybdenum, Nickel, Silicon, Vanadium, and Zinc (2000); National Academies Press: Washington, DC, USA, 2000; Available online: https://www.nap.edu/read/10026/chapter/1 (accessed on 24 May 2018).
- Food and Nutrition Information Center. Dietary Reference Intake Reports. Available online: https://www.nal.usda.gov/fnic/dietary-reference-intakes (accessed on 3 January 2020).
- Institute of Medicine. Dietary Reference Intakes for Thiamin, Riboflavin, Niacin, Vitamin B6, Folate, Vitamin B12, Pantothenic Acid, Biotin, and Choline. 1998. Available online: https://www.nap.edu/read/6015/chapter/1 (accessed on 3 January 2020).
- World Health Organization (WHO). E-Library of Evidence for Nutrition Actions (eLENA)—Nutrients. Available online: https://www.who.int/elena/about/en/ (accessed on 3 January 2020).
- US Department of Health and Human Services (DHHS) and US Department of Agriculture (USDA). Dietary Guidelines for Americans—2015–2020, 8th ed.; 2015. Available online: https://health.gov/sites/default/files/2019-09/2015-2020_Dietary_Guidelines.pdf (accessed on 3 January 2020).
- European Society for Paediatric Gastroenterology, Hepatology, and Nutrition (ESPGHAN). Position paper on assessment and Interpretation of Vitamin and Trace Element Status in Sick Children. J. Pediatr. Gastroenterol. Nutr. 2020, 70, 873–881. [Google Scholar] [CrossRef] [PubMed]
- ESPGHAN/European Society for Clinical Nutrition and Metabolism (ESPEN)/European Society of Paediatric Research (ESPR)/Chinese Society of Parenteral and Enteral Nutrition (CSPEN). Guidelines on Paediatric Parenteral Nutrition—Vitamins. Clin. Nutr. 2018, 37 Pt B, 2366–2378. [Google Scholar]
- Public Health England (PHE). Government Dietary Recommendations—Government Recommendations for Energy and Nutrients for Males and Females Aged 1–18 Years and 19+ Years. 2016. Available online: https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/618167/government_dietary_recommendations.pdf (accessed on 3 January 2020).
- Department of Health, Government of South Australia (SA Health). South Australian Perinatal Practice Guidelines—Vitamin and Mineral Supplementation in Pregnancy. 2015. Available online: https://www.sahealth.sa.gov.au/wps/wcm/connect/public+content/sa+health+internet/clinical+resources/clinical+programs+and+practice+guidelines/womens+and+babies+health/perinatal/perinatal+practice+guidelines/perinatal+practice+guidelines (accessed on 3 January 2020).
- Mehri, A. Trace Elements in Human Nutrition (II)—An Update. Int. J. Prev. Med. 2020, 11. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6993532/ (accessed on 3 January 2020).
- Kelly, C.P. Celiac disease. In Sleisenger and Fordtran’s Gastrointestinal and Liver Disease. Pathophysiology, Diagnosis, Management, 10th ed.; Feldman, M., Friedman, L., Brandt, L.J., Eds.; Elsevier-Saunders: Philadelphia, PA, USA, 2016; pp. 1849–1872. [Google Scholar]
- Guarino, M.; Gambuti, E.; Alfano, F.; Strada, A.; Ciccocioppo, R.; Lungaro, L. Life-threatening onset of coeliac disease: A case report and literature review. BMJ Open Gastroenterol. 2020, 7, e000406. [Google Scholar] [CrossRef]
- Farrell, R.J.; Kelly, C.P. Celiac sprue. N. Engl. J. Med. 2002, 346, 180–188. [Google Scholar] [CrossRef] [PubMed]
- Halfdanarson, T.R.; Litzow, M.R.; Murray, J.A. Hematological manifestations of celiac disease. Blood 2006, 109, 412–421. [Google Scholar] [CrossRef]
- Clifford, L. Micronutrient deficiencies. In Clinical Nutrition in Gastrointestinal Disease; Buchman, A.L., Ed.; SLACK Inc.: Thorofare, NJ, USA, 2006. [Google Scholar]
- Fernández-Bañares, F.; Monzón, H.; Forné, M. A short review of malabsorption and anemia. World J. Gastroenterol. 2009, 15, 4644–4652. [Google Scholar] [CrossRef] [PubMed]
- Fine, K.D. The prevalence of occult gastrointestinal bleeding in celiac sprue. N. Engl. J. Med. 1996, 334, 1163–1167. [Google Scholar] [CrossRef]
- Logan, R.F.; Howarth, G.F.; West, J.; Shepherd, K.; Robinson, M.H.E.; Hardcastle, J.D. How often is a positive faecal occult blood test the result of coeliac disease? Eur. J. Gastroenterol. Hepatol. 2003, 15, 1097–1100. [Google Scholar] [CrossRef]
- Mant, M.J.; Bain, V.G.; Maguire, C.G.; Yacyshyn, B.R. Prevalence of occult gastrointestinal bleeding in celiac disease. Clin. Gastroenterol. Hepatol. 2006, 4, 451–454. [Google Scholar] [CrossRef]
- García-Manzanares, Á.; Lucendo, A.J. Review: Nutritional and Dietary Aspects of Celiac Disease. Nutr. Clin. Pract. 2011, 26, 163–173. [Google Scholar] [CrossRef]
- Martín-Masot, R.; Nestares, M.T.; Diaz-Castro, J.; López-Aliaga, I.; Alférez, M.J.M.; Moreno-Fernandez, J.; Maldonado, J. Multifactorial Etiology of Anemia in Celiac Disease and Effect of Gluten-Free Diet: A Comprehensive Review. Nutrients 2019, 11, 2557. [Google Scholar] [CrossRef] [PubMed]
- Bode, S.; Gudmand-Hoyer, E. Symptoms and haematologic features in consecutive adult coeliac patients. Scand. J. Gastroenterol. 1996, 31, 54–60. [Google Scholar] [CrossRef]
- Dahele, A.; Ghosh, S. Vitamin B12 deficiency in untreated celiac disease. Am. J. Gastroenterol. 2001, 96, 745–750. [Google Scholar] [CrossRef]
- Wierdsma, N.; van Bokhorst-de van der Schueren, M.; Berkenpas, M.; Mulder, C.; van Bodegraven, A. Vitamin and Mineral Deficiencies Are Highly Prevalent in Newly Diagnosed Celiac Disease Patients. Nutrients 2013, 5, 3975–3992. [Google Scholar] [CrossRef] [PubMed]
- Lindenbaum, J.; Healton, E.B.; Savage, D.G.; Brust, J.C.; Garrett, T.J.; Podell, E.R.; Marcell, P.D.; Stabler, S.P.; Allen, R.H. Neuropsychiatric disorders caused by cobalamin deficiency in the absence of anemia or macrocytosis. N. Engl. J. Med. 1988, 318, 1720–1728. [Google Scholar] [CrossRef]
- Hall, J.A.; Grainger, J.R.; Spencer, S.P.; Belkaid, Y. The role of retinoic acid in tolerance and immunity. Immunity 2011, 35, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Holick, M.F. Vitamin D deficiency: What a pain it is. Mayo Clin. Proc. 2003, 78, 1457–1459. [Google Scholar] [CrossRef]
- Kozanoglu, E.; Basaran, S.; Goncu, M.K. Proximal myopathy as an unusual presenting feature of celiac disease. Clin. Rheumatol. 2005, 24, 76–78. [Google Scholar] [CrossRef]
- West, J.; Logan, R.F.; Card, T.R.; Smith, C.; Hubbard, R. Fracture risk in people with celiac disease: A population-based cohort study. Gastroenterology 2003, 125, 429–436. [Google Scholar] [CrossRef]
- Vasquez, H.; Mazure, R.; Gonzalez, D.; Flores, D.; Pedreira, S.; Niveloni, S.; Smecuol, E.; Mauriño, C.; Bai, J.C. Risk of fractures in celiac disease patients: A cross-sectional, case-control study. Am. J. Gastroenterol. 2000, 95, 183–189. [Google Scholar] [CrossRef]
- Moreno, M.L.; Vazquez, H.; Mazure, R.; Smecuol, E.; Niveloni, S.; Pedreira, S.; Sugai, E.; Mauriño, E.; Gomez, J.C.; Bai, J.C. Stratification of bone fracture risk in patients with celiac disease. Clin. Gastroenterol. Hepatol. 2004, 2, 127–134. [Google Scholar] [CrossRef]
- Zanchetta, M.B.; Longobardi, V.; Bai, J.C. Bone and Celiac Disease. Curr. Osteoporos. Rep. 2016, 14, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.W.; Joyce, A.; Ingold, K.U. Is vitamin E the only lipid-soluble, chain-breaking antioxidant in human blood plasma and erythrocyte membranes? Arch. Biochem. Biophys. 1983, 221, 281–290. [Google Scholar] [CrossRef]
- Zingg, J.M.; Azzi, A. Non-antioxidant activities of vitamin E. Curr. Med. Chem. 2004, 11, 1113–1133. [Google Scholar] [CrossRef]
- Kalra, V.; Grover, J.; Ahuja, G.K.; Kurana, D.S. Vitamin E deficiency and associated neurological deficits in children with protein-energy malnutrition. J. Trop. Pediatr. 1998, 44, 2915. [Google Scholar] [CrossRef]
- Mason, K.E. A conspectus of research on copper metabolism and requirements of man. J. Nutr. 1979, 109, 1979–2066. [Google Scholar] [CrossRef]
- Evans, G.W. The role of copper in metabolic disorders. Adv. Exp. Med. Biol. 1981, 135, 121–137. [Google Scholar]
- Wapnir, R.A. Copper absorption and bioavailability. Am. J. Clin. Nutr. 1998, 67, 1054S–1060S. [Google Scholar] [CrossRef]
- Freeman, H.J. Neurological disorders in adult celiac disease. Can. J. Gastroenterol. 2008, 22, 909–911. [Google Scholar] [CrossRef] [PubMed]
- Guevara Pacheco, G.; Chávez Cortés, E.; Castillo-Durán, C. Deficiencia de micronutrientes y enfermedad celíaca. Arch. Argent Pediatr. 2014, 112, 457–463. [Google Scholar] [PubMed]
- Halfdanarion, T.R.; Kumar, N.; Hogan, W.J.; Murray, J.A. Copper deficiency in celiac disease. J. Clin. Gastroenterol. 2009, 43, 162–164. [Google Scholar] [CrossRef]
- Cavallieri, F.; Fin, N.; Contardi, S.; Fiorini, M.; Corradini, E.; Valzania, F. Subacute copper-deficiency myelopathy in as patient with occult celiac disease. J. Spinal Cord Med. 2017, 40, 489–491. [Google Scholar] [CrossRef] [PubMed]
- Goodman, B.P.; Mistry, D.H.; Pasha, S.F.; Bosch, P.E. Copper deficiency myeloneuropathy due to occult celiac disease. Neurologist 2009, 15, 355–356. [Google Scholar] [CrossRef] [PubMed]
- Qureshi, A.; Bergbower, E.; Patel, J. Copper deficiency myeloneuropathy with a history of malabsorption: A tale of two cases. J. Community Hosp. Intern. Med. Perspect. 2021, 11, 152–155. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Prasad, A.S.; Brewer, G.J.; Owyang, C. Zinc absorption in human small intestine. Am. J. Physiol. 1989, 256, G87–G91. [Google Scholar] [CrossRef] [PubMed]
- Maares, M.; Haase, H. A Guide to Human Zinc Absorption: General Overview and Recent Advances of In Vitro Intestinal Models. Nutrients 2020, 12, 762. [Google Scholar] [CrossRef]
- Weigand, E. Absorption of trace elements: Zinc. Int. J. Vitam. Nutr. Res. Suppl. 1983, 25, 67–81. [Google Scholar]
- Sandström, B. Bioavailability of zinc. Eur. J. Clin. Nutr. 1997, 51 (Suppl. 1), S17–S19. [Google Scholar]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Montoro-Huguet, M.A.; Belloc, B.; Domínguez-Cajal, M. Small and Large Intestine (I): Malabsorption of Nutrients. Nutrients 2021, 13, 1254. https://doi.org/10.3390/nu13041254
Montoro-Huguet MA, Belloc B, Domínguez-Cajal M. Small and Large Intestine (I): Malabsorption of Nutrients. Nutrients. 2021; 13(4):1254. https://doi.org/10.3390/nu13041254
Chicago/Turabian StyleMontoro-Huguet, Miguel A., Blanca Belloc, and Manuel Domínguez-Cajal. 2021. "Small and Large Intestine (I): Malabsorption of Nutrients" Nutrients 13, no. 4: 1254. https://doi.org/10.3390/nu13041254
APA StyleMontoro-Huguet, M. A., Belloc, B., & Domínguez-Cajal, M. (2021). Small and Large Intestine (I): Malabsorption of Nutrients. Nutrients, 13(4), 1254. https://doi.org/10.3390/nu13041254