Maternal Protein Restriction in Rats Alters the Expression of Genes Involved in Mitochondrial Metabolism and Epitranscriptomics in Fetal Hypothalamus

 , , , ,
, , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. In Vitro Culture of Neurospheres from E17 Hypothalamus
2.3. Proliferation Test Using BrdU
2.4. Immunocytochemistry
2.5. Mitochondrial Membrane Potential Determination
2.6. Western Blotting
2.7. m6A RNA Methylation Assay
2.8. 3′DGE Library Preparation, Differential Gene Expression Analysis and Enrichment Analysis
2.9. Mitochondrial DNA Quantification Using qPCR
2.10. Statistics
3. Results
3.1. Fetal and Placenta Weight at E17 Did Not Differ between Control and PR Fetuses
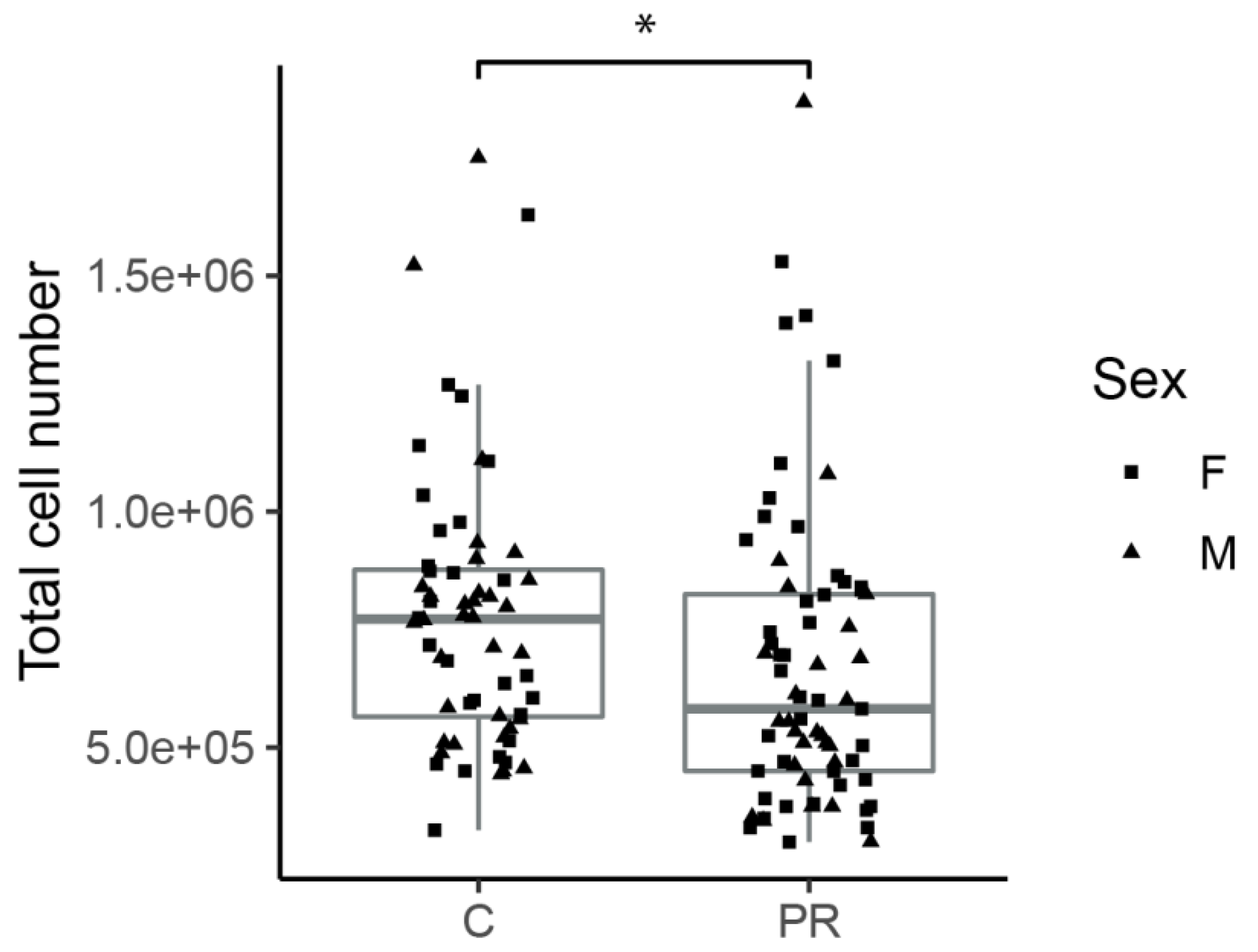
3.2. Hypothalami from PR Fetuses Contained a Lower Number of Cells
3.3. The Proportion of the Several Cell Populations Present in E17 Hypothalami Did Not Differ between Control and PR Fetuses
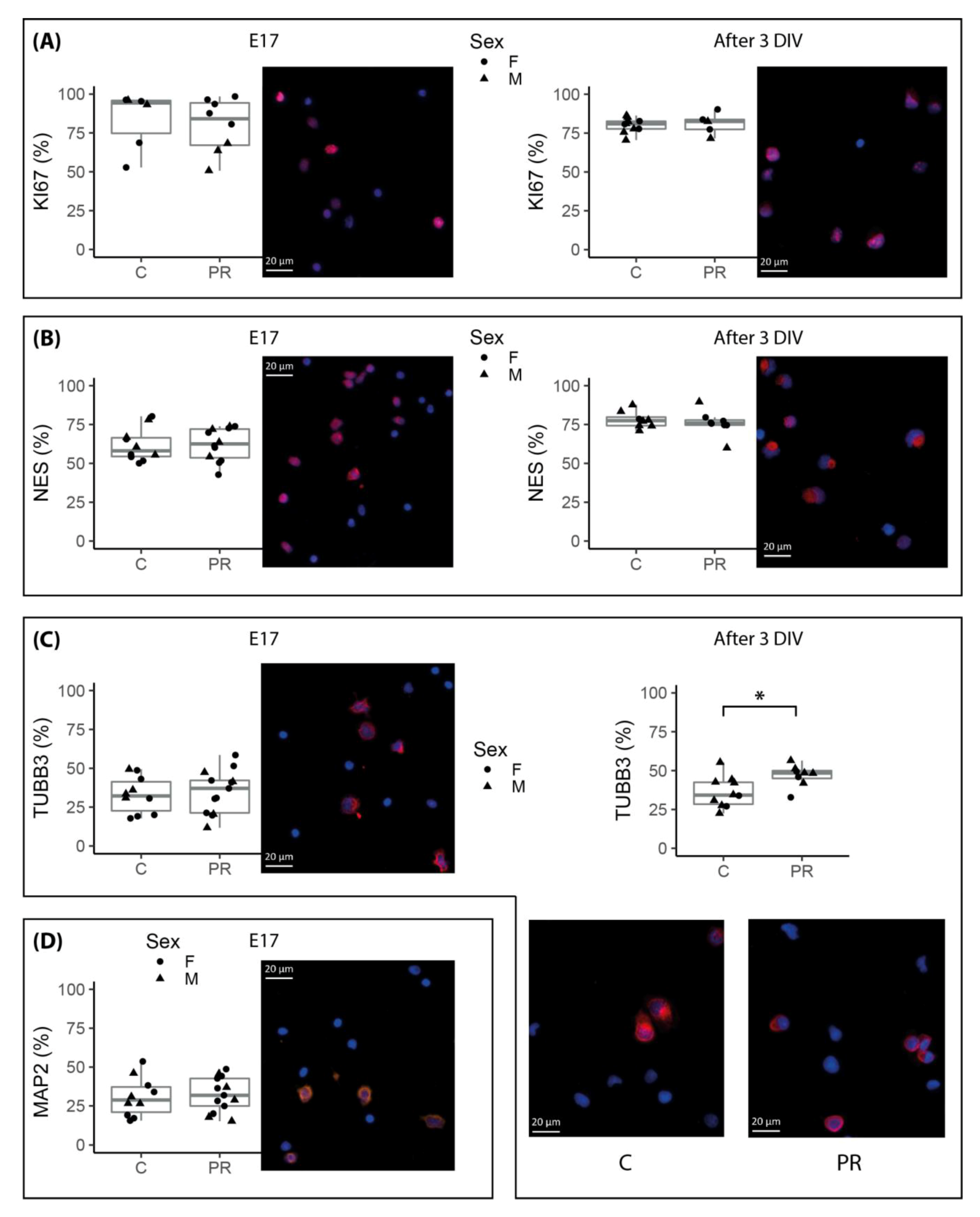
3.4. Neurosphere Cultures from PR E17 Hypothalami Contained a Higher Number of Committed Neuronal Progenitors after Three Days of Proliferation In Vitro
3.5. Neurospheres Cultures Did Not Show Any Proliferation Potential Difference between Control and PR Fetuses
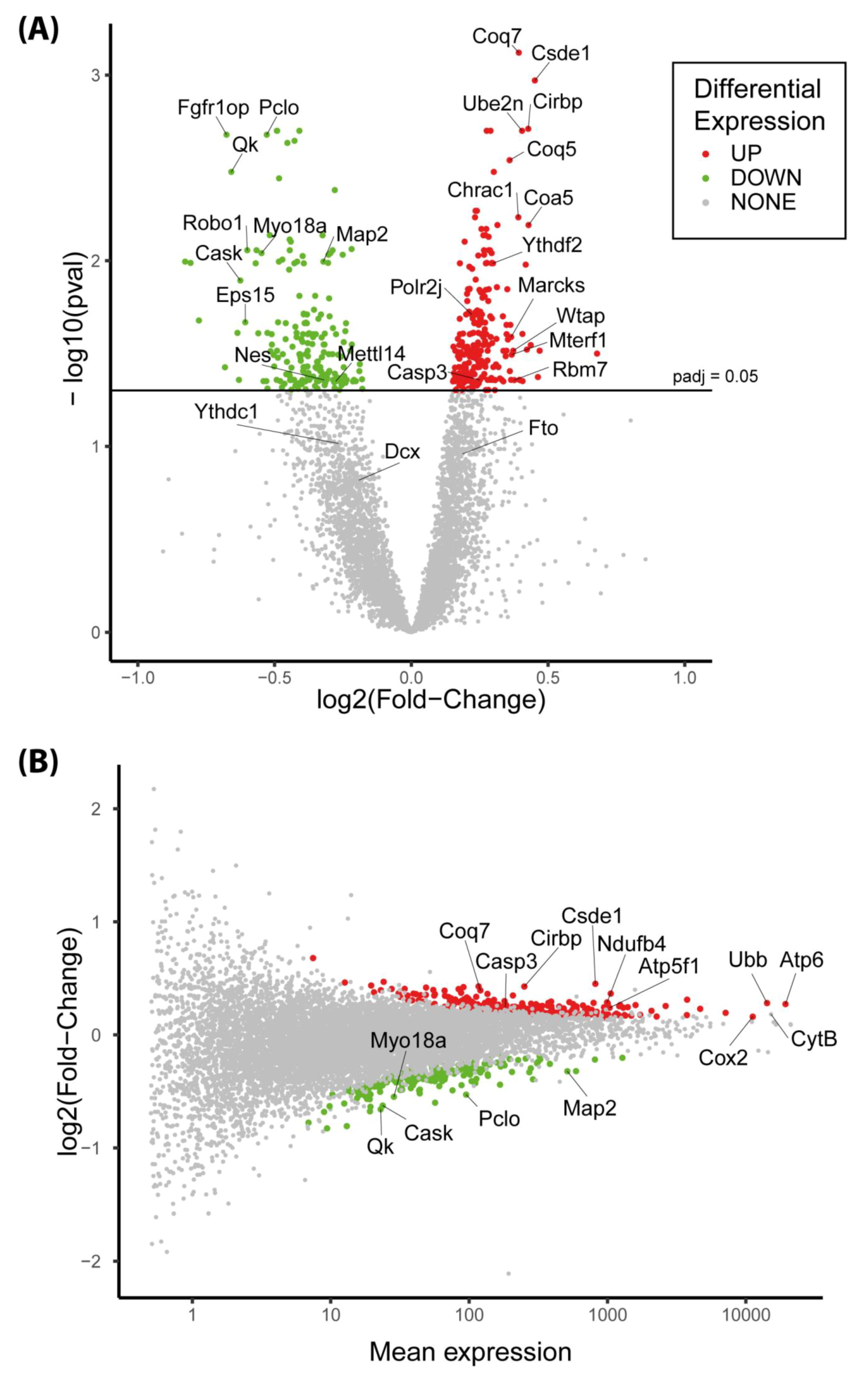
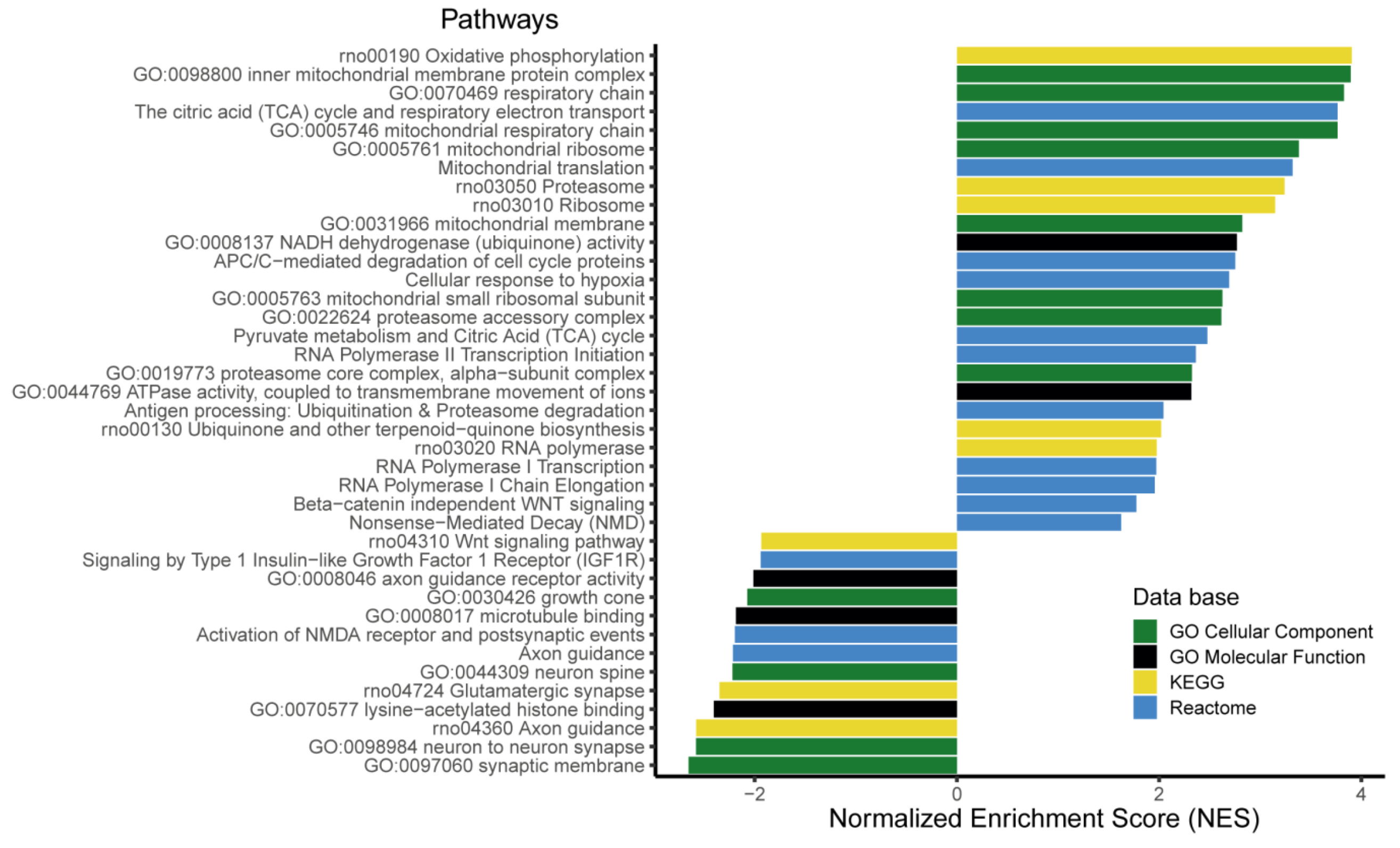
3.6. The Expression of More than 400 Genes Was Altered in PR E17 Fetal Hypothalamus
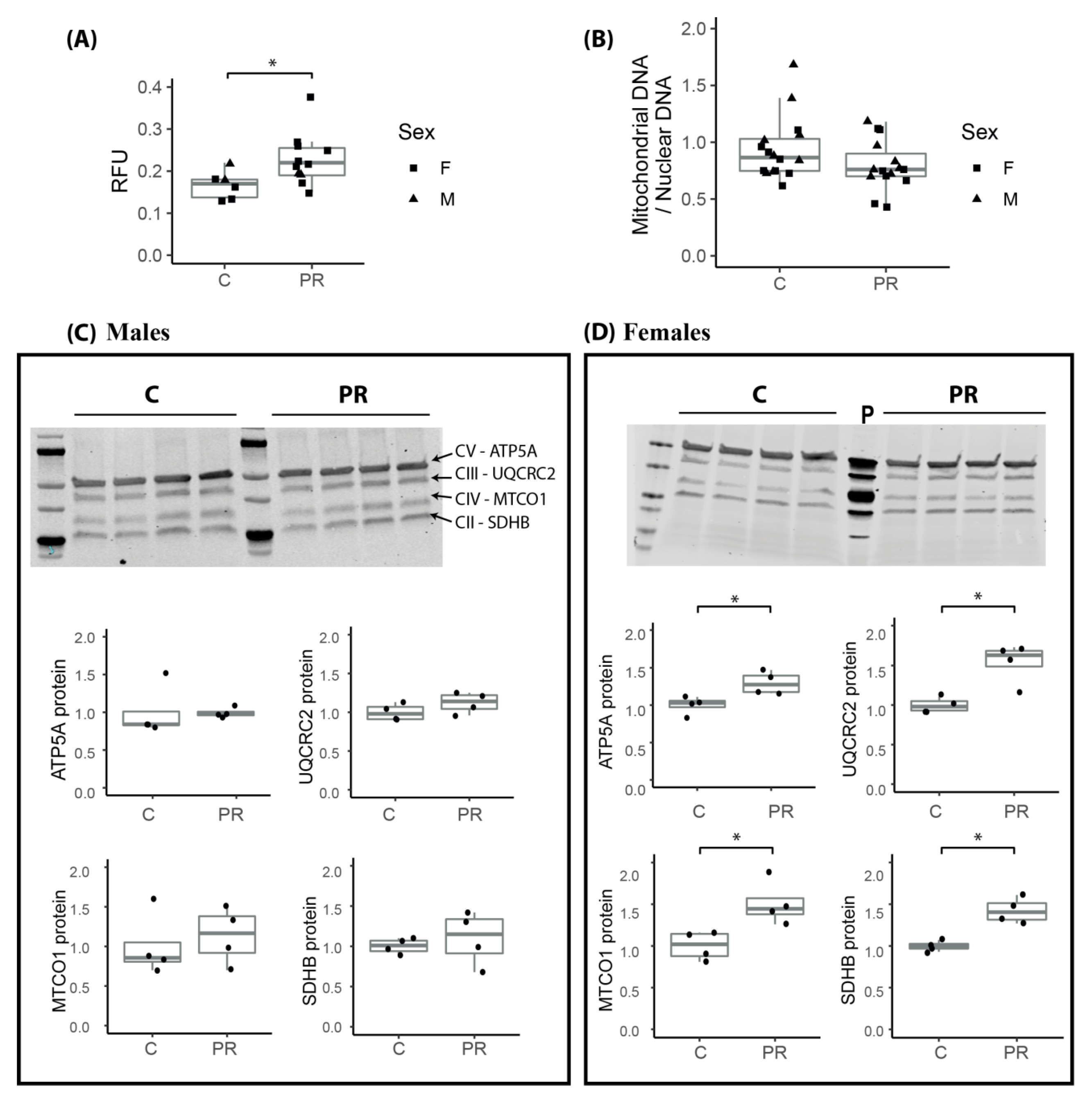
3.7. Mitochondrial Membrane Potential Was Enhanced in the Hypothalamus of E17 PR Fetuses
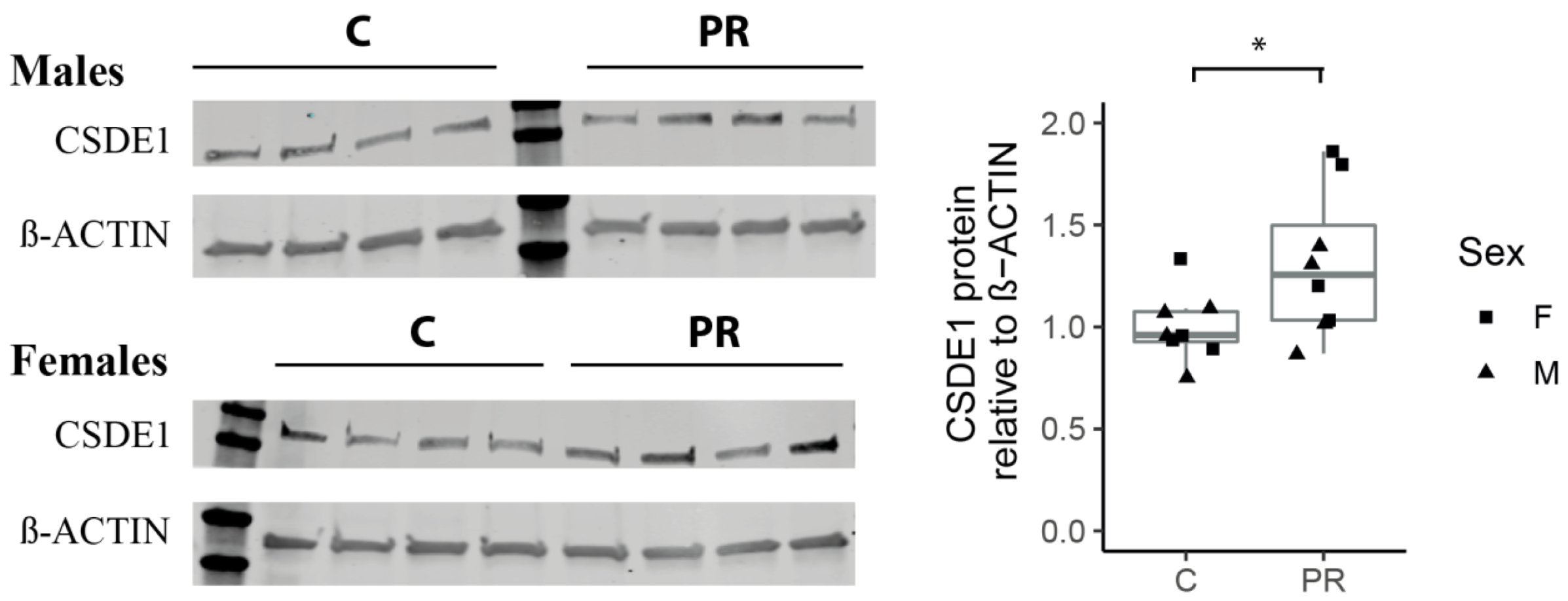
3.8. The Csde1 Gene, a Major Regulator of Neuronal Differentiation, Was Upregulated in the PR Group
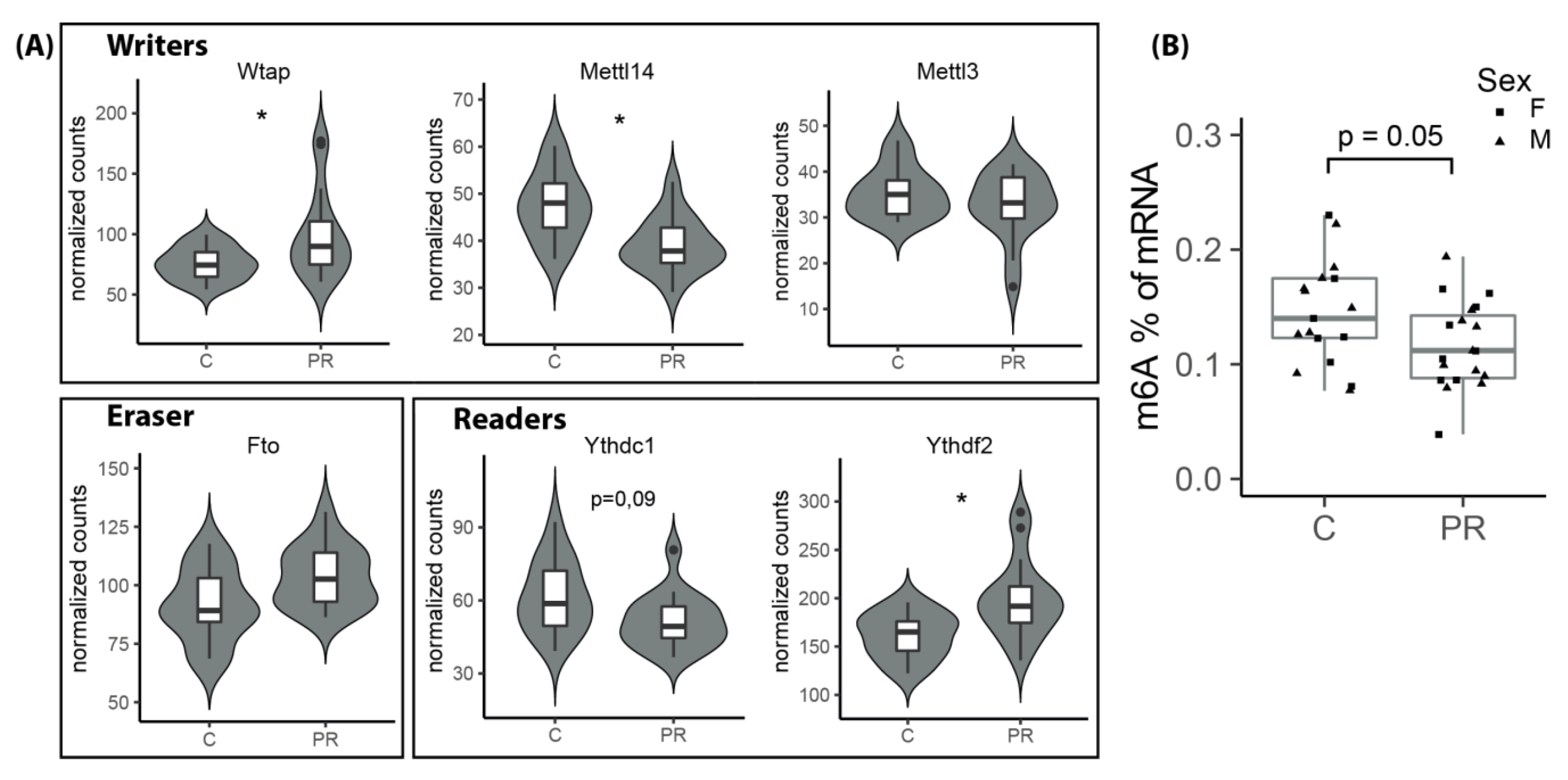
3.9. The m6A Epitranscriptomic Mark Was Altered in the PR Fetuses
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol | Log2FC | Padj | Base Mean | Full Name | Function(s) * |
|---|---|---|---|---|---|
| Slc35c2 | 0.469 | 0.031 | 24.122 | Solute carrier family 35 member C2 | May play a role in cellular response to tissue hypoxia |
| Ppcs | 0.463 | 0.042 | 12.650 | Phosphopantothenoylcysteine synthetase | Involved in the biosynthesis of coenzyme A, a precursor of Acetyl CoA |
| Csde1 | 0.452 | 0.001 | 817.685 | Cold shock domain containing E1 | RNA-binding protein—prevents neuronal differentiation in neural stem cells |
| Nipa2 | 0.437 | 0.028 | 19.597 | Nipa magnesium transporter 2 | Non-imprinted gene in Prader–Willi/Angelman syndrome region |
| Coa5 | 0.429 | 0.006 | 117.035 | Cytochrome C oxydase assembly factor 5 | Involved in the mitochondrial complex IV assembly |
| Cirbp | 0.428 | 0.002 | 250.612 | Cold inducible RNA-binding protein | Essential for embryonic gastrulation and neural development |
| Hs6st1 | 0.423 | 0.030 | 91.038 | Heparan-sulfate 6-O-sulfotransferase 1 | Critical for normal neuronal development, may play a role in neuron branching |
| LOC100174910 | 0.418 | 0.010 | 48.852 | Glutaredoxin-like protein | Involved in oxidation-reduction process. May be involved in cell redox homeostasis |
| Wdr83 | 0.407 | 0.044 | 34.218 | WD repeat domain-containing protein 83 | Involved in response to hypoxia |
| Tmem53 | 0.406 | 0.025 | 27.565 | Transmembrane protein 53 | |
| Ube2n | 0.405 | 0.002 | 83.937 | Ubiquitin conjugating enzyme E2N | Involved in protein ubiquitination. Plays a role in the control of cell cycle and differentiation |
| Tmcc2 | 0.397 | 0.044 | 55.090 | Transmembrane and coiled-coil domains protein 2 | Expressed in endoplasmic reticulum |
| Sec23b | 0.395 | 0.044 | 23.013 | COPII coat complex component | Involved in protein transport from endoplasmic reticulum |
| Coq7 | 0.393 | 0.001 | 120.626 | coenzyme Q7 hydroxylase | Involved in ubiquinone synthesis, in mitochondrial respiratory metabolism |
| Chrac1 | 0.391 | 0.006 | 66.458 | Chromatin accessibility complex protein 1 | Histone-fold protein that binds DNA. May be involved in cell growth and survival |
| Rbm7 | 0.377 | 0.044 | 20.552 | RNA-binding protein 7 | Member of the exosome targeting complex, involved in RNA degradation |
| Wtap | 0.373 | 0.031 | 87.528 | WT1 associated protein | Member of the complex that mediate m6A methylation of RNAs |
| Mterf1 | 0.367 | 0.032 | 30.933 | Mitochondrial transcription termination factor 1 | DNA-binding protein, involved in termination of mitochondrial transcription |
| Marcks | 0.365 | 0.026 | 1055.600 | Myristoylated alanine rich protein kinase C substrate | Binds protein of cytoskeleton, may be involved in cell migration |
| Pqlc1 | 0.365 | 0.044 | 39.620 | Solute carrier family 66 member 2 | Involved in phospholipid translocation |
| Gene Symbol | log2FC | Padj | Base Mean | Full Name | Function(s) * |
|---|---|---|---|---|---|
| Brsk1 | −0.807 | 0.010 | 13.066 | Serine Threonine protein kinase | Role in polarization of neurons and centrosome duplication |
| Fgfr1op | −0.676 | 0.002 | 19.199 | FGFR1 oncogen partner | Required for anchoring microtubules to the centrosomes. Involved in cell proliferation |
| Qk | −0.658 | 0.003 | 22.941 | Quaking | RNA-binding protein that regulates pre-mRNA splicing, export, stability and translation. Involved in oligodendrogenesis |
| Tcf20 | −0.636 | 0.024 | 19.373 | Transcription factor 20 | Transcriptional activator. May be involved in neurodevelopment |
| Cask | −0.625 | 0.013 | 23.749 | Calcium/calmodulin dependent serine protein kinase | Involved in synaptic membrane protein anchoring. Contributes to neurodevelopment |
| Eps15 | −0.607 | 0.021 | 12.515 | Epidermal growth factor receptor pathway substrate 15 | Involved in cell growth regulation. May be involved in the control of cell proliferation |
| Robo1 | −0.600 | 0.009 | 56.779 | Roundabout guidance receptor 1 | Involved in axon guidance and neuronal precursor cell migration |
| Fam168a | −0.569 | 0.010 | 19.145 | Also known as Tcrp1 | Involved in cancer chemotherapy resistance |
| Iqgap1 | −0.566 | 0.009 | 18.290 | IQ motif containing GTPase activating protein 1 | Regulates the assembly and dynamic if the cytoskeleton. May promote neurite outgrowth. |
| Phactr3 | −0.560 | 0.025 | 15.498 | Phosphatase and actin regulator 3 | Associated with the nuclear scaffold in proliferating cells |
| Myo18a | −0.548 | 0.009 | 28.595 | Myosin 18A | Associated with the Golgi. May be required for cell migration |
| Sesn3 | −0.541 | 0.045 | 17.734 | Sestrin 3 | Required for regulation of glucose and insulin regulation. May be involved in the protection againt oxidative stress |
| Kcnma1 | −0.530 | 0.044 | 16.245 | Potassium calcium-activated channel subfamily M alpha 1 | Involved in the control of neuron excitability |
| Pclo | −0.529 | 0.002 | 95.164 | Piccolo presynaptic cytomatrix protein | Involved in synaptogenesis |
| Mgat3 | −0.526 | 0.024 | 19.383 | beta−1,4-mannosyl-glycoprotein 4 beta-N-Acetylglucosaminyltransferase | May be involved in response to oxidative stress in brain and in cell migration regulation |
| Timp3 | −0.526 | 0.049 | 10.279 | TIMP metallopeptidase inhibitor 3 | |
| Taok1 | −0.518 | 0.007 | 43.989 | Serine Threonine protein kinase TAO1 | Acts as a regulator of cytoskeleton stability. May be involved in the induction of apoptosis |
| Nlgn1 | −0.515 | 0.031 | 15.813 | Neuroligin 1 | Neuronal cell surface protein. May be involved in the formation and remodeling of synapses |
| Kpnb1 | −0.511 | 0.025 | 18.776 | Karyopherin subunit beta 1 | Involved in nuclear protein import, including ribosomal proteins and histone H1 |
| Slmap | −0.510 | 0.028 | 14.084 | Sarcolemma associated protein | Membrane associated protein |
References
- Katz, J.; Lee, A.C.; Kozuki, N.; Lawn, J.E.; Cousens, S.; Blencowe, H.; Ezzati, M.; Bhutta, Z.A.; Marchant, T.; Willey, B.A.; et al. Mortality risk in preterm and small-for-gestational-age infants in low-income and middle-income countries: A pooled country analysis. Lancet 2013, 382, 417–425. [Google Scholar] [CrossRef]
- Sibley, C.P.; Brownbill, P.; Dilworth, M.; Glazier, J.D. Review: Adaptation in placental nutrient supply to meet fetal growth demand: Implications for programming. Placenta 2010, 31, S70–S74. [Google Scholar] [CrossRef]
- Frondas-Chauty, A.; Simon, L.; Flamant, C.; Hanf, M.; Darmaun, D.; Roze, J.C. Deficit of Fat Free Mass in Very Preterm Infants at Discharge is Associated with Neurological Impairment at Age 2 Years. J. Pediatr. 2018, 196, 301–304. [Google Scholar] [CrossRef]
- Ehrenkranz, R.A.; Dusick, A.M.; Vohr, B.R.; Wright, L.L.; Wrage, L.A.; Poole, W.K. Growth in the neonatal intensive care unit influences neurodevelopmental and growth outcomes of extremely low birth weight infants. Pediatrics 2006, 117, 1253–1261. [Google Scholar] [CrossRef]
- Barker, D.J. Adult consequences of fetal growth restriction. Clin. Obstet. Gynecol. 2006, 49, 270–283. [Google Scholar] [CrossRef]
- Delahaye, F.; Breton, C.; Risold, P.Y.; Enache, M.; Dutriez-Casteloot, I.; Laborie, C.; Lesage, J.; Vieau, D. Maternal perinatal undernutrition drastically reduces postnatal leptin surge and affects the development of arcuate nucleus proopiomelanocortin neurons in neonatal male rat pups. Endocrinology 2008, 149, 470–475. [Google Scholar] [CrossRef]
- Bouret, S.G. Role of early hormonal and nutritional experiences in shaping feeding behavior and hypothalamic development. J. Nutr. 2010, 140, 653–657. [Google Scholar] [CrossRef]
- Coupe, B.; Amarger, V.; Grit, I.; Benani, A.; Parnet, P. Nutritional programming affects hypothalamic organization and early response to leptin. Endocrinology 2010, 151, 702–713. [Google Scholar] [CrossRef]
- Coupe, B.; Grit, I.; Hulin, P.; Randuineau, G.; Parnet, P. Postnatal growth after intrauterine growth restriction alters central leptin signal and energy homeostasis. PLoS ONE 2012, 7, e30616. [Google Scholar] [CrossRef]
- Dearden, L.; Bouret, S.G.; Ozanne, S.E. Sex and gender differences in developmental programming of metabolism. Mol. Metab. 2018, 15, 8–19. [Google Scholar] [CrossRef]
- Bedont, J.L.; Newman, E.A.; Blackshaw, S. Patterning, specification, and differentiation in the developing hypothalamus. Wiley Interdiscip. Rev. Dev. Biol. 2015, 4, 445–468. [Google Scholar] [CrossRef]
- Markakis, E.A. Development of the neuroendocrine hypothalamus. Front. Neuroendocr. 2002, 23, 257–291. [Google Scholar] [CrossRef]
- Bouret, S.G. Nutritional programming of hypothalamic development: Critical periods and windows of opportunity. Int. J. Obes. Suppl. 2012, 2, S19–S24. [Google Scholar] [CrossRef]
- Gotz, M.; Huttner, W.B. The cell biology of neurogenesis. Nat. Rev. Mol. Cell Biol. 2005, 6, 777–788. [Google Scholar] [CrossRef]
- Moody, L.; Chen, H.; Pan, Y.X. Early-Life Nutritional Programming of Cognition-The Fundamental Role of Epigenetic Mechanisms in Mediating the Relation between Early-Life Environment and Learning and Memory Process. Adv. Nutr. 2017, 8, 337–350. [Google Scholar] [CrossRef]
- Val-Laillet, D.; Besson, M.; Guerin, S.; Coquery, N.; Randuineau, G.; Kanzari, A.; Quesnel, H.; Bonhomme, N.; Bolhuis, J.E.; Kemp, B.; et al. A maternal Western diet during gestation and lactation modifies offspring’s microbiota activity, blood lipid levels, cognitive responses, and hippocampal neurogenesis in Yucatan pigs. FASEB J. 2017, 31, 2037–2049. [Google Scholar] [CrossRef]
- Staples, M.C.; Fannon, M.J.; Mysore, K.K.; Dutta, R.R.; Ongjoco, A.T.; Quach, L.W.; Kharidia, K.M.; Somkuwar, S.S.; Mandyam, C.D. Dietary restriction reduces hippocampal neurogenesis and granule cell neuron density without affecting the density of mossy fibers. Brain Res. 2017, 1663, 59–65. [Google Scholar] [CrossRef]
- Gould, J.M.; Smith, P.J.; Airey, C.J.; Mort, E.J.; Airey, L.E.; Warricker, F.D.M.; Pearson-Farr, J.E.; Weston, E.C.; Gould, P.J.W.; Semmence, O.G.; et al. Mouse maternal protein restriction during preimplantation alone permanently alters brain neuron proportion and adult short-term memory. Proc. Natl. Acad. Sci. USA 2018, 115, 7398–7407. [Google Scholar] [CrossRef]
- Desai, M.; Li, T.; Ross, M.G. Fetal hypothalamic neuroprogenitor cell culture: Preferential differentiation paths induced by leptin and insulin. Endocrinology 2011, 152, 3192–3201. [Google Scholar] [CrossRef]
- Desai, M.; Ferrini, M.G.; Han, G.; Jellyman, J.K.; Ross, M.G. In vivo maternal and in vitro BPA exposure effects on hypothalamic neurogenesis and appetite regulators. Environ. Res. 2018, 164, 45–52. [Google Scholar] [CrossRef]
- Cariaga-Martinez, A.E.; Gutierrez, K.J.; Alelu-Paz, R. The Vast Complexity of the Epigenetic Landscape during Neurodevelopment: An Open Frame to Understanding Brain Function. Int. J. Mol. Sci. 2018, 19, 1333. [Google Scholar] [CrossRef]
- Tran, N.T.; Amarger, V.; Bourdon, A.; Misbert, E.; Grit, I.; Winer, N.; Darmaun, D. Maternal citrulline supplementation enhances placental function and fetal growth in a rat model of IUGR: Involvement of insulin-like growth factor 2 and angiogenic factors. J. Matern.-Fetal Neonatal Med. 2017, 30, 1906–1911. [Google Scholar] [CrossRef]
- Martin Agnoux, A.; Antignac, J.P.; Simard, G.; Poupeau, G.; Darmaun, D.; Parnet, P.; Alexandre-Gouabau, M.C. Time-window dependent effect of perinatal maternal protein restriction on insulin sensitivity and energy substrate oxidation in adult male offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R184–R197. [Google Scholar] [CrossRef]
- Sevrin, T.; Alexandre-Gouabau, M.C.; Castellano, B.; Aguesse, A.; Ouguerram, K.; Ngyuen, P.; Darmaun, D.; Boquien, C.Y. Impact of Fenugreek on Milk Production in Rodent Models of Lactation Challenge. Nutrients 2019, 11, 2571. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef]
- Soumillon, M.; Cacchiarelli, D.; Semrau, S. Characterization of directed differentiation by high-throughput single-cell RNA-Seq. BioRxiv 2014, BioRxiv:003236. Available online: https://www.biorxiv.org/content/early/2014/03/05/003236 (accessed on 26 November 2014).
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Kilens, S.; Meistermann, D.; Moreno, D.; Chariau, C.; Gaignerie, A.; Reignier, A.; Lelievre, Y.; Casanova, M.; Vallot, C.; Nedellec, S.; et al. Parallel derivation of isogenic human primed and naive induced pluripotent stem cells. Nat. Commun. 2018, 9, 360. [Google Scholar] [CrossRef]
- Korotkevich, G.; Sukhov, V.; Sergushichev, A. Fast gene set enrichment analysis. BioRxiv 2019, BioRxiv:060012. Available online: https://www.biorxiv.org/content/10.1101/060012v2 (accessed on 22 October 2019).
- The Gene Ontology, C. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019, 47, D330–D338. [Google Scholar] [CrossRef]
- Kanehisa, M.; Goto, S. KEGG: Kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Jassal, B.; Matthews, L.; Viteri, G.; Gong, C.; Lorente, P.; Fabregat, A.; Sidiropoulos, K.; Cook, J.; Gillespie, M.; Haw, R.; et al. The reactome pathway knowledgebase. Nucleic Acids Res. 2020, 48, D498–D503. [Google Scholar] [CrossRef] [PubMed]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed]
- Mulligan, K.A.; Cheyette, B.N. Wnt signaling in vertebrate neural development and function. J. Neuroimmune Pharmacol. 2012, 7, 774–787. [Google Scholar] [CrossRef] [PubMed]
- Ju Lee, H.; Bartsch, D.; Xiao, C.; Guerrero, S.; Ahuja, G.; Schindler, C.; Moresco, J.J.; Yates, J.R.; Gebauer, F.; Bazzi, H.; et al. A post-transcriptional program coordinated by CSDE1 prevents intrinsic neural differentiation of human embryonic stem cells. Nat. Commun. 2017, 8, 1456. [Google Scholar] [CrossRef] [PubMed]
- Coupe, B.; Dutriez-Casteloot, I.; Breton, C.; Lefevre, F.; Mairesse, J.; Dickes-Coopman, A.; Silhol, M.; Tapia-Arancibia, L.; Lesage, J.; Vieau, D. Perinatal undernutrition modifies cell proliferation and brain-derived neurotrophic factor levels during critical time-windows for hypothalamic and hippocampal development in the male rat. J. Neuroendocr. 2009, 21, 40–48. [Google Scholar] [CrossRef]
- Amarger, V.; Lecouillard, A.; Ancellet, L.; Grit, I.; Castellano, B.; Hulin, P.; Parnet, P. Protein content and methyl donors in maternal diet interact to influence the proliferation rate and cell fate of neural stem cells in rat hippocampus. Nutrients 2014, 6, 4200–4217. [Google Scholar] [CrossRef]
- Desai, M.; Li, T.; Ross, M.G. Hypothalamic neurosphere progenitor cells in low birth-weight rat newborns: Neurotrophic effects of leptin and insulin. Brain Res. 2011, 1378, 29–42. [Google Scholar] [CrossRef]
- Guevara, R.; Santandreu, F.M.; Valle, A.; Gianotti, M.; Oliver, J.; Roca, P. Sex-dependent differences in aged rat brain mitochondrial function and oxidative stress. Free. Radic. Biol. Med. 2009, 46, 169–175. [Google Scholar] [CrossRef]
- Gaignard, P.; Frechou, M.; Liere, P.; Therond, P.; Schumacher, M.; Slama, A.; Guennoun, R. Sex differences in brain mitochondrial metabolism: Influence of endogenous steroids and stroke. J. Neuroendocr. 2018, 30, e12497. [Google Scholar] [CrossRef]
- Acaz-Fonseca, E.; Ortiz-Rodriguez, A.; Lopez-Rodriguez, A.B.; Garcia-Segura, L.M.; Astiz, M. Developmental Sex Differences in the Metabolism of Cardiolipin in Mouse Cerebral Cortex Mitochondria. Sci. Rep. 2017, 7, 43878. [Google Scholar] [CrossRef]
- Khacho, M.; Harris, R.; Slack, R.S. Mitochondria as central regulators of neural stem cell fate and cognitive function. Nat. Rev. Neurosci. 2019, 20, 34–48. [Google Scholar] [CrossRef] [PubMed]
- Khacho, M.; Clark, A.; Svoboda, D.S.; Azzi, J.; MacLaurin, J.G.; Meghaizel, C.; Sesaki, H.; Lagace, D.C.; Germain, M.; Harper, M.E.; et al. Mitochondrial Dynamics Impacts Stem Cell Identity and Fate Decisions by Regulating a Nuclear Transcriptional Program. Cell Stem Cell 2016, 19, 232–247. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.J.S.; Pedroza, A.A.; Braz, G.R.F.; Fernandes, M.P.; Lagranha, C.J. Mitochondrial dysfunction: Maternal protein restriction as a trigger of reactive species overproduction and brainstem energy failure in male offspring brainstem. Nutr. Neurosci. 2018, 22, 778–788. [Google Scholar] [CrossRef]
- Alexandre-Gouabau, M.C.; Bailly, E.; Moyon, T.L.; Grit, I.C.; Coupe, B.; Le Drean, G.; Rogniaux, H.J.; Parnet, P. Postnatal growth velocity modulates alterations of proteins involved in metabolism and neuronal plasticity in neonatal hypothalamus in rats born with intrauterine growth restriction. J. Nutr. Biochem. 2011, 23, 140–152. [Google Scholar] [CrossRef]
- Guitart-Mampel, M.; Juarez-Flores, D.L.; Youssef, L.; Moren, C.; Garcia-Otero, L.; Roca-Agujetas, V.; Catalan-Garcia, M.; Gonzalez-Casacuberta, I.; Tobias, E.; Milisenda, J.C.; et al. Mitochondrial implications in human pregnancies with intrauterine growth restriction and associated cardiac remodelling. J. Cell. Mol. Med. 2019, 23, 3962–3973. [Google Scholar] [CrossRef]
- Martin-Gronert, M.S.; Ozanne, S.E. Mechanisms underlying the developmental origins of disease. Rev. Endocr. Metab. Disord. 2012, 13, 85–92. [Google Scholar] [CrossRef]
- Eckert, J.J.; Porter, R.; Watkins, A.J.; Burt, E.; Brooks, S.; Leese, H.J.; Humpherson, P.G.; Cameron, I.T.; Fleming, T.P. Metabolic induction and early responses of mouse blastocyst developmental programming following maternal low protein diet affecting life-long health. PLoS ONE 2012, 7, e52791. [Google Scholar] [CrossRef]
- Liesa, M.; Shirihai, O.S. Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 2013, 17, 491–506. [Google Scholar] [CrossRef]
- Gomes, L.C.; Di Benedetto, G.; Scorrano, L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol. 2011, 13, 589–598. [Google Scholar] [CrossRef]
- Ross, J.M.; Olson, L.; Coppotelli, G. Mitochondrial and Ubiquitin Proteasome System Dysfunction in Ageing and Disease: Two Sides of the Same Coin? Int. J. Mol. Sci. 2015, 16, 19458–19476. [Google Scholar] [CrossRef]
- Ekstrand, M.I.; Terzioglu, M.; Galter, D.; Zhu, S.; Hofstetter, C.; Lindqvist, E.; Thams, S.; Bergstrand, A.; Hansson, F.S.; Trifunovic, A.; et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 1325–1330. [Google Scholar] [CrossRef] [PubMed]
- Cook, C.; Petrucelli, L. A critical evaluation of the ubiquitin-proteasome system in Parkinson’s disease. Biochim. Biophys. Acta-Mol. Basis Dis. 2009, 1792, 664–675. [Google Scholar] [CrossRef] [PubMed]
- Sahu, I.; Nanaware, P.; Mane, M.; Mulla, S.W.; Roy, S.; Venkatraman, P. Role of a 19S Proteasome Subunit- PSMD10(Gankyrin) in Neurogenesis of Human Neuronal Progenitor Cells. Int. J. Stem Cells 2019, 12, 463. [Google Scholar] [CrossRef]
- Niu, X.; Zhao, Y.; Yang, N.; Zhao, X.; Zhang, W.; Bai, X.; Li, A.; Yang, W.; Lu, L. Proteasome activation by insulin-like growth factor-1/nuclear factor erythroid 2-related factor 2 signaling promotes exercise-induced neurogenesis. Stem Cells 2019, 38, 246–260. [Google Scholar] [CrossRef]
- Adegoke, O.A.J.; Beatty, B.E.; Kimball, S.R.; Wing, S.S. Interactions of the super complexes: When mTORC1 meets the proteasome. Int. J. Biochem. Cell Biol. 2019, 117, 105638. [Google Scholar] [CrossRef]
- Chantranupong, L.; Sabatini, D.M. Cell biology: The TORC1 pathway to protein destruction. Nature 2016, 536, 155–156. [Google Scholar] [CrossRef][Green Version]
- Lee, K.M.; Hwang, S.K.; Lee, J.A. Neuronal autophagy and neurodevelopmental disorders. Exp. Neurobiol. 2013, 22, 133–142. [Google Scholar] [CrossRef]
- Sanosaka, T.; Namihira, M.; Nakashima, K. Epigenetic mechanisms in sequential differentiation of neural stem cells. Epigenetics 2009, 4, 89–92. [Google Scholar] [CrossRef]
- Juliandi, B.; Abematsu, M.; Nakashima, K. Epigenetic regulation in neural stem cell differentiation. Dev. Growth Differ. 2010, 52, 493–504. [Google Scholar] [CrossRef]
- Tsujimura, K.; Abematsu, M.; Kohyama, J.; Namihira, M.; Nakashima, K. Neuronal differentiation of neural precursor cells is promoted by the methyl-CpG-binding protein MeCP2. Exp. Neurol. 2009, 219, 104–111. [Google Scholar] [CrossRef]
- Saito, A.; Kanemoto, S.; Kawasaki, N.; Asada, R.; Iwamoto, H.; Oki, M.; Miyagi, H.; Izumi, S.; Sanosaka, T.; Nakashima, K.; et al. Unfolded protein response, activated by OASIS family transcription factors, promotes astrocyte differentiation. Nat. Commun. 2012, 3, 967. [Google Scholar] [CrossRef] [PubMed]
- Imayoshi, I.; Kageyama, R. bHLH factors in self-renewal, multipotency, and fate choice of neural progenitor cells. Neuron 2014, 82, 9–23. [Google Scholar] [CrossRef] [PubMed]
- Imayoshi, I.; Isomura, A.; Harima, Y.; Kawaguchi, K.; Kori, H.; Miyachi, H.; Fujiwara, T.; Ishidate, F.; Kageyama, R. Oscillatory control of factors determining multipotency and fate in mouse neural progenitors. Science 2013, 342, 1203–1208. [Google Scholar] [CrossRef] [PubMed]
- Berger, S.L.; Sassone-Corsi, P. Metabolic Signaling to Chromatin. Cold Spring Harb. Perspect. Biol. 2016, 8, a019463. [Google Scholar] [CrossRef]
- Baser, A.; Skabkin, M.; Kleber, S.; Dang, Y.; Gulculer Balta, G.S.; Kalamakis, G.; Gopferich, M.; Ibanez, D.C.; Schefzik, R.; Lopez, A.S.; et al. Onset of differentiation is post-transcriptionally controlled in adult neural stem cells. Nature 2019, 566, 100–104. [Google Scholar] [CrossRef]
- Widagdo, J.; Anggono, V. The m6A-epitranscriptomic signature in neurobiology: From neurodevelopment to brain plasticity. J. Neurochem. 2018, 147, 137–152. [Google Scholar] [CrossRef]
- Chang, M.; Lv, H.; Zhang, W.; Ma, C.; He, X.; Zhao, S.; Zhang, Z.W.; Zeng, Y.X.; Song, S.; Niu, Y.; et al. Region-specific RNA m(6)A methylation represents a new layer of control in the gene regulatory network in the mouse brain. Open Biol. 2017, 7, 170166. [Google Scholar] [CrossRef]
- Yoon, K.J.; Ringeling, F.R.; Vissers, C.; Jacob, F.; Pokrass, M.; Jimenez-Cyrus, D.; Su, Y.; Kim, N.S.; Zhu, Y.; Zheng, L.; et al. Temporal Control of Mammalian Cortical Neurogenesis by m(6)A Methylation. Cell 2017, 171, 877–889.e17. [Google Scholar] [CrossRef]
- Li, X.C.; Song, M.F.; Sun, F.; Tian, F.J.; Wang, Y.M.; Wang, B.Y.; Chen, J.H. Fragile X-related protein 1 (FXR1) regulates cyclooxygenase-2 (COX-2) expression at the maternal-fetal interface. Reprod. Fertil. Dev. 2018, 30, 1566–1574. [Google Scholar] [CrossRef]
- Gulati, P.; Cheung, M.K.; Antrobus, R.; Church, C.D.; Harding, H.P.; Tung, Y.C.; Rimmington, D.; Ma, M.; Ron, D.; Lehner, P.J.; et al. Role for the obesity-related FTO gene in the cellular sensing of amino acids. Proc. Natl. Acad. Sci. USA 2013, 110, 2557–2562. [Google Scholar] [CrossRef]
- UniProt, C. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Res. 2019, 47, D506–D515. [Google Scholar] [CrossRef]
- Coordinators, N.R. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2018, 46, D8–D13. [Google Scholar] [CrossRef] [PubMed]







| Controls | Protein Restricted | p-Value | |||
|---|---|---|---|---|---|
| Litter Size | 13.00 ± 3.39 (n = 13) | 14.13 ± 2.61 (n = 15) | 0.50 * | ||
| Maternal Weight Gain (g) | 112.39 ± 17.77 (n = 13) | 99.47 ± 21.12 (n = 13) | 0.16 * | ||
| Fetal Weight (g) | ♀ (n = 32) | ♂ (n = 38) | ♀ (n = 54) | ♂ (n = 36) | Group: 0.41 # |
| 0.76 ± 0.05 | 0.79 ± 0.05 | 0.77 ± 0.07 | 0.81 ± 0.06 | Sex: <0.0001 # | |
| Placenta Weight (g) | 0.39 ± 0.06 (n = 70) | 0.40 ± 0.07 (n = 81) | 0.85 * | ||
| PFR (Placenta–Fetus Ratio) | 0.51 ± 0.09 (n = 70) | 0.52 ± 0.10 (n = 81) | 0.83 * | ||
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Frapin, M.; Guignard, S.; Meistermann, D.; Grit, I.; Moullé, V.S.; Paillé, V.; Parnet, P.; Amarger, V. Maternal Protein Restriction in Rats Alters the Expression of Genes Involved in Mitochondrial Metabolism and Epitranscriptomics in Fetal Hypothalamus. Nutrients 2020, 12, 1464. https://doi.org/10.3390/nu12051464
Frapin M, Guignard S, Meistermann D, Grit I, Moullé VS, Paillé V, Parnet P, Amarger V. Maternal Protein Restriction in Rats Alters the Expression of Genes Involved in Mitochondrial Metabolism and Epitranscriptomics in Fetal Hypothalamus. Nutrients. 2020; 12(5):1464. https://doi.org/10.3390/nu12051464
Chicago/Turabian StyleFrapin, Morgane, Simon Guignard, Dimitri Meistermann, Isabelle Grit, Valentine S. Moullé, Vincent Paillé, Patricia Parnet, and Valérie Amarger. 2020. "Maternal Protein Restriction in Rats Alters the Expression of Genes Involved in Mitochondrial Metabolism and Epitranscriptomics in Fetal Hypothalamus" Nutrients 12, no. 5: 1464. https://doi.org/10.3390/nu12051464
APA StyleFrapin, M., Guignard, S., Meistermann, D., Grit, I., Moullé, V. S., Paillé, V., Parnet, P., & Amarger, V. (2020). Maternal Protein Restriction in Rats Alters the Expression of Genes Involved in Mitochondrial Metabolism and Epitranscriptomics in Fetal Hypothalamus. Nutrients, 12(5), 1464. https://doi.org/10.3390/nu12051464