Familial Aggregation of Endemic Congenital Hypothyroidism Syndrome in Congo (DR): Historical Data


Abstract
1. Introduction
- The myxedematous phenotype is associated with hypothyroidism around birth, persisting during the first years of life in the absence of iodine deficiency correction. Infantile and juvenile hypothyroidism in severe iodine deficient areas were quite common. For unknown reasons, some hypothyroid children lose progressively their thyroid responsiveness to iodine, and evolve through irreversible hypothyroidism, even after correction of iodine deficiency [13,14].
2. Materials and Methods
2.1. Epidemiological Context
2.2. Clinical Classification
- Myxedematous phenotype: moderate to severe intellectual deficiency, stunted growth with disproportionate nanism, persistent myxedema with variable severity, and including mixed form (myxedematous phenotype associated with signs of neurological phenotype);
- Neurological phenotype: profound intellectual deficiency, no evidence of persistent myxedema, a variable combination of clinical evidence of spasticity, diplopia, abnormal movements, deaf-mutism, and moderately decreased cranial volume.
2.3. Basic Statistical Approach
2.4. Statistical Development
2.5. Ethical Aspect
3. Results
3.1. Epidemiological Description
3.2. Basic Statistical Approach
3.3. Statistical Development
4. Discussion
Major Limits of the Investigation
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bunuel, L. Las Hurdes Tierra Sin Pan [Motion Picture]. Available online: https://www.youtube.com (accessed on 30 April 2020).
- Vanderpas, J.; Moreno-Reyes, R. Historical aspects of iodine deficiency control. Minerva 2017, 108, 124–135. [Google Scholar]
- Quervain, F. Endemic Cretinism (Translation of Der Endemische Kretinismus 1936); Dennison, J., Oxnard, C., Obendorf, P., Eds.; Springer: New York, NY, USA, 2019; p. 186. [Google Scholar]
- Trimarchi, F.; Vermiglio, F.; Finocchiaro, M.; Battiato, S.; Lo Presti, V.; La Torre, N.; Vigneri, R. Epidemiology and clinical characteristics of endemic cretinism in Sicily. J. Endocrinol. Invest. 1990, 13, 543–548. [Google Scholar] [CrossRef] [PubMed]
- Stanbury, J.; Delange, F.; Ermans, A.; Fierro-Benitez, R. The varied manifestations of endemic cretinism. Trans. Am. Clin. Climatol. Assoc. 1974, 85, 6–17. Available online: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2441327/ (accessed on 30 September 2020). [PubMed]
- Lagasse, R.; Luvivila, K.; Yunga, Y.; Gerard, M.; Hanson, A.; Bourdoux, P.; Delange, F.; Thilly, C.H. Endemic goitre and cretinism in ubangi (chapter 4). In Role of Cassava in the Etiology of Endemic Goitre and Cretinism; Ermans, A., Mbulamoko, N., Delange, F., Ahluwalia, R., Eds.; International Development Research Centre: Ottawa, ON, Canada, 1980; pp. 45–60. Available online: https://idl-bnc-idrc.dspacedirect.org/handle/10625/4287 (accessed on 30 September 2020).
- Held, K.; Cruz, M.; Moncayo, F. Clinical pattern and the genetics of the fetal iodine deficiency disorder (endemic cretinism): Results of a field study in highland ecuador. Am. J. Med. Genet. 1990, 35, 85–90. [Google Scholar] [CrossRef] [PubMed]
- McCarrison, R. Observations on endemic cretinism in the chitral and gilgit valleys. Lancet 1908, 2, 1275–1280. [Google Scholar] [CrossRef]
- Sankar, R.; Pulger, T.; Rai, B.; Gomathi, S.R.; Gyatso, T.R.; Pandav, C.S. Epidemiology of Endemic Cretinism in Sikkim, India. Indian J. Pediatr. 1998, 65, 303–309. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.Y.; Jiang, X.M.; Dou, Z.H.; Rakeman, M.A.; Zhang, M.L.; O’Donnell, J.; Ma, T.; Amette, K.; DeLong, N.; DeLong, G.R. Timing of vulnerability of the brain to iodine deficiency in endemic cretinism. N. Engl. J. Med. 1994, 331, 1739–1744. [Google Scholar] [CrossRef] [PubMed]
- Boyages, S.; Helpern, J. Endemic cretinism: Toward a unifying hypothesis. Thyroid 1993, 3, 59–69. [Google Scholar] [CrossRef]
- Thorpe-Beeston, J.; Nicolaides, K.; Felton, C.; Butler, J.; McGregor, A. Maturation of the secretion of thyroid hormone and thyroid-stimulating hormone in the fetus. N. Engl. J. Med. 1991, 324, 532–536. [Google Scholar] [CrossRef]
- Vanderpas, J.; Rivera-Vanderpas, M.T.; Bourdoux, P.; Luvivila, K.; Lagasse, R.; Perlmutter-Cremer, N.; Thilly, C.H. Reversibility of severe hypothyroidism with supplementary iodine in patients with endemic cretinism. N. Engl. J. Med. 1986, 315, 791–795. [Google Scholar] [CrossRef]
- Contempre, B.; Duale, L.; Gervy, C.; Alexandre, J.; Vanovervelt, N.; Dumont, J.E. Hypothyroid patients showing shortened responsiveness to oral Iodized oil have paradoxically low serum thyroglobulin and low thyroid reserve. Thyroglobulin/thyrotropin ratio as a measure of thyroid Damage. Eur. J. Endocrinol. 1996, 134, 342–351. [Google Scholar] [CrossRef] [PubMed]
- Wemeau, J.L.; Kopp, P. Pendred syndrome. Best Pract. Res. Clin. Endocrinol. 2017, 31, 213–224. [Google Scholar] [CrossRef] [PubMed]
- Friesema, E.; Grueters, A.; Biebermann, H.; Krude, H.; von Moers, A.; Reeser, M.; Visser, T.J. Association between mutations in a thyroid hormone transporter and severe X-linked psychomotor retardation. Lancet 2004, 364, 1435–1437. [Google Scholar] [CrossRef]
- Pharoah, P.; Buttfield, I.; Hetzel, B. Neurological damage to the fetus resulting from severe iodine deficiency during pregnancy. Lancet 1971, 297, 308–310. [Google Scholar] [CrossRef]
- Thilly, C.H.; Delange, F.; Lagasse, R.; Bourdoux, P.; Ramioul, L.; Berquist, H.; Ermans, A. Fetal hypothyroidism and maternal thyroid status in severe endemic goiter. J. Clin. Endocrinol. Metab. 1978, 47, 354–360. [Google Scholar] [CrossRef]
- Vanderpas, J. Endemic neonatal, infantile and juvenile hypothyroidism in Ubangi, Northen Zaire: Clinical consequences and prevention. In The Damaged Brain of Iodine Deficiency; Stanbury, J.B., Ed.; Cognizant Communication Corporation: Philadelphia, PA, USA, 1994; pp. 209–223, Accessible as Supplementary material to this article. [Google Scholar]
- Perez, C.; Scrimshaw, N.; Munoz, J. Classification of goitre and technique of endemic goitre surveys. Bull. World Health Organ. 1958, 18, 217–232. [Google Scholar]
- Rainer, J.; Taliun, D.; D’Elia, Y.; Pattaro, C.; Domingues, F.S.; Weichenberger, C.X. Famagg: An R package to evaluate familial aggregation of traits in large pedigrees. Bioinformatics 2016, 32, 1583–1585. [Google Scholar] [CrossRef]
- Yu, C.; Zelterman, D. Statistical inference for familial disease clusters. Biometrics 2002, 58, 481–491. [Google Scholar] [CrossRef]
- Malécot, G. Les Mathématiques de L’hérédité; Masson et Cie: Paris, France, 1958. [Google Scholar]
- Hill, J. A survey of cancer sites by kinship in the utah mormon population. In Banbury Report 4: Cancer Incidence in Defined Population; Cairns, J., Lyon, J.L., Skolnick, M., Eds.; Cold Spring Harbor Laboratories: Suffolk, NY, USA, 1980; pp. 299–318. [Google Scholar]
- Weichenberger, C.; Rainer, J.; Pattaro, C.; Pramstaller, P.; Domingues, F. Comparative assessment of different familial aggregation methods in the context of large and unstructured pedigrees. Bioinformatics 2019, 35, 69–76. [Google Scholar] [CrossRef]
- Khoury, M.J.; Beaty, T.H.; Cohen, B.H. Chapter 8. Genetic approaches to familial aggregation. Section II. Segregation analysis. In Fundamentals of Genetic Epidemiology; Khoury, M.J., Beaty, T.H., Cohen, B.H., Eds.; Oxford University Press: Oxford, UK, 1993; pp. 233–283. [Google Scholar]
- Delange, F.; Ermans, A.M.; Vis, H.L.; Stanbury, J.B. Endemic cretinism in Idjwi Island (Kivu Lake, Republic of Congo). J. Clin. Endocrinol. Metab. 1972, 34, 1059–1066. [Google Scholar] [CrossRef]
- Vassart, G.; Dumont, J.E. Thyroid dysgenesis: Multigenic or epigenetic… or both? Endocrinol 2005, 146, 5035–5037. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, G.; Deladoeÿ, J. Diagnosis, treatment and outcome of congenital hypothyroidism. Endocr. Dev. 2014, 26, 50–59. [Google Scholar] [PubMed]
- Taylor, P.N.; Porcu, E.; Chew, S.; Campbell, P.J.; Traglia, M.; Brown, S.J.; Mullin, B.H.; Shihab, H.A.; Min, J.; Walter, K.; et al. Whole-genome sequence-based analysis of thyroid function. Nat. Commun. 2015, 6, 5681–5691. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Long, W.; Yang, Y.; Wang, Y.; Jiang, L.; Cai, Z.; Wang, H. Newborn screening and molecular profile of congenital hypothyroidism in a Chinese population. Front. Genet. 2018, 9, 509. [Google Scholar] [CrossRef]
- Vanderpas, J.B.; Chapitrer, V. Histoire Naturelle de la Fonction Thyroidienne de la Naissance à 7 ans en Ubangi. L’hypothyroïdie Juvénile en Ubangi, Zaïre. Ph.D. Thesis, Université Libre de Bruxelles, Brussels, Belgium, 1991; pp. 42–55. Available online: https://difusion.ulb.ac.be (accessed on 30 April 2020).
- Lazarus, J. Thyroid Regulation and Dysfunction in the Pregnant Woman. Available online: https://www.thyroidmanager.org/chapter/thyroid-regulation-and-dysfunction-in-the-pregnant-patient/ (accessed on 15 April 2020).
- Moreno, J.C.; Bikker, H.; Kempers, M.J.E.; Van Trotsenburg, A.S.P.; Baas, F.; De Vijlder, J.J.M.; Vulsma, T.; Ris-Stalpers, C. Inactivating mutations in the gene for thyroid oxidase 2 (THOX2) and congenital hypothyroidism. N. Engl. J. Med. 2002, 347, 95–102. [Google Scholar] [CrossRef]
- Fugazzola, L.; Muzza, A.; Weber, G.; Beck-Peccoza, P.P. DUOXS defects: Genotype-phenotype correlations. Ann. Endocrinol. 2011, 72, 82–86. [Google Scholar] [CrossRef]
- De Deken, X.; Miot, F. Hypothyroidism, DUOX defects and their roles in congenital hypothyroidism. Meth. Mol. Biol. 2019, 1982, 667–693. [Google Scholar]
- Thambwe Kibambe, T. From severe endemic cretinism to iodine sufficiency: An IDD success story in the Democratic Republic of Congo. IDD Newsl. 2007, 26, 1–4. Available online: https://www.ign.org/idd-newsletter-dashboard.htm (accessed on 2 April 2020).
- Mayambu, B.; Kalonji Kamune, B. Evaluation de la lutte contre les troubles dus à la carence en iode en République Démocratique du Congo. J. Epidémiol. Santé Publ. 2013, 10, 51–57. [Google Scholar]
- Jacobsen, B.; Brandt, N. Congenital hypothyroidism in Denmark. Arch. Dis. Child. 1981, 56, 134–136. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Normal | Myxedematous (n = 44) | Neurological (n = 2) | Dead (n = 162) | Unknown (n = 4) | |||
|---|---|---|---|---|---|---|---|
| 422 | Grade Severe | 20 | Neurological | 1 | Without signs 1 | 159 | 4 2 |
| Moderate | 17 | Deaf/mute | 1 | With signs 1 | 3 | ||
| Unknown grade | 3 | ||||||
| Mixed phenotype | 4 | ||||||
| Endemic CH (n = 46) | No Endemic CH (n = 422) | Test and 2-Tailed p-Value 3 | ||
|---|---|---|---|---|
| Gender | Number of M/F (ratio M/F) | 24/22 (1.09) | 185/237 (0.78) | Fisher’s p = 0.35 |
| Age | Missing values, n/total (%) | 2/46 (4.3%) | 136/422 (32.23%) | Fisher’s p = 1.3 × 10−5 |
| Median, years (IQR) (n obs 1) | 15.5 (6.5, 23.0) (44) | 16.0 (6.0, 30.0)(286) | Mann–Whitney’s p = 0.33 | |
| <5 years, n/n obs (%) | 5/44 (11.36%) 2 | 51/286 (17.83%) | ||
| 5–18 years, n/n obs (%) | 19/44 (43.18%) | 99/286 (34.62%) | ||
| ≥18 years, n/n obs (%) | 20/44 (45.46%) | 136/286 (47.55%) | Fisher’s p = 0.46 | |
| Goitre | Missing values, n/total (%) | 4/46 (8.7%) | 180/422 (42.65%) | Fisher’s p = 1.9 × 10−6 |
| Any form, n/n obs (%) | 22/42 (52.38%) | 187/242 (77.27%) | Fisher’s p = 0.002 | |
| Visible form, n/n obs (%) | 18/42 (42.86%) | 161/242 (66.53%) | Fisher’s p = 0.005 | |
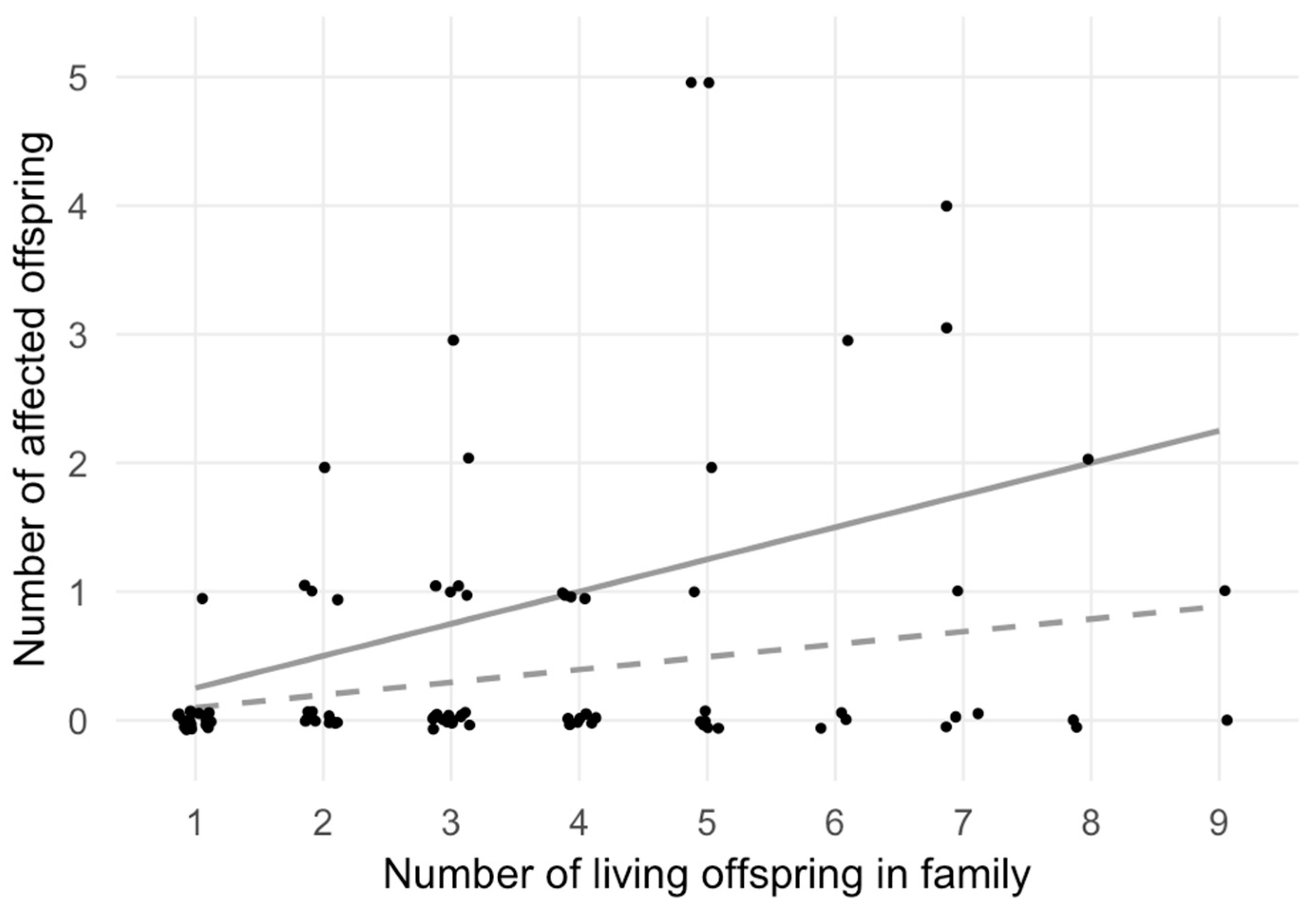
| Sibship size | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
| Total no. families 1 | 19 | 15 | 19 | 11 | 10 | 4 | 6 | 3 | 2 |
| Affected no. families 1 | 1 | 5 | 6 | 4 | 4 | 1 | 3 | 1 | 1 |
| Observed no. 2 | 1 | 5 | 9 | 4 | 13 | 3 | 8 | 2 | 1 |
| Expected no. 3 | 1.87 | 2.95 | 5.60 | 4.32 | 4.91 | 2.36 | 4.13 | 2.36 | 1.77 |
| χ2-test statistics | χ2 = 21.41, df = 8, p = 6.13 × 10−3 | ||||||||
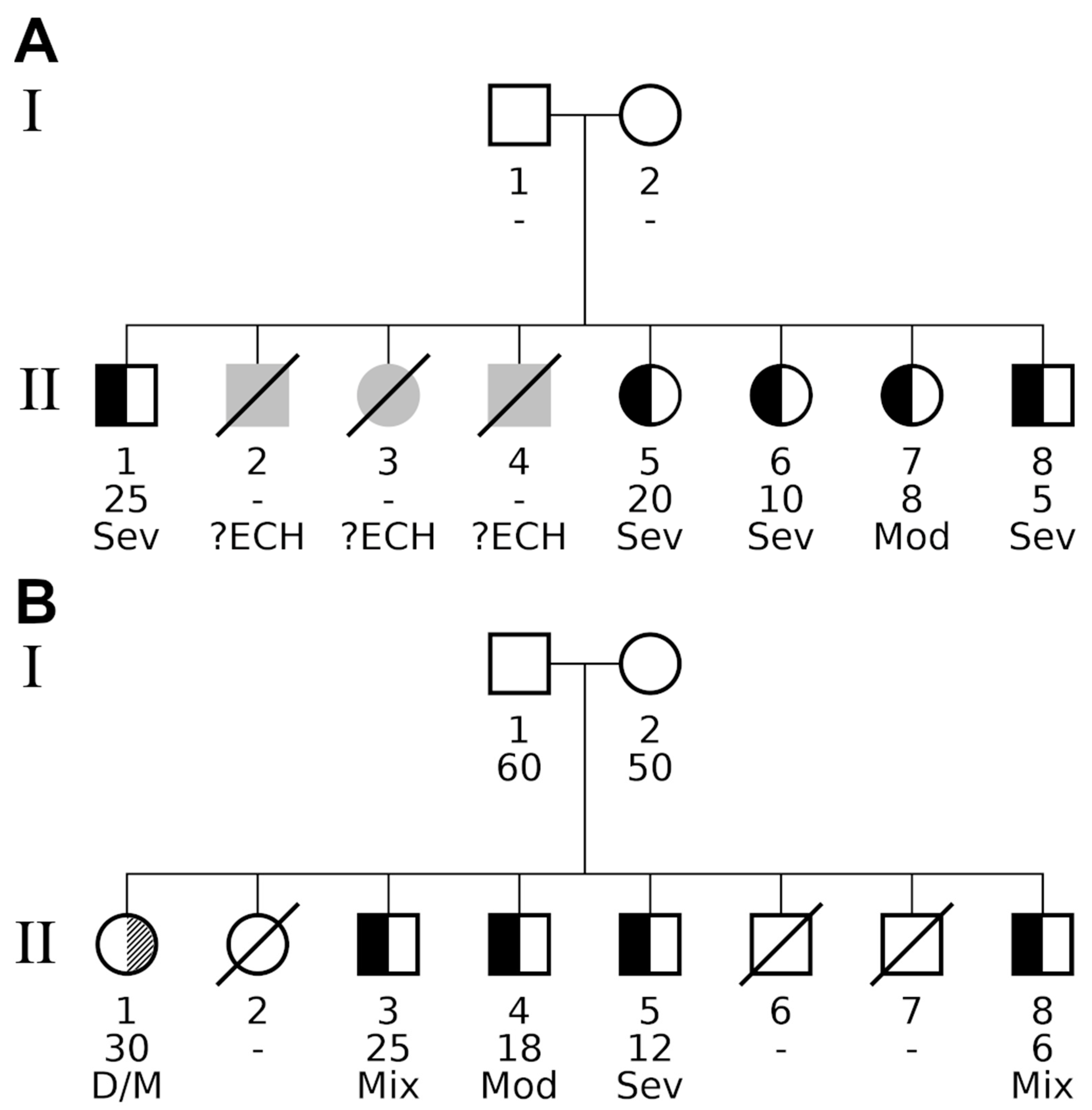
| Family A | Family B | ||||
|---|---|---|---|---|---|
| Phenotype | Test | p | padj | p | padj |
| Combined (neurological and myxedematous) | Binom. Prob. 1 | 1.62 × 10−4 | 5.04 × 10−3 | 1.62 × 10−4 | 5.04 × 10−3 |
| Fam. Genealog. Index | 0.523 | 1 | 0.523 | 1 | |
| Kinship Sum | 1.95 × 10−3 | 8.98 × 10−3 | 1.95 × 10−3 | 8.98 × 10−3 | |
| Population based distribution of endemic CH within all families | |||||
| Genealog. Index 2 | p < 1.00 × 10−6 | ||||
| Prob. Famil. Cluster. 2 | p = 5.05 × 10−21 | ||||
| Myxedematous | Binom. Prob. | 9.31 × 10−5 | 5.77 × 10−3 | 1.66 × 10−3 | 5.14 × 10−2 |
| Fam. Genealog. Index | 0.524 | 1 | 0.714 | 1 | |
| Kinship Sum | 1.23 × 10−3 | 1.01 × 10−2 | 1.14 × 10−2 | 5.20 × 10−2 | |
| Population based distribution of endemic CH within all families | |||||
| Genealog. Index | 5.00 × 10−6 | ||||
| Prob. Famil. Cluster. | 4.32 × 10−7 | ||||
| Observed | Tested Scenarios | |||
|---|---|---|---|---|
| Disease prevalence | 10% 1 | 7% | 5% | 2% |
| Number of observed families with endemic CH aggregation at padj < 0.05 | ||||
| Combined phenotype | 2 (families A, B) | 3 | 3 | 9 |
| Myxedematous phenotype | 1 (family A) | 2 | 2 | 9 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Weichenberger, C.X.; Rivera, M.T.; Vanderpas, J. Familial Aggregation of Endemic Congenital Hypothyroidism Syndrome in Congo (DR): Historical Data. Nutrients 2020, 12, 3021. https://doi.org/10.3390/nu12103021
Weichenberger CX, Rivera MT, Vanderpas J. Familial Aggregation of Endemic Congenital Hypothyroidism Syndrome in Congo (DR): Historical Data. Nutrients. 2020; 12(10):3021. https://doi.org/10.3390/nu12103021
Chicago/Turabian StyleWeichenberger, Christian X., Maria Teresa Rivera, and Jean Vanderpas. 2020. "Familial Aggregation of Endemic Congenital Hypothyroidism Syndrome in Congo (DR): Historical Data" Nutrients 12, no. 10: 3021. https://doi.org/10.3390/nu12103021
APA StyleWeichenberger, C. X., Rivera, M. T., & Vanderpas, J. (2020). Familial Aggregation of Endemic Congenital Hypothyroidism Syndrome in Congo (DR): Historical Data. Nutrients, 12(10), 3021. https://doi.org/10.3390/nu12103021