Role of Resveratrol and Selenium on Oxidative Stress and Expression of Antioxidant and Anti-Aging Genes in Immortalized Lymphocytes from Alzheimer’s Disease Patients


 ,
,  , ,
, , 
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Characterization of Oxidative Stress by DCFH-DA (2′7′-dichloro-dihydro-fluorescein diacetate) Assay
2.3. DCFH-DA Assay to Study Se (IV), Se (VI,) and RV Antioxidant Effects
2.4. Gene Expression Analysis
2.5. mRNA Purification
2.6. cDNA Reverse Transcription
2.7. Real-Time Quantitative PCR
2.8. Statistical Analysis
3. Results
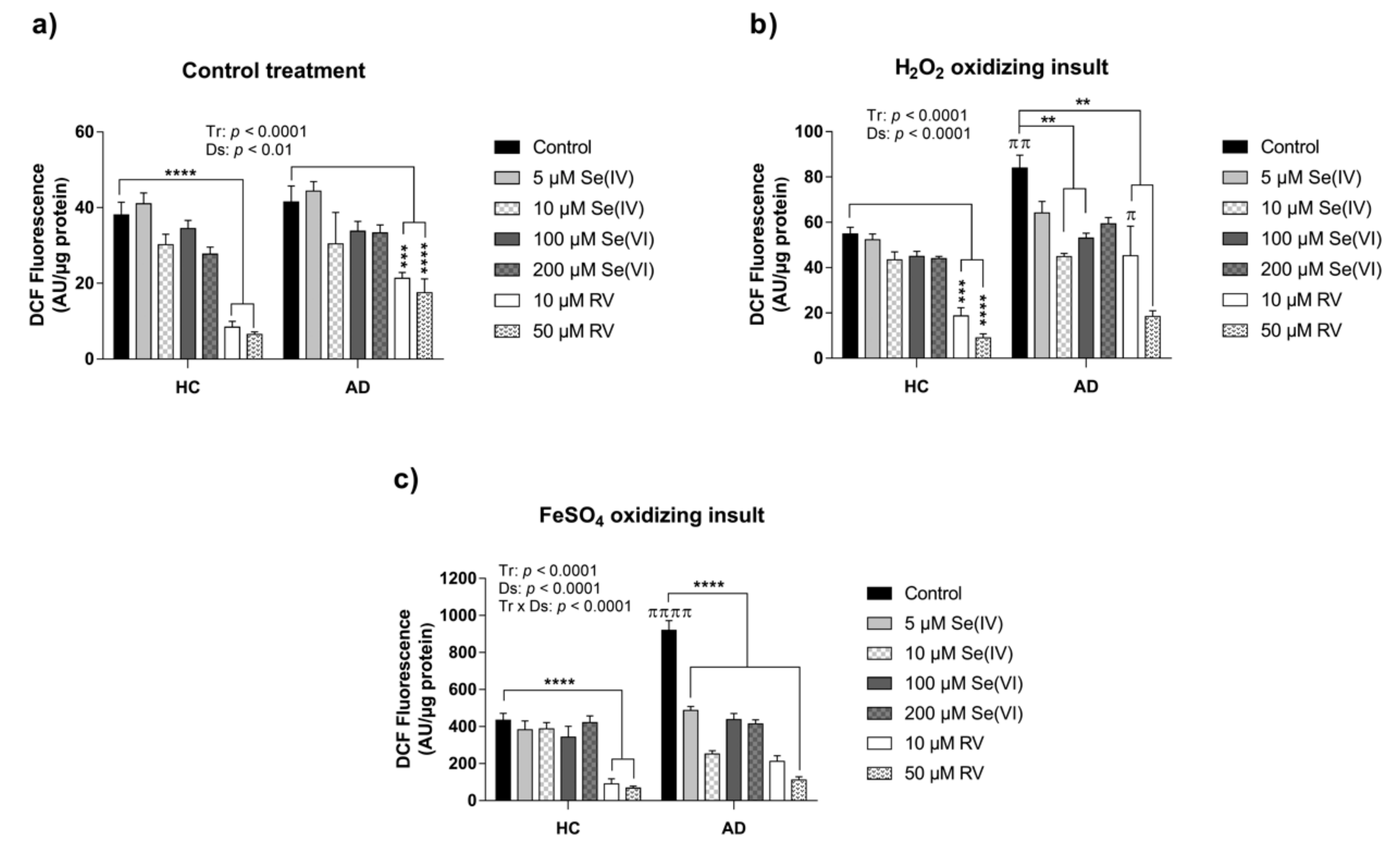
3.1. Characterization of HC and AD LCLs in Response to Acute Exposure of Oxidizing Agents
3.2. Protective Effect of RV and Se against ROS Production in HC and AD LCLs
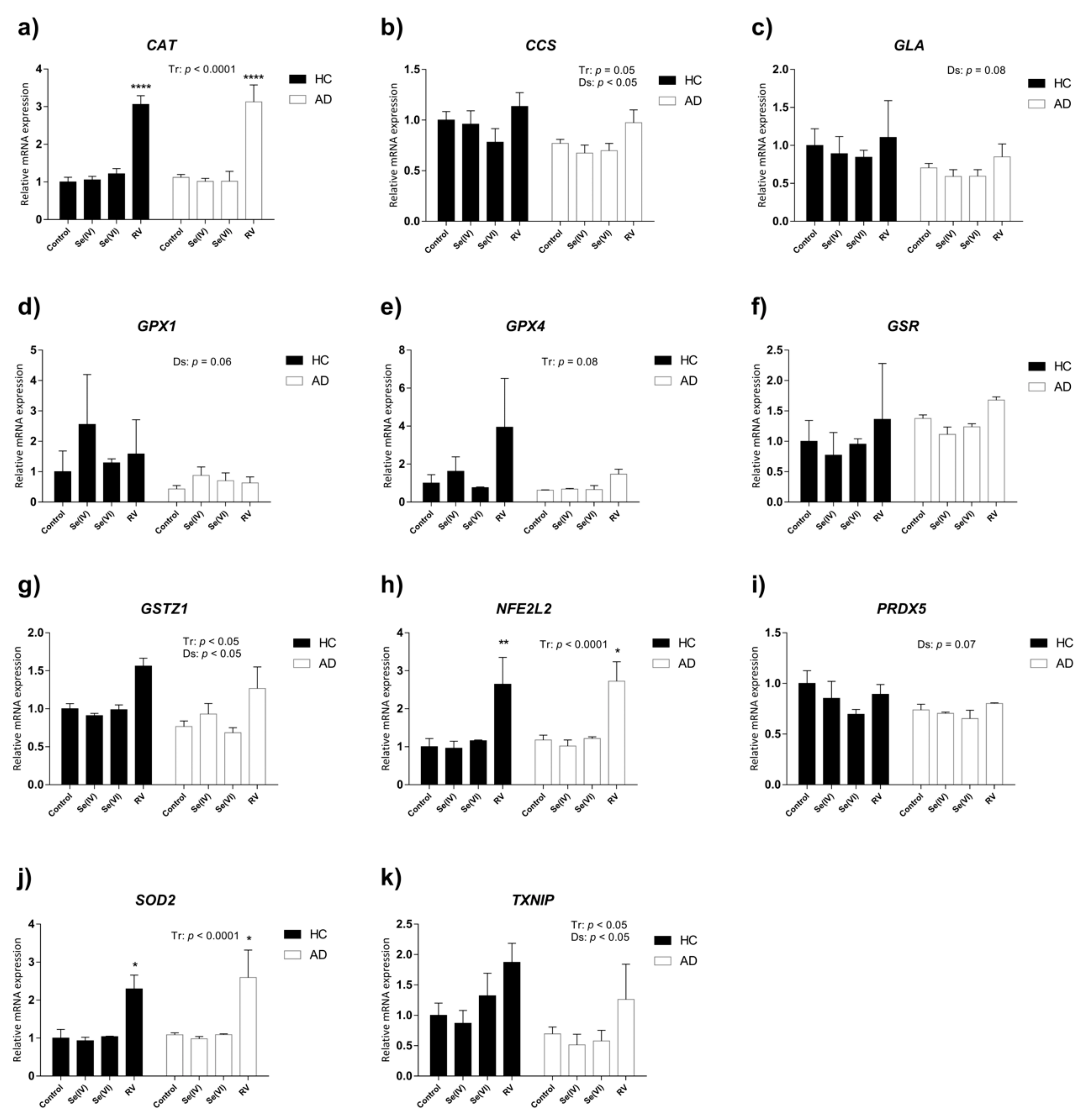
3.3. Transcriptional Changes in Oxidative Stress-Related Genes Induced by Se (IV), Se (VI), and RV in HC and AD LCLs
3.4. Transcriptional Changes in Age-Related Genes Induced by Se (IV), Se (VI), and RV in HC and AD LCLs
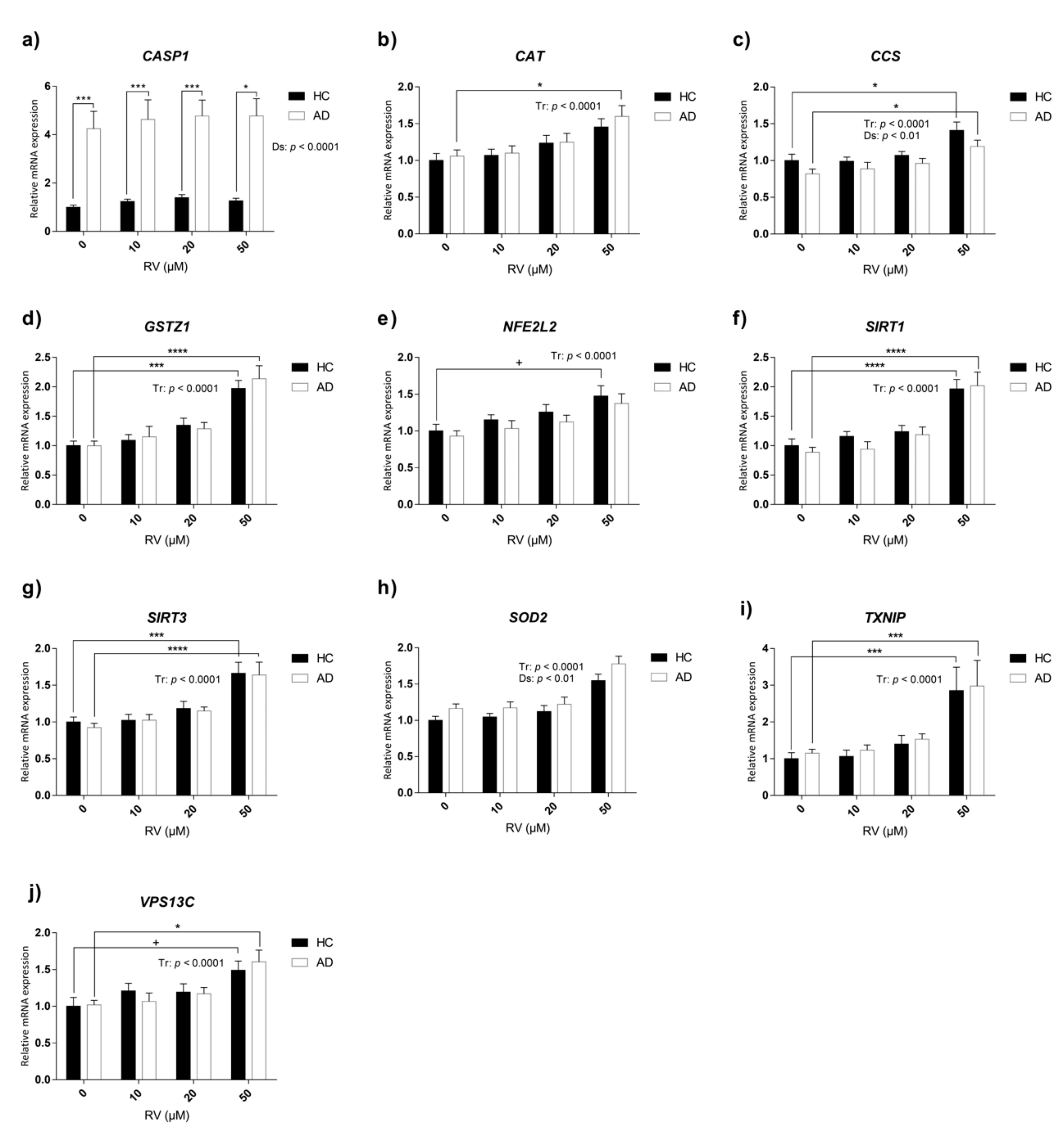
3.5. Characterization of Transcriptional Changes Induced by RV at Different Concentrations on Selected Candidate Genes in HC and AD LCLs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bondi, M.W.; Edmonds, E.C.; Salmon, D.P. Alzheimer’s Disease: Past, Present, and Future. J. Int. Neuropsychol. Soc. JINS 2017, 23, 818–831. [Google Scholar] [CrossRef]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimers Dement. J. Alzheimer’s Assoc. 2016, 12, 292–323. [Google Scholar] [CrossRef]
- Jack, C.R.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA Research Framework: Toward a biological definition of Alzheimer’s disease. Alzheimer’s Dement. J. Alzheimers Assoc. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Wojsiat, J.; Zoltowska, K.M.; Laskowska-Kaszub, K.; Wojda, U. Oxidant/Antioxidant Imbalance in Alzheimer’s Disease: Therapeutic and Diagnostic Prospects. Oxid. Med. Cell. Longev. 2018, 2018, 16. [Google Scholar] [CrossRef]
- Sutherland, G.T.; Chami, B.; Youssef, P.; Witting, P.K. Oxidative stress in Alzheimer’s disease: Primary villain or physiological by-product? Redox Rep. Commun. Free Radic. Res. 2013, 18, 134–141. [Google Scholar] [CrossRef]
- García-Mesa, Y.; Colie, S.; Corpas, R.; Cristòfol, R.; Comellas, F.; Nebreda, A.R.; Giménez-Llort, L.; Sanfeliu, C. Oxidative Stress Is a Central Target for Physical Exercise Neuroprotection Against Pathological Brain Aging. J. Gerontol. A. Biol. Sci. Med. Sci. 2016, 71, 40–49. [Google Scholar] [CrossRef]
- García-Matas, S.; Paul, R.K.; Molina-Martínez, P.; Palacios, H.; Gutierrez, V.M.; Corpas, R.; Pallas, M.; Cristòfol, R.; de Cabo, R.; Sanfeliu, C. In vitro caloric restriction induces protective genes and functional rejuvenation in senescent SAMP8 astrocytes. Aging Cell 2015, 14, 334–344. [Google Scholar] [CrossRef]
- Sebastià, J.; Pertusa, M.; Vílchez, D.; Planas, A.M.; Verbeek, R.; Rodríguez-Farré, E.; Cristòfol, R.; Sanfeliu, C. Carboxyl-terminal fragment of amyloid precursor protein and hydrogen peroxide induce neuronal cell death through different pathways. J. Neural Transm. Vienna Austria 1996 2006, 113, 1837–1845. [Google Scholar] [CrossRef][Green Version]
- Kelsey, N.A.; Wilkins, H.M.; Linseman, D.A. Nutraceutical Antioxidants as Novel Neuroprotective Agents. Molecules 2010, 15, 7792–7814. [Google Scholar] [CrossRef]
- Evans, H.M.; Howe, P.R.C.; Wong, R.H.X. Effects of Resveratrol on Cognitive Performance, Mood and Cerebrovascular Function in Post-Menopausal Women; A 14-Week Randomised Placebo-Controlled Intervention Trial. Nutrients 2017, 9, 27. [Google Scholar] [CrossRef]
- Hubbard, B.P.; Sinclair, D.A. Small molecule SIRT1 activators for the treatment of aging and age-related diseases. Trends Pharmacol. Sci. 2014, 35, 146–154. [Google Scholar] [CrossRef]
- Price, N.L.; Gomes, A.P.; Ling, A.J.Y.; Duarte, F.V.; Martin-Montalvo, A.; North, B.J.; Agarwal, B.; Ye, L.; Ramadori, G.; Teodoro, J.S.; et al. SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab. 2012, 15, 675–690. [Google Scholar] [CrossRef]
- Park, S.-J.; Ahmad, F.; Philp, A.; Baar, K.; Williams, T.; Luo, H.; Ke, H.; Rehmann, H.; Taussig, R.; Brown, A.L.; et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 2012, 148, 421–433. [Google Scholar] [CrossRef]
- Kim, D.; Nguyen, M.D.; Dobbin, M.M.; Fischer, A.; Sananbenesi, F.; Rodgers, J.T.; Delalle, I.; Baur, J.A.; Sui, G.; Armour, S.M.; et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J. 2007, 26, 3169–3179. [Google Scholar] [CrossRef]
- Corpas, R.; Griñán-Ferré, C.; Rodríguez-Farré, E.; Pallàs, M.; Sanfeliu, C. Resveratrol Induces Brain Resilience against Alzheimer Neurodegeneration through Proteostasis Enhancement. Mol. Neurobiol. 2019, 56, 1502–1516. [Google Scholar] [CrossRef]
- Porquet, D.; Griñán-Ferré, C.; Ferrer, I.; Camins, A.; Sanfeliu, C.; Del Valle, J.; Pallàs, M. Neuroprotective role of trans-resveratrol in a murine model of familial Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2014, 42, 1209–1220. [Google Scholar] [CrossRef]
- Porquet, D.; Casadesús, G.; Bayod, S.; Vicente, A.; Canudas, A.M.; Vilaplana, J.; Pelegrí, C.; Sanfeliu, C.; Camins, A.; Pallàs, M.; et al. Dietary resveratrol prevents Alzheimer’s markers and increases life span in SAMP8. Age Dordr. Neth. 2013, 35, 1851–1865. [Google Scholar] [CrossRef]
- Cristòfol, R.; Porquet, D.; Corpas, R.; Coto-Montes, A.; Serret, J.; Camins, A.; Pallàs, M.; Sanfeliu, C. Neurons from senescence-accelerated SAMP8 mice are protected against frailty by the sirtuin 1 promoting agents melatonin and resveratrol. J. Pineal Res. 2012, 52, 271–281. [Google Scholar] [CrossRef]
- Turner, R.S.; Thomas, R.G.; Craft, S.; van Dyck, C.H.; Mintzer, J.; Reynolds, B.A.; Brewer, J.B.; Rissman, R.A.; Raman, R.; Aisen, P.S.; et al. A randomized, double-blind, placebo-controlled trial of resveratrol for Alzheimer disease. Neurology 2015, 85, 1383–1391. [Google Scholar] [CrossRef]
- Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Turner, R.S. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J. Neuroinflamm. 2017, 14, 1. [Google Scholar] [CrossRef]
- Fairweather-Tait, S.J.; Bao, Y.; Broadley, M.R.; Collings, R.; Ford, D.; Hesketh, J.E.; Hurst, R. Selenium in human health and disease. Antioxid. Redox Signal. 2011, 14, 1337–1383. [Google Scholar] [CrossRef]
- Zheng, L.; Zhu, H.-Z.; Wang, B.-T.; Zhao, Q.-H.; Du, X.-B.; Zheng, Y.; Jiang, L.; Ni, J.-Z.; Zhang, Y.; Liu, Q. Sodium selenate regulates the brain ionome in a transgenic mouse model of Alzheimer’s disease. Sci. Rep. 2016, 6, 39290. [Google Scholar] [CrossRef]
- Haratake, M.; Yoshida, S.; Mandai, M.; Fuchigami, T.; Nakayama, M. Elevated amyloid-β plaque deposition in dietary selenium-deficient Tg2576 transgenic mice. Met. Integr. Biomet. Sci. 2013, 5, 479–483. [Google Scholar] [CrossRef]
- Van Eersel, J.; Ke, Y.D.; Liu, X.; Delerue, F.; Kril, J.J.; Götz, J.; Ittner, L.M. Sodium selenate mitigates tau pathology, neurodegeneration, and functional deficits in Alzheimer’s disease models. Proc. Natl. Acad. Sci. USA 2010, 107, 13888–13893. [Google Scholar] [CrossRef]
- Varikasuvu, S.R.; Prasad, V.S.; Kothapalli, J.; Manne, M. Brain Selenium in Alzheimer’s Disease (BRAIN SEAD Study): A Systematic Review and Meta-Analysis. Biol. Trace Elem. Res. 2019, 189, 361–369. [Google Scholar] [CrossRef]
- Cardoso, B.R.; Hare, D.J.; Bush, A.I.; Li, Q.-X.; Fowler, C.J.; Masters, C.L.; Martins, R.N.; Ganio, K.; Lothian, A.; Mukherjee, S.; et al. Selenium Levels in Serum, Red Blood Cells, and Cerebrospinal Fluid of Alzheimer’s Disease Patients: A Report from the Australian Imaging, Biomarker & Lifestyle Flagship Study of Ageing (AIBL). J. Alzheimer’s Dis. JAD 2017, 57, 183–193. [Google Scholar]
- Wang, J.; Gu, B.J.; Masters, C.L.; Wang, Y.J. A systemic view of Alzheimer disease—Insights from amyloid-β metabolism beyond the brain. Nat. Rev. Neurol. 2017, 13, 612–623. [Google Scholar] [CrossRef]
- Wojda, U. Alzheimer’s disease lymphocytes: potential for biomarkers? Biomark. Med. 2016, 10, 1–4. [Google Scholar] [CrossRef]
- Hui-Yuen, J.; McAllister, S.; Koganti, S.; Hill, E.; Bhaduri-McIntosh, S. Establishment of Epstein-Barr virus growth-transformed lymphoblastoid cell lines. J. Vis. Exp. JoVE 2011, 57, e3321. [Google Scholar] [CrossRef]
- De las Cuevas, N.; Urcelay, E.; Hermida, O.G.; Saíz-Diaz, R.A.; Bermejo, F.; Ayuso, M.S.; Martín-Requero, A. Ca2+/calmodulin-dependent modulation of cell cycle elements pRb and p27kip1 involved in the enhanced proliferation of lymphoblasts from patients with Alzheimer dementia. Neurobiol. Dis. 2003, 13, 254–263. [Google Scholar] [CrossRef]
- Del Cerro, P.; Alquézar, C.; Bartolomé, F.; González-Naranjo, P.; Pérez, C.; Carro, E.; Páez, J.A.; Campillo, N.E.; Martín-Requero, Á. Activation of the Cannabinoid Type 2 Receptor by a Novel Indazole Derivative Normalizes the Survival Pattern of Lymphoblasts from Patients with Late-Onset Alzheimer’s Disease. CNS Drugs 2018, 32, 579–591. [Google Scholar] [CrossRef]
- Esteras, N.; Alquézar, C.; Bermejo-Pareja, F.; Bialopiotrowicz, E.; Wojda, U.; Martín-Requero, A. Downregulation of extracellular signal-regulated kinase 1/2 activity by calmodulin KII modulates p21Cip1 levels and survival of immortalized lymphocytes from Alzheimer’s disease patients. Neurobiol. Aging 2013, 34, 1090–1100. [Google Scholar] [CrossRef]
- Lahiri, D.K.; Xu, Y.; Klaunig, J.; Baiyewu, O.; Ogunniyi, A.; Hall, K.; Hendrie, H.; Sahota, A. Effect of oxidative stress on DNA damage and beta-amyloid precursor proteins in lymphoblastoid cell lines from a Nigerian population. Ann. N. Y. Acad. Sci. 1999, 893, 331–336. [Google Scholar] [CrossRef]
- Johnston, J.A.; Lannfelt, L.; Wiehager, B.; O’Neill, C.; Cowburn, R.F. Amyloid precursor protein heat shock response in lymphoblastoid cell lines bearing presenilin-1 mutations. Biochim. Biophys. Acta 1997, 1362, 183–192. [Google Scholar] [CrossRef][Green Version]
- Matsumoto, A.; Fujiwara, Y. Abnormal and deficient processing of beta-amyloid precursor protein in familial Alzheimer’s disease lymphoblastoid cells. Biochem. Biophys. Res. Commun. 1991, 175, 361–365. [Google Scholar] [CrossRef]
- Matsumoto, A.; Fujiwara, Y. Aberrant proteolysis of the beta-amyloid precursor protein in familial Alzheimer’s disease lymphoblastoid cells. Eur. J. Biochem. 1993, 217, 21–27. [Google Scholar] [CrossRef]
- Abe, K.; St George-Hyslop, P.H.; Tanzi, R.E.; Kogure, K. Induction of amyloid precursor protein mRNA after heat shock in cultured human lymphoblastoid cells. Neurosci. Lett. 1991, 125, 169–171. [Google Scholar] [CrossRef]
- Ounanian, A.; Guilbert, B.; Seigneurin, J.M. Characteristics of Epstein-Barr virus transformed B cell lines from patients with Alzheimer’s disease and age-matched controls. Mech. Ageing Dev. 1992, 63, 105–116. [Google Scholar] [CrossRef]
- Bryant, E.M.; Bird, T.D.; Ogburn, C.E.; Traylor, G.H.; Lampe, T.H.; Martin, G.M. Lack of detectable radiation hypersensitivity in lymphoblastoid cells from multiple pedigrees of familial Alzheimer disease. Alzheimer Dis. Assoc. Disord. 1993, 7, 88–97. [Google Scholar] [CrossRef]
- Kalyanaraman, B.; Darley-Usmar, V.; Davies, K.J.A.; Dennery, P.A.; Forman, H.J.; Grisham, M.B.; Mann, G.E.; Moore, K.; Roberts, L.J.; Ischiropoulos, H. Measuring reactive oxygen and nitrogen species with fluorescent probes: challenges and limitations. Free Radic. Biol. Med. 2012, 52, 1–6. [Google Scholar] [CrossRef]
- Spyrou, G.; Björnstedt, M.; Skog, S.; Holmgren, A. Selenite and selenate inhibit human lymphocyte growth via different mechanisms. Cancer Res. 1996, 56, 4407–4412. [Google Scholar]
- Pedram, S.; Mohammadirad, A.; Rezvanfar, M.A.; Navaei-Nigjeh, M.; Baeeri, M.; Abdollahi, M. On The Protection by The Combination of CeO2 Nanoparticles and Sodium Selenite on Human Lymphocytes against Chlorpyrifos-Induced Apoptosis In Vitro. Cell J. 2015, 17, 361–371. [Google Scholar]
- Lee, L.-C.; Weng, Y.-T.; Wu, Y.-R.; Soong, B.-W.; Tseng, Y.-C.; Chen, C.-M.; Lee-Chen, G.-J. Downregulation of proteins involved in the endoplasmic reticulum stress response and Nrf2-ARE signaling in lymphoblastoid cells of spinocerebellar ataxia type 17. J. Neural Transm. Vienna Austria 1996 2014, 121, 601–610. [Google Scholar] [CrossRef]
- Hosein Poor Feyzi, A.A.; Farshdousti Hagh, M.; Ebadi, T.; Shams Asanjan, K.; Movasagpoor Akbari, A.; Talebi, M.; Emadi, B. The effect of resveratrol on the expression of MDR1 gene in leukemic lymphoblast’s of acute lymphoblastic leukemia patients. Casp. J. Intern. Med. 2015, 6, 113–115. [Google Scholar]
- Gambini, J.; Inglés, M.; Olaso, G.; Lopez-Grueso, R.; Bonet-Costa, V.; Gimeno-Mallench, L.; Mas-Bargues, C.; Abdelaziz, K.M.; Gomez-Cabrera, M.C.; Vina, J.; et al. Properties of Resveratrol: In Vitro and In Vivo Studies about Metabolism, Bioavailability, and Biological Effects in Animal Models and Humans. Oxid. Med. Cell. Longev. 2015, 2015. [Google Scholar] [CrossRef]
- Zoidis, E.; Seremelis, I.; Kontopoulos, N.; Danezis, G.P. Selenium-Dependent Antioxidant Enzymes: Actions and Properties of Selenoproteins. Antioxidants 2018, 7, 66. [Google Scholar] [CrossRef]
- Al-Abrash, A.S.; Al-Quobaili, F.A.; Al-Akhras, G.N. Catalase evaluation in different human diseases associated with oxidative stress. Saudi Med. J. 2000, 21, 826–830. [Google Scholar]
- Brown, N.M.; Torres, A.S.; Doan, P.E.; O’Halloran, T.V. Oxygen and the copper chaperone CCS regulate posttranslational activation of Cu,Zn superoxide dismutase. Proc. Natl. Acad. Sci. USA. 2004, 101, 5518–5523. [Google Scholar] [CrossRef]
- Shen, J.-S.; Meng, X.-L.; Moore, D.F.; Quirk, J.M.; Shayman, J.A.; Schiffmann, R.; Kaneski, C.R. Globotriaosylceramide induces oxidative stress and up-regulates cell adhesion molecule expression in Fabry disease endothelial cells. Mol. Genet. Metab. 2008, 95, 163–168. [Google Scholar] [CrossRef]
- Mohammedi, K.; Patente, T.A.; Bellili-Muñoz, N.; Driss, F.; Le Nagard, H.; Fumeron, F.; Roussel, R.; Hadjadj, S.; Corrêa-Giannella, M.L.; Marre, M.; et al. Glutathione peroxidase-1 gene (GPX1) variants, oxidative stress and risk of kidney complications in people with type 1 diabetes. Metabolism 2016, 65, 12–19. [Google Scholar] [CrossRef]
- Seiler, A.; Schneider, M.; Förster, H.; Roth, S.; Wirth, E.K.; Culmsee, C.; Plesnila, N.; Kremmer, E.; Rådmark, O.; Wurst, W.; et al. Glutathione Peroxidase 4 Senses and Translates Oxidative Stress into 12/15-Lipoxygenase Dependent- and AIF-Mediated Cell Death. Cell Metab. 2008, 8, 237–248. [Google Scholar] [CrossRef]
- Cheng, S.B.; Liu, H.T.; Chen, S.Y.; Lin, P.T.; Lai, C.Y.; Huang, Y.C. Changes of Oxidative Stress, Glutathione, and Its Dependent Antioxidant Enzyme Activities in Patients with Hepatocellular Carcinoma before and after Tumor Resection. PLoS ONE 2017, 12, e0170016. [Google Scholar] [CrossRef]
- Blackburn, A.C.; Matthaei, K.I.; Lim, C.; Taylor, M.C.; Cappello, J.Y.; Hayes, J.D.; Anders, M.W.; Board, P.G. Deficiency of glutathione transferase zeta causes oxidative stress and activation of antioxidant response pathways. Mol. Pharmacol. 2006, 69, 650–657. [Google Scholar] [CrossRef]
- Wen, J.J.; Porter, C.; Garg, N.J. Inhibition of NFE2L2-Antioxidant Response Element Pathway by Mitochondrial Reactive Oxygen Species Contributes to Development of Cardiomyopathy and Left Ventricular Dysfunction in Chagas Disease. Antioxid. Redox Signal. 2017, 27, 550–566. [Google Scholar] [CrossRef]
- Yuan, J.; Murrell, G.A.C.; Trickett, A.; Landtmeters, M.; Knoops, B.; Wang, M.-X. Overexpression of antioxidant enzyme peroxiredoxin 5 protects human tendon cells against apoptosis and loss of cellular function during oxidative stress. Biochim. Biophys. Acta 2004, 1693, 37–45. [Google Scholar] [CrossRef]
- Velarde, M.C.; Flynn, J.M.; Day, N.U.; Melov, S.; Campisi, J. Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging 2012, 4, 3–12. [Google Scholar] [CrossRef]
- Oberacker, T.; Bajorat, J.; Ziola, S.; Schroeder, A.; Röth, D.; Kastl, L.; Edgar, B.A.; Wagner, W.; Gülow, K.; Krammer, P.H. Enhanced expression of thioredoxin-interacting-protein regulates oxidative DNA damage and aging. Febs Lett. 2018, 592, 2297–2307. [Google Scholar] [CrossRef]
- Cai, Z.; Zhang, J.; Li, H. Selenium, aging and aging-related diseases. Aging Clin. Exp. Res. 2018, 31, 1035–1047. [Google Scholar] [CrossRef]
- Gemma, C.; Bickford, P.C. Interleukin-1beta and caspase-1: players in the regulation of age-related cognitive dysfunction. Rev. Neurosci. 2007, 18, 137–148. [Google Scholar] [CrossRef]
- Bedford, D.C.; Brindle, P.K. Is histone acetylation the most important physiological function for CBP and p300? Aging 2012, 4, 247–255. [Google Scholar] [CrossRef]
- Martins, R.; Lithgow, G.J.; Link, W. Long live FOXO: Unraveling the role of FOXO proteins in aging and longevity. Aging Cell 2016, 15, 196–207. [Google Scholar] [CrossRef]
- Grabowska, W.; Sikora, E.; Bielak-Zmijewska, A. Sirtuins, a promising target in slowing down the ageing process. Biogerontology 2017, 18, 447–476. [Google Scholar] [CrossRef]
- Berchtold, N.C.; Coleman, P.D.; Cribbs, D.H.; Rogers, J.; Gillen, D.L.; Cotman, C.W. Synaptic genes are extensively downregulated across multiple brain regions in normal human aging and Alzheimer’s disease. Neurobiol. Aging 2013, 34, 1653–1661. [Google Scholar] [CrossRef]
- McCulloch, V.; Shadel, G.S. Human Mitochondrial Transcription Factor B1 Interacts with the C-Terminal Activation Region of h-mtTFA and Stimulates Transcription Independently of Its RNA Methyltransferase Activity. Mol. Cell. Biol. 2003, 23, 5816–5824. [Google Scholar] [CrossRef]
- Savage, S.A.; Giri, N.; Baerlocher, G.M.; Orr, N.; Lansdorp, P.M.; Alter, B.P. TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am. J. Hum. Genet. 2008, 82, 501–509. [Google Scholar] [CrossRef]
- Chen, K.; Yuan, R.; Geng, S.; Zhang, Y.; Ran, T.; Kowalski, E.; Liu, J.; Li, L. Toll-interacting protein deficiency promotes neurodegeneration via impeding autophagy completion in high-fat diet-fed ApoE−/− mouse model. Brain Behav. Immun. 2017, 59, 200–210. [Google Scholar] [CrossRef]
- Kumar, N.; Leonzino, M.; Hancock-Cerutti, W.; Horenkamp, F.A.; Li, P.; Lees, J.A.; Wheeler, H.; Reinisch, K.M.; De Camilli, P. VPS13A and VPS13C are lipid transport proteins differentially localized at ER contact sites. J. Cell Biol. 2018, 217, 3625–3639. [Google Scholar] [CrossRef]
- Wojsiat, J.; Prandelli, C.; Laskowska-Kaszub, K.; Martín-Requero, A.; Wojda, U. Oxidative Stress and Aberrant Cell Cycle in Alzheimer’s Disease Lymphocytes: Diagnostic Prospects. J. Alzheimer’s Dis. JAD 2015, 46, 329–350. [Google Scholar] [CrossRef]
- Sarroca, S.; Molina-Martínez, P.; Aresté, C.; Etzrodt, M.; García de Frutos, P.; Gasa, R.; Antonell, A.; Molinuevo, J.L.; Sánchez-Valle, R.; Saura, C.A.; et al. Preservation of cell-survival mechanisms by the presenilin-1 K239N mutation may cause its milder clinical phenotype. Neurobiol. Aging 2016, 46, 169–179. [Google Scholar] [CrossRef]
- Wang, H.; Jiang, T.; Li, W.; Gao, N.; Zhang, T. Resveratrol attenuates oxidative damage through activating mitophagy in an in vitro model of Alzheimer’s disease. Toxicol. Lett. 2018, 282, 100–108. [Google Scholar] [CrossRef]
- Dong, Y.T.; Cao, K.; Tan, L.C.; Wang, X.L.; Qi, X.L.; Xiao, Y.; Guan, Z.Z. Stimulation of SIRT1 Attenuates the Level of Oxidative Stress in the Brains of APP/PS1 Double Transgenic Mice and in Primary Neurons Exposed to Oligomers of the Amyloid-β Peptide. J. Alzheimer’s Dis. JAD 2018, 63, 283–301. [Google Scholar] [CrossRef]
- Ramírez-Garza, S.L.; Laveriano-Santos, E.P.; Marhuenda-Muñoz, M.; Storniolo, C.E.; Tresserra-Rimbau, A.; Vallverdú-Queralt, A.; Lamuela-Raventós, R.M. Health Effects of Resveratrol: Results from Human Intervention Trials. Nutrients 2018, 10, 1892. [Google Scholar] [CrossRef]
- Cardoso, B.R.; Szymlek-Gay, E.A.; Roberts, B.R.; Formica, M.; Gianoudis, J.; O’Connell, S.; Nowson, C.A.; Daly, R.M. Selenium Status Is Not Associated with Cognitive Performance: A Cross-Sectional Study in 154 Older Australian Adults. Nutrients 2018, 10, 1847. [Google Scholar] [CrossRef]
- Kryscio, R.J.; Abner, E.L.; Caban-Holt, A.; Lovell, M.; Goodman, P.; Darke, A.K.; Yee, M.; Crowley, J.; Schmitt, F.A. Association of Antioxidant Supplement Use and Dementia in the Prevention of Alzheimer’s Disease by Vitamin E and Selenium Trial (PREADViSE). JAMA Neurol. 2017, 74, 567–573. [Google Scholar] [CrossRef]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef]
- Han, J.; Liu, X.; Li, Y.; Zhang, J.; Yu, H. Sirt1/Nrf2 signalling pathway prevents cognitive impairment in diabetic rats through anti-oxidative stress induced by miRNA-23b-3p expression. Mol. Med. Rep. 2018, 17, 8414–8422. [Google Scholar] [CrossRef]
- Huang, K.; Gao, X.; Wei, W. The crosstalk between Sirt1 and Keap1/Nrf2/ARE anti-oxidative pathway forms a positive feedback loop to inhibit FN and TGF-β1 expressions in rat glomerular mesangial cells. Exp. Cell Res. 2017, 361, 63–72. [Google Scholar] [CrossRef]
- Gao, J.; Liu, S.; Xu, F.; Liu, Y.; Lv, C.; Deng, Y.; Shi, J.; Gong, Q. Trilobatin Protects Against Oxidative Injury in Neuronal PC12 Cells Through Regulating Mitochondrial ROS Homeostasis Mediated by AMPK/Nrf2/Sirt3 Signaling Pathway. Front. Mol. Neurosci. 2018, 11, 267. [Google Scholar] [CrossRef]
- Satterstrom, F.K.; Swindell, W.R.; Laurent, G.; Vyas, S.; Bulyk, M.L.; Haigis, M.C. Nuclear respiratory factor 2 induces SIRT3 expression. Aging Cell 2015, 14, 818–825. [Google Scholar] [CrossRef]
- Da Cunha, M.D.S.B.; Arruda, S.F. Tucum-do-Cerrado (Bactris setosa Mart.) May Promote Anti-Aging Effect by Upregulating SIRT1-Nrf2 Pathway and Attenuating Oxidative Stress and Inflammation. Nutrients 2017, 9, 1243. [Google Scholar] [CrossRef]
- Chai, D.; Zhang, L.; Xi, S.; Cheng, Y.; Jiang, H.; Hu, R. Nrf2 Activation Induced by Sirt1 Ameliorates Acute Lung Injury After Intestinal Ischemia/Reperfusion Through NOX4-Mediated Gene Regulation. Cell. Physiol. Biochem. Int. J. Exp. Cell. Physiol. Biochem. Pharmacol. 2018, 46, 781–792. [Google Scholar] [CrossRef]
- Sun, Y.; Yang, T.; Leak, R.K.; Chen, J.; Zhang, F. Preventive and protective roles of dietary Nrf2 activators against central nervous system diseases. CNS Neurol. Disord. Drug Targets 2017, 16, 326–338. [Google Scholar] [CrossRef]
- Kim, E.N.; Lim, J.H.; Kim, M.Y.; Ban, T.H.; Jang, I.-A.; Yoon, H.E.; Park, C.W.; Chang, Y.S.; Choi, B.S. Resveratrol, an Nrf2 activator, ameliorates aging-related progressive renal injury. Aging 2018, 10, 83–99. [Google Scholar] [CrossRef]
- Gomes, B.A.Q.; Silva, J.P.B.; Romeiro, C.F.R.; dos Santos, S.M.; Rodrigues, C.A.; Gonçalves, P.R.; Sakai, J.T.; Mendes, P.F.S.; Varela, E.L.P.; Monteiro, M.C. Neuroprotective Mechanisms of Resveratrol in Alzheimer’s Disease: Role of SIRT1. Available online: https://www.hindawi.com/journals/omcl/2018/8152373/ (accessed on 6 May 2019).
- Cheng, Y.; Takeuchi, H.; Sonobe, Y.; Jin, S.; Wang, Y.; Horiuchi, H.; Parajuli, B.; Kawanokuchi, J.; Mizuno, T.; Suzumura, A. Sirtuin 1 attenuates oxidative stress via upregulation of superoxide dismutase 2 and catalase in astrocytes. J. Neuroimmunol. 2014, 269, 38–43. [Google Scholar] [CrossRef]
- Lin, C.; Zeng, H.; Lu, J.; Xie, Z.; Sun, W.; Luo, C.; Ding, J.; Yuan, S.; Geng, M.; Huang, M. Acetylation at lysine 71 inactivates superoxide dismutase 1 and sensitizes cancer cells to genotoxic agents. Oncotarget 2015, 6, 20578–20591. [Google Scholar] [CrossRef]
- Lee, J.; Kim, Y.; Liu, T.; Hwang, Y.J.; Hyeon, S.J.; Im, H.; Lee, K.; Alvarez, V.E.; McKee, A.C.; Um, S.J.; et al. SIRT3 deregulation is linked to mitochondrial dysfunction in Alzheimer’s disease. Aging Cell 2018, 17, e12679. [Google Scholar] [CrossRef]
- Fukui, M.; Choi, H.J.; Zhu, B.T. Mechanism for the Protective Effect of Resveratrol against Oxidative Stress-Induced Neuronal Death. Free Radic. Biol. Med. 2010, 49, 800–813. [Google Scholar] [CrossRef]
- Liang, Y.; Che, X.; Zhao, Q.; Darwazeh, R.; Zhang, H.; Jiang, D.; Zhao, J.; Xiang, X.; Qin, W.; Liu, L.; et al. Thioredoxin-interacting protein mediates mitochondrion-dependent apoptosis in early brain injury after subarachnoid hemorrhage. Mol. Cell. Biochem. 2019, 450, 149–158. [Google Scholar] [CrossRef]
- Meda, S.A.; Narayanan, B.; Liu, J.; Perrone-Bizzozero, N.I.; Stevens, M.C.; Calhoun, V.D.; Glahn, D.C.; Shen, L.; Risacher, S.L.; Saykin, A.J.; et al. A large scale multivariate parallel ICA method reveals novel imaging-genetic relationships for Alzheimer’s disease in the ADNI cohort. NeuroImage 2012, 60, 1608–1621. [Google Scholar] [CrossRef]
- Shamim, A.; Mahmood, T.; Ahsan, F.; Kumar, A.; Bagga, P. Lipids: An insight into the neurodegenerative disorders. Clin. Nutr. Exp. 2018, 20, 1–19. [Google Scholar] [CrossRef]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 23. [Google Scholar] [CrossRef]
- Flores, J.; Noël, A.; Foveau, B.; Lynham, J.; Lecrux, C.; LeBlanc, A.C. Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat. Commun. 2018, 9, 3916. [Google Scholar] [CrossRef]
- Massaad, C.A. Neuronal and Vascular Oxidative Stress in Alzheimer’s Disease. Curr. Neuropharmacol. 2011, 9, 662–673. [Google Scholar] [CrossRef]
- Angeletti, B.; Waldron, K.J.; Freeman, K.B.; Bawagan, H.; Hussain, I.; Miller, C.C.J.; Lau, K.-F.; Tennant, M.E.; Dennison, C.; Robinson, N.J.; et al. BACE1 cytoplasmic domain interacts with the copper chaperone for superoxide dismutase-1 and binds copper. J. Biol. Chem. 2005, 280, 17930–17937. [Google Scholar] [CrossRef]
- Gray, E.H.; De Vos, K.J.; Dingwall, C.; Perkinton, M.S.; Miller, C.C.J. Deficiency of the Copper Chaperone for Superoxide Dismutase Increases Amyloid-β Production. J. Alzheimer’s Dis. JAD 2010, 21, 1101–1105. [Google Scholar] [CrossRef]
- Greenough, M.A.; Volitakis, I.; Li, Q.-X.; Laughton, K.; Evin, G.; Ho, M.; Dalziel, A.H.; Camakaris, J.; Bush, A.I. Presenilins promote the cellular uptake of copper and zinc and maintain copper chaperone of SOD1-dependent copper/zinc superoxide dismutase activity. J. Biol. Chem. 2011, 286, 9776–9786. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Association with Oxidative Stress | Ref | Gene Symbol | Interaction | Treatment Effect | Disease Effect | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F | (Dfn, DFd) | p-Value | F | (Dfn, DFd) | p-Value | F | (Dfn, DFd) | p-Value | ||||
| Catalase | Catalase is an enzyme that protects aerobic cells from oxidative stress by catalyzing the rapid decomposition of hydrogen peroxide. | [47] | CAT | 0.368 | (3, 12) | 0.7775 | 59.07 | (3, 12) | <0.0001 | 0.00215 | (1, 12) | 0.9638 |
| Copper chaperone for SOD1 | CCS is involved in physiological SOD1 activation (one of the three superoxide dismutases responsible for metabolizing free superoxide radicals in the body), and its primary function is thought to be the delivery of copper to the enzyme. | [48] | CCS | 0.3915 | (3, 11) | 0.7616 | 3.52 | (3, 11) | 0.0524 | 7.828 | (1, 11) | 0.0173 |
| Alpha-galactosidase | GLA is an enzyme that hydrolyses the terminal alpha-galactosyl moieties from glycolipids and glycoproteins. Insufficient activity of GLA leads to accumulation of ROS. | [49] | GLA | 0.0085 | (3, 11) | 0.9988 | 0.6197 | (3, 11) | 0.6167 | 3.679 | (1, 11) | 0.0814 |
| Glutathione peroxidase 1 | Glutathione peroxidase (GPX) is a class of antioxidant enzymes that catalyze the reduction of hydrogen peroxide to water. GPX1 overexpression is associated with enhanced protection against oxidative stress. GPX4 is the only glutathione peroxidase that accepts phospholipid hydroperoxides in membranes as an oxidizing substrate, and under conditions of glutathione deprivation, protein-thiol groups as the reducing substrate. | [50] | GPX1 | 0.3812 | (3, 12) | 0.7684 | 1.062 | (3, 12) | 0.4014 | 4.048 | (1, 12) | 0.0672 |
| Glutathione peroxidase 4 | [51] | GPX4 | 0.99 | (3, 12) | 0.4304 | 2.928 | (3, 12) | 0.0770 | 2.928 | (1, 12) | 0.1128 | |
| Glutathione reductase | GSR is an enzyme involved in the glutathione-dependent antioxidant system by reducing oxidized glutathione. | [52] | GSR | 0.0076 | (3, 11) | 0.9990 | 0.953 | (3, 11) | 0.4487 | 1.935 | (1, 11) | 0.1917 |
| Glutathione S-transferase zeta 1 | GSTZ1 catalyzes glutathione-dependent isomerization of maleylacetoacetate to fumarylacetoacetate, which is the second-to-last step in the vital phenylalanine and tyrosine degradation pathway. Deficiency of this enzyme causes oxidative stress and activation of antioxidant response pathways. | [53] | GSTZ1 | 0.7762 | (3, 11) | 0.5313 | 10.16 | (3, 11) | 0.0017 | 6.139 | (1, 11) | 0.0307 |
| Nuclear factor (erythroid-derived 2)-like 2 | NFE2L2 is a transcription factor involved in the intracellular antioxidant machinery. This enzyme transactivates genes with antioxidant response elements (AREs), and it coordinates the expression of cytoprotective genes to counteract endogenously or exogenously generated oxidative stress | [54] | NFE2L2 | 0.05632 | (3, 12) | 0.9816 | 15.56 | (3, 12) | 0.0002 | 0.2812 | (1, 12) | 0.6056 |
| Peroxiredoxin 5 | PRDX5 is a novel thioredoxin peroxidase which directly promotes the elimination of hydrogen peroxide and neutralization of other reactive oxygen species. | [55] | PRDX5 | 0.5956 | (3, 11) | 0.6309 | 1.719 | (3, 11) | 0.2206 | 4.015 | (1, 11) | 0.0704 |
| Superoxide dismutase 2 | This gene is a member of the iron/manganese superoxide dismutase family. It encodes an antioxidant mitochondrial protein that binds to the superoxide byproducts of oxidative phosphorylation and converts them to hydrogen peroxide and diatomic oxygen. | [56] | SOD2 | 0.09583 | (3, 12) | 0.9609 | 14.93 | (3, 12) | 0.0002 | 0.6395 | (1, 12) | 0.4394 |
| Thioredoxin interacting protein | TXNIP is a negative regulator of TRX, which plays a major role in maintaining the redox status. It is upregulated with aging; its overexpression shortens lifespan due to elevated oxidative DNA damage, whereas its downregulation enhances oxidative stress resistance and extends lifespan. | [57] | TXNIP | 0.3262 | (3, 11) | 0.8065 | 3.863 | (3, 11) | 0.0413 | 7.142 | (1, 11) | 0.0217 |
| Gene Name | Association with Aging | Ref | Gene Symbol | Interaction | Treatment Effect | Disease Effect | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| F | (DFn, DFd) | p-Value | F | (DFn, DFd) | p-Value | F | (DFn, DFd) | p-Value | ||||
| Caspase 1 | CASP1 is an inflammatory/apoptotic caspase involved in age-related cognitive impairment. | [59] | CASP1 | 0.02562 | (3, 11) | 0.9941 | 0.04521 | (3, 11) | 0.9865 | 8.679 | (1, 11) | 0.0133 |
| E1A binding protein p300 | EP300 is a transcriptional coactivator that mediates many transcriptional events including DNA repair. It also acts as a histone acetyltransferase to regulate transcription through chromatin structural changes. EP300 activity is attenuated in ageing mice. | [60] | EP300 | 0.06183 | (3, 11) | 0.9789 | 0.5095 | (3, 11) | 0.6838 | 1.135 | (1, 11) | 0.3096 |
| Forkhead box O1 | FOXO proteins represent a subfamily of transcription factors that act as key regulators of longevity downstream of insulin and insulin-like growth factor signaling. They are involved in stress resistance, metabolism, cell cycle arrest, and apoptosis. | [61] | FOXO1 | 0.05955 | (3, 11) | 0.9800 | 0.1451 | (3, 11) | 0.9307 | 1.252 | (1, 11) | 0.2870 |
| Sirtuin 1 | Sirtuins are nicotinamide adenine dinucleotide (NAD)-dependent protein deacetylases involved in oxidative stress, metabolism, inflammation, and other aging-related cellular processes. Lifestyle factors, including physical activity and diet, can influence healthspan via modifying the level of sirtuins. | [62] | SIRT1 | 0.5235 | (3, 11) | 0.675 | 4.022 | (3, 11) | 0.0371 | 0.7906 | (1, 11) | 0.3930 |
| Sirtuin 3 | SIRT3 | 0.07819 | (3, 11) | 0.9705 | 7.481 | (3, 11) | 0.0053 | 1.058 | (1, 11) | 0.3258 | ||
| Sirtuin 6 | SIRT6 | 0.2715 | (3, 11) | 0.8447 | 0.5264 | (3, 11) | 0.6732 | 0.04267 | (1, 11) | 0.5270 | ||
| Synaptosome associated protein 23 | SNAP23 regulates synaptic vesicle trafficking and fusion, and it is increased with aging and in AD patients. | [63] | SNAP23 | 0.04866 | (3, 11) | 0.985 | 1.97 | (3, 11) | 0.1771 | 0.7959 | (1, 11) | 0.3914 |
| Transcription factor B1, mitochondrial | TFB1M is a dimethyltransferase involved in mitochondrial transcription. It is thought that this protein plays a role on the loss of mitochondrial function encountered in numerous disease states and the aging process. | [64] | TFB1M | 0.7357 | (3, 11) | 0.5523 | 0.3503 | (3, 11) | 0.7898 | 2.256 | (1, 11) | 0.1612 |
| TERF1 interacting nuclear factor 2 | TINF2 is a component of the shelterin complex (telosome) that is involved in the regulation of telomere length and protection. | [65] | TINF2 | 0.1374 | (3, 11) | 0.9356 | 0.31 | (3, 11) | 0.8178 | 1.166 | (1, 11) | 0.3033 |
| Toll interacting protein | TOLLIP is an adaptor molecule within the toll-like receptor (TLR) signaling pathway. It is involved in autophagy and clearance of protein aggregates and it is decreased in AD models. | [66] | TOLLIP | 22.59 | (3, 11) | 0.200 | 46.49 | (3, 11) | 0.0516 | 0.009 | (1, 11) | 0.962 |
| Vacuolar protein sorting 13 homolog C | VPS13A is a lipid transport protein. Its dysfunction in the nervous system is described to shorten life span and trigger age-associated neurodegeneration in animal models. Mutations in the human VPS13 genes are responsible for neurodevelopmental and neurodegenerative disorders. | [67] | VPS13C | 0.2332 | (3, 11) | 0.8713 | 1.327 | (3, 11) | 0.3153 | 9.003 | (1, 11) | 0.0121 |
| Gene Symbol | Interaction | Treatment Effect | Disease Effect | ||||||
|---|---|---|---|---|---|---|---|---|---|
| F | (DFn, DFd) | P Value | F | (DFn, DFd) | p-Value | F | (DFn, DFd) | p-Value | |
| CASP1 | 0.01959 | (3, 84) | 0.9962 | 0.2699 | (3, 84) | 0.8469 | 73.8 | (1, 84) | <0.0001 |
| CAT | 0.1504 | (3, 84) | 0.9292 | 8.508 | (3, 84) | <0.0001 | 0.6411 | (1, 84) | 0.4256 |
| CCS | 0.2515 | (3, 84) | 0.8601 | 10.34 | (3, 84) | <0.0001 | 7.605 | (1, 84) | 0.0071 |
| GSTZ1 | 0.2779 | (3, 84) | 0.8412 | 26.42 | (3, 84) | <0.0001 | 0.1917 | (1, 84) | 0.6626 |
| NFE2L2 | 0.03848 | (3, 84) | 0.9898 | 7.453 | (3, 84) | <0.0001 | 2.202 | (1, 84) | 0.1416 |
| SIRT1 | 0.4004 | (3, 84) | 0.7531 | 27.86 | (3, 84) | <0.0001 | 0.8486 | (1, 84) | 0.3596 |
| SIRT3 | 0.06445 | (3, 84) | 0.9785 | 23.87 | (3, 84) | <0.0001 | 0.2658 | (1, 84) | 0.6075 |
| SOD2 | 0.2123 | (3, 82) | 0.8876 | 20 | (3, 82) | <0.0001 | 0.4858 | (1, 82) | 0.0132 |
| TXNIP | 0.002772 | (3, 84) | 0.9998 | 21.07 | (3, 84) | <0.0001 | 0.5962 | (1, 84) | 0.4422 |
| VPS13C | 0.452 | (3, 84) | 0.7165 | 8.398 | (3, 84) | <0.0001 | 0.01005 | (1, 84) | 0.9204 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cosín-Tomàs, M.; Senserrich, J.; Arumí-Planas, M.; Alquézar, C.; Pallàs, M.; Martín-Requero, Á.; Suñol, C.; Kaliman, P.; Sanfeliu, C. Role of Resveratrol and Selenium on Oxidative Stress and Expression of Antioxidant and Anti-Aging Genes in Immortalized Lymphocytes from Alzheimer’s Disease Patients. Nutrients 2019, 11, 1764. https://doi.org/10.3390/nu11081764
Cosín-Tomàs M, Senserrich J, Arumí-Planas M, Alquézar C, Pallàs M, Martín-Requero Á, Suñol C, Kaliman P, Sanfeliu C. Role of Resveratrol and Selenium on Oxidative Stress and Expression of Antioxidant and Anti-Aging Genes in Immortalized Lymphocytes from Alzheimer’s Disease Patients. Nutrients. 2019; 11(8):1764. https://doi.org/10.3390/nu11081764
Chicago/Turabian StyleCosín-Tomàs, Marta, Júlia Senserrich, Marta Arumí-Planas, Carolina Alquézar, Mercè Pallàs, Ángeles Martín-Requero, Cristina Suñol, Perla Kaliman, and Coral Sanfeliu. 2019. "Role of Resveratrol and Selenium on Oxidative Stress and Expression of Antioxidant and Anti-Aging Genes in Immortalized Lymphocytes from Alzheimer’s Disease Patients" Nutrients 11, no. 8: 1764. https://doi.org/10.3390/nu11081764
APA StyleCosín-Tomàs, M., Senserrich, J., Arumí-Planas, M., Alquézar, C., Pallàs, M., Martín-Requero, Á., Suñol, C., Kaliman, P., & Sanfeliu, C. (2019). Role of Resveratrol and Selenium on Oxidative Stress and Expression of Antioxidant and Anti-Aging Genes in Immortalized Lymphocytes from Alzheimer’s Disease Patients. Nutrients, 11(8), 1764. https://doi.org/10.3390/nu11081764