Genetic Permissiveness and Dietary Glycemic Load Interact to Predict Type-II Diabetes in the Nile rat (Arvicanthis niloticus)


Abstract
1. Introduction
2. Materials and Methods
2.1. Animal Model (Nile rat)
2.2. Semipurified diets
2.3. Procedures
2.3.1. Experiment 1: Early Diabetes Onset in both Male and Female Rats Fed Diet 73MBS (70:10:20, CHO:Fat:Protein %energy) for 2 Weeks or 4 Weeks
2.3.2. Experiment 2: Diabetes Progression Tracked 90 Male Nile Rats for 10 Weeks into Sexual Maturity while Fed Diet 133 (60:20:20, CHO:Fat:Protein %Energy)
2.3.3. Experiment 3: Diabetes Progression Followed 8 Weeks in 32 Male Nile Rats Fed Diet 73MB/73MBS (70:10:20, CHO:Fat:Protein %Energy) at Increased GLoad
2.3.4. Experiment 4: Compared Diabetes in 30 Female Nile Rats after 3 Weeks while Fed Diet 133
3. Results
3.1. Experiment 1: Diabetes Detected Early between 5–7 Weeks Old with Diet 73MBS (70:10:20)
3.2. Experiment 2. T2DM Allowed to Develop in Males to 13 Weeks of Age
3.3. Experiment 3: Diet 70:10:20 for 8 Weeks in Males
3.4. Experiment 4: Females Fed 60:20:20
4. Discussion
4.1. Early Age of Diabetes Onset
4.1.1. Weanling rats
4.1.2. FBG Less Sensitive Than RBG. OGTT More Sensitive
4.1.3. Similarity to hiCHO in Childhood
4.2. Genetic Permissiveness
4.3. Diet CHO Quality and GLoad
4.4. Cumulative GLoad, Growth Rate and T2DM
4.4.1. Muscle Clock Gene
4.4.2. Bile Acids
5. Conclusions
Author Contributions
Acknowledgments
Funding Support
Conflicts of Interest
References
- Hu, F.B. Globalization of Diabetes: The Role of Diet, Lifestyle, and Genes. Diabetes Care 2011, 34, 1249–1257. [Google Scholar] [CrossRef] [PubMed]
- Herman, W.H.; Zimmet, P. Type 2 Diabetes: An Epidemic Requiring Global Attention and Urgent Action. Diabetes Care 2012, 35, 943–944. [Google Scholar] [CrossRef] [PubMed]
- Golden, S.H.; Brown, A.; Cauley, J.A.; Chin, M.H.; Gary-Webb, T.L.; Kim, C.; Sosa, J.A.; Sumner, A.E.; Anton, B. Health Disparities in Endocrine Disorders: Biological, Clinical, and Nonclinical Factors—An Endocrine Society Scientific Statement. J. Clin. Endocrinol. Metab. 2012, 97, E1579–E1639. [Google Scholar] [CrossRef] [PubMed]
- The Lancet. Beat Diabetes: An Urgent Call for Global Action. Lancet 2016, 387, 1483. [Google Scholar] [CrossRef]
- Krug, E.G. Trends in Diabetes: Sounding the Alarm. Lancet 2016, 387, 1485–1486. [Google Scholar] [CrossRef]
- Ezzati, M. Worldwide Trends in Diabetes since 1980: A Pooled Analysis of 751 Population-Based Studies with 4.4 Million Participants. Lancet 2016, 387, 1513–1530. [Google Scholar] [CrossRef]
- Lilienfeld, A.M. Practical Limitations of Epidemiologic Methods. Environ. Health Perspect. 1983, 52, 3–8. [Google Scholar] [CrossRef]
- Maki, K.C.; Slavin, J.L.; Rains, T.M.; Kris-Etherton, P.M. Limitations of Observational Evidence: Implications for Evidence-Based Dietary Recommendations. Adv. Nutr. 2014, 5, 7–15. [Google Scholar] [CrossRef]
- Satija, A.; Yu, E.; Willett, W.C.; Hu, F.B. Understanding Nutritional Epidemiology and Its Role in Policy. Adv. Nutr. 2015, 6, 5–18. [Google Scholar] [CrossRef]
- Greenwood, D.C.; Threapleton, D.E.; Evans, C.E.L.; Cleghorn, C.L.; Nykjaer, C.; Woodhead, C.; Burley, V.J. Glycemic Index, Glycemic Load, Carbohydrates, and Type 2 Diabetes: Systematic Review and Dose-Response Meta-Analysis of Prospective Studies. Diabetes Care 2013, 36, 4166–4171. [Google Scholar] [CrossRef]
- Livesey, G.; Taylor, R.; Livesey, H.F.; Buyken, A.E.; Jenkins, D.J.A.; Augustin, L.S.A.; Sievenpiper, J.L.; Barclay, A.W.; Liu, S.; Wolever, T.M.S.; et al. Dietary Glycemic Index and Load and the Risk of 2 Type 2 Diabetes: A Systematic Review and Updated Meta-Analyses of Prospective Cohort Studies. Nutrients 2019, 11, 1280. [Google Scholar] [CrossRef] [PubMed]
- Bolsinger, J.; Pronczuk, A.; Hayes, K.C. Dietary Carbohydrate Dictates Development of Type 2 Diabetes in the Nile Rat. J. Nutr. Biochem. 2013, 24, 1945–1952. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, A.; Landstrom, M.; Luu, A.; Hayes, K.C. The Nile Rat (Arvicanthis niloticus) as a Superior Carbohydrate-Sensitive Model for Type 2 Diabetes Mellitus (T2DM). Nutrients 2018, 10, 235. [Google Scholar] [CrossRef] [PubMed]
- Bolsinger, J.; Landstrom, M.; Pronczuk, A.; Auerbach, A.; Hayes, K.C. Low Glycemic Load Diets Protect against Metabolic Syndrome and Type 2 Diabetes Mellitus in the Male Nile Rat. J. Nutr. Biochem. 2017, 42, 134–148. [Google Scholar] [CrossRef] [PubMed]
- Bryant, C.D.; Zhang, N.N.; Sokoloff, G.; Fanselow, M.S.; Ennes, H.S.; Palmer, A.A.; McRoberts, J.A. Behavioral Differences among C57BL/6 Substrains: Implications for Transgenic and Knockout Studies. J. Neurogenet. 2008, 22, 315–331. [Google Scholar] [CrossRef] [PubMed]
- Cummings, B.P.; Digitale, E.K.; Stanhope, K.L.; Graham, J.L.; Baskin, D.G.; Reed, B.J.; Sweet, I.R.; Griffen, S.C.; Havel, P.J. Development and Characterization of a Novel Rat Model of Type 2 Diabetes Mellitus: The UC Davis Type 2 Diabetes Mellitus UCD-T2DM Rat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1782–R1793. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Ridaura, V.K.; Faith, J.J.; Rey, F.E.; Knight, R.; Gordon, J.I. The Effect of Diet on the Human Gut Microbiome: A Metagenomic Analysis in Humanized Gnotobiotic Mice. Sci. Transl. Med. 2009, 1, 6ra14. [Google Scholar] [CrossRef]
- Guo, J.; Jou, W.; Gavrilova, O.; Hall, K.D. Persistent Diet-Induced Obesity in Male C57BL/6 Mice Resulting from Temporary Obesigenic Diets. PLoS ONE 2009, 4, e5370. [Google Scholar] [CrossRef]
- Mekada, K.; Abe, K.; Murakami, A.; Nakamura, S.; Nakata, H.; Moriwaki, K.; Obata, Y.; Yoshiki, A. Genetic Differences among C57BL/6 Substrains. Exp. Anim. 2009, 58, 141–149. [Google Scholar] [CrossRef]
- Cummings, B.P.; Stanhope, K.L.; Graham, J.L.; Evans, J.L.; Baskin, D.G.; Griffen, S.C.; Havel, P.J. Dietary Fructose Accelerates the Development of Diabetes in UCD-T2DM Rats: Amelioration by the Antioxidant, Alpha-Lipoic Acid. AJP Regul. Integr. Comp. Physiol. 2010, 298, R1343–R1350. [Google Scholar] [CrossRef]
- Bryant, C.D. The Blessings and Curses of C57BL/6 Substrains in Mouse Genetic Studies. Ann. N. Y. Acad. Sci. 2011, 1245, 31–33. [Google Scholar] [CrossRef] [PubMed]
- Borghjid, S.; Feinman, R. Response of C57Bl/6 Mice to a Carbohydrate-Free Diet. Nutr. Metab. 2012, 9, 69. [Google Scholar] [CrossRef] [PubMed]
- Feinman, R.D.; Pogozelski, W.K.; Astrup, A.; Bernstein, R.K.; Fine, E.J.; Westman, E.C.; Accurso, A.; Frassetto, L.; Gower, B.A.; McFarlane, S.I.; et al. Dietary Carbohydrate Restriction as the First Approach in Diabetes Management: Critical Review and Evidence Base. Nutrition 2015, 31, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Ussar, S.; Griffin, N.W.; Bezy, O.; Fujisaka, S.; Vienberg, S.; Softic, S.; Deng, L.; Bry, L.; Gordon, J.I.; Kahn, C.R. Interactions between Gut Microbiota, Host Genetics and Diet Modulate the Predisposition to Obesity and Metabolic Syndrome. Cell Metab. 2015, 22, 516–530. [Google Scholar] [CrossRef] [PubMed]
- Surwit, R.S.; Kuhn, C.M.; Cochrane, C.; McCubbin, J.A.; Feinglos, M.N. Diet-Induced Type II Diabetes in C57BL/6J Mice. Diabetes 1988, 37, 1163–1167. [Google Scholar] [CrossRef] [PubMed]
- West, D.B.; Boozer, C.N.; Moody, D.L.; Atkinson, R.L. Dietary Obesity in Nine Inbred Mouse Strains. Am. J. Physiol. 1992, 262, R1025–R1032. [Google Scholar] [CrossRef] [PubMed]
- Surwit, R.S.; Feinglos, M.N.; Rodin, J.; Sutherland, A.; Petro, A.E.; Opara, E.C.; Kuhn, C.M.; Rebuffé-Scrive, M. Differential Effects of Fat and Sucrose on the Development of Obesity and Diabetes in C57BL/6J and A/J Mice. Metabolism 1995, 44, 645–651. [Google Scholar] [CrossRef]
- Schreyer, S.A.; Wilson, D.L.; Leboeuf, R.C. C57BL/6 Mice Fed High Fat Diets as Models for Diabetes-Accelerated Atherosclerosis. Atherosclerosis 1998, 136, 17–24. [Google Scholar] [CrossRef]
- Collins, S.; Martin, T.L.; Surwit, R.S.; Robidoux, J. Genetic Vulnerability to Diet-Induced Obesity in the C57BL/6J Mouse: Physiological and Molecular Characteristics. Physiol. Behav. 2004, 81, 243–248. [Google Scholar] [CrossRef]
- Feinman, R.D.; Volek, J.S. Low Carbohydrate Diets Improve Atherogenic Dyslipidemia Even in the Absence of Weight Loss. Nutr. Metab. 2006, 3, 24. [Google Scholar] [CrossRef]
- Mao, H.Z.; Roussos, E.T.; Péterfy, M. Genetic Analysis of the Diabetes-Prone C57BLKS/J Mouse Strain Reveals Genetic Contribution from Multiple Strains. Biochim. Biophys. Acta 2006, 1762, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Pellizzon, M.A.; Ricci, M.R. The Common Use of Improper Control Diets in Diet-Induced Metabolic Disease Research Confounds Data Interpretation: The Fiber Factor. Nutr. Metab. 2018, 15, 3. [Google Scholar] [CrossRef] [PubMed]
- Rucker, R.; Watkins, B. Inadequate Diet Descriptions: A Conundrum for Animal Model Research. Nutr. Res. 2019, 65, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Hu, F.B. Dietary Pattern Analysis: A New Direction in Nutritional Epidemiology. Curr. Opin. Lipidol. 2002, 13, 3–9. [Google Scholar] [CrossRef]
- Yang, K.; Gotzmann, J.; Kuny, S.; Huang, H.; Sauvé, Y.; Chan, C.B. Five Stages of Progressive β-Cell Dysfunction in the Laboratory Nile Rat Model of Type 2 Diabetes. J. Endocrinol. 2016, 229, 343–356. [Google Scholar] [CrossRef]
- Salmerón, J.; Ascherio, A.; Rimm, E.B.; Colditz, G.A.; Spiegelman, D.; Jenkins, D.J.; Stampfer, M.J.; Wing, A.L.; Willett, W.C. Dietary Fiber, Glycemic Load, and Risk of NIDDM in Men. Diabetes Care 1997, 20, 545–550. [Google Scholar] [CrossRef]
- Salmerón, J. Dietary Fiber, Glycemic Load, and Risk of Non—Insulin-Dependent Diabetes Mellitus in Women. JAMA J. Am. Med. Assoc. 1997, 277, 472–477. [Google Scholar] [CrossRef]
- Willett, W.; Manson, J.; Liu, S. Glycemic Index, Glycemic Load, and Risk of Type 2 Diabetes. Am. J. Clin. Nutr. 2002, 76, 274S–280S. [Google Scholar] [CrossRef]
- Willett, W.C. Dietary Fat Plays a Major Role in Obesity: No. Obes. Rev. 2002, 3, 59–68. [Google Scholar] [CrossRef]
- Schulze, M.B.; Liu, S.; Rimm, E.B.; Manson, J.A.E.; Willett, W.C.; Hu, F.B. Glycemic Index, Glycemic Load, and Dietary Fiber Intake and Incidence of Type 2 Diabetes in Younger and Middle-Aged Women. Am. J. Clin. Nutr. 2004, 80, 348–356. [Google Scholar] [CrossRef]
- Halton, T.L.; Liu, S.; Manson, J.E.; Hu, F.B. Low-Carbohydrate-Diet Score and Risk of Type 2 Diabetes in Women. Am. J. Clin. Nutr. 2008, 87, 339–346. [Google Scholar] [CrossRef] [PubMed]
- De Koning, L.; Fung, T.T.; Liao, X.; Chiuve, S.E.; Rimm, E.B.; Willett, W.C.; Spiegelman, D.; Hu, F.B. Low-Carbohydrate Diet Scores and Risk of Type 2 Diabetes in Men. Am. J. Clin. Nutr. 2011, 93, 844–850. [Google Scholar] [CrossRef] [PubMed]
- Seidelmann, S.B.; Claggett, B.; Cheng, S.; Henglin, M.; Shah, A.; Steffen, L.M.; Folsom, A.R.; Rimm, E.B.; Willett, W.C.; Solomon, S.D. Dietary Carbohydrate Intake and Mortality: A Prospective Cohort Study and Meta-Analysis. Lancet Public Health 2018, 3, e419–e428. [Google Scholar] [CrossRef]
- Forsythe, C.E.; Phinney, S.D.; Fernandez, M.L.; Quann, E.E.; Wood, R.J.; Bibus, D.M.; Kraemer, W.J.; Feinman, R.D.; Volek, J.S. Comparison of Low Fat and Low Carbohydrate Diets on Circulating Fatty Acid Composition and Markers of Inflammation. Lipids 2008, 43, 65–77. [Google Scholar] [CrossRef] [PubMed]
- Leite, J.O.; DeOgburn, R.; Ratliff, J.C.; Su, R.; Volek, J.S.; McGrane, M.M.; Dardik, A.; Fernandez, M.L. Low-Carbohydrate Diet Disrupts the Association between Insulin Resistance and Weight Gain. Metabolism 2009, 58, 1116–1122. [Google Scholar] [CrossRef] [PubMed]
- Hussain, T.A.; Mathew, T.C.; Dashti, A.A.; Asfar, S.; Al-Zaid, N.; Dashti, H.M. Effect of Low-Calorie versus Low-Carbohydrate Ketogenic Diet in Type 2 Diabetes. Nutrition 2012, 28, 1016–1021. [Google Scholar] [CrossRef]
- Hall, K.D.; Chen, K.Y.; Guo, J.; Lam, Y.Y.; Leibel, R.L.; Mayer, L.E.; Reitman, M.L.; Rosenbaum, M.; Smith, S.R.; Walsh, B.T.; et al. Energy Expenditure and Body Composition Changes after an Isocaloric Ketogenic Diet in Overweight and Obese Men. Am. J. Clin. Nutr. 2016, 104, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, D.S.; Willett, W.C.; Volek, J.S.; Neuhouser, M.L. Dietary Fat: From Foe to Friend? Science 2018, 362, 764–770. [Google Scholar] [CrossRef]
- Mozaffarian, D. Effects of Dietary Fats versus Carbohydrates on Coronary Heart Disease: A Review of the Evidence. Curr. Atheroscler. Rep. 2005, 7, 435–445. [Google Scholar] [CrossRef]
- Mozaffarian, D.; Rimm, E.B.; Herrington, D.M. Dietary Fats, Carbohydrate, and Progression of Coronary Atherosclerosis in Postmenopausal Women. Am. J. Clin. Nutr. 2004, 80, 1175–1184. [Google Scholar] [CrossRef]
- Ludwig, D.S.; Hu, F.B.; Tappy, L.; Brand-Miller, J. Dietary Carbohydrates: Role of Quality and Quantity in Chronic Disease. BMJ 2018, 361, k2340. [Google Scholar] [CrossRef] [PubMed]
- Volek, J.S.; Sharman, M.J.; Love, D.M.; Avery, N.G.; Gómez, A.L.; Scheett, T.P.; Kraemer, W.J. Body Composition and Hormonal Responses to a Carbohydrate-Restricted Diet. Metabolism 2002, 51, 864–870. [Google Scholar] [CrossRef] [PubMed]
- Westman, E.C.; Yancy, W.S.; Olsen, M.K.; Dudley, T.; Guyton, J.R. Effect of a Low-Carbohydrate, Ketogenic Diet Program Compared to a Low-Fat Diet on Fasting Lipoprotein Subclasses. Int. J. Cardiol. 2006, 110, 212–216. [Google Scholar] [CrossRef]
- Tendler, D.; Lin, S.; Yancy, W.S.; Mavropoulos, J.; Sylvestre, P.; Rockey, D.C.; Westman, E.C. The Effect of a Low-Carbohydrate, Ketogenic Diet on Nonalcoholic Fatty Liver Disease: A Pilot Study. Dig. Dis. Sci. 2007, 52, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Senzota, R. The Habitat and Food Habits of the Grass Rats (Arvicanthis niloticus) in the Serengeti National Park, Tanzania. Afr. J. Ecol. 1982, 20, 241–252. [Google Scholar] [CrossRef]
- Suliman, S.; Shumake, S.; Jackson, W. Food Preference in the Nile Rat Arvicanthis niloticus. Trop. Pest. Manag. 1984, 30, 151–158. [Google Scholar] [CrossRef]
- Delany, M.J.; Monro, R.H. Growth and Development of Wild and Captive Nile Rats, Arvicanthis niloticus (Rodentia: Muridae). Afr. J. Ecol. 1985, 23, 121–131. [Google Scholar] [CrossRef]
- Rabiu, S.; Rose, R.K. A Quantitative Study of Diet in Three Species of Rodents in Natural and Irrigated Savanna Fields. Acta Theriol. 1997, 42, 55–70. [Google Scholar] [CrossRef]
- Chaabo, F.; Pronczuk, A.; Maslova, E.; Hayes, K. Nutritional Correlates and Dynamics of Diabetes in the Nile Rat (Arvicanthis niloticus): A Novel Model for Diet-Induced Type 2 Diabetes and the Metabolic Syndrome. Nutr. Metab. 2010, 7, 29. [Google Scholar] [CrossRef]
- Noda, K.; Melhorn, M.I.; Zandi, S.; Frimmel, S.; Tayyari, F.; Hisatomi, T.; Almulki, L.; Pronczuk, A.; Hayes, K.C.; Hafezi-Moghadam, A. An Animal Model of Spontaneous Metabolic Syndrome: Nile Grass Rat. FASEB J. 2010, 24, 2443–2453. [Google Scholar] [CrossRef]
- Singh, J.; Yousuf, M.S.; Jones, K.E.; Shelemey, P.T.M.; Joy, T.; Macandili, H.; Kerr, B.J.; Zochodne, D.W.; Sauvé, Y.; Ballanyi, K.; et al. Characterization of the Nile Grass Rat as a Unique Model for Type 2 Diabetic Polyneuropathy. J. Neuropathol. Exp. Neurol. 2018, 77, 469–478. [Google Scholar] [CrossRef] [PubMed]
- Schneider, J.; Kuny, S.; Beker, D.; Sauve, Y. Cardiovascular Sexual Dimorphism in a Diet-Induced Type 2 Diabetes Rodent Model, the Nile Rat (Arvicanthis niloticus). PLoS ONE 2018, 13, e0208987. [Google Scholar] [CrossRef] [PubMed]
- Noda, K.; Nakao, S.; Zandi, S.; Sun, D.; Hayes, K.C.; Hafezi-Moghadam, A. Retinopathy in a Novel Model of Metabolic Syndrome and Type 2 Diabetes: New Insight on the Inflammatory Paradigm. FASEB J. 2014, 28, 2038–2046. [Google Scholar] [CrossRef] [PubMed]
- Han, W.H.; Gotzmann, J.; Kuny, S.; Huang, H.; Chan, C.B.; Lemieux, H.; Sauvé, Y. Modifications in Retinal Mitochondrial Respiration Precede Type 2 Diabetes and Protracted Microvascular Retinopathy. Investig. Ophthalmol. Vis. Sci. 2017, 58, 3826–3839. [Google Scholar] [CrossRef] [PubMed]
- Mizutani, M.; Kern, T.S.; Lorenzi, M. Accelerated Death of Retinal Microvascular Cells in Human and Experimental Diabetic Retinopathy. J. Clin. Investig. 1996, 97, 2883–2890. [Google Scholar] [CrossRef] [PubMed]
- Toh, H.; Smolentsev, A.; Bozadjian, R.V.; Keeley, P.W.; Lockwood, M.D.; Sadjadi, R.; Clegg, D.O.; Blodi, B.A.; Coffey, P.J.; Reese, B.E.; et al. Vascular Changes in Diabetic Retinopathy—A Longitudinal Study in the Nile Rat. Lab. Investig. 2019. [Google Scholar] [CrossRef] [PubMed]
- Lyons, J.; Brown, F.; Remillard, D.E.; Bolsinger, J.; Hayes, K.C. Pathology of the Nile Rat Developing Type 2 Diabetes. FASEB J. 2013, 27, 874-13. [Google Scholar]
- Leow, S.-S.; Bolsinger, J.; Pronczuk, A.; Hayes, K.C.; Sambanthamurthi, R. Hepatic Transcriptome Implications for Palm Fruit Juice Deterrence of Type 2 Diabetes Mellitus in Young Male Nile Rats. Genes Nutr. 2016, 11, 29. [Google Scholar] [CrossRef]
- Kang, Y.; Park, S.; Kim, S.; Koh, H. Estimated Prevalence of Adolescents with Nonalcoholic Fatty Liver Disease in Korea. J. Korean Med. Sci. 2018, 33, e109. [Google Scholar] [CrossRef]
- D’Souza, S.S.; Sri Charan Bindu, B.; Mohammed Ali, M.; Tisha, A.; Deepthi, K.V.; Siona, S.; Santy, F.; Abraham, A. Nutritional Profile of High Fat Simple Carbohydrate Diet Used to Induce Metabolic Syndrome in C57BL/6J Mice. J. Nutr. Intermed. Metab. 2016, 3, 41–49. [Google Scholar] [CrossRef]
- Ble-Castillo, J.L.; Aparicio-Trapala, M.A.; Juárez-Rojop, I.E.; Torres-Lopez, J.E.; Mendez, J.D.; Aguilar-Mariscal, H.; Olvera-Hernández, V.; Palma-Cordova, L.C.; Diaz-Zagoya, J.C. Differential Effects of High-Carbohydrate and High-Fat Diet Composition on Metabolic Control and Insulin Resistance in Normal Rats. Int. J. Environ. Res. Public Health 2012, 9, 1663–1676. [Google Scholar] [CrossRef] [PubMed]
- Leibowitz, G.; Yuli, M.; Donath, M.Y.; Nesher, R.; Melloul, D.; Cerasi, E.; Gross, D.J.; Kaiser, N. Beta-Cell Glucotoxicity in the Psammomys obesus Model of Type 2 Diabetes. Diabetes 2001, 50, S113–S117. [Google Scholar] [CrossRef] [PubMed]
- Nesher, R.; Gross, D.J.; Donath, M.Y.; Cerasi, E.; Kaiser, N. Interaction between Genetic and Dietary Factors Determines Beta-Cell Function in Psammomys obesus, an Animal Model of Type 2 Diabetes. Diabetes 1999, 48, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, N.; Nesher, R.; Donath, M.Y.; Fraenkel, M.; Behar, V.; Magnan, C.; Ktorza, A.; Cerasi, E.; Leibowitz, G. Psammomys obesus, a Model for Environment-Gene Interactions in Type 2 Diabetes. Diabetes 2005, 54, S137–S144. [Google Scholar] [CrossRef] [PubMed]
- Ziv, E.; Shafrir, E.; Kalman, R.; Galer, S.; Bar-On, H. Changing Pattern of Prevalence of Insulin Resistance in Psammomys obesus, a Model of Nutritionally Induced Type 2 Diabetes. Metabolism 1999, 48, 1549–1554. [Google Scholar] [CrossRef]
- Hillel, J.; Gefel, D.; Kalman, R.; Ben-Ari, G.; David, L.; Orion, O.; Feldman, M.W.; Bar-On, H.; Blum, S.; Raz, I.; et al. Evidence for a Major Gene Affecting the Transition from Normoglycaemia to Hyperglycaemia in Psammomys obesus. Heredity 2005, 95, 158–165. [Google Scholar] [CrossRef]
- Kalman, R.; Adler, J.H.; Lazarovici, G.; Bar-On, H.; Ziv, E. The Efficiency of Sand Rat Metabolism Is Responsible for Development of Obesity and Diabetes. J. Basic Clin. Physiol. Pharmacol. 1993, 4, 57–68. [Google Scholar] [CrossRef]
- Carter, S.; Clifton, P.M.; Keogh, J.B. Effect of Intermittent Compared With Continuous Energy Restricted Diet on Glycemic Control in Patients With Type 2 Diabetes: A Randomized Noninferiority Trial. JAMA Netw. Open 2018, 1, E180756. [Google Scholar] [CrossRef]
- Schwimmer, J.B.; Ugalde-Nicalo, P.; Welsh, J.A.; Angeles, J.E.; Cordero, M.; Harlow, K.E.; Alarazaki, A.; Durelle, J.; Knight-Scott, J.; Newton, K.P.; et al. Effect of a Low Free Sugar Diet vs Usual Diet on Nonalcoholic Fatty Liver Disease in Adolescent Boys: A Randomized Clinical Trial. JAMA J. Am. Med. Assoc. 2019, 321, 256–265. [Google Scholar] [CrossRef]
- Parry, S.A.; Hodson, L. Influence of Dietary Macronutrients on Liver Fat Accumulation and Metabolism. J. Investig. Med. 2017, 65, 1102–1115. [Google Scholar] [CrossRef]
- Ma, J.; Zhou, Q.; Li, H. Gut Microbiota and Nonalcoholic Fatty Liver Disease: Insights on Mechanisms and Therapy. Nutrients 2017, 9, 1124. [Google Scholar] [CrossRef] [PubMed]
- Softic, S.; Cohen, D.E.; Kahn, C.R. Role of Dietary Fructose and Hepatic De Novo Lipogenesis in Fatty Liver Disease. Dig. Dis. Sci. 2016, 61, 1282–1293. [Google Scholar] [CrossRef] [PubMed]
- Mai, B.; Yan, L. The Negative and Detrimental Effects of High Fructose on the Liver, with Special Reference to Metabolic Disorders. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 821–826. [Google Scholar] [CrossRef] [PubMed]
- Kopp, W. Diet-Induced Hyperinsulinemia as a Key Factor in the Etiology of Both Benign Prostatic Hyperplasia and Essential Hypertension? Nutr. Metab. Insights 2018, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Harvey, L.; Martin, R.; van der Beek, E.M.; Knol, J.; Cryan, J.F.; Renes, I.B. Targeting the Gut Microbiota to Influence Brain Development and Function in Early Life. Neurosci. Biobehav. Rev. 2018, 95, 191–201. [Google Scholar] [CrossRef]
- Friedman, J.E. Developmental Programming of Obesity and Diabetes in Mouse, Monkey, and Man in 2018: Where Are We Headed? Diabetes 2018, 67, 2137–2151. [Google Scholar] [CrossRef] [PubMed]
- Symonds, M.E.; Mendez, M.A.; Meltzer, H.M.; Koletzko, B.; Godfrey, K.; Forsyth, S.; Van Der Beek, E.M. Early Life Nutritional Programming of Obesity: Mother-Child Cohort Studies. Ann. Nutr. Metab. 2013, 62, 137–145. [Google Scholar] [CrossRef]
- Toftlund, L.H.; Halken, S.; Agertoft, L.; Zachariassen, G. Early Nutrition and Signs of Metabolic Syndrome at 6 y of Age in Children Born Very Preterm. Am. J. Clin. Nutr. 2018, 107, 717–724. [Google Scholar] [CrossRef]
- Toftlund, L.H.; Halken, S.; Agertoft, L.; Zachariassen, G. Catch-Up Growth, Rapid Weight Growth, and Continuous Growth from Birth to 6 Years of Age in Very-Preterm-Born Children. Neonatology 2018, 114, 285–293. [Google Scholar] [CrossRef]
- Khalangot, M.D.; Kovtun, V.A.; Okhrimenko, N.V.; Gurianov, V.G.; Kravchenko, V.I. Glucose Tolerance Testing and Anthropometric Comparisons Among Rural Residents of Kyiv Region: Investigating the Possible Effect of Childhood Starvation—A Community-Based Study. Nutr. Metab. Insights 2017, 10, 1–5. [Google Scholar] [CrossRef]
- Nauta, A.J.; Ben Amor, K.; Knol, J.; Garssen, J.; Van Der Beek, E.M. Relevance of Pre- and Postnatal Nutrition to Development and Interplay between the Microbiota and Metabolic and Immune Systems. Am. J. Clin. Nutr. 2013, 98, 586S–593S. [Google Scholar] [CrossRef] [PubMed]
- Rothschild, D.; Weissbrod, O.; Barkan, E.; Kurilshikov, A.; Korem, T.; Zeevi, D.; Costea, P.I.; Godneva, A.; Kalka, I.N.; Bar, N.; et al. Environment Dominates over Host Genetics in Shaping Human Gut Microbiota. Nature 2018, 555, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Ussar, S.; Fujisaka, S.; Kahn, C.R. Interactions between Host Genetics and Gut Microbiome in Diabetes and Metabolic Syndrome. Mol. Metab. 2016, 5, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Chagwedera, D.N.; Ang, Q.Y.; Bisanz, J.E.; Leong, Y.A.; Ganeshan, K.; Cai, J.; Patterson, A.D.; Turnbaugh, P.J.; Chawla, A. Nutrient Sensing in CD11c Cells Alters the Gut Microbiota to Regulate Food Intake and Body Mass. Cell Metab. 2019, 30, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Bolsinger, J.; Pronczuk, A.; Sambanthamurthi, R.; Hayes, K.C. Anti-Diabetic Effects of Palm Fruit Juice in the Nile Rat (Arvicanthis niloticus). J. Nutr. Sci. 2014, 3, e5. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.K.; Chang, H.W.; Yan, D.; Lee, K.M.; Ucmak, D.; Wong, K.; Abrouk, M.; Farahnik, B.; Nakamura, M.; Zhu, T.H.; et al. Influence of Diet on the Gut Microbiome and Implications for Human Health. J. Transl. Med. 2017, 15, 73. [Google Scholar] [CrossRef] [PubMed]
- Silva, F.M.; Kramer, C.K.; de Almeida, J.C.; Steemburgo, T.; Gross, J.L.; Azevedo, M.J. Fiber Intake and Glycemic Control in Patients with Type 2 Diabetes Mellitus: A Systematic Review with Meta-Analysis of Randomized Controlled Trials. Nutr. Rev. 2013, 71, 790–801. [Google Scholar] [CrossRef]
- Jovanovski, E.; Khayyat, R.; Zurbau, A.; Komishon, A.; Mazhar, N.; Sievenpiper, J.L.; Mejia, S.B.; Ho, H.V.T.; Li, D.; Jenkins, A.L.; et al. Should Viscous Fiber Supplements Be Considered in Diabetes Control? Results From a Systematic Review and Meta-Analysis of Randomized Controlled Trials. Diabetes Care 2019, 42, 755–766. [Google Scholar] [CrossRef]
- Perry, R.J.; Peng, L.; Barry, N.A.; Cline, G.W.; Zhang, D.; Cardone, R.L.; Petersen, K.F.; Kibbey, R.G.; Goodman, A.L.; Shulman, G.I. Acetate Mediates a Microbiome-Brain-β-Cell Axis to Promote Metabolic Syndrome. Nature 2016, 534, 213–217. [Google Scholar] [CrossRef]
- Hodge, B.A.; Wen, Y.; Riley, L.A.; Zhang, X.; England, J.H.; Harfmann, B.D.; Schroder, E.A.; Esser, K.A. The Endogenous Molecular Clock Orchestrates the Temporal Separation of Substrate Metabolism in Skeletal Muscle. Skelet. Muscle 2015, 5, 17. [Google Scholar] [CrossRef]
- Dyar, K.A.; Hubert, M.J.; Mir, A.A.; Ciciliot, S.; Lutter, D.; Greulich, F.; Quagliarini, F.; Kleinert, M.; Fischer, K.; Eichmann, T.O.; et al. Transcriptional Programming of Lipid and Amino Acid Metabolism by the Skeletal Muscle Circadian Clock. PLoS Biol. 2018, 16, e2005886. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.; Patti, M.E. Bile Acids, Obesity, and the Metabolic Syndrome. Best Pract. Res. Clin. Gastroenterol. 2014, 28, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, S.; Loo, T.M.; Atarashi, K.; Kanda, H.; Sato, S.; Oyadomari, S.; Iwakura, Y.; Oshima, K.; Morita, H.; Hattori, M.; et al. Obesity-Induced Gut Microbial Metabolite Promotes Liver Cancer through Senescence Secretome. Nature 2013, 499, 97–101. [Google Scholar] [CrossRef] [PubMed]
- Sayin, S.I.; Wahlström, A.; Felin, J.; Jäntti, S.; Marschall, H.U.; Bamberg, K.; Angelin, B.; Hyötyläinen, T.; Orešič, M.; Bäckhed, F. Gut Microbiota Regulates Bile Acid Metabolism by Reducing the Levels of Tauro-Beta-Muricholic Acid, a Naturally Occurring FXR Antagonist. Cell Metab. 2013, 17, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Islam, K.B.M.S.; Fukiya, S.; Hagio, M.; Fujii, N.; Ishizuka, S.; Ooka, T.; Ogura, Y.; Hayashi, T.; Yokota, A. Bile Acid Is a Host Factor That Regulates the Composition of the Cecal Microbiota in Rats. Gastroenterology 2011, 141, 1773–1781. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Brandeis Diet # | Diet 133 | Diet 73MBS | Diet 73MB |
|---|---|---|---|
| Experiment # | 2,4 | 1,3 | 3 |
| CHO:Fat:Protein %Energy | 60:20:20 | 70:10:20 | 70:10:20 |
| (Fat/Protein % Energy ratio) | 1.0 | 0.5 | 0.5 |
| kcal/g | 4.22 | 3.98 | 3.98 |
| GLoad per 2000 kcal | 224 | 259 | 295 |
| Ingredients | g/kg diet | ||
| Casein | 106 | 100 | 100 |
| Lactalbumin | 106 | 100 | 100 |
| Dextrose | 186 | 200 | 350 |
| Sucrose | 186 | 200 | 0 |
| Cornstarch * | 200 (+60 gel) | 238 (+60 gel) | 288 (+60 gel) |
| Fat [average American Fat Blend] | |||
| Margarine B (94% fat w butter) ** | 100 (94 as fat) | 47 (44 as fat) | 47 (44 as fat) |
| butter (fat component only) | 21 | 6 | 6 |
| tallow | 46 | 15 | 15 |
| lard | 15 | 0 | 0 |
| soybean oil | 18 | 23 | 23 |
| SFA:MUFA:PUFA ratio ## | 45:40:15 | 45:40:15 | 45:40:15 |
| P/S ratio # | 0.33 | 0.33 | 0.33 |
| Mineral mix, Ausman-Hayes a | 46 | 44 | 44 |
| Vitamin mix, Hayes-Cathcart b | 12 | 11 | 11 |
| Choline chloride (75% choline) | 3 | 3 | 3 |
| Cholesterol (0.06%) | 0.6 | 0.6 | 0.6 |
| Experiment | Table | Diet (CHO:Fat:Protein %Energy Ratio) | Length of Study (Weeks) | Sex of Nile Rat | Energy Intake (Week on Study) | RBG (Week on Study) | FBG (Week on Study) | OGTT | OGTT | Necropsy (Weeks on Study) | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Kcal/day (Weeks on Study) | Body Weight Gain/Day (Weeks on Study) | 30 min (Week on Study) | 60 min (Week on Study) | Organ Weights | Plasma Lipids | |||||||
| 1 | 3 | 73MBS (70:10:20) | 2,4 | M | na | na | na | ✓ (2,4) | ✓ (2,4) | na | na | na |
| 73MBS (70:10:20) | 2,4 | F | na | na | na | ✓ (2,4) | ✓ (2,4) | na | na | na | ||
| 2 | 4 | 133 (60:20:20) | 10 | M | ✓ (1–9) | ✓ (1–9) | ✓ (6,10) | ✓ (6,10) | ✓ (6,10) | ✓ (6,10) | ✓ (10) | ✓ (10) |
| 3 | 5 | 73MB/73MBS (70:10:20) | 8 | M | ✓ (1–7) | ✓ (1–7) | ✓ (4,8) | ✓ (4,8) | ✓ (4,8) | ✓ (4,8) | ✓ (8) | ✓ (8) |
| 4 | 6 | 133 (60:20:20) | 3 | F | na | na | ✓ (3) | ✓ (3) | ✓ (3) | ✓ (3) | ✓ (3) | na |
| CHO:Fat:Protein%energy | Diet 73MBS (70:10:20) * | Diet 73MBS (70:10:20) * | ||||||||
| (a) Males | ||||||||||
| Housed together for 2 weeks (n = 165) | Housed together for 4 weeks (n = 90) | |||||||||
| Quintile | 1 | 2 | 3 | 4 | 5 | 1 | 2 | 3 | 4 | 5 |
| 30’-OGTT range (mg/dl) | (59–61) | (59–161) | (214–241) | (245–292) | (296–435) | (44–98) | (103–149) | (150–192) | (193–257) | (260–525) |
| Fasting Body Weight (g) | 56 ± 9abc | 57 ± 7 | 60 ± 10a | 61 ± 7b | 61 ± 7c | 67 ± 11 | 65 ± 7ab | 67 ± 7 | 71 ± 10a | 72 ± 9b |
| Oral Glucose Tolerance Test (mg/dl) after 2 weeks | Oral Glucose Tolerance Test (mg/dl) after 4 weeks | |||||||||
| FBG, 0 min | 55 ± 20a | 54 ± 19b | 55 ± 16c | 62 ± 23 | 65 ± 21abc | 41 ± 10ab | 51 ± 27c | 47 ± 11d | 59 ± 23a | 67 ± 34bcd |
| 30 min | 121 ± 28abcd | 193 ± 15aefg | 227 ± 9behi | 270 ± 14cfhj | 337 ± 37dgij | 82 ± 15abcd | 127 ± 15aefg | 174 ± 13behi | 236 ± 17cfhj | 336 ± 79dgij |
| (b) Females | ||||||||||
| Housed together for 2 weeks (n = 138) | Housed together for 4 weeks (n = 109) | |||||||||
| Quintile | 1 | 2 | 3 | 4 | 5 | 1 | 2 | 3 | 4 | 5 |
| 30’-OGTT range (mg/dl) | (86–138) | (140–175) | (178–216) | (216–270) | (272–427) | (40–105) | (106–139) | (141–174) | (175–216) | (223–398) |
| Fasting Body Weight (g) | 44 ± 7ab | 47 ± 7 | 46 ± 6c | 51 ± 8ac | 49 ± 8b | 47 ± 9a | 48 ± 10b | 51 ± 12 | 55 ± 11ab | 51 ± 10 |
| Oral Glucose Tolerance Test (mg/dl) after 2 weeks | Oral Glucose Tolerance Test (mg/dl) after 4 weeks | |||||||||
| FBG, 0 min | 56 ± 18a | 58 ± 17b | 61 ± 18c | 68 ± 20 | 74 ± 34abc | 52 ± 16ab | 60 ± 21 | 68 ± 40a | 64 ± 15 | 76 ± 27b |
| 30 min | 111 ± 14abcd | 159 ± 11aefg | 197 ± 11behi | 246 ± 18cfhj | 318 ± 47dhij | 82 ± 18abcd | 124 ± 9aefg | 156 ± 11behi | 196 ± 15cfhj | 279 ± 43dgij |
| Diet (CHO:Fat:Protein%energy) | Diet 133 (60:20:20) * | ||||
| Quintiles by RBG | Q1 | Q2 | Q3 | Q4 | Q5 |
| T2DM ’genetic permissiveness’ ranked by quintiles | Resistant | Resistant | Susceptible–preDiabetic | Susceptible–moderate | Susceptible–severe |
| n = | 18 | 18 | 18 | 18 | 18 |
| RBG (range) after 10 weeks on diet (mg/dl) | (43–58) | (58–66) | (66–82) | (82–237) | (270–600) |
| after 6 weeks diet | 66 ± 16a | 73 ± 19b | 70 ± 19c | 68 ± 18d | 295 ± 179abcd |
| after 10 weeks diet | 53 ± 4ab | 62 ± 3cd | 72 ± 5ef | 158 ± 54aceg | 439 ± 119bdfg |
| Body Weight (g) | |||||
| Initial (3 weeks old) | 31 ± 5 | 30 ± 4 | 32 ± 3 | 31 ± 4 | 32 ± 5 |
| after 6 weeks diet | 82 ± 9abcd | 88 ± 5a | 87 ± 7b | 90 ± 6c | 92 ± 8d |
| after 10 weeks diet | 94 ± 10abcd | 101 ± 8ae | 103 ± 8b | 107 ± 6cef | 100 ± 11df |
| Body Weight Gain per day (g/day for 9 weeks) | |||||
| 1–6 week average | 1.18 ± 0.22abc | 1.34 ± 0.19a | 1.26 ± 0.15d | 1.37 ± 0.15b | 1.39 ± 0.19cd |
| 7–10 week average | 0.46 ± 0.25a | 0.45 ± 0.23b | 0.54 ± 0.12cd | 0.72 ± 0.49abce | 0.31 ± 0.17de |
| Total average (after 9 weeks) | 1.00 ± 0.22a | 1.09 ± 0.18b | 1.09 ± 0.11c | 1.22 ± 0.18abcd | 1.06 ± 0.19d |
| Water Intake per day in the 9th week (ml/day) | 5 ± 2a | 4 ± 1b | 4 ± 1c | 5 ± 2d | 22 ± 27abcd |
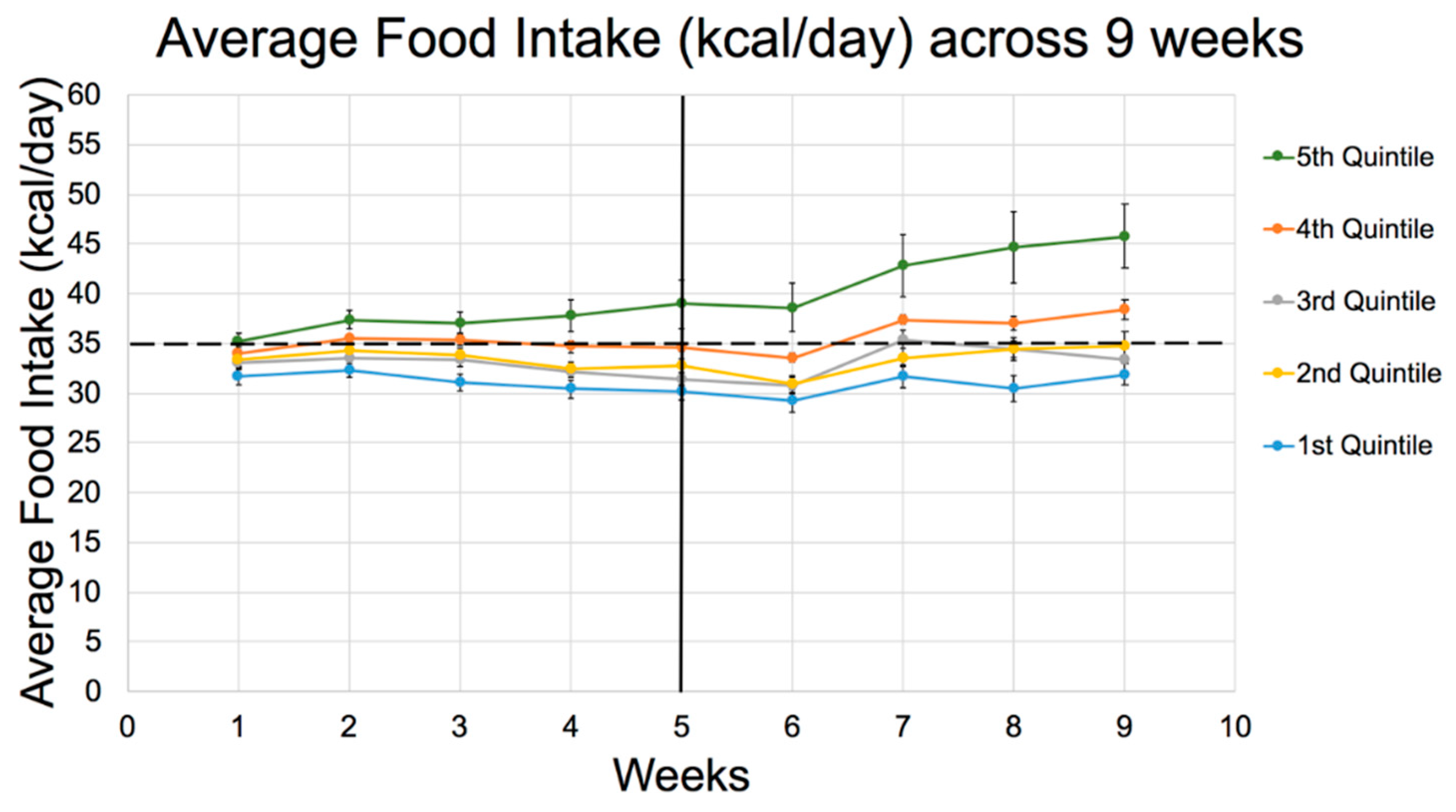
| Energy Intake (kcal/day) | |||||
| 1st week | 32 ± 4ab | 33 ± 3 | 33 ± 3c | 34 ± 3a | 35 ± 4bc |
| 2nd week | 32 ± 3abc | 34 ± 3ad | 34 ± 3e | 36 ± 2b | 37 ± 4cde |
| 6th week | 29 ± 5ab | 31 ± 4c | 31 ± 3d | 34 ± 2ae | 39 ± 10bcde |
| 8th week | 31 ± 6ab | 35 ± 5c | 34 ± 4d | 37 ± 3ae | 45 ± 15bcde |
| 9th week | 32 ± 5ab | 35 ± 6c | 33 ± 5d | 38 ± 4ae | 46 ± 13bcde |
| Total average (after 9 weeks) | 31 ± 3ab | 33 ± 3c | 33 ± 2d | 35 ± 1ae | 40 ± 8bcde |
| Glycemic Load | |||||
| per kg body weight at 10 weeks | 37 ± 4a | 37 ± 3b | 36 ± 2c | 37 ± 3d | 46 ± 15abcd |
| per rat/day | 3.48 ± 0.30ab | 3.74 ± 0.29c | 3.70 ± 0.25d | 3.97 ± 0.15ae | 4.46 ± 0.92bcde |
| per BMI/rat/day | 0.66 ± 0.07a | 0.70 ± 0.06b | 0.67 ± 0.04c | 0.71 ± 0.04d | 0.88 ± 0.32abcd |
| Cumulative GLoad /rat for 10 weeks | 243 ± 21ab | 262 ± 20c | 259 ± 17d | 278 ± 11ae | 312 ± 64bcde |
| Food Efficiency (g gained /1000 kcal) | |||||
| 1–6 week Average | 38 ± 6 | 41 ± 5 | 39 ± 4 | 40 ± 4 | 38 ± 8 |
| 7–10 week Average | 15 ± 7ab | 13 ± 7cd | 16 ± 3e | 20 ± 14acf | 8 ± 5bdef |
| Total average (after 9 weeks) | 32 ± 6a | 33 ± 5b | 33 ± 3c | 35 ± 6d | 28 ± 7abcd |
| Oral Glucose Tolerance Test (mg/dl) after 6 weeks | |||||
| FBG, 0 min | 44 ± 10ab | 49 ± 9c | 51 ± 9d | 57 ± 12a | 62 ± 23bcd |
| 30 min | 147 ± 39abc | 181 ± 58de | 214 ± 62af | 242 ± 67bdg | 366 ± 91cefg |
| Oral Glucose Tolerance Test (mg/dl) after 10 weeks | |||||
| FBG, 0 min | 49 ± 14a | 49 ± 9b | 45 ± 10c | 53 ± 14d | 76 ± 47abcd |
| 30 min | 147 ± 47abc | 171 ± 49de | 221 ± 46af | 264 ± 114bdg | 432 ± 123cefg |
| 60 min | 113 ± 56ab | 103 ± 62cd | 136 ± 48e | 173 ± 93acf | 331 ± 143bdef |
| Organ weight (%BW) | |||||
| Liver | 3.56 ± 0.47a | 3.49 ± 0.50b | 3.50 ± 0.24c | 3.60 ± 0.33d | 5.16 ± 1.23abcd |
| Kidney | 0.70 ± 0.06a | 0.69 ± 0.06b | 0.68 ± 0.04c | 0.73 ± 0.08d | 1.02 ± 0.28abcd |
| Cecum | 1.17 ± 0.17a | 1.08 ± 0.33b | 1.12 ± 0.18c | 1.00 ± 0.15d | 1.57 ± 0.69abcd |
| Adipose (%BW) | |||||
| Epididymal | 2.85 ± 0.67a | 2.96 ± 0.69b | 2.90 ± 0.58c | 3.02 ± 0.56d | 2.24 ± 0.70abcd |
| Perirenal | 1.36 ± 0.47ab | 1.72 ± 0.48c | 1.95 ± 0.53ad | 1.97 ± 0.37be | 1.33 ± 0.85cde |
| Brown fat | 1.77 ± 0.44ab | 2.09 ± 0.48c | 2.37 ± 0.52ad | 2.59 ± 0.41bce | 1.73 ± 0.78de |
| Total fat | 5.97 ± 1.31ab | 6.57 ± 1.37c | 7.03 ± 1.16ad | 7.18 ± 1.29be | 5.16 ± 2.12cde |
| Carcass (%BW) | 75 ± 2ab | 74 ± 2c | 74 ± 1d | 73 ± 2a | 72 ± 2bcd |
| Length (cm) | 13.2 ± 0.4abc | 13.5 ± 0.5a | 13.3 ± 0.4 | 13.6 ± 0.3b | 13.6 ± 0.6c |
| Body Mass Index (kg/m2) at 10 weeks | 5.27 ± 0.48a | 5.40 ± 0.52 | 5.57 ± 0.34b | 5.65 ± 0.26ac | 5.23 ± 0.66bc |
| Plasma (mg/dl) | |||||
| Total Cholesterol | 123 ± 23a | 113 ± 26b | 116 ± 13c | 131 ± 39d | 322 ± 410abcd |
| Total Triglycerides | 70 ± 26a | 74 ± 25b | 68 ± 25c | 96 ± 20d | 213 ± 298abcd |
| Diet (CHO:Fat:Protein %energy) | Diet 73 (70:10:20) * | ||||
| Quintiles by RBG | Q1 | Q2 | Q3 | Q4 | Q5 |
| T2DM ’genetic permissiveness’ ranked by quintiles | Resistant | Resistant | Susceptible | Susceptible | Susceptible |
| n = | 6 | 6 | 6 | 7 | 7 |
| RBG (range) after 8 weeks on diet (mg/dl) | (43–63) | (65–86) | (88–286) | (294–444) | (469–600) |
| after 4 weeks on diet | 65 ± 9ab | 68 ± 6cd | 107 ± 33ef | 217 ± 111ace | 283 ± 128bdf |
| after 8 weeks on diet | 53 ± 7abc | 78 ± 10def | 185 ± 85adgh | 369 ± 50begi | 545 ± 58cfhi |
| Body Weight (g) | |||||
| Initial (3 weeks of age) | 36 ± 6 | 35 ± 10 | 32 ± 6 | 35 ± 8 | 36 ± 9 |
| after 4 weeks on diet | 76 ± 5a | 74 ± 6b | 78 ± 6 | 85 ± 8abc | 77 ± 7c |
| estimated at 6 weeks on diet | 85 ± 4 | 83 ± 7a | 88 ± 5 | 92 ± 8ab | 83 ± 4b |
| after 8 weeks on diet | 93 ± 5 | 92 ± 7a | 97 ± 6 | 99 ± 9b | 89 ± 6ab |
| Body Weight Gain per day (g/day for 7 weeks) | |||||
| 1–4 week average | 1.64 ± 0.66 | 1.32 ± 0.16a | 2.02 ± 1.03ab | 1.84 ± 0.23 | 1.38 ± 0.20b |
| 5–7 week average | 0.69 ± 0.22a | 0.70 ± 0.12b | 0.62 ± 0.21 | 0.53 ± 0.13 | 0.38 ± 0.38ab |
| Total average (after 7 weeks) | 1.26 ± 0.11a | 1.15 ± 0.16b | 1.39 ± 0.33bc | 1.34 ± 0.11d | 1.01 ± 0.22acd |
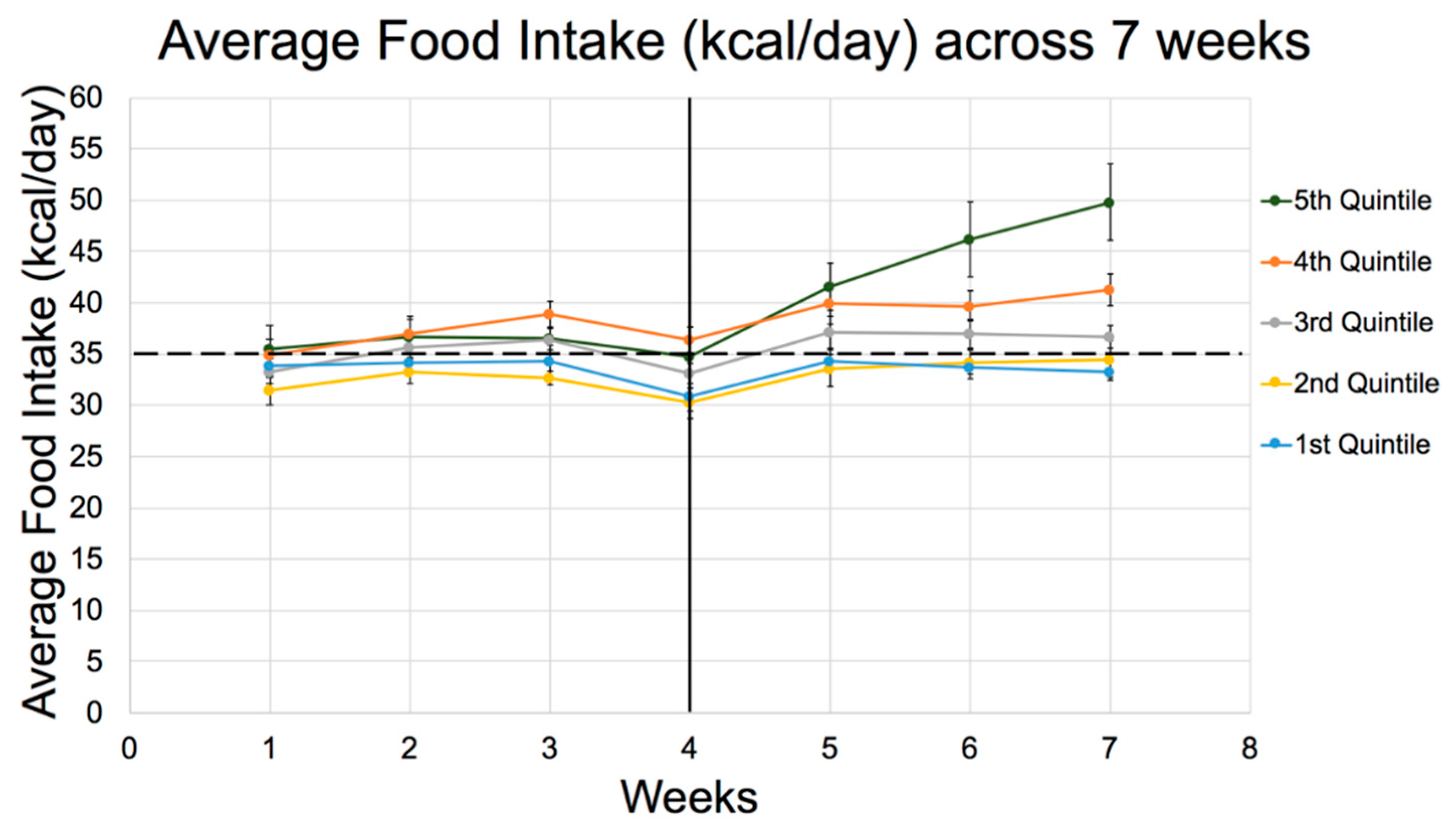
| Energy Intake (kcal/day) | |||||
| 1st week | 34 ± 3 | 32 ± 3 | 33 ± 3 | 35 ± 4 | 36 ± 6 |
| 2nd week | 34 ± 2 | 33 ± 3 | 36 ± 2 | 37 ± 4 | 37 ± 6 |
| 3rd week | 34 ± 4a | 33 ± 2b | 36 ± 3 | 39 ± 3ab | 37 ± 6 |
| 4th week | 31 ± 3a | 30 ± 4bc | 33 ± 2 | 36 ± 4ab | 35 ± 5c |
| 5th week | 34 ± 2ab | 34 ± 4cd | 37 ± 4 | 40 ± 5ac | 42 ± 6bd |
| 6th week | 34 ± 2a | 34 ± 4b | 37 ± 4c | 40 ± 4d | 46 ± 10abcd |
| 7th week | 33 ± 2ab | 35 ± 4cd | 37 ± 3e | 41 ± 4acf | 50 ± 10bdef |
| Total average (After 7 weeks) | 35 ± 1a | 34 ± 3bc | 37 ± 3 | 40 ± 3ab | 39 ± 6c |
| Glycemic Load | |||||
| per kg body weight at 8 weeks | 54 ± 2a | 52 ± 3b | 56 ± 3c | 59 ± 6d | 67 ± 11abcd |
| per rat/day | 5.03 ± 0.22ab | 4.81 ± 0.46cd | 5.42 ± 0.41 | 5.81 ± 0.46ac | 5.96 ± 0.96bd |
| per BMI/rat/day | 1.01 ± 0.56ab | 0.97 ± 0.09cd | 1.08 ± 0.12e | 1.19 ± 0.07ac | 1.26 ± 0.23bde |
| Cumulative GLoad/rat for 8 weeks | 282 ± 12ab | 269 ± 26cd | 304 ± 23 | 326 ± 26ac | 334 ± 54bd |
| Food Efficiency (g gained /1000 kcal) | |||||
| 1–4 week average | 48 ± 15 | 42 ± 7 | 57 ± 26a | 50 ± 7 | 39 ± 7a |
| estimated 1–6 week average | 37 ± 5a | 37 ± 6 | 40 ± 7b | 37 ± 3c | 30 ± 8abc |
| 5–7 week average | 21 ± 7a | 22 ± 5bc | 17 ± 6 | 14 ± 3b | 10 ± 12c |
| Total average (After 7 weeks) | 37 ± 2a | 36 ± 6b | 38 ± 7c | 34 ± 3d | 26 ± 10abcd |
| Oral Glucose Tolerance Test (mg/dl) at 4 weeks | |||||
| FBG, 0 min | 51 ± 11 | 47 ± 8a | 61 ± 12abc | 43 ± 7b | 48 ± 14c |
| 30 min | 164 ± 40abc | 182 ± 68de | 240 ± 74a | 258 ± 71bd | 269 ± 57ce |
| Oral Glucose Tolerance Test (mg/dl) at 8 weeks | |||||
| FBG, 0 min | 43 ± 5a | 55 ± 16 | 50 ± 12b | 56 ± 27 | 90 ± 64ab |
| 30 min | 162 ± 64abc | 199 ± 40de | 265 ± 72afg | 364 ± 95bdf | 405 ± 86ceg |
| Organ weight (%BW) | |||||
| Liver | 3.24 ± 0.23abc | 3.56 ± 0.27de | 3.85 ± 0.43af | 4.34 ± 0.43bd | 4.67 ± 0.84cef |
| Kidney | 0.71 ± 0.03a | 0.70 ± 0.06bc | 0.75 ± 0.06de | 0.92 ± 0.15bdf | 1.07 ± 0.11acef |
| Cecum | 1.17 ± 0.24a | 1.15 ± 0.20b | 1.03 ± 0.19c | 1.39 ± 0.31d | 2.20 ± 0.91abcd |
| Adipose (%BW) | |||||
| Epididymal | 3.29 ± 0.74a | 3.19 ± 0.80b | 3.01 ± 0.49c | 3.12 ± 0.50d | 2.10 ± 0.53abcd |
| Perirenal | 1.59 ± 0.35a | 1.38 ± 0.34b | 1.72 ± 0.53c | 1.28 ± 0.20d | 0.72 ± 0.72abcd |
| Brown fat | 1.86 ± 0.37a | 2.28 ± 0.24b | 2.27 ± 0.44c | 1.97 ± 0.28d | 1.19 ± 0.53abcd |
| Total fat | 4.21 ± 2.74 | 5.26 ± 2.59 | 5.55 ± 2.88 | 4.42 ± 2.09 | 3.33 ± 2.18 |
| Carcass (%BW) | 72 ± 3 | 73 ± 2 | 73 ± 1 | 73 ± 1 | 73 ± 1 |
| Length (cm) | 13.6 ± 0.6 | 13.5 ± 0.4a | 13.7 ± 0.6 | 14.1 ± 0.4ab | 13.3 ± 0.5b |
| Body Mass Index (kg/m2) based on 8 week FBW | 4.97 ± 0.39 | 4.97 ± 0.54 | 5.05 ± 0.37 | 4.90 ± 0.35 | 4.77 ± 0.53 |
| Plasma (mg/dl) | |||||
| Total Cholesterol | 124 ± 25a | 119 ± 17b | 75 ± 12c | 117 ± 42d | 250 ± 83abcd |
| Total Triglycerides | 65 ± 9a | 93 ± 15 | 105 ± 46 | 127 ± 24a | 88 ± 30 |
| Diet (CHO:Fat:Protein %energy) | Diet 133 (60:20:20) * | ||||
| Quintiles by RBG | Q1 | Q2 | Q3 | Q4 | Q5 |
| T2DM ’genetic permissiveness’ ranked by quintiles | Resistant | Resistant | Susceptible | Susceptible | Susceptible |
| n = | 6 | 6 | 6 | 6 | 6 |
| RBG (range) after 3 weeks (mg/dl) | (51–64) | (65–74) | (74–80) | (80–88) | (89–215) |
| Ave after 3 weeks | 60 ± 5a | 69 ± 4b | 77 ± 3c | 82 ± 3d | 123 ± 49abcd |
| Body Weight (g) | |||||
| Initial (3 weeks of age) | 27 ± 3 | 30 ± 6 | 30 ± 4 | 28 ± 4 | 28 ± 2 |
| after 3 weeks on diet | 57 ± 9 | 57 ± 5 | 61 ± 4 | 59 ± 4 | 56 ± 10 |
| Oral Glucose Tolerance Test (mg/dl) after 3 weeks | |||||
| FBG, 0 min | 64 ± 25 | 51 ± 11 | 51 ± 11 | 69 ± 25 | 70 ± 18 |
| 30 min | 159 ± 48a | 193 ± 67 | 243 ± 56a | 168 ± 41 | 225 ± 109 |
| Organ weight (%BW) | |||||
| Liver | 3.55 ± 0.52 | 3.37 ± 0.34 | 3.88 ± 0.6a | 3.23 ± 0.29ab | 3.93 ± 0.56b |
| Kidney | 0.7 ± 0.09 | 0.73 ± 0.06 | 0.67 ± 0.06 | 0.66 ± 0.05 | 0.67 ± 0.09 |
| Cecum | 1.42 ± 0.16 | 1.61 ± 0.53 | 1.44 ± 0.26 | 1.36 ± 0.36 | 1.56 ± 0.4 |
| Adipose | |||||
| Epididymal | 1.97 ± 0.56 | 2.16 ± 0.84 | 2.33 ± 0.48 | 2.54 ± 0.49 | 2.06 ± 0.6 |
| Perirenal | 1.16 ± 0.6 | 0.98 ± 0.27a | 1.26 ± 0.47 | 1.5 ± 0.46a | 1.25 ± 0.27 |
| Brown fat | 2.07 ± 0.58 | 2.16 ± 0.18 | 2.47 ± 0.45 | 2.42 ± 0.27 | 2.24 ± 0.62 |
| Total fat | 4.34 ± 1.64 | 4.65 ± 2.04 | 4.73 ± 2.04 | 5.65 ± 1.66 | 4.65 ± 1.72 |
| Carcass (%BW) | 73 ± 10 | 77 ± 1 | 76 ± 1 | 76 ± 1 | 77 ± 2 |
| Length (cm) | 11.4 ± 0.4 | 11.5 ± 0.5 | 11.9 ± 0.2 | 11.6 ± 0.4 | 11.5 ± 0.7 |
| Body Mass Index (kg/m2) based on 3 week FBW | 4.42 ± 0.46 | 4.32 ± 0.34 | 4.31 ± 0.27 | 4.38 ± 0.52 | 4.24 ± 0.54 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subramaniam, A.; Landstrom, M.; Hayes, K.C. Genetic Permissiveness and Dietary Glycemic Load Interact to Predict Type-II Diabetes in the Nile rat (Arvicanthis niloticus). Nutrients 2019, 11, 1538. https://doi.org/10.3390/nu11071538
Subramaniam A, Landstrom M, Hayes KC. Genetic Permissiveness and Dietary Glycemic Load Interact to Predict Type-II Diabetes in the Nile rat (Arvicanthis niloticus). Nutrients. 2019; 11(7):1538. https://doi.org/10.3390/nu11071538
Chicago/Turabian StyleSubramaniam, Avinaash, Michelle Landstrom, and K. C. Hayes. 2019. "Genetic Permissiveness and Dietary Glycemic Load Interact to Predict Type-II Diabetes in the Nile rat (Arvicanthis niloticus)" Nutrients 11, no. 7: 1538. https://doi.org/10.3390/nu11071538
APA StyleSubramaniam, A., Landstrom, M., & Hayes, K. C. (2019). Genetic Permissiveness and Dietary Glycemic Load Interact to Predict Type-II Diabetes in the Nile rat (Arvicanthis niloticus). Nutrients, 11(7), 1538. https://doi.org/10.3390/nu11071538