Dietary Care for ADPKD Patients: Current Status and Future Directions

 and
and 
Abstract
1. Background
2. Autosomal Dominant Polycystic Kidney Disease (ADPKD)
3. Diet in CKD
3.1. Dietary Patterns
3.2. Protein Intake
3.3. Sodium
3.4. Phosphate
3.5. Potassium
3.6. Fruits and Vegetables
3.7. Dietary Fiber
3.8. Caloric Intake
3.9. Fluid Intake
4. Diet in ADPKD: Current Guidelines
4.1. Protein
4.2. Water and Fluid Intake
4.3. Salt
4.4. Osmole Intake
4.5. Caffeine
4.6. Other Dietary Components
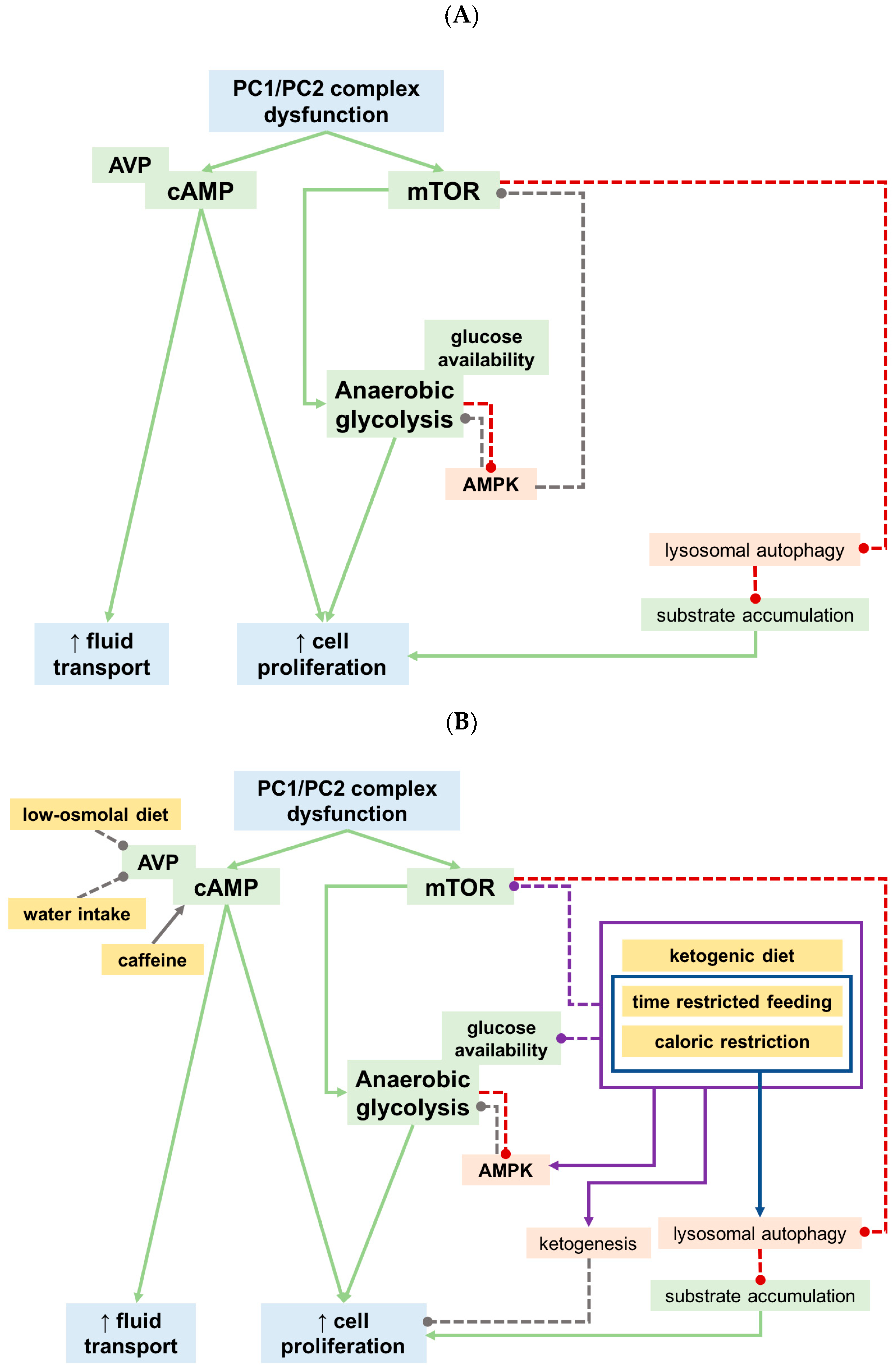
5. What Did We Learn from Recent Preclinical Studies?
5.1. Calorie Restricted and Ketogenic Diets
5.2. Phosphate
5.3. Oxalate
5.4. Other Dietary Approaches
6. Conclusions and Future Directions
Authors Contributions
Funding
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
| Intervention | Spanish 2014 [94] | Japanese 2014 [154] | KDIGO 2015 [86] | KHA-CARI 2015 [85] | Canadian 2018 [95] | The European ADPKD Forum Multidisciplinary Position Statement on ADPKD Care. [155] |
|---|---|---|---|---|---|---|
| Protein | NR | Limited evidence. It may be considered. | 0.8 g/kg/day if eGFR <30 mL/min/1.73 m2 (same as non PKD-CKD), especially if total renal and liver volume is high. | (0.75–1.0 g/kg/day) | NR | Low protein diet, when appropriate (similar to KDIGO). |
| Water intake | High free water intake (2–3 L/d) recommended for CKD G1–3 | 2.5–4 L/d | Increase water intake | Drink fluid to satisfy thirst | Adjusted water intake if tolvaptan. (Goal uOSM <250 mOsm/kg.) | NR |
| Sodium-Salt | <6 g per day. | NR | Sodium-restricted diet. | <100 mmol/day (or 2.3 g sodium or 6 g salt per day). | According to Canadian Hypertension Guidelines: <5 g/d salt or 87 mmol/d sodium. If tolvaptan: ≤2.4 g/d (≤100 mmol/d). | Low salt diet |
| Osmolyte intake | NR | NR | NR | NR | Osmolyte restriction if tolvaptan to achieve a uOSM <250 mOsm/kg. | NR |
| Phosphate | NR | NR | NR | NR | NR | NR |
| Caloric Intake | Maintenance of ideal body weight. | NR | NR | Maintain healthy weight | NR | NR |
| Fruits/vegetables | NR | NR | NR | NR | NR | NR |
| Caffeine | Avoid caffeine | NR | Avoid high caffeine intake | ≤200 mg/d caffeine (≤2 cups of coffee or ≤4 cups of tea per day | NR. (Coffee intake does not seem to affect adversely kidney size or function). | Control caffeine intake. |
| Oxalate | NR | Medical prevention not recommended because of lack of studies on efficacy. | Treat oxalate nephrolithiasis with potassium citrate. | NR | NR | NR |
| Ketogenic diet or fasting | NR | NR | NR | NR | NR | NR |
| Blood pressure target | Similar to other CKD patients. | Similar to other CKD patients. | ≤140/90 mmHg. If heart disease, diabetes, proteinuria: ≤130/80 mmHg. | ≤130/80 mmHg. | Target ≤110/75 mmHg if >60 mL/min/1.73 m2 and no significant cardiovascular comorbidity | 95/60–110/75 mmHg |
| Acid–base | NR | NR | NR | NR | NR | NR |
| Intervention | Chebib et al. [89] | Di Ioro. [91] |
|---|---|---|
| Protein | 0.8–1.0 g/kg of ideal body weight. Intervention: Dietitian counseling, monitoring protein intake: 6.253 (urine urea nitrogen in g/d1(0.033 weight in kg)). | No clear evidence on low-protein diet delaying ADPKD progression. Any recommendations based on non–ADPKD CKD patients. If prescribed, also should be accompanied with adequate energy intake. |
| Water intake | Enhanced hydration over 24 h. Water prescription according to urine osmolality. (24 h urine solute load (mOsm)/280) + 1 insensible loss (~0:5 L). Measure 24 h-urine sodium and first morning urine osmolality, plasma copeptin if available. | No evidence about efficacy No studies on deterioration of residual renal function. Useful in preventing renal calculus. Could be recommended in early stages of the disease, but not if moderate to severe reduction of renal function. |
| Sodium-salt | Guided by counseling and/or dietitian follow-up. Moderate restriction (2.3–3 g/d), adjusted for renal losses if appropriate (hot climate, runners, sauna, bowel disease) | Dietary sodium restriction. |
| Osmolyte intake | Patients adherent to moderate sodium and protein restrictions will have lower osmolar loads. | NR |
| Phosphate | Goal: Moderate intake. 1. Phosphate restriction (800 mg/d), with dietitian counseling 2. Read food labels and watch for foods additives with phosphates. | Follow dietary recommendations that patients with non-ADPKD CKD. |
| Caloric Intake | Goal: Normal BMI (19–24.9 kg/m2). Moderate caloric intake. Intervention: Dietitian follow-up and regular exercise. | Adequate caloric intake if low protein diet. |
| Fruits and vegetables | Increase fruits/vegetables (2–4 cups/day) | If protein restriction, increase fruit and vegetables intake, and limit animal protein. |
| Caffeine | NR | NR |
| Oxalate | NR | NR |
| Ketogenic diet or fasting | NR | NR |
| Hypertension | Blood pressure control: DASH-like diets in early stages | NR |
| Acid-base | Goal: Plasma HCO3 in normal range. >22 mEq/L. Increase fruits/vegetables (2–4 cups/d) | Correction of metabolic acidosis. A vegetarian low-protein diet makes possible a 50% decrease of administered sodium bicarbonate. |
| Lipid control | Goal: LDL ≤ 100 mg/dl. Intervention: 1. Dietician. 2. Regular exercise. | NR |
Appendix B


References
- Spithoven, E.M.; Kramer, A.; Meijer, E.; Orskov, B.; Wanner, C.; Caskey, F.; Collart, F.; Finne, P.; Fogarty, D.G.; Groothoff, J.W.; et al. Analysis of data from the ERA-EDTA Registry indicates that conventional treatments for chronic kidney disease do not reduce the need for renal replacement therapy in autosomal dominant polycystic kidney disease. Kidney Int. 2014, 86, 1244–1252. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Osorio, L.; Vanessa Perez-Gomez, M.; Ortiz, A. Decreasing incidence of renal replacement therapy over time at the critical 50–59-year age range suggests a role for nephroprotective therapy in ADPKD. Kidney Int. 2015, 88, 194. [Google Scholar] [CrossRef] [PubMed]
- Ikizler, T.A.; Cano, N.J.; Franch, H.; Fouque, D.; Himmelfarb, J.; Kalantar-Zadeh, K.; Kuhlmann, M.K.; Stenvinkel, P.; TerWee, P.; Teta, D.; et al. Prevention and treatment of protein energy wasting in chronic kidney disease patients: A consensus statement by the International Society of Renal Nutrition and Metabolism. Kidney Int. 2013, 84, 1096–1107. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.L.; Rangan, G.K.; Lopez-Vargas, P.; Tong, A. KHA-CARI Autosomal Dominant Polycystic Kidney Disease Guideline: Diet and Lifestyle Management. Semin. Nephrol. 2015, 35, 572–581.e517. [Google Scholar] [CrossRef] [PubMed]
- Pippias, M.; Kramer, A.; Noordzij, M.; Afentakis, N.; Alonso de la Torre, R.; Ambühl, P.M.; Aparicio Madre, M.I.; Arribas Monzón, F.; Åsberg, A.; Bonthuis, M.; et al. The European Renal Association—European Dialysis and Transplant Association Registry Annual Report 2014: A summary. Clin. Kidney J. 2017, 10, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Ong, A.C.M.; Devuyst, O.; Knebelmann, B.; Walz, G.; ERA-EDTA Working Group. Autosomal dominant polycystic kidney disease: The changing face of clinical management. Lancet 2015, 385, 1993–2002. [Google Scholar] [CrossRef]
- Solazzo, A.; Testa, F.; Giovanella, S.; Busutti, M.; Furci, L.; Carrera, P.; Ferrari, M.; Ligabue, G.; Mori, G.; Leonelli, M.; et al. The prevalence of autosomal dominant polycystic kidney disease (ADPKD): A meta-analysis of European literature and prevalence evaluation in the Italian province of Modena suggest that ADPKD is a rare and underdiagnosed condition. PLoS ONE 2018, 13, e0190430. [Google Scholar] [CrossRef]
- Lanktree, M.B.; Haghighi, A.; Guiard, E.; Iliuta, I.A.; Song, X.; Harris, P.C.; Paterson, A.D.; Pei, Y. Prevalence Estimates of Polycystic Kidney and Liver Disease by Population Sequencing. J. Am. Soc. Nephrol. 2018, 29, 2593–2600. [Google Scholar] [CrossRef]
- Kidney Disease: Improving Global Outcomes (KDIGO) CKD Work Group. KDIGO 2012 clinical practice guideline for the evaluation and management of chronic kidney disease. Kidney Int. Suppl. 2013, 3, 5–14. [Google Scholar]
- Perez-Gomez, M.V.; Bartsch, L.A.; Castillo-Rodriguez, E.; Fernandez-Prado, R.; Fernandez-Fernandez, B.; Martin-Cleary, C.; Gracia-Iguacel, C.; Ortiz, A. Clarifying the concept of chronic kidney disease for non-nephrologists. Clin. Kidney J. 2019, 12, 258–261. [Google Scholar] [CrossRef]
- Müller, R.U.; Benzing, T. Management of autosomal-dominant polycystic kidney disease-state-of-the-art. Clin. Kidney J. 2018, 11, i2–i13. [Google Scholar] [CrossRef] [PubMed]
- Gansevoort, R.T.; van Gastel, M.D.A.; Chapman, A.B.; Blais, J.D.; Czerwiec, F.S.; Higashihara, E.; Lee, J.; Ouyang, J.; Perrone, R.D.; Stade, K.; et al. Plasma copeptin levels predict disease progression and tolvaptan efficacy in autosomal dominant polycystic kidney disease. Kidney Int. 2019. [Google Scholar] [CrossRef] [PubMed]
- The polycystic kidney disease 1 gene encodes a 14 kb transcript and lies within a duplicated region on chromosome 16. The European Polycystic Kidney Disease Consortium. Cell 1994, 77, 881–894. [CrossRef]
- Mochizuki, T.; Wu, G.; Hayashi, T.; Xenophontos, S.L.; Veldhuisen, B.; Saris, J.J.; Reynolds, D.M.; Cai, Y.; Gabow, P.A.; Pierides, A.; et al. PKD2, a gene for polycystic kidney disease that encodes an integral membrane protein. Science 1996, 272, 1339–1342. [Google Scholar] [CrossRef] [PubMed]
- Porath, B.; Gainullin, V.G.; Cornec-Le Gall, E.; Dillinger, E.K.; Heyer, C.M.; Hopp, K.; Edwards, M.E.; Madsen, C.D.; Mauritz, S.R.; Banks, C.J.; et al. Mutations in GANAB, Encoding the Glucosidase IIα Subunit, Cause Autosomal-Dominant Polycystic Kidney and Liver Disease. Am. J. Hum. Genet. 2016, 98, 1193–1207. [Google Scholar] [CrossRef] [PubMed]
- Cornec-Le Gall, E.; Olson, R.J.; Besse, W.; Heyer, C.M.; Gainullin, V.G.; Smith, J.M.; Audrézet, M.P.; Hopp, K.; Porath, B.; Shi, B.; et al. Monoallelic Mutations to DNAJB11 Cause Atypical Autosomal-Dominant Polycystic Kidney Disease. Am. J. Hum. Genet. 2018, 102, 832–844. [Google Scholar] [CrossRef]
- Torres, V.E.; Harris, P.C. Autosomal dominant polycystic kidney disease: The last 3 years. Kidney Int. 2009, 76, 149–168. [Google Scholar] [CrossRef]
- Yamaguchi, T.; Nagao, S.; Kasahara, M.; Takahashi, H.; Grantham, J.J. Renal accumulation and excretion of cyclic adenosine monophosphate in a murine model of slowly progressive polycystic kidney disease. Am. J. Kidney Dis. 1997, 30, 703–709. [Google Scholar] [CrossRef]
- Belibi, F.A.; Reif, G.; Wallace, D.P.; Yamaguchi, T.; Olsen, L.; Li, H.; Helmkamp, G.M.; Grantham, J.J. Cyclic AMP promotes growth and secretion in human polycystic kidney epithelial cells. Kidney Int. 2004, 66, 964–973. [Google Scholar] [CrossRef]
- Hanaoka, K.; Guggino, W.B. cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells. J. Am. Soc. Nephrol. 2000, 11, 1179–1187. [Google Scholar]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Grantham, J.J.; Higashihara, E.; Perrone, R.D.; Krasa, H.B.; Ouyang, J.; Czerwiec, F.S.; et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2012, 367, 2407–2418. [Google Scholar] [CrossRef] [PubMed]
- Meijer, E.; Visser, F.W.; van Aerts, R.M.M.; Blijdorp, C.J.; Casteleijn, N.F.; D’Agnolo, H.M.A.; Dekker, S.E.I.; Drenth, J.P.H.; de Fijter, J.W.; van Gastel, M.D.A.; et al. Effect of Lanreotide on Kidney Function in Patients With Autosomal Dominant Polycystic Kidney Disease: The DIPAK 1 Randomized Clinical Trial. JAMA 2018, 320, 2010–2019. [Google Scholar] [CrossRef] [PubMed]
- Perico, N.; Ruggenenti, P.; Perna, A.; Caroli, A.; Trillini, M.; Sironi, S.; Pisani, A.; Riccio, E.; Imbriaco, M.; Dugo, M.; et al. Octreotide-LAR in later-stage autosomal dominant polycystic kidney disease (ALADIN 2): A randomized, double-blind, placebo-controlled, multicenter trial. PLoS Med. 2019, 16, e1002777. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.; Happé, H.; Veraar, K.; Scharpfenecker, M.; Peters, D.J.; Consortium, D. The expression of somatostatin receptor 2 decreases during cyst growth in mice with polycystic kidney disease. Exp. Biol. Med. (Maywood) 2018, 243, 1092–1098. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.C.; Maggo, J.K.; Campbell, K.L.; Craig, J.C.; Johnson, D.W.; Sutanto, B.; Ruospo, M.; Tong, A.; Strippoli, G.F. Dietary interventions for adults with chronic kidney disease. Cochrane Database Syst. Rev. 2017, 4, CD011998. [Google Scholar] [CrossRef] [PubMed]
- Collaborators, G.M. Global, regional, and national age-sex-specific mortality and life expectancy, 1950-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1684–1735. [Google Scholar]
- Collaborators, G.R.F. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks for 195 countries and territories, 1990-2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet 2018, 392, 1923–1994. [Google Scholar]
- Estruch, R.; Ros, E.; Salas-Salvadó, J.; Covas, M.I.; Corella, D.; Arós, F.; Gómez-Gracia, E.; Ruiz-Gutiérrez, V.; Fiol, M.; Lapetra, J.; et al. Primary Prevention of Cardiovascular Disease with a Mediterranean Diet Supplemented with Extra-Virgin Olive Oil or Nuts. N. Engl. J. Med. 2018, 378, e34. [Google Scholar] [CrossRef] [PubMed]
- KDOQI. K/DOQI clinical practice guidelines on hypertension and antihypertensive agents in chronic kidney disease. Am. J. Kidney Dis. 2004, 43, S1–S290. [Google Scholar]
- Johnson, D.W.; Atai, E.; Chan, M.; Phoon, R.K.; Scott, C.; Toussaint, N.D.; Turner, G.L.; Usherwood, T.; Wiggins, K.J. KHA-CARI guideline: Early chronic kidney disease: Detection, prevention and management. Nephrol. (Carlton) 2013, 18, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Mitch, W.E.; Remuzzi, G. Diets for patients with chronic kidney disease, should we reconsider? BMC Nephrol. 2016, 17, 80. [Google Scholar] [CrossRef]
- Cupisti, A.; Brunori, G.; Di Iorio, B.R.; D’Alessandro, C.; Pasticci, F.; Cosola, C.; Bellizzi, V.; Bolasco, P.; Capitanini, A.; Fantuzzi, A.L.; et al. Nutritional treatment of advanced CKD: Twenty consensus statements. J. Nephrol. 2018, 31, 457–473. [Google Scholar] [CrossRef] [PubMed]
- Clegg, D.J.; Hill Gallant, K.M. Plant-Based Diets in CKD. Clin. J. Am. Soc. Nephrol. 2019, 14, 141–143. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Caulfield, L.E.; Garcia-Larsen, V.; Steffen, L.M.; Grams, M.E.; Coresh, J.; Rebholz, C.M. Plant-Based Diets and Incident CKD and Kidney Function. Clin. J. Am. Soc. Nephrol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Passey, C. Reducing the Dietary Acid Load: How a More Alkaline Diet Benefits Patients With Chronic Kidney Disease. J. Ren. Nutr. 2017, 27, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, T.; Crews, D.C.; Wesson, D.E.; Tilea, A.M.; Saran, R.; Ríos-Burrows, N.; Williams, D.E.; Powe, N.R.; Team, C.f.D.C.; Surveillance, P.C.K.D. High Dietary Acid Load Predicts ESRD among Adults with CKD. J. Am. Soc. Nephrol. 2015, 26, 1693–1700. [Google Scholar] [CrossRef] [PubMed]
- Khatri, M.; Moon, Y.P.; Scarmeas, N.; Gu, Y.; Gardener, H.; Cheung, K.; Wright, C.B.; Sacco, R.L.; Nickolas, T.L.; Elkind, M.S. The association between a Mediterranean-style diet and kidney function in the Northern Manhattan Study cohort. Clin. J. Am. Soc. Nephrol. 2014, 9, 1868–1875. [Google Scholar] [CrossRef]
- Goraya, N.; Simoni, J.; Jo, C.H.; Wesson, D.E. A comparison of treating metabolic acidosis in CKD stage 4 hypertensive kidney disease with fruits and vegetables or sodium bicarbonate. Clin. J. Am. Soc. Nephrol. 2013, 8, 371–381. [Google Scholar] [CrossRef]
- Brunori, G.; Viola, B.F.; Parrinello, G.; De Biase, V.; Como, G.; Franco, V.; Garibotto, G.; Zubani, R.; Cancarini, G.C. Efficacy and safety of a very-low-protein diet when postponing dialysis in the elderly: A prospective randomized multicenter controlled study. Am. J. Kidney Dis. 2007, 49, 569–580. [Google Scholar] [CrossRef]
- Mancini, M.; Parfitt, V.J.; Rubba, P. Antioxidants in the Mediterranean diet. Can. J. Cardiol. 1995, 11 (Suppl. G), 105G–109G. [Google Scholar]
- Billingsley, H.E.; Carbone, S. The antioxidant potential of the Mediterranean diet in patients at high cardiovascular risk: An in-depth review of the PREDIMED. Nutr. Diabetes 2018, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Foundation, N.K. K/DOQI clinical practice guidelines for chronic kidney disease: Evaluation, classification, and stratification. Am. J. Kidney Dis. 2002, 39, S1–S266. [Google Scholar]
- Menon, V.; Kopple, J.D.; Wang, X.; Beck, G.J.; Collins, A.J.; Kusek, J.W.; Greene, T.; Levey, A.S.; Sarnak, M.J. Effect of a very low-protein diet on outcomes: Long-term follow-up of the Modification of Diet in Renal Disease (MDRD) Study. Am. J. Kidney Dis. 2009, 53, 208–217. [Google Scholar] [CrossRef] [PubMed]
- Klahr, S.; Levey, A.S.; Beck, G.J.; Caggiula, A.W.; Hunsicker, L.; Kusek, J.W.; Striker, G. The effects of dietary protein restriction and blood-pressure control on the progression of chronic renal disease. Modification of Diet in Renal Disease Study Group. N. Engl. J. Med. 1994, 330, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Akbari, A.; Clase, C.M.; Acott, P.; Battistella, M.; Bello, A.; Feltmate, P.; Grill, A.; Karsanji, M.; Komenda, P.; Madore, F.; et al. Canadian Society of Nephrology commentary on the KDIGO clinical practice guideline for CKD evaluation and management. Am. J. Kidney Dis. 2015, 65, 177–205. [Google Scholar] [CrossRef] [PubMed]
- Scottish Intercollegiate Guidelines Network (SIGN). Diagnosis and Managemnt of Chronic Kidney Disease; (SIGN publication no. 103); SIGN: Edinburgh, UK, 2008. [Google Scholar]
- Torres, V.E.; Chapman, A.B.; Devuyst, O.; Gansevoort, R.T.; Perrone, R.D.; Koch, G.; Ouyang, J.; McQuade, R.D.; Blais, J.D.; Czerwiec, F.S.; et al. Tolvaptan in Later-Stage Autosomal Dominant Polycystic Kidney Disease. N. Engl. J. Med. 2017, 377, 1930–1942. [Google Scholar] [CrossRef] [PubMed]
- Messchendorp, A.L.; van Londen, M.; Taylor, J.M.; de Borst, M.H.; Navis, G.; Casteleijn, N.F.; Gaillard, C.A.J.M.; Bakker, S.J.L.; Gansevoort, R.T.; Consortium, D. Kidney Function Reserve Capacity in Early and Later Stage Autosomal Dominant Polycystic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2018, 13, 1680–1692. [Google Scholar] [CrossRef] [PubMed]
- Kopple, J.D.; Monteon, F.J.; Shaib, J.K. Effect of energy intake on nitrogen metabolism in nondialyzed patients with chronic renal failure. Kidney Int. 1986, 29, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Kalantar-Zadeh, K.; Fouque, D. Nutritional Management of Chronic Kidney Disease. N. Engl. J. Med. 2017, 377, 1765–1776. [Google Scholar] [CrossRef] [PubMed]
- Sacks, F.M.; Svetkey, L.P.; Vollmer, W.M.; Appel, L.J.; Bray, G.A.; Harsha, D.; Obarzanek, E.; Conlin, P.R.; Miller, E.R.; Simons-Morton, D.G.; et al. Effects on blood pressure of reduced dietary sodium and the Dietary Approaches to Stop Hypertension (DASH) diet. DASH-Sodium Collaborative Research Group. N. Engl. J. Med. 2001, 344, 3–10. [Google Scholar] [CrossRef] [PubMed]
- McMahon, E.J.; Campbell, K.L.; Bauer, J.D.; Mudge, D.W. Altered dietary salt intake for people with chronic kidney disease. Cochrane Database Syst. Rev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Suckling, R.J.; He, F.J.; Macgregor, G.A. Altered dietary salt intake for preventing and treating diabetic kidney disease. Cochrane Database Syst. Rev. 2010. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, C.; Borrelli, S.; Provenzano, M.; De Stefano, T.; Vita, C.; Chiodini, P.; Minutolo, R.; De Nicola, L.; Conte, G. Dietary Salt Restriction in Chronic Kidney Disease: A Meta-Analysis of Randomized Clinical Trials. Nutrients 2018, 10, 732. [Google Scholar] [CrossRef] [PubMed]
- Imanishi, M.; Yoshioka, K.; Okumura, M.; Konishi, Y.; Okada, N.; Morikawa, T.; Sato, T.; Tanaka, S.; Fujii, S. Sodium sensitivity related to albuminuria appearing before hypertension in type 2 diabetic patients. Diabetes Care 2001, 24, 111–116. [Google Scholar] [CrossRef]
- Gunn, J.P.; Barron, J.L.; Bowman, B.A.; Merritt, R.K.; Cogswell, M.E.; Angell, S.Y.; Bauer, U.E.; Frieden, T.R. Sodium reduction is a public health priority: Reflections on the Institute of Medicine’s report, sodium intake in populations: Assessment of evidence. Am. J. Hypertens. 2013, 26, 1178–1180. [Google Scholar] [CrossRef] [PubMed]
- Parvanova, A.; Trillini, M.; Podestà, M.A.; Iliev, I.P.; Ruggiero, B.; Abbate, M.; Perna, A.; Peraro, F.; Diadei, O.; Rubis, N.; et al. Moderate salt restriction with or without paricalcitol in type 2 diabetes and losartan-resistant macroalbuminuria (PROCEED): A randomised, double-blind, placebo-controlled, crossover trial. Lancet Diabetes Endocrinol. 2018, 6, 27–40. [Google Scholar] [CrossRef]
- Fernandez-Fernandez, B.; Ortiz, A. Paricalcitol and albuminuria: Tread carefully. Lancet Diabetes Endocrinol. 2018, 6, 3–5. [Google Scholar] [CrossRef]
- González-Parra, E.; Gracia-Iguacel, C.; Egido, J.; Ortiz, A. Phosphorus and nutrition in chronic kidney disease. Int. J. Nephrol. 2012, 2012, 597605. [Google Scholar] [CrossRef]
- Palmer, S.C.; Hayen, A.; Macaskill, P.; Pellegrini, F.; Craig, J.C.; Elder, G.J.; Strippoli, G.F. Serum levels of phosphorus, parathyroid hormone, and calcium and risks of death and cardiovascular disease in individuals with chronic kidney disease: A systematic review and meta-analysis. JAMA 2011, 305, 1119–1127. [Google Scholar] [CrossRef]
- Chang, A.R.; Anderson, C. Dietary Phosphorus Intake and the Kidney. Annu. Rev. Nutr. 2017, 37, 321–346. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, S.; Trivedi, B.K.; Kalantar-Zadeh, K.; Kovesdy, C.P. Association of disorders in mineral metabolism with progression of chronic kidney disease. Clin. J. Am. Soc. Nephrol. 2006, 1, 825–831. [Google Scholar] [CrossRef] [PubMed]
- Voormolen, N.; Noordzij, M.; Grootendorst, D.C.; Beetz, I.; Sijpkens, Y.W.; van Manen, J.G.; Boeschoten, E.W.; Huisman, R.M.; Krediet, R.T.; Dekker, F.W.; et al. High plasma phosphate as a risk factor for decline in renal function and mortality in pre-dialysis patients. Nephrol. Dial. Transpl. 2007, 22, 2909–2916. [Google Scholar] [CrossRef]
- O’Seaghdha, C.M.; Hwang, S.J.; Muntner, P.; Melamed, M.L.; Fox, C.S. Serum phosphorus predicts incident chronic kidney disease and end-stage renal disease. Nephrol. Dial. Transpl. 2011, 26, 2885–2890. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Parra, E.; Tuñón, J.; Egido, J.; Ortiz, A. Phosphate: A stealthier killer than previously thought? Cardiovasc. Pathol. 2012, 21, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Fernandez, B.; Izquierdo, M.C.; Valiño-Rivas, L.; Nastou, D.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Albumin downregulates Klotho in tubular cells. Nephrol. Dial. Transpl. 2018, 33, 1712–1722. [Google Scholar] [CrossRef] [PubMed]
- De Seigneux, S.; Courbebaisse, M.; Rutkowski, J.M.; Wilhelm-Bals, A.; Metzger, M.; Khodo, S.N.; Hasler, U.; Chehade, H.; Dizin, E.; Daryadel, A.; et al. Proteinuria Increases Plasma Phosphate by Altering Its Tubular Handling. J. Am. Soc. Nephrol. 2015, 26, 1608–1618. [Google Scholar] [CrossRef]
- Block, G.A.; Hulbert-Shearon, T.E.; Levin, N.W.; Port, F.K. Association of serum phosphorus and calcium x phosphate product with mortality risk in chronic hemodialysis patients: A national study. Am. J. Kidney Dis. 1998, 31, 607–617. [Google Scholar] [CrossRef]
- Ketteler, M.; Block, G.A.; Evenepoel, P.; Fukagawa, M.; Herzog, C.A.; McCann, L.; Moe, S.M.; Shroff, R.; Tonelli, M.A.; Toussaint, N.D.; et al. Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease-Mineral and Bone Disorder: Synopsis of the Kidney Disease: Improving Global Outcomes 2017 Clinical Practice Guideline Update. Ann. Intern. Med. 2018, 168, 422–430. [Google Scholar] [CrossRef]
- Montford, J.R.; Linas, S. How Dangerous Is Hyperkalemia? J. Am. Soc. Nephrol. 2017, 28, 3155–3165. [Google Scholar] [CrossRef]
- Aaron, K.; Sanders, A. Role of Dietary Salt and Potassium Intake in Cardiovascular Health and Disease: A Review of the Evidence. Mayo Clin. Proc. 2013, 88, 987–995. [Google Scholar] [CrossRef] [PubMed]
- Leonberg-Yoo, A.; Tighiouart, H.A.S.L.; Levey, A.S.; Beck, G.J.; Sarnak, M.J. Urine Potassium Excretion, Kidney Failure, and Mortality in CKD. Am. J. Kidney Dis. 2017, 69, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wei, G.; Jalili, T.; Metos, J.; Giri, A.; Cho, M.E.; Boucher, R.; Greene, T.; Beddhu, S. The Associations of Plant Protein Intake With All-Cause Mortality in CKD. Am. J. Kidney Dis. 2016, 67, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, A.; Mann, J.; Cummings, J.; Winter, N.; Mete, E.; Te Morenga, L. Carbohydrate quality and human health: A series of systematic reviews and meta-analyses. Lancet 2019, 393, 434–445. [Google Scholar] [CrossRef]
- Krishnamurthy, V.M.; Wei, G.; Baird, B.C.; Murtaugh, M.; Chonchol, M.B.; Raphael, K.L.; Greene, T.; Beddhu, S. High dietary fiber intake is associated with decreased inflammation and all-cause mortality in patients with chronic kidney disease. Kidney Int. 2012, 81, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Hennig, B.; Toborek, M.; McClain, C.J. High-energy diets, fatty acids and endothelial cell function: Implications for atherosclerosis. J. Am. Coll. Nutr. 2001, 20, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Foods With Added Fiber Lower Serum Creatinine Levels in Patients With Chronic Kidney Disease. J. Ren. Nutr. 2013, 23, e29–e32. [CrossRef] [PubMed]
- Cupisti, A.; D’Alessandro, C.; Gesualdo, L.; Cosola, C.; Gallieni, M.; Egidi, M.F.; Fusaro, M. Non-Traditional Aspects of Renal Diets: Focus on Fiber, Alkali and Vitamin K1 Intake. Nutrients 2017, 9, 444. [Google Scholar] [CrossRef] [PubMed]
- Kramer, H.; Shoham, D.; McClure, L.A.; Durazo-Arvizu, R.; Howard, G.; Judd, S.; Muntner, P.; Safford, M.; Warnock, D.G.; McClellan, W. Association of waist circumference and body mass index with all-cause mortality in CKD: The REGARDS (Reasons for Geographic and Racial Differences in Stroke) Study. Am. J. Kidney Dis. 2011, 58, 177–185. [Google Scholar] [CrossRef] [PubMed]
- Iseki, K.; Ikemiya, Y.; Kinjo, K.; Inoue, T.; Iseki, C.; Takishita, S. Body mass index and the risk of development of end-stage renal disease in a screened cohort. Kidney Int. 2004, 65, 1870–1876. [Google Scholar] [CrossRef]
- Yamagata, K.; Ishida, K.; Sairenchi, T.; Takahashi, H.; Ohba, S.; Shiigai, T.; Narita, M.; Koyama, A. Risk factors for chronic kidney disease in a community-based population: A 10-year follow-up study. Kidney Int. 2007, 71, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Kopple, J.D. National kidney foundation K/DOQI clinical practice guidelines for nutrition in chronic renal failure. Am. J. Kidney Dis. 2001, 37, S66–S70. [Google Scholar] [CrossRef] [PubMed]
- Strippoli, G.F.; Craig, J.C.; Rochtchina, E.; Flood, V.M.; Wang, J.J.; Mitchell, P. Fluid and nutrient intake and risk of chronic kidney disease. Nephrol. (Carlton) 2011, 16, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Clark, W.F.; Sontrop, J.M.; Huang, S.H.; Gallo, K.; Moist, L.; House, A.A.; Cuerden, M.S.; Weir, M.A.; Bagga, A.; Brimble, S.; et al. Effect of Coaching to Increase Water Intake on Kidney Function Decline in Adults With Chronic Kidney Disease: The CKD WIT Randomized Clinical Trial. JAMA 2018, 319, 1870–1879. [Google Scholar] [CrossRef] [PubMed]
- Rangan, G.K.; Alexander, S.I.; Campbell, K.L.; Dexter, M.A.; Lee, V.W.; Lopez-Vargas, P.; Mai, J.; Mallett, A.; Patel, C.; Patel, M.; et al. KHA-CARI guideline recommendations for the diagnosis and management of autosomal dominant polycystic kidney disease. Nephrol. (Carlton) 2016, 21, 705–716. [Google Scholar] [CrossRef] [PubMed]
- Chapman, A.B.; Devuyst, O.; Eckardt, K.U.; Gansevoort, R.T.; Harris, T.; Horie, S.; Kasiske, B.L.; Odland, D.; Pei, Y.; Perrone, R.D.; et al. Autosomal-dominant polycystic kidney disease (ADPKD): Executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 2015, 88, 17–27. [Google Scholar] [CrossRef]
- Sagar, P.S.; Zhang, J.; Luciuk, M.; Mannix, C.; Wong, A.T.Y.; Rangan, G.K. Increased water intake reduces long-term renal and cardiovascular disease progression in experimental polycystic kidney disease. PLoS ONE 2019, 14, e0209186. [Google Scholar] [CrossRef]
- Nagao, S.; Nishii, K.; Katsuyama, M.; Kurahashi, H.; Marunouchi, T.; Takahashi, H.; Wallace, D.P. Increased water intake decreases progression of polycystic kidney disease in the PCK rat. J. Am. Soc. Nephrol. 2006, 17, 2220–2227. [Google Scholar] [CrossRef]
- Chebib, F.T.; Torres, V.E. Recent Advances in the Management of Autosomal Dominant Polycystic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2018, 13, 1765–1776. [Google Scholar] [CrossRef]
- Perrier, E.T.; Buendia-Jimenez, I.; Vecchio, M.; Armstrong, L.E.; Tack, I.; Klein, A. Twenty-four-hour urine osmolality as a physiological index of adequate water intake. Dis. Mark. 2015, 2015, 231063. [Google Scholar] [CrossRef]
- Di Iorio, B.R.; Cupisti, A.; D’Alessandro, C.; Bellasi, A.; Barbera, V.; Di Lullo, L. Nutritional therapy in autosomal dominant polycystic kidney disease. J. Nephrol. 2018, 31, 635–643. [Google Scholar] [CrossRef] [PubMed]
- El-Damanawi, R.; Lee, M.; Harris, T.; Mader, L.B.; Bond, S.; Pavey, H.; Sandford, R.N.; Wilkinson, I.B.; Burrows, A.; Woznowski, P.; et al. Randomised controlled trial of high versus ad libitum water intake in patients with autosomal dominant polycystic kidney disease: Rationale and design of the DRINK feasibility trial. BMJ Open 2018, 8, e022859. [Google Scholar] [PubMed]
- Wong, A.T.Y.; Mannix, C.; Grantham, J.J.; Allman-Farinelli, M.; Badve, S.V.; Boudville, N.; Byth, K.; Chan, J.; Coulshed, S.; Edwards, M.E.; et al. Randomised controlled trial to determine the efficacy and safety of prescribed water intake to prevent kidney failure due to autosomal dominant polycystic kidney disease (PREVENT-ADPKD). BMJ Open 2018, 8, e018794. [Google Scholar] [PubMed]
- Ars, E.; Bernis, C.; Fraga, G.; Martínez, V.; Martins, J.; Ortiz, A.; Rodríguez-Pérez, J.C.; Sans, L.; Torra, R.; Disease, S.W.G.o.I.K. Spanish guidelines for the management of autosomal dominant polycystic kidney disease. Nephrol. Dial. Transpl. 2014, 29 (Suppl. 4), iv95–iv105. [Google Scholar] [CrossRef]
- Soroka, S.; Alam, A.; Bevilacqua, M.; Girard, L.P.; Komenda, P.; Loertscher, R.; McFarlane, P.; Pandeya, S.; Tam, P.; Bichet, D.G. Updated Canadian Expert Consensus on Assessing Risk of Disease Progression and Pharmacological Management of Autosomal Dominant Polycystic Kidney Disease. Can. J. Kidney Health Dis. 2018, 5. [Google Scholar] [CrossRef] [PubMed]
- Nerenberg, K.A.; Zarnke, K.B.; Leung, A.A.; Dasgupta, K.; Butalia, S.; McBrien, K.; Harris, K.C.; Nakhla, M.; Cloutier, L.; Gelfer, M.; et al. Hypertension Canada’s 2018 Guidelines for Diagnosis, Risk Assessment, Prevention, and Treatment of Hypertension in Adults and Children. Can. J. Cardiol. 2018, 34, 506–525. [Google Scholar] [CrossRef] [PubMed]
- Torres, V.E.; Grantham, J.J.; Chapman, A.B.; Mrug, M.; Bae, K.T.; King, B.F.; Wetzel, L.H.; Martin, D.; Lockhart, M.E.; Bennett, W.M.; et al. Potentially modifiable factors affecting the progression of autosomal dominant polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 2011, 6, 640–647. [Google Scholar] [CrossRef]
- Torres, V.E.; Abebe, K.Z.; Schrier, R.W.; Perrone, R.D.; Chapman, A.B.; Yu, A.S.; Braun, W.E.; Steinman, T.I.; Brosnahan, G.; Hogan, M.C.; et al. Dietary salt restriction is beneficial to the management of autosomal dominant polycystic kidney disease. Kidney Int. 2017, 91, 493–500. [Google Scholar] [CrossRef]
- Amro, O.W.; Paulus, J.K.; Noubary, F.; Perrone, R.D. Low-Osmolar Diet and Adjusted Water Intake for Vasopressin Reduction in Autosomal Dominant Polycystic Kidney Disease: A Pilot Randomized Controlled Trial. Am. J. Kidney Dis. 2016, 68, 882–891. [Google Scholar] [CrossRef]
- Belibi, F.A.; Wallace, D.P.; Yamaguchi, T.; Christensen, M.; Reif, G.; Grantham, J.J. The effect of caffeine on renal epithelial cells from patients with autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2002, 13, 2723–2729. [Google Scholar] [CrossRef]
- Meca, R.; Balbo, B.E.; Ormanji, M.S.; Fonseca, J.M.; Iannuzzi, L.R.; Santana Costa, E.; Onuchic, L.F.; Heilberg, I.P. Caffeine Accelerates Cystic Kidney Disease in a Pkd1-Deficient Mouse Model. Cell. Physiol. Biochem. 2019, 52, 1061–1074. [Google Scholar] [PubMed]
- Tanner, G.A.; Tanner, J.A. Chronic caffeine consumption exacerbates hypertension in rats with polycystic kidney disease. Am. J. Kidney Dis. 2001, 38, 1089–1095. [Google Scholar] [CrossRef] [PubMed]
- Vendramini, L.C.; Nishiura, J.L.; Baxmann, A.C.; Heilberg, I.P. Caffeine intake by patients with autosomal dominant polycystic kidney disease. Braz. J. Med. Biol. Res. 2012, 45, 834–840. [Google Scholar] [CrossRef] [PubMed]
- McKenzie, K.A.; El Ters, M.; Torres, V.E.; Harris, P.C.; Chapman, A.B.; Mrug, M.; Rahbari-Oskoui, F.F.; Bae, K.T.; Landsittel, D.P.; Bennett, W.M.; et al. Relationship between caffeine intake and autosomal dominant polycystic kidney disease progression: A retrospective analysis using the CRISP cohort. BMC Nephrol. 2018, 19, 378. [Google Scholar] [CrossRef] [PubMed]
- Girardat-Rotar, L.; Puhan, M.A.; Braun, J.; Serra, A.L. Long-term effect of coffee consumption on autosomal dominant polycystic kidneys disease progression: Results from the Suisse ADPKD, a Prospective Longitudinal Cohort Study. J. Nephrol. 2018, 31, 87–94. [Google Scholar] [CrossRef] [PubMed]
- Nowak, K.L.; You, Z.; Gitomer, B.; Brosnahan, G.; Torres, V.E.; Chapman, A.B.; Perrone, R.D.; Steinman, T.I.; Abebe, K.Z.; Rahbari-Oskoui, F.F.; et al. Overweight and Obesity Are Predictors of Progression in Early Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 571–578. [Google Scholar] [CrossRef] [PubMed]
- Pavik, I.; Jaeger, P.; Kistler, A.D.; Poster, D.; Krauer, F.; Cavelti-Weder, C.; Rentsch, K.M.; Wüthrich, R.P.; Serra, A.L. Patients with autosomal dominant polycystic kidney disease have elevated fibroblast growth factor 23 levels and a renal leak of phosphate. Kidney Int. 2011, 79, 234–240. [Google Scholar] [CrossRef] [PubMed]
- Chonchol, M.; Gitomer, B.; Isakova, T.; Cai, X.; Salusky, I.; Pereira, R.; Abebe, K.; Torres, V.; Steinman, T.I.; Grantham, J.J.; et al. Fibroblast Growth Factor 23 and Kidney Disease Progression in Autosomal Dominant Polycystic Kidney Disease. Clin. J. Am. Soc. Nephrol. 2017, 12, 1461–1469. [Google Scholar] [CrossRef] [PubMed]
- Rowe, I.; Chiaravalli, M.; Mannella, V.; Ulisse, V.; Quilici, G.; Pema, M.; Song, X.W.; Xu, H.; Mari, S.; Qian, F.; et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat. Med. 2013, 19, 488–493. [Google Scholar] [CrossRef]
- Warner, G.; Hein, K.Z.; Nin, V.; Edwards, M.; Chini, C.C.; Hopp, K.; Harris, P.C.; Torres, V.E.; Chini, E.N. Food Restriction Ameliorates the Development of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2016, 27, 1437–1447. [Google Scholar] [CrossRef]
- Chiaravalli, M.; Rowe, I.; Mannella, V.; Quilici, G.; Canu, T.; Bianchi, V.; Gurgone, A.; Antunes, S.; D’Adamo, P.; Esposito, A.; et al. 2-Deoxy-d-Glucose Ameliorates PKD Progression. J. Am. Soc. Nephrol. 2016, 27, 1958–1969. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Wang, M.T.; Tang, Y.; Chen, Y.; Jiang, H.; Jones, T.T.; Rao, K.; Brewer, G.J.; Singh, K.K.; Nie, D. Impairment of mitochondrial respiration in mouse fibroblasts by oncogenic H-RAS(Q61L). Cancer Biol. 2010, 9, 122–133. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, T.; Schuster, S.; Bonhoeffer, S. Cooperation and competition in the evolution of ATP-producing pathways. Science 2001, 292, 504–507. [Google Scholar] [CrossRef] [PubMed]
- Lunt, S.Y.; Vander Heiden, M.G. Aerobic glycolysis: Meeting the metabolic requirements of cell proliferation. Annu. Rev. Cell Dev. Biol. 2011, 27, 441–464. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Liu, M.; Li, L.; Chen, L. Involvement of the Warburg effect in non-tumor diseases processes. J. Cell. Physiol. 2018, 233, 2839–2849. [Google Scholar] [CrossRef]
- Beck Gooz, M.; Maldonado, E.N.; Dang, Y.; Amria, M.Y.; Higashiyama, S.; Abboud, H.E.; Lemasters, J.J.; Bell, P.D. ADAM17 promotes proliferation of collecting duct kidney epithelial cells through ERK activation and increased glycolysis in polycystic kidney disease. Am. J. Physiol. Ren. Physiol. 2014, 307, F551–F559. [Google Scholar] [CrossRef]
- Riwanto, M.; Kapoor, S.; Rodriguez, D.; Edenhofer, I.; Segerer, S.; Wüthrich, R.P. Inhibition of Aerobic Glycolysis Attenuates Disease Progression in Polycystic Kidney Disease. PLoS ONE 2016, 11, e0146654. [Google Scholar] [CrossRef]
- Testa, F.; Busutti, M.; Chiaravalli, M.; Leonelli, M.; Capistrano, M.; Scolari, F.; Spotti, D.; Boletta, A.; Magistroni, R. Design of a Phase I Clinical Trial with 2-Deoxy -D-Glucose in ADPKD. J. Am. Soc. Nephrol. 2016, 27, 769A. [Google Scholar]
- Ruggenenti, P.; Gentile, G.; Perico, N.; Perna, A.; Barcella, L.; Trillini, M.; Cortinovis, M.; Ferrer Siles, C.P.; Reyes Loaeza, J.A.; Aparicio, M.C.; et al. Effect of Sirolimus on Disease Progression in Patients with Autosomal Dominant Polycystic Kidney Disease and CKD Stages 3b-4. Clin. J. Am. Soc. Nephrol. 2016, 11, 785–794. [Google Scholar] [CrossRef]
- Walz, G.; Budde, K.; Mannaa, M.; Nürnberger, J.; Wanner, C.; Sommerer, C.; Kunzendorf, U.; Banas, B.; Hörl, W.H.; Obermüller, N.; et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2010, 363, 830–840. [Google Scholar] [CrossRef] [PubMed]
- Serra, A.L.; Poster, D.; Kistler, A.D.; Krauer, F.; Raina, S.; Young, J.; Rentsch, K.M.; Spanaus, K.S.; Senn, O.; Kristanto, P.; et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N. Engl. J. Med. 2010, 363, 820–829. [Google Scholar] [CrossRef]
- Shillingford, J.M.; Piontek, K.B.; Germino, G.G.; Weimbs, T. Rapamycin ameliorates PKD resulting from conditional inactivation of Pkd1. J. Am. Soc. Nephrol. 2010, 21, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Kipp, K.R.; Rezaei, M.; Lin, L.; Dewey, E.C.; Weimbs, T. A mild reduction of food intake slows disease progression in an orthologous mouse model of polycystic kidney disease. Am. J. Physiol. Ren. Physiol. 2016, 310, F726–F731. [Google Scholar] [CrossRef]
- Torres, J.; Broderick, C.; Kruger, S.; Schimmel, M.; Weimbs, T. A Ketogenic Diet Slows Disease Progression in a Rat Model of Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 297. [Google Scholar]
- Kossoff, E.H.; Zupec-Kania, B.A.; Auvin, S.; Ballaban-Gil, K.R.; Christina Bergqvist, A.G.; Blackford, R.; Buchhalter, J.R.; Caraballo, R.H.; Cross, J.H.; Dahlin, M.G.; et al. Optimal clinical management of children receiving dietary therapies for epilepsy: Updated recommendations of the International Ketogenic Diet Study Group. Epilepsia Open 2018, 3, 175–192. [Google Scholar] [CrossRef]
- DeCampo, D.M.; Kossoff, E.H. Ketogenic dietary therapies for epilepsy and beyond. Curr. Opin. Clin. Nutr. Metab. Care 2019, 22, 264–268. [Google Scholar] [CrossRef]
- LaFountain, R.A.; Miller, V.J.; Barnhart, E.C.; Hyde, P.N.; Crabtree, C.D.; McSwiney, F.T.; Beeler, M.K.; Buga, A.; Sapper, T.N.; Short, J.A.; et al. Extended Ketogenic Diet and Physical Training Intervention in Military Personnel. Mil. Med. 2019. [Google Scholar] [CrossRef]
- Sarafidis, P.; Ferro, C.J.; Morales, E.; Ortiz, A.; Malyszko, J.; Hojs, R.; Khazim, K.; Ekart, R.; Valdivielso, J.; Fouque, D.; et al. SGLT-2 inhibitors and GLP-1 receptor agonists for nephroprotection and cardioprotection in patients with diabetes mellitus and chronic kidney disease. A consensus statement by the EURECA-m and the DIABESITY working groups of the ERA-EDTA. Nephrol. Dial. Transpl. 2019, 34, 208–230. [Google Scholar] [CrossRef]
- Ekinci, I.; Erkoc, R.; Gursu, M.; Dogan, E.E.; Kilic, E.; Cebeci, E.; Ozturk, S.; Kazancioglu, R. Effects of fasting during the month of Ramadan on renal function in patients with autosomal dominant polycystic kidney disease. Clin. Nephrol. 2018, 89, 103–112. [Google Scholar] [CrossRef]
- Gansevoort, R.T.; Meijer, E.; Chapman, A.B.; Czerwiec, F.S.; Devuyst, O.; Grantham, J.J.; Higashihara, E.; Krasa, H.B.; Ouyang, J.; Perrone, R.D.; et al. Albuminuria and tolvaptan in autosomal-dominant polycystic kidney disease: Results of the TEMPO 3:4 Trial. Nephrol. Dial. Transpl. 2016, 31, 1887–1894. [Google Scholar] [CrossRef] [PubMed]
- Thoreen, C.C.; Sabatini, D.M. Rapamycin inhibits mTORC1, but not completely. Autophagy 2009, 5, 725–726. [Google Scholar] [CrossRef] [PubMed]
- Flowers, E.M.; Sudderth, J.; Zacharias, L.; Mernaugh, G.; Zent, R.; DeBerardinis, R.J.; Carroll, T.J. Lkb1 deficiency confers glutamine dependency in polycystic kidney disease. Nat. Commun. 2018, 9, 814. [Google Scholar] [CrossRef] [PubMed]
- Soomro, I.; Sun, Y.; Li, Z.; Diggs, L.; Hatzivassiliou, G.; Thomas, A.G.; Rais, R.; Slusher, B.S.; Somlo, S.; Skolnik, E.Y. Glutamine metabolism via glutaminase 1 in autosomal-dominant polycystic kidney disease. Nephrol. Dial. Transpl. 2018, 33, 1343–1353. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.M. Chunyu.Chapman, Arlene. Preferential Utilization of Histidine as a Glucogenic Amino Acid for PKD Cyst Growth. J. Am. Soc. Nephrol. 2017, 28, 246. [Google Scholar]
- Yamamoto, J.; Nishio, S.; Hattanda, F.; Nakazawa, D.; Kimura, T.; Sata, M.; Makita, M.; Ishikawa, Y.; Atsumi, T. Branched-chain amino acids enhance cyst development in autosomal dominant polycystic kidney disease. Kidney Int. 2017, 92, 377–387. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.; Omede, O.; Daniel, E.; Wallace, D.; Stubbs, J. Dietary Phosphate Restriction Attenuates Renal Cystic Disease in pcy/pcy Mice. J. Am. Soc. Nephrol. 2018, 29, 107. [Google Scholar]
- Rezaei, M.; Torres, J.; Lin, L.; Broderick, C.; Cowley, B.; Torres, V. Calcium oxalate crystals promote cystogenesis and aggravate polycystic kidney disease. Nephrol. Dial. Transplant. 2017, 32 (Suppl. 3), iii94. [Google Scholar] [CrossRef]
- Li, X.; Bu, L.; Zhou, X.; Agborsbesong, E.; Li, X. Protection Effect of Klotho on Cyst Growth in Autosomal Dominant Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2018, 29, 671. [Google Scholar]
- Ohnishi, M.; Razzaque, M.S. Dietary and genetic evidence for phosphate toxicity accelerating mammalian aging. Faseb. J. 2010, 24, 3562–3571. [Google Scholar] [CrossRef]
- Han, H.; Segal, A.M.; Seifter, J.L.; Dwyer, J.T. Nutritional Management of Kidney Stones (Nephrolithiasis). Clin. Nutr. Res. 2015, 4, 137–152. [Google Scholar] [CrossRef] [PubMed]
- Mulay, S.R.; Anders, H.J. Crystal nephropathies: Mechanisms of crystal-induced kidney injury. Nat. Rev. Nephrol. 2017, 13, 226–240. [Google Scholar] [CrossRef] [PubMed]
- Makkapati, S.; D’Agati, V.D.; Balsam, L. “Green Smoothie Cleanse” Causing Acute Oxalate Nephropathy. Am. J. Kidney Dis. 2018, 71, 281–286. [Google Scholar] [CrossRef] [PubMed]
- Getting, J.E.; Gregoire, J.R.; Phul, A.; Kasten, M.J. Oxalate nephropathy due to ‘juicing’: Case report and review. Am. J. Med. 2013, 126, 768–772. [Google Scholar] [CrossRef] [PubMed]
- Syed, F.; Mena-Gutierrez, A.; Ghaffar, U. A case of iced-tea nephropathy. N. Engl. J. Med. 2015, 372, 1377–1378. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Devassy, J.G.; Monirujjaman, M.; Gabbs, M.; Aukema, H.M. Lack of Benefit of Early Intervention with Dietary Flax and Fish Oil and Soy Protein in Orthologous Rodent Models of Human Hereditary Polycystic Kidney Disease. PLoS ONE 2016, 11, e0155790. [Google Scholar] [CrossRef] [PubMed]
- Menezes, L.F.; Lin, C.C.; Zhou, F.; Germino, G.G. Fatty Acid Oxidation is Impaired in An Orthologous Mouse Model of Autosomal Dominant Polycystic Kidney Disease. EBioMedicine 2016, 5, 183–192. [Google Scholar] [CrossRef]
- Simms, M. Informed dissent: The views of some mothers of severely mentally handicapped young adults. J. Med. Ethics 1986, 12, 72–76. [Google Scholar] [CrossRef]
- Taylor, J.M.; Hamilton-Reeves, J.M.; Sullivan, D.K.; Gibson, C.A.; Creed, C.; Carlson, S.E.; Wesson, D.E.; Grantham, J.J. Diet and polycystic kidney disease: A pilot intervention study. Clin. Nutr. 2017, 36, 458–466. [Google Scholar] [CrossRef]
- Ingram, D.K.; Roth, G.S. Calorie restriction mimetics: Can you have your cake and eat it, too? Ageing Res. Rev. 2015, 20, 46–62. [Google Scholar] [CrossRef]
- Boletta, A. Slowing Polycystic Kidney Disease by Fasting. J. Am. Soc. Nephrol. 2016, 27, 1268–1270. [Google Scholar] [CrossRef] [PubMed]
- Magistroni, R.; Boletta, A. Defective glycolysis and the use of 2-deoxy-D-glucose in polycystic kidney disease: From animal models to humans. J. Nephrol. 2017, 30, 511–519. [Google Scholar] [CrossRef] [PubMed]
- Tong, A.; Tunnicliffe, D.J.; Lopez-Vargas, P.; Mallett, A.; Patel, C.; Savige, J.; Campbell, K.; Patel, M.; Tchan, M.C.; Alexander, S.I.; et al. Identifying and integrating consumer perspectives in clinical practice guidelines on autosomal-dominant polycystic kidney disease. Nephrol. (Carlton) 2016, 21, 122–132. [Google Scholar] [CrossRef] [PubMed]
- Horie, S.; Mochizuki, T.; Muto, S.; Hanaoka, K.; Fukushima, Y.; Narita, I.; Nutahara, K.; Tsuchiya, K.; Tsuruya, K.; Kamura, K.; et al. Evidence-based clinical practice guidelines for polycystic kidney disease 2014. Clin. Exp. Nephrol. 2016, 20, 493–509. [Google Scholar] [CrossRef] [PubMed]
- Harris, T.; Sandford, R.; de Coninck, B.; Devuyst, O.; Drenth, J.P.; Ecder, T.; Kent, A.; Gansevoort, R.T.; Górriz, J.L.; Ong, A.C.M.; et al. European ADPKD Forum multidisciplinary position statement on autosomal dominant polycystic kidney disease care: European ADPKD Forum and Multispecialist Roundtable participants. Nephrol. Dial. Transpl. 2018, 33, 563–573. [Google Scholar]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carriazo, S.; Perez-Gomez, M.V.; Cordido, A.; García-González, M.A.; Sanz, A.B.; Ortiz, A.; Sanchez-Niño, M.D. Dietary Care for ADPKD Patients: Current Status and Future Directions. Nutrients 2019, 11, 1576. https://doi.org/10.3390/nu11071576
Carriazo S, Perez-Gomez MV, Cordido A, García-González MA, Sanz AB, Ortiz A, Sanchez-Niño MD. Dietary Care for ADPKD Patients: Current Status and Future Directions. Nutrients. 2019; 11(7):1576. https://doi.org/10.3390/nu11071576
Chicago/Turabian StyleCarriazo, Sol, Maria Vanessa Perez-Gomez, Adrian Cordido, Miguel Angel García-González, Ana Belen Sanz, Alberto Ortiz, and Maria Dolores Sanchez-Niño. 2019. "Dietary Care for ADPKD Patients: Current Status and Future Directions" Nutrients 11, no. 7: 1576. https://doi.org/10.3390/nu11071576
APA StyleCarriazo, S., Perez-Gomez, M. V., Cordido, A., García-González, M. A., Sanz, A. B., Ortiz, A., & Sanchez-Niño, M. D. (2019). Dietary Care for ADPKD Patients: Current Status and Future Directions. Nutrients, 11(7), 1576. https://doi.org/10.3390/nu11071576