Crescent-Like Lesions as an Early Signature of Nephropathy in a Rat Model of Prediabetes Induced by a Hypercaloric Diet

 , , , ,
, , , ,  ,
,  ,
, 

Abstract
:1. Introduction
2. Methods
2.1. Animals and Experimental Design
2.2. Blood and Tissues Collection
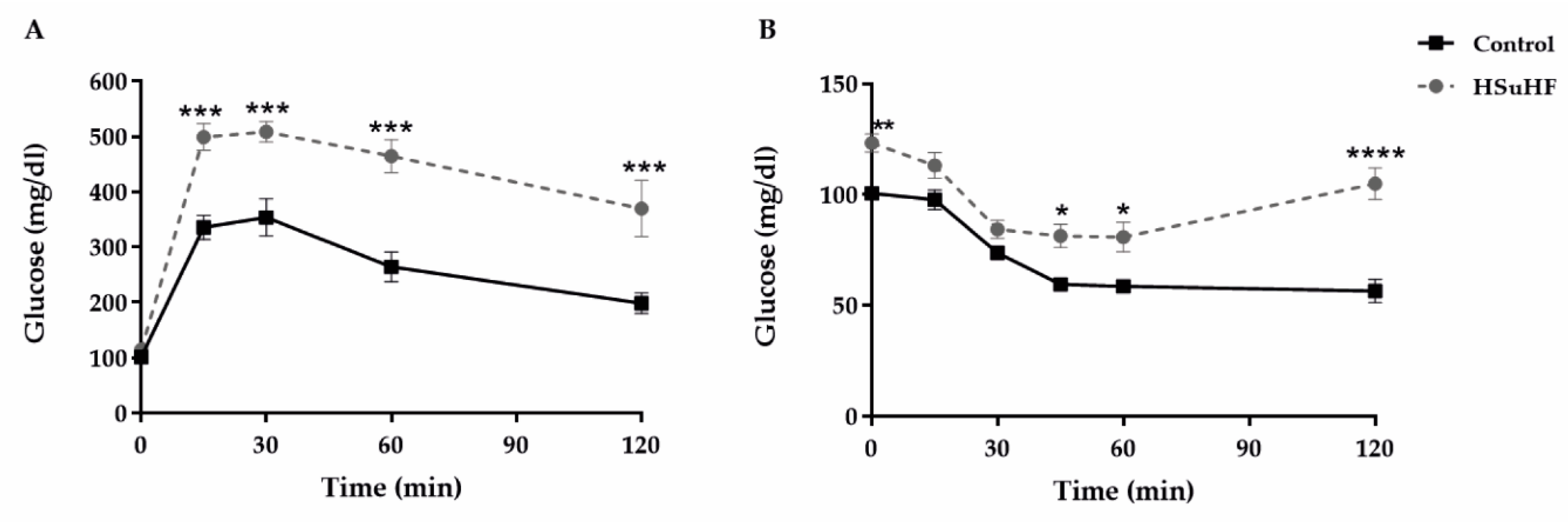
2.3. Metabolic Characterization
2.3.1. Glycemic Profile
2.3.2. Insulinemic Profile
2.3.3. Lipidic Profile
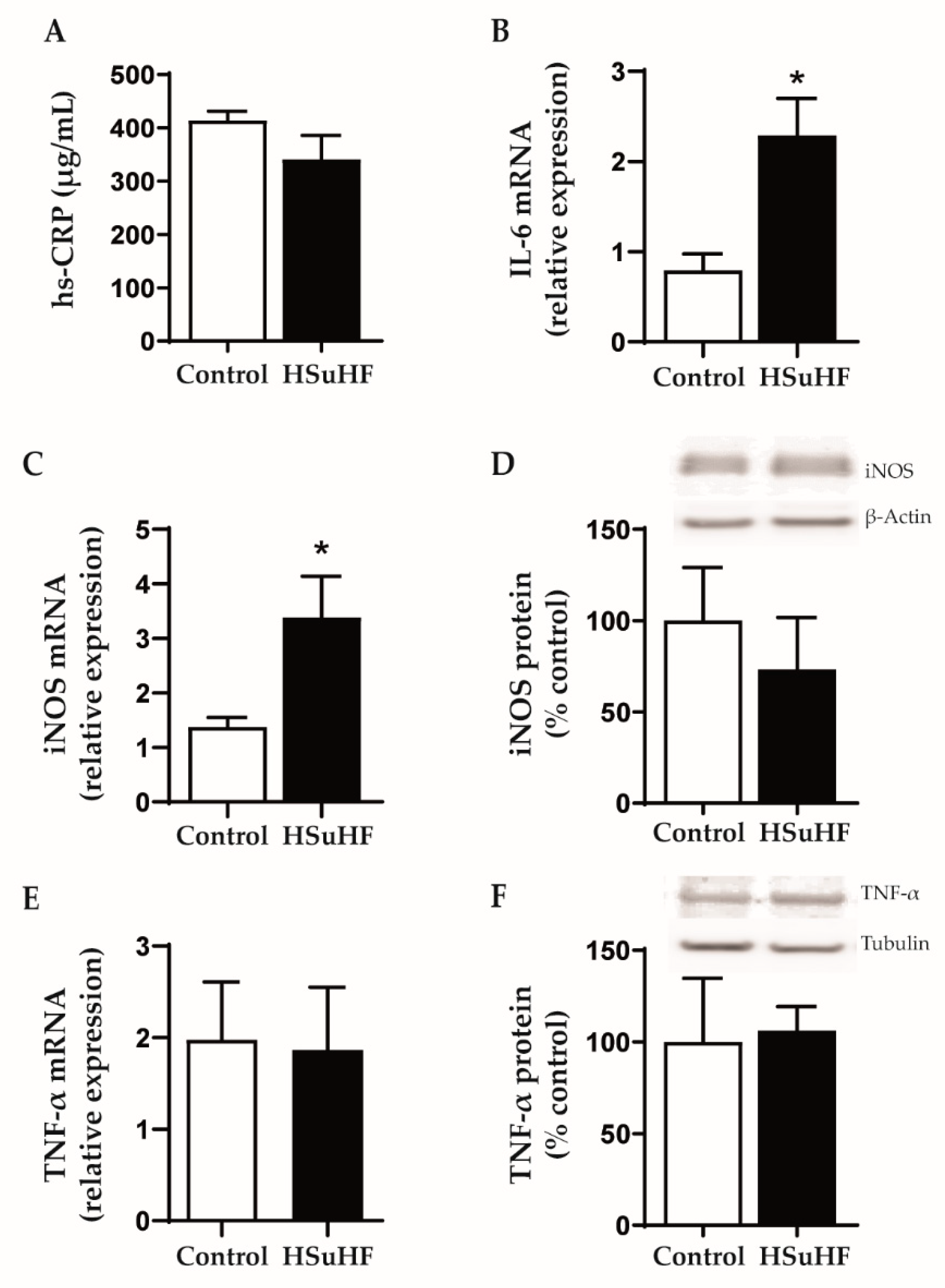
2.4. Serum Markers of Redox Status and Inflammation
2.5. Renal Function Data
2.5.1. Serum Biochemical Parameters
2.5.2. 24-h Urine Parameters
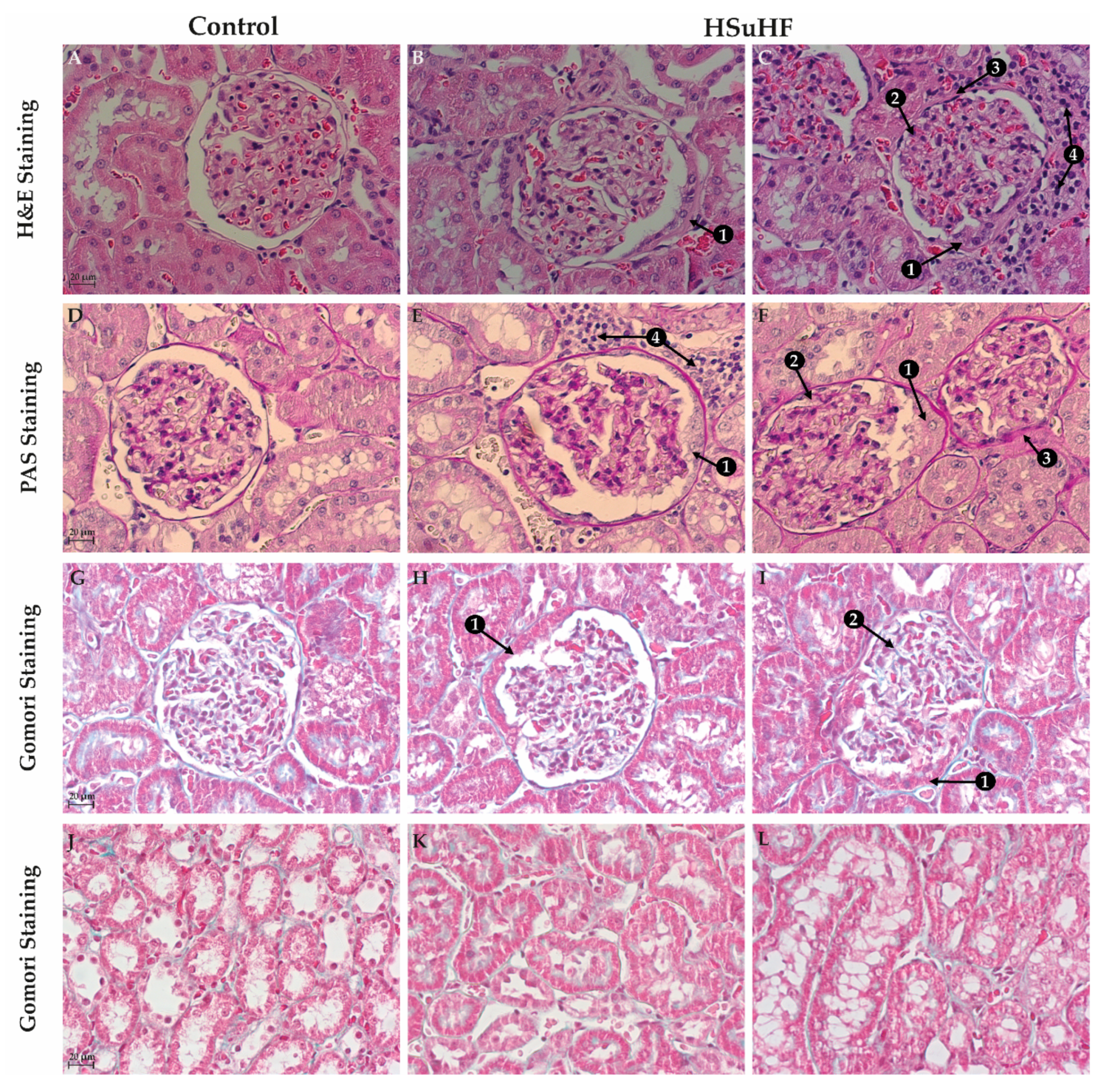
2.6. Renal Histomorphology
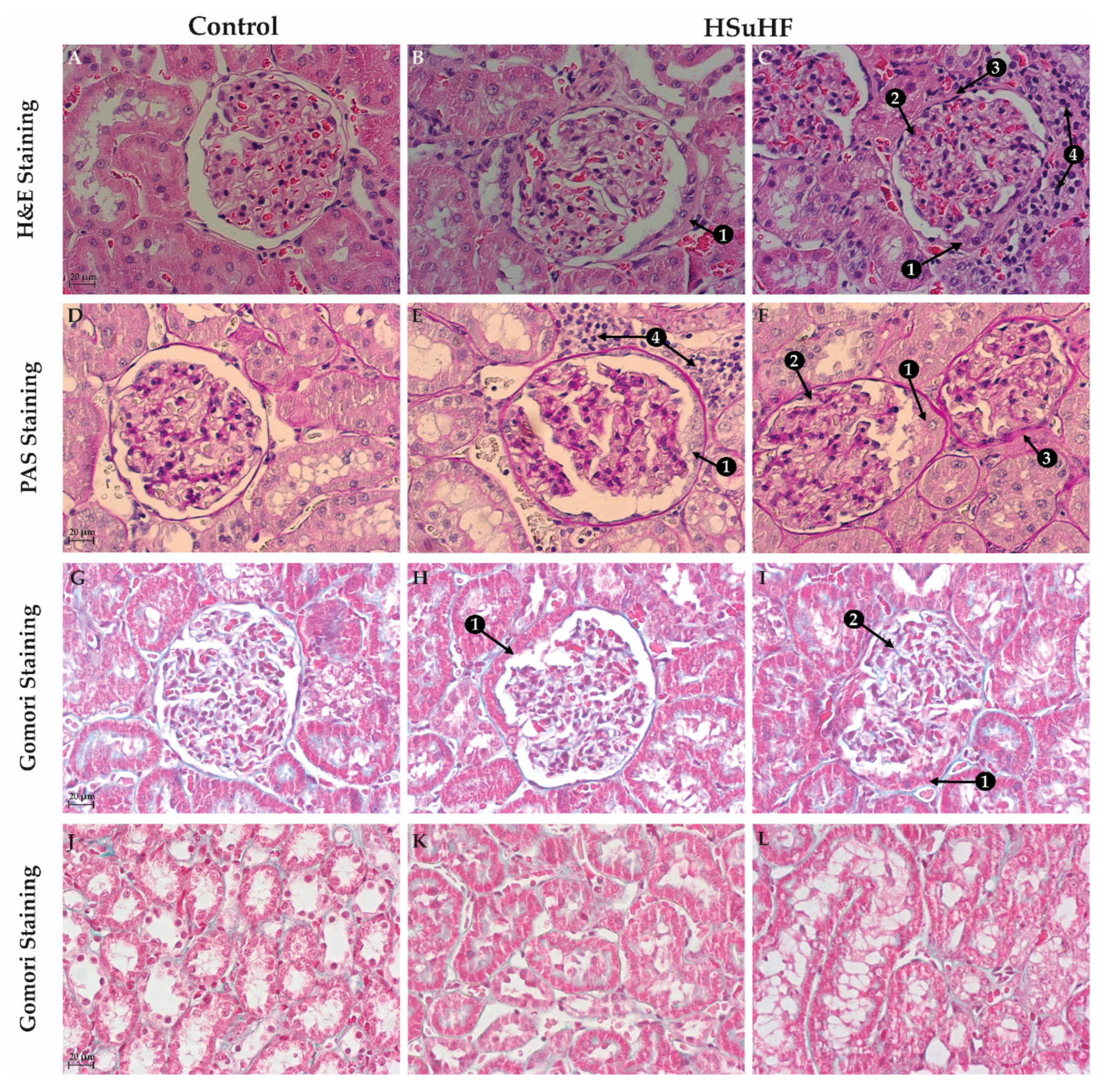
2.6.1. Hematoxylin and Eosin (H&E) Staining
2.6.2. Periodic Acid of Shiff (PAS) and PAS-Diastase Staining
2.6.3. Gomori’s Green Trichrome Staining
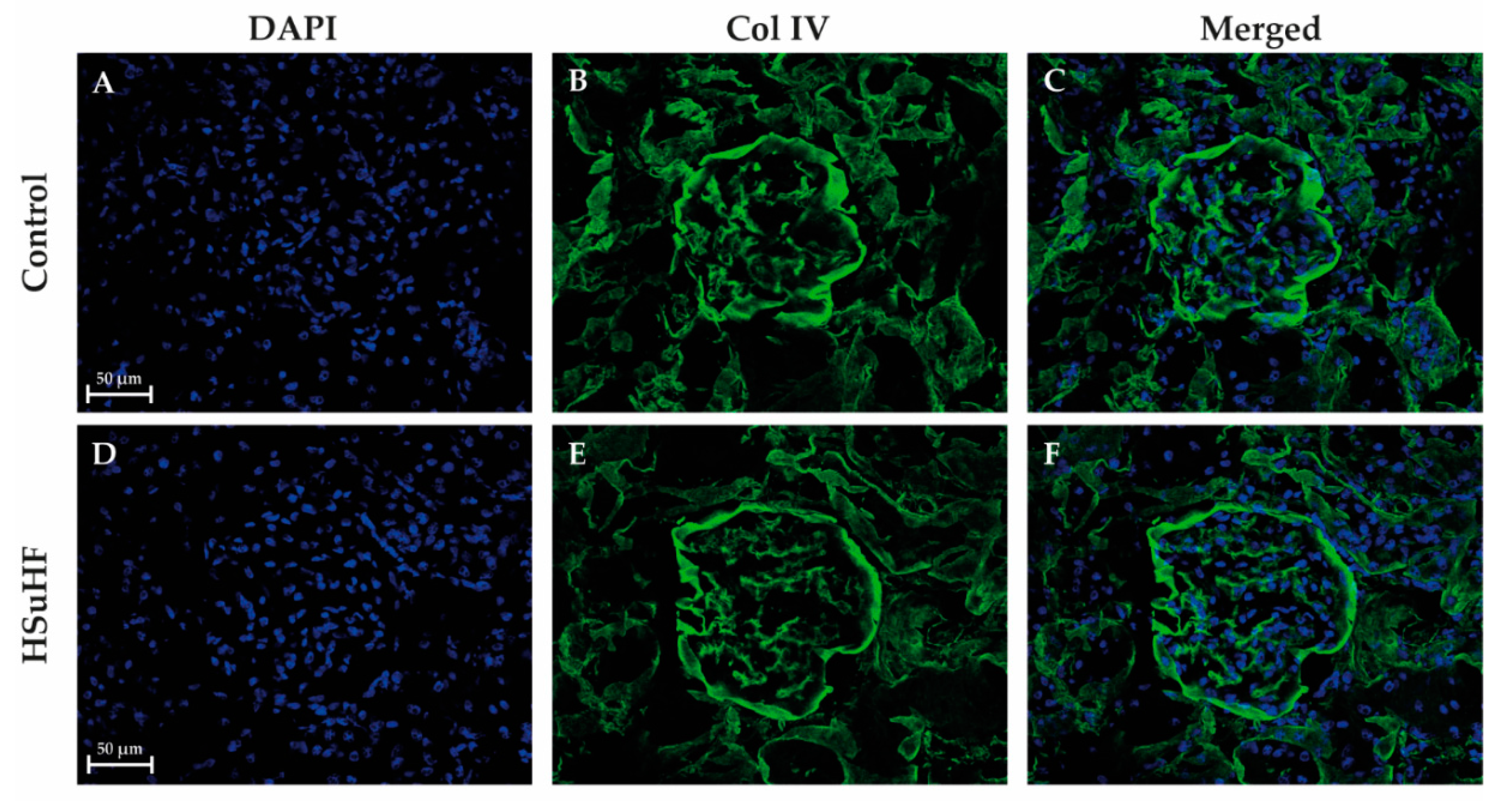
2.6.4. Collagen IV Immunohistochemistry
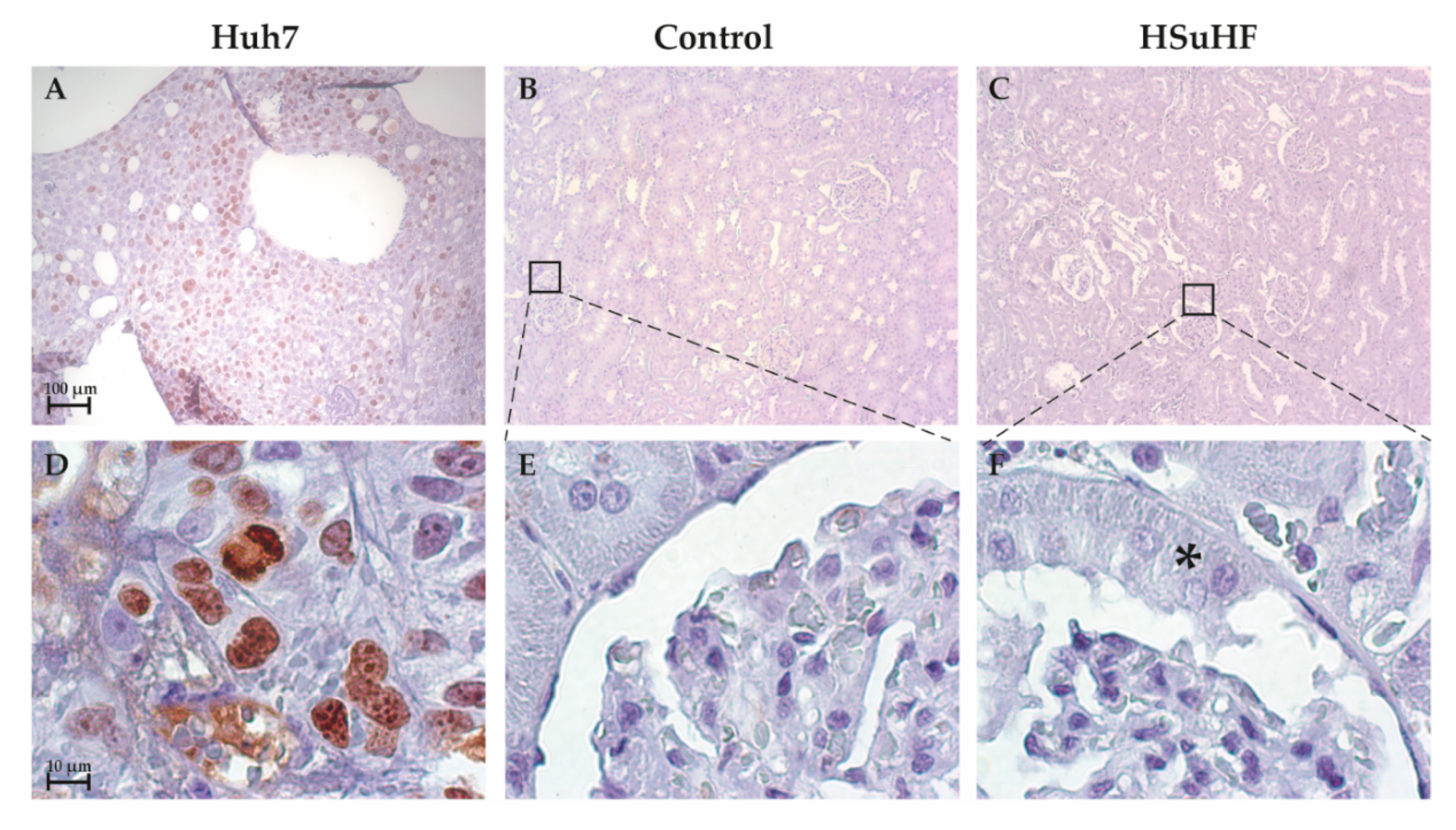
2.6.5. Ki-67 Immunostaining
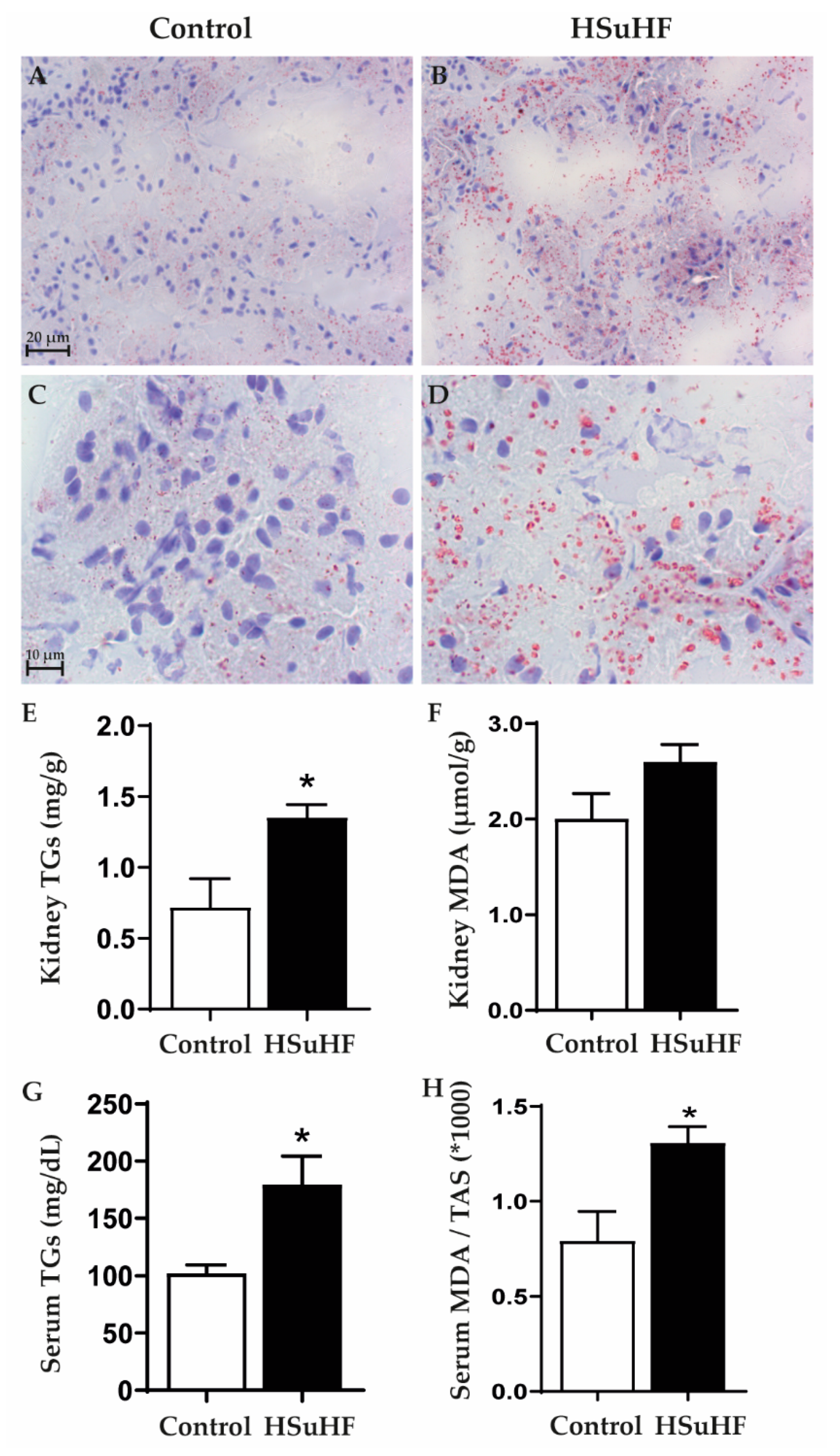
2.7. Renal Lipid Deposition
2.7.1. Oil Red O Staining
2.7.2. Kidney Triglycerides Levels
2.8. Kidney Gene and Protein Expression
2.8.1. qRT-PCR Gene Expression Analysis
2.8.2. Protein Content Analysis by Western Blotting
2.9. Statistical Analysis
3. Results
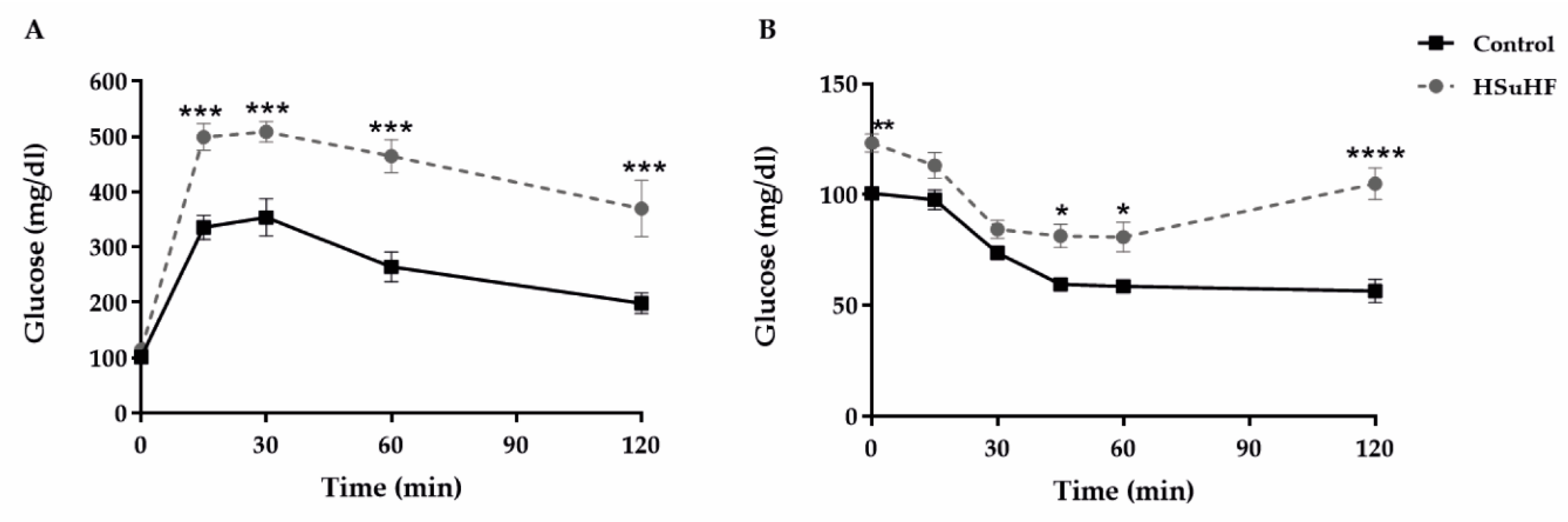
3.1. Body Weight, Caloric Intake and Glycemic and Insulinemic Profile
3.2. Renal Function Data
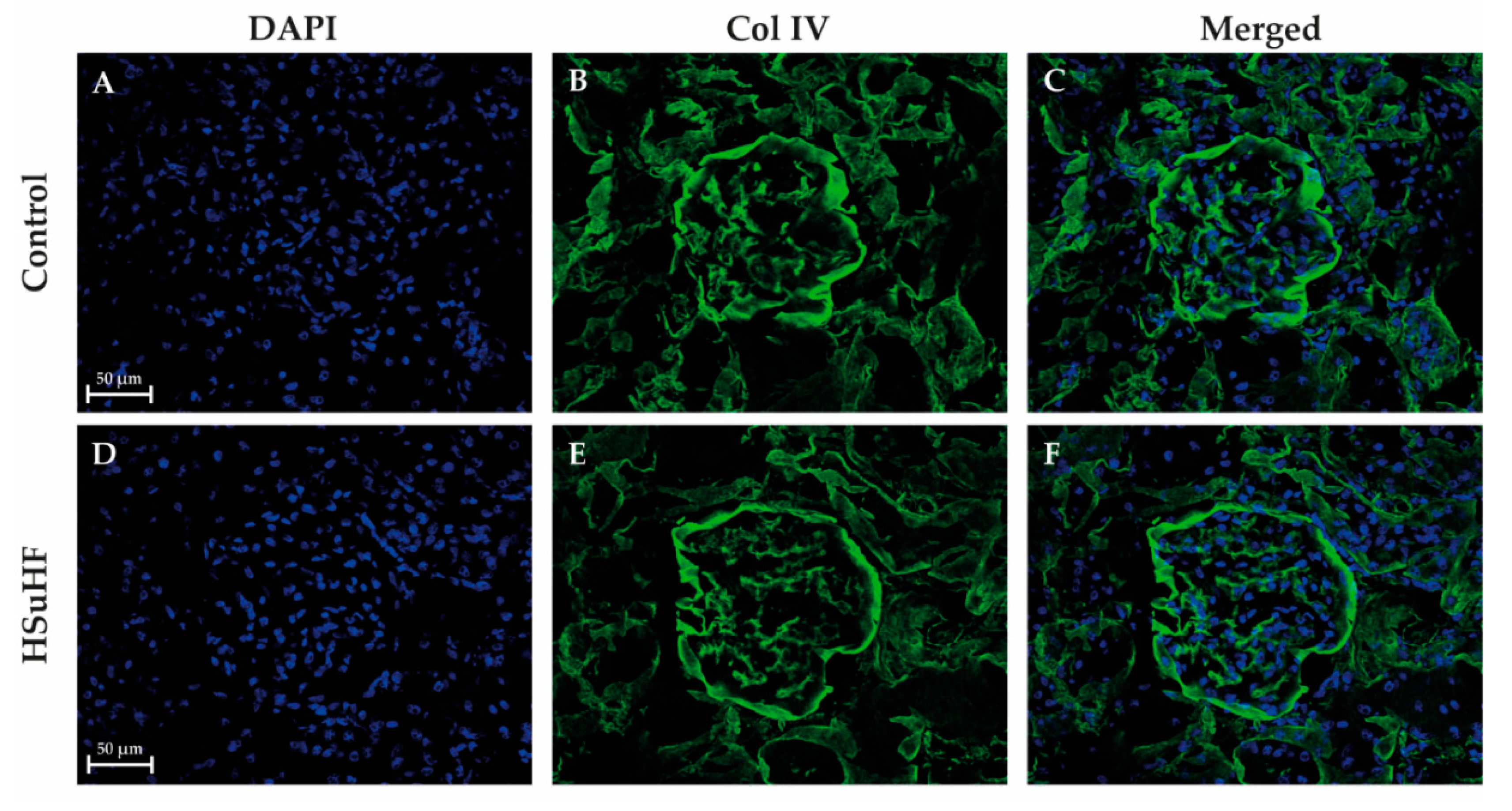
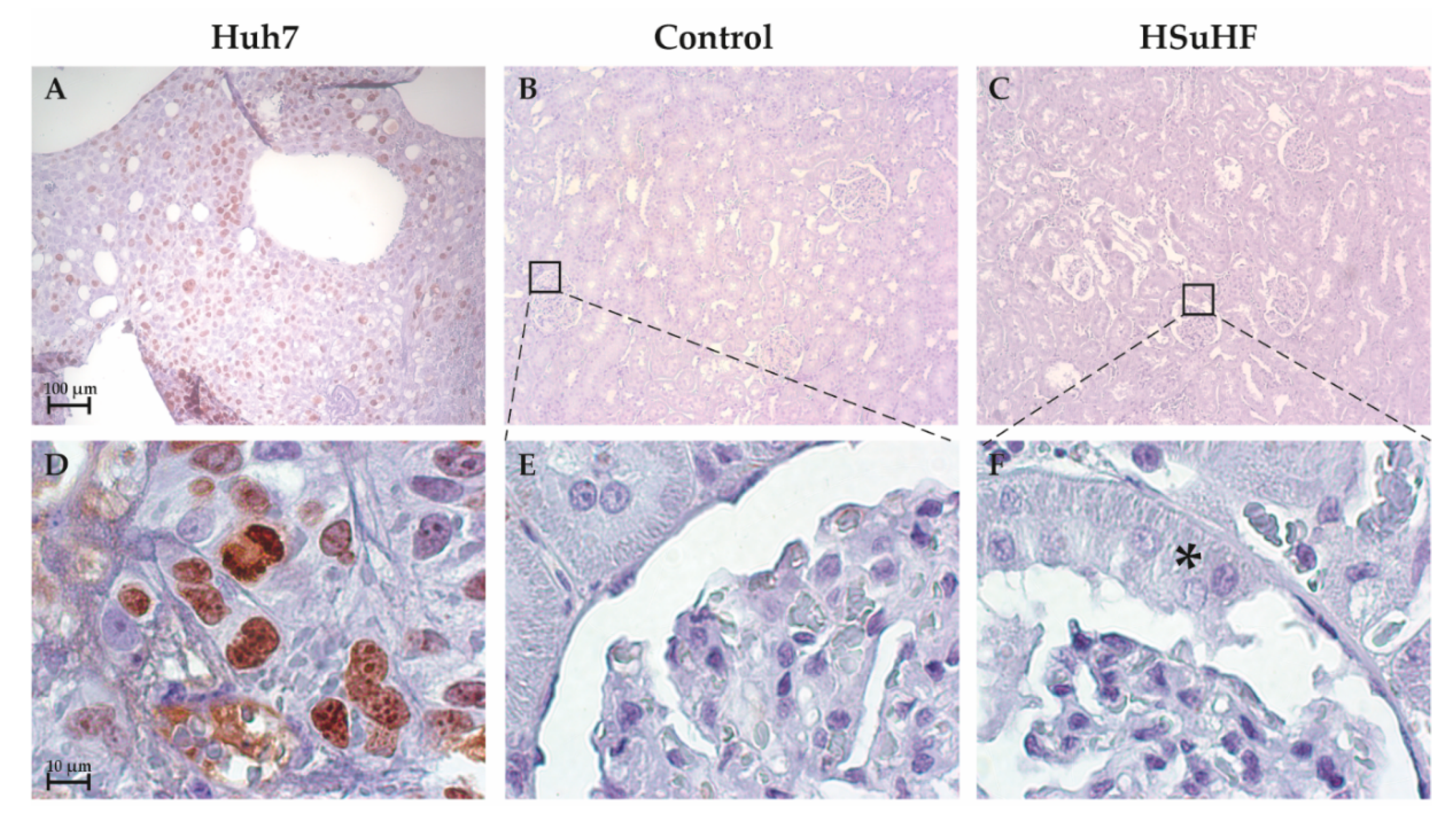
3.3. Kidney Histomorphology
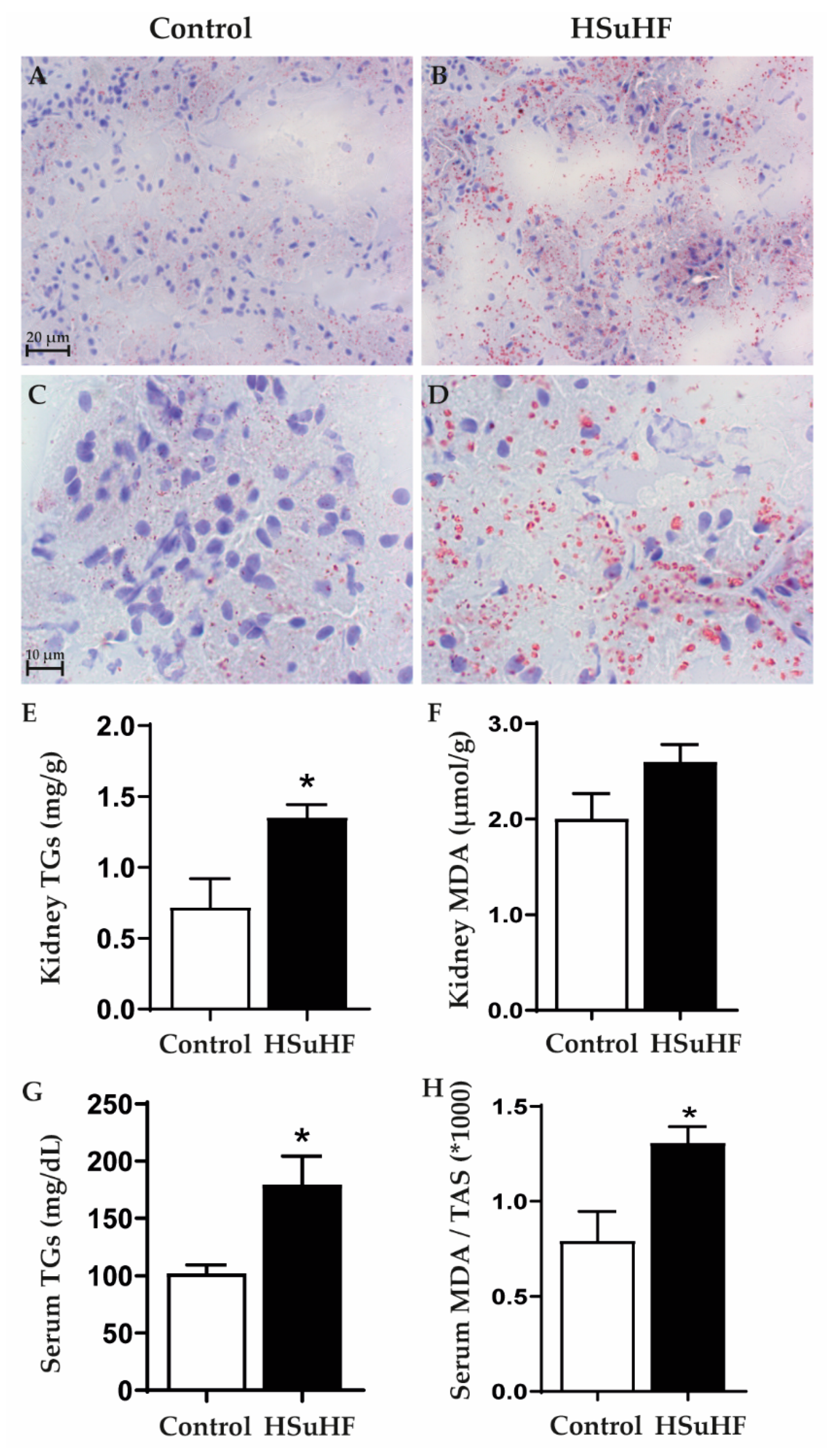
3.4. Serum and Renal Lipids
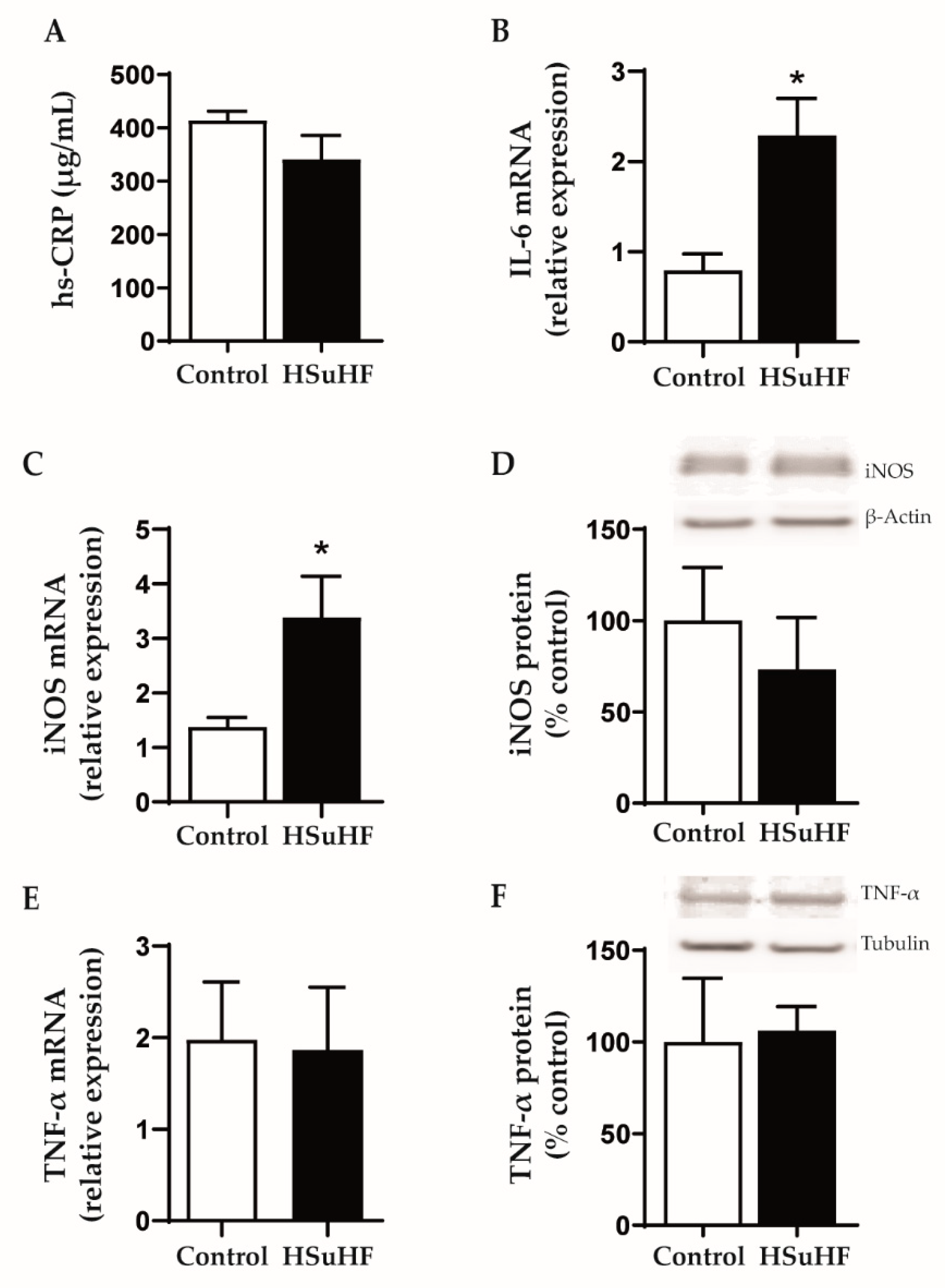
3.5. Serum and Renal Inflammatory Markers
4. Discussion
5. Conclusions and Future Directions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Federation, I.D. IDF Diabetes Atlas, 9th ed.; International Diabetes Federation: Brussels, Belgium, 2019. [Google Scholar]
- Reutens, A.T.; Atkins, R.C. Epidemiology of diabetic nephropathy. Contrib. Nephrol. 2011, 170, 1–7. [Google Scholar] [CrossRef]
- Spijkerman, A.M.; Dekker, J.M.; Nijpels, G.; Adriaanse, M.C.; Kostense, P.J.; Ruwaard, D.; Stehouwer, C.D.; Bouter, L.M.; Heine, R.J. Microvascular complications at time of diagnosis of type 2 diabetes are similar among diabetic patients detected by targeted screening and patients newly diagnosed in general practice: The hoorn screening study. Diabetes Care 2003, 26, 2604–2608. [Google Scholar] [CrossRef] [Green Version]
- Stehouwer, C.D.A. Microvascular Dysfunction and Hyperglycemia: A Vicious Cycle With Widespread Consequences. Diabetes 2018, 67, 1729–1741. [Google Scholar] [CrossRef] [Green Version]
- Arora, M.K.; Singh, U.K. Molecular mechanisms in the pathogenesis of diabetic nephropathy: An update. Vasc. Pharmacol. 2013, 58, 259–271. [Google Scholar] [CrossRef]
- Valencia, W.M.; Florez, H. How to prevent the microvascular complications of type 2 diabetes beyond glucose control. BMJ 2017, 356, i6505. [Google Scholar] [CrossRef] [Green Version]
- Barrett, E.J.; Liu, Z.; Khamaisi, M.; King, G.L.; Klein, R.; Klein, B.E.K.; Hughes, T.M.; Craft, S.; Freedman, B.I.; Bowden, D.W.; et al. Diabetic Microvascular Disease: An Endocrine Society Scientific Statement. J. Clin. Endocrinol. Metab. 2017, 102, 4343–4410. [Google Scholar] [CrossRef]
- Tabak, A.G.; Herder, C.; Rathmann, W.; Brunner, E.J.; Kivimaki, M. Prediabetes: A high-risk state for diabetes development. Lancet 2012, 379, 2279–2290. [Google Scholar] [CrossRef] [Green Version]
- Melsom, T.; Schei, J.; Stefansson, V.T.; Solbu, M.D.; Jenssen, T.G.; Mathisen, U.D.; Wilsgaard, T.; Eriksen, B.O. Prediabetes and Risk of Glomerular Hyperfiltration and Albuminuria in the General Nondiabetic Population: A Prospective Cohort Study. Am. J. Kidney Dis. 2016, 67, 841–850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Markus, M.R.P.; Ittermann, T.; Baumeister, S.E.; Huth, C.; Thorand, B.; Herder, C.; Roden, M.; Siewert-Markus, U.; Rathmann, W.; Koenig, W.; et al. Prediabetes is associated with microalbuminuria, reduced kidney function and chronic kidney disease in the general population: The KORA (Cooperative Health Research in the Augsburg Region) F4-Study. Nutr. Metab. Cardiovasc. Dis. 2018, 28, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Echouffo-Tcheugui, J.B.; Narayan, K.M.; Weisman, D.; Golden, S.H.; Jaar, B.G. Association between prediabetes and risk of chronic kidney disease: A systematic review and meta-analysis. Diabet. Med. 2016, 33, 1615–1624. [Google Scholar] [CrossRef] [PubMed]
- Ferguson, M.A.; Waikar, S.S. Established and emerging markers of kidney function. Clin. Chem. 2012, 58, 680–689. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waikar, S.S.; Betensky, R.A.; Bonventre, J.V. Creatinine as the gold standard for kidney injury biomarker studies? Nephrol. Dial. Transpl. 2009, 24, 3263–3265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Currie, G.; McKay, G.; Delles, C. Biomarkers in diabetic nephropathy: Present and future. World J. Diabetes 2014, 5, 763–776. [Google Scholar] [CrossRef]
- Soler, M.J.; Riera, M.; Batlle, D. New experimental models of diabetic nephropathy in mice models of type 2 diabetes: Efforts to replicate human nephropathy. Exp. Diabetes Res. 2012, 2012, 616313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaur, M.; Bedi, O.; Sachdeva, S.; Reddy, B.V.; Kumar, P. Rodent animal models: From mild to advanced stages of diabetic nephropathy. Inflammopharmacology 2014, 22, 279–293. [Google Scholar] [CrossRef] [PubMed]
- Betz, B.; Conway, B.R. An Update on the Use of Animal Models in Diabetic Nephropathy Research. Curr. Diabetes Rep. 2016, 16, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tervaert, T.W.; Mooyaart, A.L.; Amann, K.; Cohen, A.H.; Cook, H.T.; Drachenberg, C.B.; Ferrario, F.; Fogo, A.B.; Haas, M.; de Heer, E.; et al. Pathologic classification of diabetic nephropathy. J. Am. Soc. Nephrol. 2010, 21, 556–563. [Google Scholar] [CrossRef] [Green Version]
- Brosius, F.C., III; Alpers, C.E.; Bottinger, E.P.; Breyer, M.D.; Coffman, T.M.; Gurley, S.B.; Harris, R.C.; Kakoki, M.; Kretzler, M.; Leiter, E.H.; et al. Mouse models of diabetic nephropathy. J. Am. Soc. Nephrol. 2009, 20, 2503–2512. [Google Scholar] [CrossRef] [Green Version]
- Kong, L.L.; Wu, H.; Cui, W.P.; Zhou, W.H.; Luo, P.; Sun, J.; Yuan, H.; Miao, L.N. Advances in murine models of diabetic nephropathy. J. Diabetes Res. 2013, 2013, 797548. [Google Scholar] [CrossRef] [Green Version]
- Kitada, M.; Ogura, Y.; Koya, D. Rodent models of diabetic nephropathy: Their utility and limitations. Int. J. Nephrol. Renovasc. Dis. 2016, 9, 279–290. [Google Scholar] [CrossRef] [Green Version]
- Preguica, I.; Alves, A.; Nunes, S.; Gomes, P.; Fernandes, R.; Viana, S.D.; Reis, F. Diet-Induced Rodent Models of Diabetic Peripheral Neuropathy, Retinopathy and Nephropathy. Nutrients 2020, 12, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgeiro, A.; Cerqueira, M.G.; Varela-Rodriguez, B.M.; Nunes, S.; Neto, P.; Pereira, F.C.; Reis, F.; Carvalho, E. Glucose and Lipid Dysmetabolism in a Rat Model of Prediabetes Induced by a High-Sucrose Diet. Nutrients 2017, 9, 638. [Google Scholar] [CrossRef] [PubMed]
- Nunes, S.; Soares, E.; Fernandes, J.; Viana, S.; Carvalho, E.; Pereira, F.C.; Reis, F. Early cardiac changes in a rat model of prediabetes: Brain natriuretic peptide overexpression seems to be the best marker. Cardiovasc. Diabetol. 2013, 12, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin-Cordero, L.; Galvez, I.; Hinchado, M.D.; Ortega, E. beta2 Adrenergic Regulation of the Phagocytic and Microbicide Capacity of Macrophages from Obese and Lean Mice: Effects of Exercise. Nutrients 2019, 11, 2721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galvez, I.; Martin-Cordero, L.; Hinchado, M.D.; Alvarez-Barrientos, A.; Ortega, E. Obesity Affects beta2 Adrenergic Regulation of the Inflammatory Profile and Phenotype of Circulating Monocytes from Exercised Animals. Nutrients 2019, 11, 2630. [Google Scholar] [CrossRef] [Green Version]
- Chang, A.R.; Grams, M.E.; Ballew, S.H.; Bilo, H.; Correa, A.; Evans, M.; Gutierrez, O.M.; Hosseinpanah, F.; Iseki, K.; Kenealy, T.; et al. Adiposity and risk of decline in glomerular filtration rate: Meta-analysis of individual participant data in a global consortium. BMJ 2019, 364, k5301. [Google Scholar] [CrossRef] [Green Version]
- Whaley-Connell, A.; Sowers, J.R. Obesity and kidney disease: From population to basic science and the search for new therapeutic targets. Kidney Int. 2017, 92, 313–323. [Google Scholar] [CrossRef]
- Tziomalos, K.; Athyros, V.G. Diabetic Nephropathy: New Risk Factors and Improvements in Diagnosis. Rev. Diabet. Stud. 2015, 12, 110–118. [Google Scholar] [CrossRef] [Green Version]
- Gutwein, P.; Abdel-Bakky, M.S.; Doberstein, K.; Schramme, A.; Beckmann, J.; Schaefer, L.; Amann, K.; Doller, A.; Kampfer-Kolb, N.; Abdel-Aziz, A.A.; et al. CXCL16 and oxLDL are induced in the onset of diabetic nephropathy. J. Cell Mol. Med. 2009, 13, 3809–3825. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.C.; Feng, Y.; Lu, C.Q.; Ju, S. Renal fat fraction and diffusion tensor imaging in patients with early-stage diabetic nephropathy. Eur. Radiol. 2018, 28, 3326–3334. [Google Scholar] [CrossRef]
- Moreno, J.A.; Gomez-Guerrero, C.; Mas, S.; Sanz, A.B.; Lorenzo, O.; Ruiz-Ortega, M.; Opazo, L.; Mezzano, S.; Egido, J. Targeting inflammation in diabetic nephropathy: A tale of hope. Expert Opin. Investig. Drugs 2018, 27, 917–930. [Google Scholar] [CrossRef]
- Ferreira, L.; Teixeira-de-Lemos, E.; Pinto, F.; Parada, B.; Mega, C.; Vala, H.; Pinto, R.; Garrido, P.; Sereno, J.; Fernandes, R.; et al. Effects of sitagliptin treatment on dysmetabolism, inflammation, and oxidative stress in an animal model of type 2 diabetes (ZDF rat). Mediators Inflamm. 2010, 2010, 592760. [Google Scholar] [CrossRef]
- Pestel, S.; Krzykalla, V.; Weckesser, G. Measurement of glomerular filtration rate in the conscious rat. J. Pharmacol. Toxicol. Methods 2007, 56, 277–289. [Google Scholar] [CrossRef]
- Viana, S.D.; Fernandes, R.C.; Canas, P.M.; Silva, A.M.; Carvalho, F.; Ali, S.F.; Fontes Ribeiro, C.A.; Pereira, F.C. Presymptomatic MPTP Mice Show Neurotrophic S100B/mRAGE Striatal Levels. CNS Neurosci. Ther. 2016, 22, 396–403. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.J.; Lin, Y.C.; Lee, J.C.; Kuo, S.C.; Ho, C.T.; Huang, L.J.; Kuo, D.H.; Way, T.D. CCT327 enhances TRAIL-induced apoptosis through the induction of death receptors and downregulation of cell survival proteins in TRAIL-resistant human leukemia cells. Oncol. Rep. 2014, 32, 1257–1264. [Google Scholar] [CrossRef] [Green Version]
- Glassock, R.J.; Warnock, D.G.; Delanaye, P. The global burden of chronic kidney disease: Estimates, variability and pitfalls. Nat. Rev. Nephrol. 2017, 13, 104–114. [Google Scholar] [CrossRef] [PubMed]
- Jha, V.; Garcia-Garcia, G.; Iseki, K.; Li, Z.; Naicker, S.; Plattner, B.; Saran, R.; Wang, A.Y.; Yang, C.W. Chronic kidney disease: Global dimension and perspectives. Lancet 2013, 382, 260–272. [Google Scholar] [CrossRef]
- Marques, C.; Meireles, M.; Norberto, S.; Leite, J.; Freitas, J.; Pestana, D.; Faria, A.; Calhau, C. High-fat diet-induced obesity Rat model: A comparison between Wistar and Sprague-Dawley Rat. Adipocyte 2016, 5, 11–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangel Silvares, R.; Nunes Goulart da Silva Pereira, E.; Eduardo Ilaquita Flores, E.; Lino Rodrigues, K.; Ribeiro Silva, A.; Goncalves-de-Albuquerque, C.F.; Daliry, A. High-fat diet-induced kidney alterations in rats with metabolic syndrome: Endothelial dysfunction and decreased antioxidant defense. Diabetes Metab. Syndr. Obes. 2019, 12, 1773–1781. [Google Scholar] [CrossRef] [Green Version]
- Jennette, J.C. Rapidly progressive crescentic glomerulonephritis. Kidney Int. 2003, 63, 1164–1177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, M.; Verhave, J.C.; Liu, Z.H.; Alpers, C.E.; Barratt, J.; Becker, J.U.; Cattran, D.; Cook, H.T.; Coppo, R.; Feehally, J.; et al. A Multicenter Study of the Predictive Value of Crescents in IgA Nephropathy. J. Am. Soc. Nephrol. 2017, 28, 691–701. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, N.; Takeda, S.; Imai, T.; Akimoto, T.; Nagata, D. Unexpected observation of glomerular crescents in a patient with diabetes who developed drug-induced acute tubulointerstitial nephritis: A possible feature of diabetic nephropathy? Nephrology 2015, 20, 438–439. [Google Scholar] [CrossRef] [PubMed]
- Toth, T. Epithelial crescent in diabetic glomeruli. A case report. Int. Urol. Nephrol. 1987, 19, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Otani, N.; Akimoto, T.; Yumura, W.; Matsubara, D.; Iwazu, Y.; Numata, A.; Miki, T.; Takemoto, F.; Fukushima, N.; Muto, S.; et al. Is there a link between diabetic glomerular injury and crescent formation? A case report and literature review. Diagn. Pathol. 2012, 7, 46. [Google Scholar] [CrossRef] [Green Version]
- Mottl, A.K.; Gasim, A.; Schober, F.P.; Hu, Y.; Dunnon, A.K.; Hogan, S.L.; Jennette, J.C. Segmental Sclerosis and Extracapillary Hypercellularity Predict Diabetic ESRD. J. Am. Soc. Nephrol. 2018, 29, 694–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elfenbein, I.B.; Reyes, J.W. Crescents in diabetic glomerulopathy. Incidence and clinical significance. Lab. Investig. 1975, 33, 687–695. [Google Scholar]
- Nasr, S.H.; D’Agati, V.D.; Said, S.M.; Stokes, M.B.; Appel, G.B.; Valeri, A.M.; Markowitz, G.S. Pauci-immune crescentic glomerulonephritis superimposed on diabetic glomerulosclerosis. Clin. J. Am. Soc. Nephrol. 2008, 3, 1282–1288. [Google Scholar] [CrossRef] [Green Version]
- Mazzucco, G.; Bertani, T.; Fortunato, M.; Bernardi, M.; Leutner, M.; Boldorini, R.; Monga, G. Different patterns of renal damage in type 2 diabetes mellitus: A multicentric study on 393 biopsies. Am. J. Kidney Dis. 2002, 39, 713–720. [Google Scholar] [CrossRef]
- Marques, C.; Mega, C.; Goncalves, A.; Rodrigues-Santos, P.; Teixeira-Lemos, E.; Teixeira, F.; Fontes-Ribeiro, C.; Reis, F.; Fernandes, R. Sitagliptin prevents inflammation and apoptotic cell death in the kidney of type 2 diabetic animals. Mediators Inflamm. 2014, 2014, 538737. [Google Scholar] [CrossRef] [Green Version]
- Mega, C.; de Lemos, E.T.; Vala, H.; Fernandes, R.; Oliveira, J.; Mascarenhas-Melo, F.; Teixeira, F.; Reis, F. Diabetic nephropathy amelioration by a low-dose sitagliptin in an animal model of type 2 diabetes (Zucker diabetic fatty rat). Exp. Diabetes Res. 2011, 2011, 162092. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Zhang, Y.; Li, L.; Mann, D.; Imig, J.D.; Emmett, N.; Gibbons, G.; Jin, L.M. Glomerular expression of kidney injury molecule-1 and podocytopenia in diabetic glomerulopathy. Am. J. Nephrol. 2011, 34, 268–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaut, J.P.; Hoshi, M.; Jain, S.; Liapis, H. Claudin 1 and nephrin label cellular crescents in diabetic glomerulosclerosis. Hum. Pathol. 2014, 45, 628–635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaissling, B.; Le Hir, M. The renal cortical interstitium: Morphological and functional aspects. Histochem. Cell Biol. 2008, 130, 247–262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Farris, A.B.; Alpers, C.E. What is the best way to measure renal fibrosis?: A pathologist’s perspective. Kidney Int. Suppl. 2014, 4, 9–15. [Google Scholar] [CrossRef] [Green Version]
- Roeder, S.S.; Stefanska, A.; Eng, D.G.; Kaverina, N.; Sunseri, M.W.; McNicholas, B.A.; Rabinovitch, P.; Engel, F.B.; Daniel, C.; Amann, K.; et al. Changes in glomerular parietal epithelial cells in mouse kidneys with advanced age. Am. J. Physiol. Renal Physiol. 2015, 309, F164–F178. [Google Scholar] [CrossRef] [Green Version]
- Miller, I.; Min, M.; Yang, C.; Tian, C.; Gookin, S.; Carter, D.; Spencer, S.L. Ki67 is a Graded Rather than a Binary Marker of Proliferation versus Quiescence. Cell Rep. 2018, 24, 1105–1112. [Google Scholar] [CrossRef] [Green Version]
- Zambon, A.C. Use of the Ki67 promoter to label cell cycle entry in living cells. Cytom. A 2010, 77, 564–570. [Google Scholar] [CrossRef] [Green Version]
- Geraci, G.; Zammuto, M.M.; Mattina, A.; Zanoli, L.; Geraci, C.; Granata, A.; Nardi, E.; Fatuzzo, P.M.; Cottone, S.; Mule, G. Para-perirenal distribution of body fat is associated with reduced glomerular filtration rate regardless of other indices of adiposity in hypertensive patients. J. Clin. Hypertens. 2018, 20, 1438–1446. [Google Scholar] [CrossRef] [Green Version]
- Markova, I.; Miklankova, D.; Huttl, M.; Kacer, P.; Skibova, J.; Kucera, J.; Sedlacek, R.; Kacerova, T.; Kazdova, L.; Malinska, H. The Effect of Lipotoxicity on Renal Dysfunction in a Nonobese Rat Model of Metabolic Syndrome: A Urinary Proteomic Approach. J. Diabetes Res. 2019, 2019, 8712979. [Google Scholar] [CrossRef]
- Izquierdo-Lahuerta, A.; Martinez-Garcia, C.; Medina-Gomez, G. Lipotoxicity as a trigger factor of renal disease. J. Nephrol. 2016, 29, 603–610. [Google Scholar] [CrossRef]
- Feigerlova, E.; Battaglia-Hsu, S.F. IL-6 signaling in diabetic nephropathy: From pathophysiology to therapeutic perspectives. Cytokine Growth Factor Rev. 2017, 37, 57–65. [Google Scholar] [CrossRef] [PubMed]
- De Vries, A.P.; Ruggenenti, P.; Ruan, X.Z.; Praga, M.; Cruzado, J.M.; Bajema, I.M.; D’Agati, V.D.; Lamb, H.J.; Pongrac Barlovic, D.; Hojs, R.; et al. Fatty kidney: Emerging role of ectopic lipid in obesity-related renal disease. Lancet Diabetes Endocrinol. 2014, 2, 417–426. [Google Scholar] [CrossRef]
- Dijkman, H.B.; Weening, J.J.; Smeets, B.; Verrijp, K.C.; van Kuppevelt, T.H.; Assmann, K.K.; Steenbergen, E.J.; Wetzels, J.F. Proliferating cells in HIV and pamidronate-associated collapsing focal segmental glomerulosclerosis are parietal epithelial cells. Kidney Int. 2006, 70, 338–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward Primer Sequence | Reverse | T (°C) |
|---|---|---|---|
| iNOS | AGAGACAGAAGTGCGATC | AGATTCAGTAGTCCACAATAGTA | 60 |
| IL-6 | GGAGAAGTTAGAGTCACAGA | GCCGAGTAGACCTCATAG | 60 |
| TNF-α | CCCAATCTGTGTCCTTCT | TTCTGAGCATCGTAGTTGT | 60 |
| GAPDH | GACTTCAACAGCAACTCC | GCCATATTCATTGTCATACCA | 60 |
| HPRT | ATGGGAGGCCATCACATTGT | ATGTAATCCAGCAGGTCAGCAA | 58 |
| Antibody | Dilution | Catalog No.-Company |
|---|---|---|
| iNOS | 1:1000 | Ab178945-Abcam |
| TNF-α | 1:500 | Ab66579-Abcam |
| β-actin | 1:1000 | A5316-Sigma-Aldrich |
| Tubulin | 1:1000 | AB0046-200-SICGEN |
| Goat anti-mouse IgG-HRP | 1:10,000 | R-05071-Advansta |
| Goat anti-rabbit IgG-HRP | 1:10,000 | R-05072-Advansta |
| Parameters | Control | HSuHF |
|---|---|---|
| Body Weight (BW) | ||
| Initial (g) | 300.00 ± 10.88 | 297.11 ± 9.71 |
| Final (g) | 507.57 ± 18.15 | 578.62 ± 32.89 |
| Delta BW (g) | 228.50 ± 10.04 | 311.20 ± 21.11* |
| Caloric Data | ||
| Food Intake (g/week) | 163.60 ± 2.20 | 77.11 ± 0.54 *** |
| Beverage Intake (mL/week) | 211.80 ± 3.69 | 326.70 ± 50.89 *** |
| Calories from Carbohydrates (kcal/rat/week) | 350.40 ± 3.89 | 574.50 ± 13.84 *** |
| Calories from Lipids (kcal/rat/week) | 44.22 ± 0.47 | 97.74 ± 8.71 |
| Calories from Proteins (kcal/rat/week) | 121.20 ± 1.29 | 51.68 ± 1.60 *** |
| ∑ Total calories (kcal/rat/week) | 515.80 ± 5.49 | 713.40 ± 10.17 *** |
| Glycemic Profile | ||
| Fasting glucose(mg/dL) | 100.50 ± 1.37 | 115.77 ± 1.42 |
| Fed glucose (mg/dL) | 149.14 ± 7.95 | 273.14 ± 33.53 *** |
| GTT (AUC) | 31856 ± 2565 | 54974.2 ± 3413 *** |
| HbA1c (%) | 3.73 ± 0.05 | 4.00 ± 0.15 |
| Insulinemic Profile | ||
| Fasting insulin (µg/L) | 0.577 ± 0.082 | 1.454 ± 0.197 |
| Fed insulin (µg/L) | 0.899 ± 0.159 | 2.426 ± 0.535 * |
| ITT (AUC) | 8107 ± 299 | 11175 ± 587 ** |
| Parameters | Control | HSuHF |
|---|---|---|
| Kidney Weight (KW) | ||
| Absolute (g) | 2.86 ± 0.13 | 2.90 ± 0.12 |
| KW/BW (g/kg) | 5.63 ± 0.22 | 4.92 ± 0.07 |
| Creatinine | ||
| Serum (mg/dL) | 0.36 ± 0.01 | 0.38 ± 0.02 |
| Urine (mg/dL) | 100.40 ± 14.78 | 61.15 ± 7.84 |
| Clearance (mL/h) | 200.40 ± 18.55 | 184.4 ± 11.57 |
| BUN | ||
| Serum (mg/dL) | 14.19 ± 0.58 | 8.57 ± 0.60 *** |
| Urine (mg/dL) | 1554.00 ± 132.80 | 493.00 ± 82.90 *** |
| Clearance (mL/h) | 92.53 ± 7.24 | 35.59 ± 4.29 ** |
| Uric Acid | ||
| Serum (mg/dL) | 1.79 ± 0.33 | 2.33 ± 0.52 |
| Urine (mg/dL) | 12.80 ± 1.74 | 9.86 ± 0.69 |
| Clearance (mL/h) | 6.50 ± 1.10 | 6.00 ± 1.02 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nunes, S.; Alves, A.; Preguiça, I.; Barbosa, A.; Vieira, P.; Mendes, F.; Martins, D.; Viana, S.D.; Reis, F. Crescent-Like Lesions as an Early Signature of Nephropathy in a Rat Model of Prediabetes Induced by a Hypercaloric Diet. Nutrients 2020, 12, 881. https://doi.org/10.3390/nu12040881
Nunes S, Alves A, Preguiça I, Barbosa A, Vieira P, Mendes F, Martins D, Viana SD, Reis F. Crescent-Like Lesions as an Early Signature of Nephropathy in a Rat Model of Prediabetes Induced by a Hypercaloric Diet. Nutrients. 2020; 12(4):881. https://doi.org/10.3390/nu12040881
Chicago/Turabian StyleNunes, Sara, André Alves, Inês Preguiça, Adelaide Barbosa, Pedro Vieira, Fernando Mendes, Diana Martins, Sofia D. Viana, and Flávio Reis. 2020. "Crescent-Like Lesions as an Early Signature of Nephropathy in a Rat Model of Prediabetes Induced by a Hypercaloric Diet" Nutrients 12, no. 4: 881. https://doi.org/10.3390/nu12040881
APA StyleNunes, S., Alves, A., Preguiça, I., Barbosa, A., Vieira, P., Mendes, F., Martins, D., Viana, S. D., & Reis, F. (2020). Crescent-Like Lesions as an Early Signature of Nephropathy in a Rat Model of Prediabetes Induced by a Hypercaloric Diet. Nutrients, 12(4), 881. https://doi.org/10.3390/nu12040881