Over Restriction of Dietary Protein Allowance: The Importance of Ongoing Reassessment of Natural Protein Tolerance in Phenylketonuria





Abstract
:1. Introduction
2. Materials and Methods
2.1. Project Design
2.2. Data Collection
2.3. Anthropometry
2.4. Dietary Assessment
2.5. Blood Phe Levels
2.6. Reassessment of NP Tolerance
2.7. Ethical Statement
2.8. Statistics
3. Results
3.1. Subjects
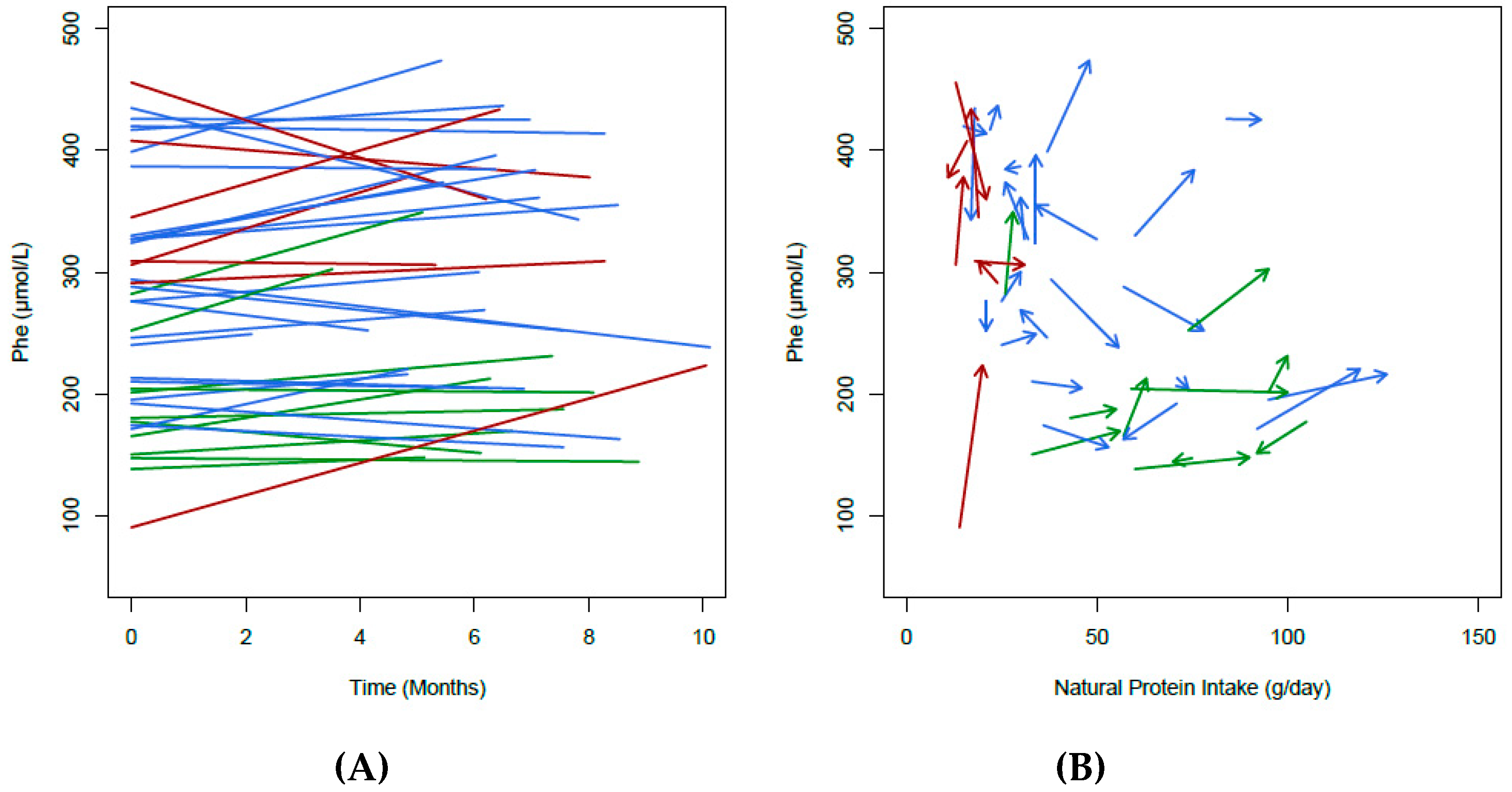
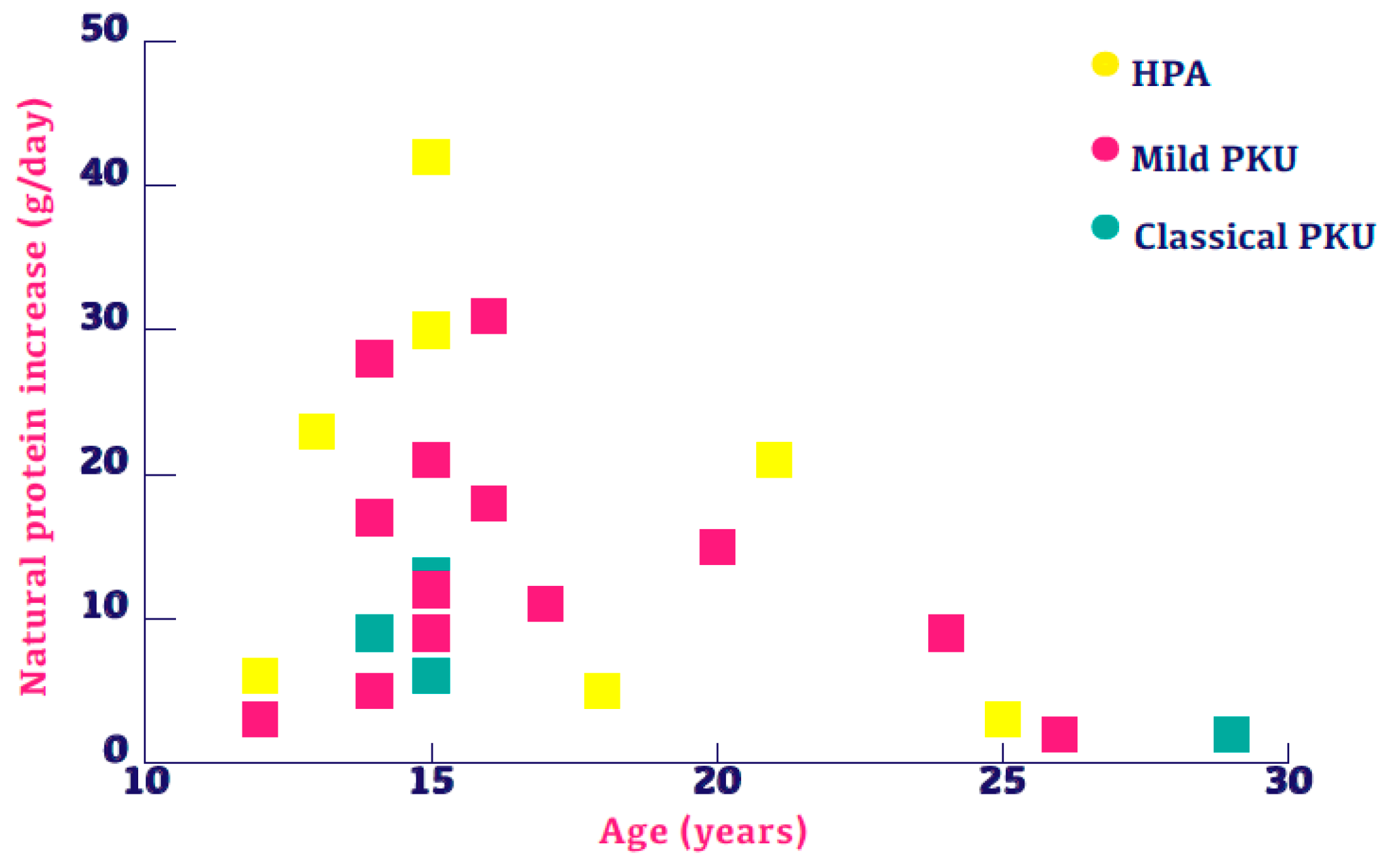
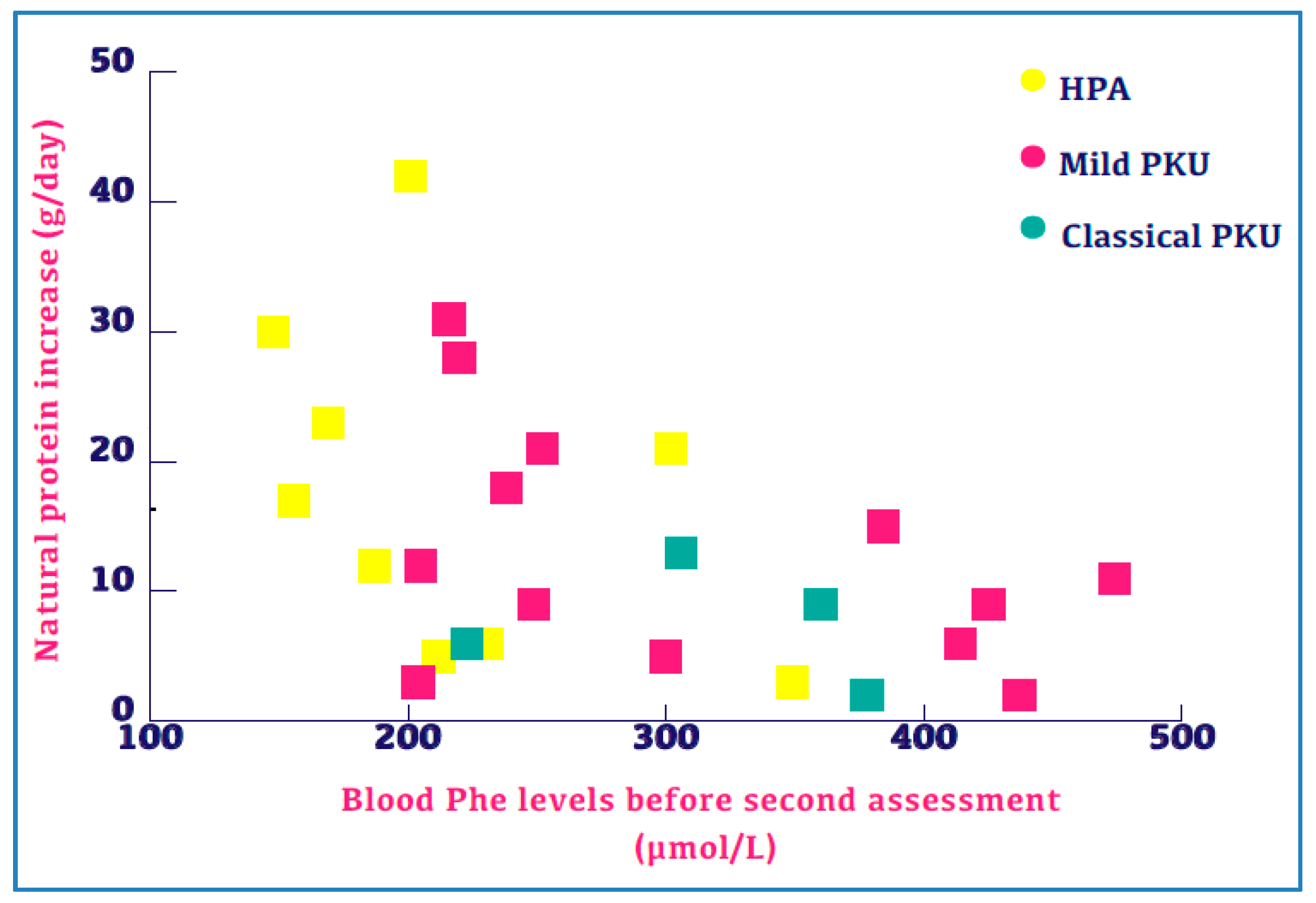
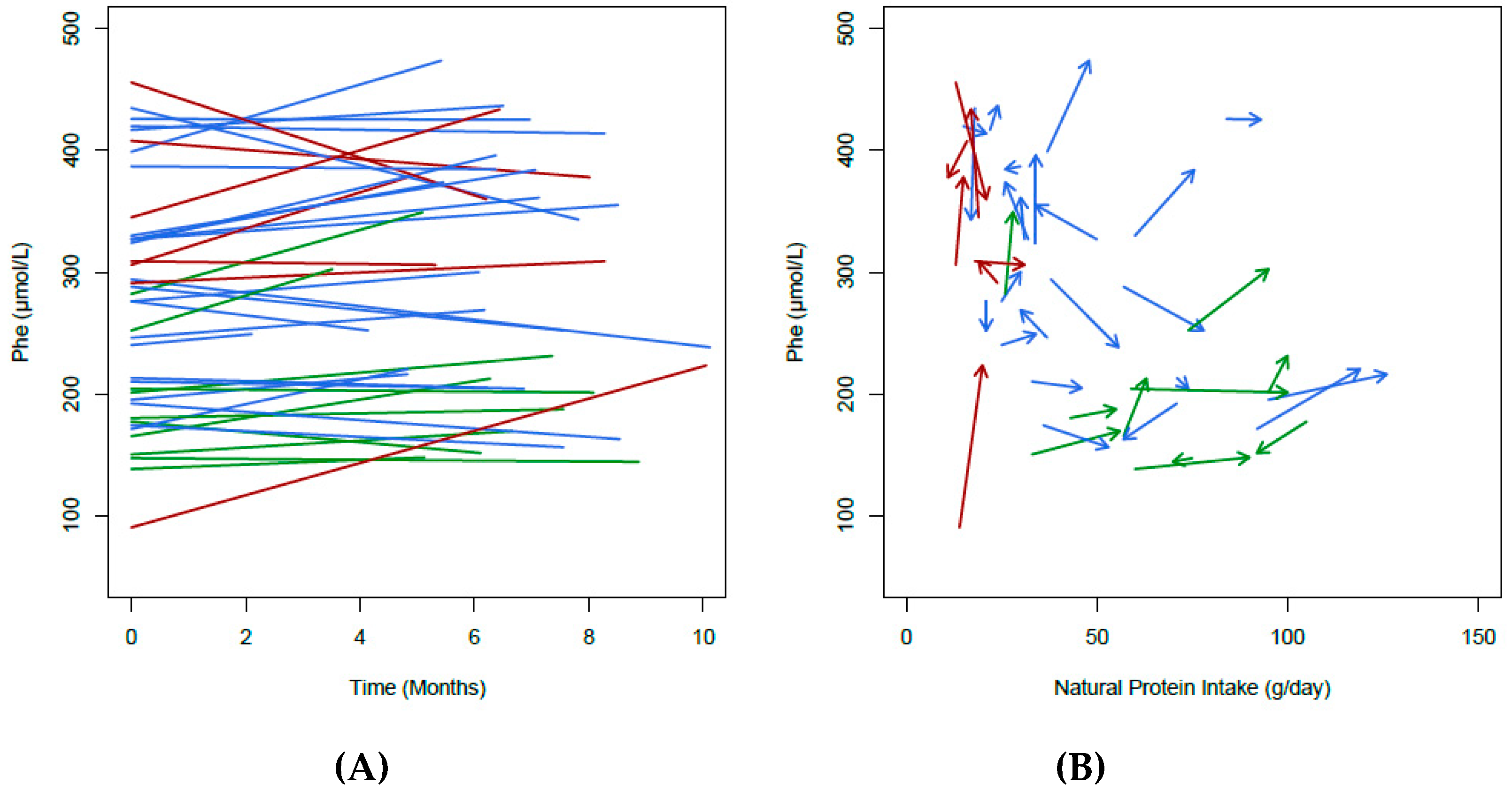
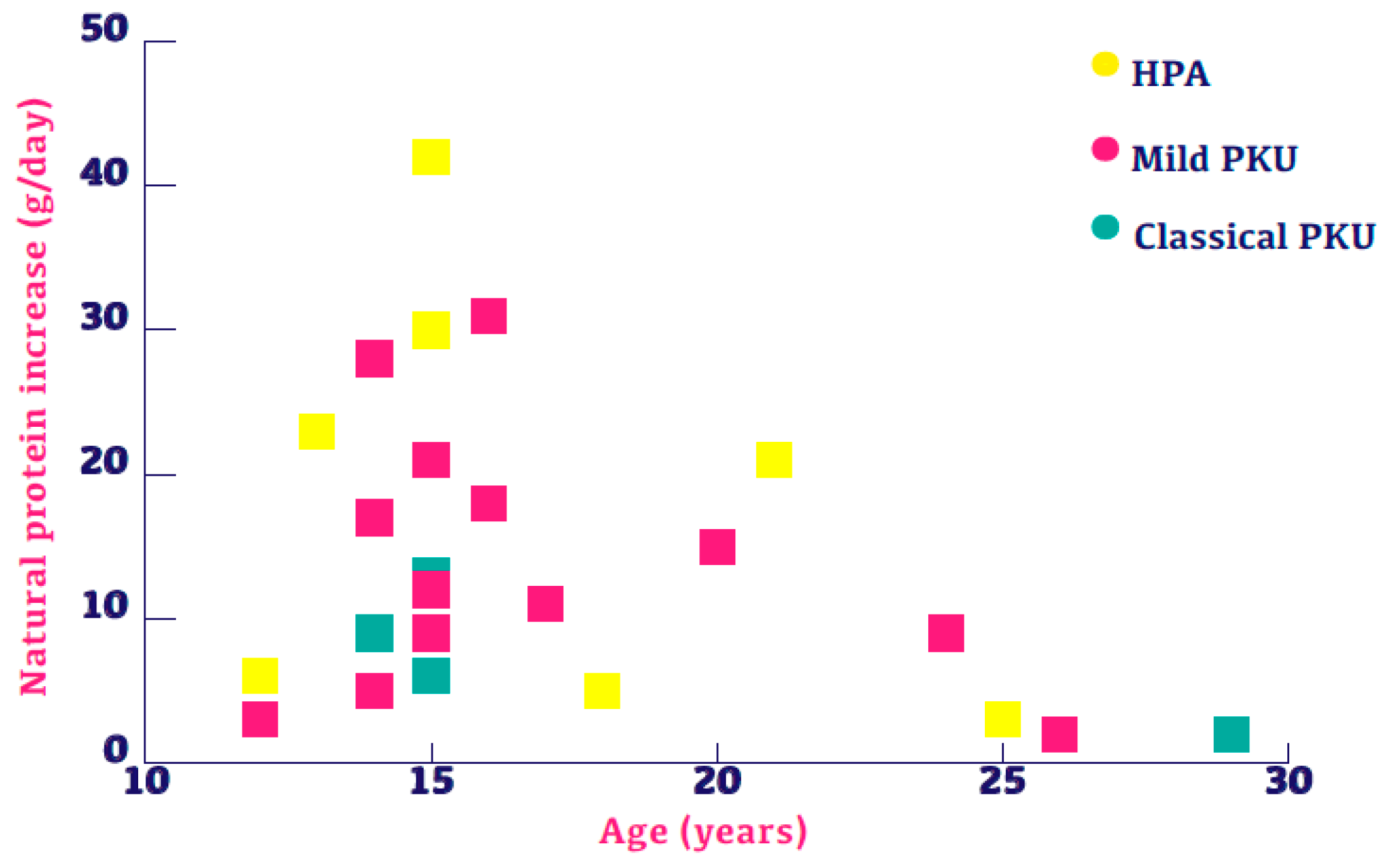
3.2. Change in Natural Protein Intake
3.3. Source of Natural Protein
3.4. Protein Substitute
3.5. Energy Intake
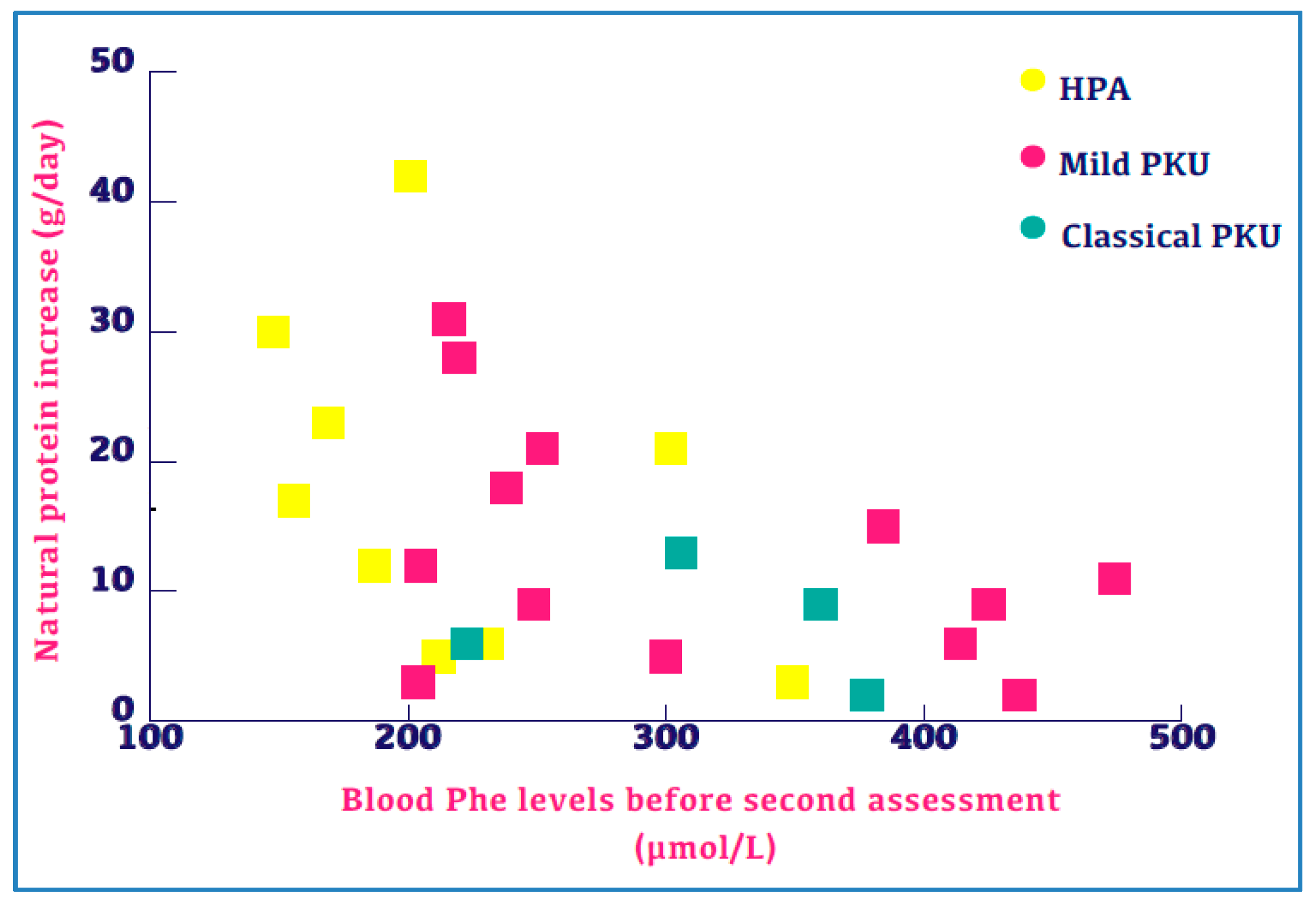
3.6. Blood Phe Control
3.7. Changes in Anthropometric Measurements
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Blau, N.; van Spronsen, F.J.; Levy, H.L. Phenylketonuria. Lancet 2010, 376, 1417–1427. [Google Scholar] [CrossRef]
- Rocha, J.C.; MacDonald, A. Dietary intervention in the management of phenylketonuria: Current perspectives. Pediatric Health Med. Ther. 2016, 7, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Rocha, J.C.; Vilarinho, L.; Cabral, A.; Osório, R.V.; Almeida, M.F.d. Consensus for the nutritional treatment of phenylketonuria. Acta Pediatr. Port. 2007, 38, 44–54. [Google Scholar]
- Rocha, J.C.; MacDonald, A. Treatment options and dietary supplements for patients with phenylketonuria. Expert Opin. Orphan Drugs 2018, 6, 667–681. [Google Scholar] [CrossRef]
- Guttler, F.; Guldberg, P. The influence of mutations of enzyme activity and phenylalanine tolerance in phenylalanine hydroxylase deficiency. Eur. J. Pediatr. 1996, 155, S6–S10. [Google Scholar] [CrossRef] [PubMed]
- Van Wegberg, A.M.J.; MacDonald, A.; Ahring, K.; Belanger-Quintana, A.; Blau, N.; Bosch, A.M.; Burlina, A.; Campistol, J.; Feillet, F.; Gizewska, M.; et al. The complete European guidelines on phenylketonuria: Diagnosis and treatment. Orphanet J. Rare Dis. 2017, 12, 162. [Google Scholar] [CrossRef] [PubMed]
- Van Spronsen, F.J.; van Rijn, M.; Dorgelo, B.; Hoeksma, M.; Bosch, A.M.; Mulder, M.F.; de Klerk, J.B.; de Koning, T.; Rubio-Gozalbo, M.E.; de Vries, M.; et al. Phenylalanine tolerance can already reliably be assessed at the age of 2 years in patients with PKU. J. Inherit. Metab. Dis. 2009, 32, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Van Rijn, M.; Hoeksma, M.; Sauer, P.J.; Modderman, P.; Reijngoud, D.J.; van Spronsen, F.J. Adult patients with well-controlled phenylketonuria tolerate incidental additional intake of phenylalanine. Ann. Nutr. Metab. 2011, 58, 94–100. [Google Scholar] [CrossRef] [PubMed]
- MacLeod, E.L.; Gleason, S.T.; van Calcar, S.C.; Ney, D.M. Reassessment of phenylalanine tolerance in adults with phenylketonuria is needed as body mass changes. Mol. Genet. Metab. 2009, 98, 331–337. [Google Scholar] [CrossRef] [PubMed]
- Aldamiz-Echevarria, L.; Bueno, M.A.; Couce, M.L.; Lage, S.; Dalmau, J.; Vitoria, I.; Andrade, F.; Blasco, J.; Alcalde, C.; Gil, D.; et al. Anthropometric characteristics and nutrition in a cohort of PAH-deficient patients. Clin. Nutr. (Edinburgh, Scotland) 2014, 33, 702–717. [Google Scholar] [CrossRef] [PubMed]
- Thiele, A.G.; Gausche, R.; Lindenberg, C.; Beger, C.; Arelin, M.; Rohde, C.; Mutze, U.; Weigel, J.F.; Mohnike, K.; Baerwald, C.; et al. Growth and Final Height Among Children With Phenylketonuria. Pediatrics 2017, 140. [Google Scholar] [CrossRef] [PubMed]
- Hoeksma, M.; Van Rijn, M.; Verkerk, P.H.; Bosch, A.M.; Mulder, M.F.; de Klerk, J.B.; de Koning, T.J.; Rubio-Gozalbo, E.; de Vries, M.; Sauer, P.J.; et al. The intake of total protein, natural protein and protein substitute and growth of height and head circumference in Dutch infants with phenylketonuria. J. Inherit. Metab. Dis. 2005, 28, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.; Truby, H.; Boneh, A. The relationship between dietary intake, growth and body composition in Phenylketonuria. Mol. Genet. Metab. 2017, 122, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Huemer, M.; Huemer, C.; Moslinger, D.; Huter, D.; Stockler-Ipsiroglu, S. Growth and body composition in children with classical phenylketonuria: Results in 34 patients and review of the literature. J. Inherit. Metab. Dis. 2007, 30, 694–699. [Google Scholar] [CrossRef]
- Vockley, J.; Andersson, H.C.; Antshel, K.M.; Braverman, N.E.; Burton, B.K.; Frazier, D.M.; Mitchell, J.; Smith, W.E.; Thompson, B.H.; Berry, S.A. Phenylalanine hydroxylase deficiency: Diagnosis and management guideline. Genet. Med. 2014, 16, 188–200. [Google Scholar]
- Vilarinho, L.; Rocha, H.; Sousa, C.; Marcao, A.; Fonseca, H.; Bogas, M.; Osorio, R.V. Four years of expanded newborn screening in Portugal with tandem mass spectrometry. J. Inherit. Metab. Dis. 2010, 33, S133–S138. [Google Scholar] [CrossRef] [PubMed]
- Sousa Barbosa, C.; Almeida, M.F.; Sousa, C.; Rocha, S.; Guimas, A.; Ribeiro, R.; Martins, E.; Bandeira, A.; Oliveira, B.M.P.M.; Borges, N.; et al. Metabolic Control in Patients With Phenylketonuria Pre- and Post-Sapropterin Loading Test. J. Inborn Errors Metab. Screen. 2018, 6, 1–6. [Google Scholar] [CrossRef]
- Rocha, J.C.; van Rijn, M.; van Dam, E.; Ahring, K.; Bélanger-Quintana, A.; Dokoupil, K.; Gokmen Ozel, H.; Lammardo, A.M.; Robert, M.; Heidenborg, C.; et al. Weight Management in Phenylketonuria: What Should Be Monitored. Ann. Nutr. Metab. 2016, 68, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Salgado, P.; Montagne, L.; Freire, J.P.; Ferreira, R.B.; Teixeira, A.; Bento, O.; Abreu, M.C.; Toullec, R.; Lalles, J.P. Legume grains enhance ileal losses of specific endogenous serine-protease proteins in weaned pigs. J. Nutr. 2002, 132, 1913–1920. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
| Covariate | Level | Classical PKU | HPA | Mild PKU | Total |
|---|---|---|---|---|---|
| Total | 7 | 10 | 23 | 40 | |
| Gender | F | 2 (12%) | 3 (19%) | 11 (69%) | 16 |
| M | 5 (21%) | 7 (29%) | 12 (50%) | 24 | |
| Age (months) | median (IQR) | 190 (169.0, 243.5) | 185.5 (168.3, 214.5) | 190 (179.5, 270.5) | 190 (175.5, 256.0) |
| Diagnostic level (blood Phe in mg/dl) | median (IQR) | 25.2 (21.65, 27.75) | 4.65 (4.05, 5.18) | 11 (7.65, 15.00) | 9.95 (5.88, 16.00) |
| Months between assessments | median (IQR) | 6 (5.5, 8.0) | 6.5 (5.3, 7.0) | 7 (5.5, 7.0) | 6.5 (5, 7.3) |
| Covariate | Level | Visit 1 | Visit 2 | Total |
|---|---|---|---|---|
| Weight (kg) | median (IQR) | 52.50 (44.86, 68.63) | 58 (47.75, 69.88) | 54.25 (45.38, 69.00) |
| Height (cm) | median (IQR) | 161.0 (155.5, 167.8) | 163.1 (157.6, 169.1) | 162 (156.0, 169.0) |
| Height Z score | median (IQR) | −0.33 (−0.84, 0.29) | −0.07 (−0.88, 0.2) | −0.27 (−0.88, 0.27) |
| BMI (kg/m2) | median (IQR) | 21.05 (17.98, 23.63) | 21.60 (18.28, 24.23) | 21.30 (18.15, 24.03) |
| BMI Z score | median (IQR) | 0.00 (−0.74, 1.05) | 0.37 (−0.91, 1.43) | 0.28 (−0.76, 1.20) |
| Natural Protein (g/day) | median (IQR) | 35.0 (23.5, 60.0) | 40.0 (25.5, 74.4) | 35.0 (24.0, 70.3) |
| Natural Protein (g/kg/day) | median (IQR) | 0.66 (0.44, 1.04) | 0.71 (0.40, 1.14) | 0.70 (0.42, 1.06) |
| Phe * (mg/kg/day) | median (IQR) | 32.8 (21.9, 51.9) | 35.3 (20.0, 57.1) | 35.0 (20.9, 53.1) |
| Protein equivalent intake from low-Phe/Phe-free protein substitute (g/day) | median (IQR) | 45.0 (27.0, 55.4) | 41.5 (19.5, 54.8) | 44.8 (24.0, 55.1) |
| Protein equivalent intake from low-Phe/Phe-free protein substitute (g/kg/day) | median (IQR) | 0.82 (0.51, 1.07) | 0.75 (0.34, 0.99) | 0.82 (0.48, 1.04) |
| Energy intake (Kcal/day) | median (IQR) | 2371 (2200, 2536) | 2365 (2210, 2603) | 2365 (2202, 2575) |
| Blood Phe Levels (μmol/L) | median (IQR) | 279 (194, 328) | 284 (210, 375) | 279 (203, 360) |
| Number of blood spots | median (IQR) | 6 (4, 9) | 11 (7, 14) | 9 (5, 12) |
| % Nutritional Protein Increase | Total | HPA | Mild PKU | Classical PKU |
| Minimum 20% natural protein increase | 20/40 (50%) | 5/10 (50%) | 11/23 (48%) | 4/7 (57%) |
| Minimum 50% natural protein increase | 6/40 (15%) | 3/10 (30%) | 1/23 (4%) | 2/7 (29%) |
| Source of extra natural protein | Total | HPA | Mild PKU | Classical PKU |
| Animal foods: Dairy products, fish and meat | 20/26 (77%) | 8/8 (100%) | 11/14 (79%) | 1/4(25%) |
| Plant foods: Bread, pasta and potatoes | 6/26 (23%) | 0 | 3/14 (21%) | 3/4 (75%) |
| Sample | Height (cm) (1st vs. 2nd Assessment, p-Value) | Height z-Scores (1st vs. 2nd Assessment, p-Value) | BMI (kg/m2) (1st vs. 2nd Assessment, p-Value) | BMI z-Scores (1st vs. 2nd Assessment, p-Value) |
|---|---|---|---|---|
| Patients <19 years (n = 29) | 159.2 vs. 163.0, p < 0.001 | −0.33 vs. −0.07, p = 0.933 | 19.6 vs. 21.3, p = 0.06 | 0.00 vs. 0.37, p = 0.405 |
| Patients <19 years that increased natural protein intake (n = 19) | 161.0 vs. 163.0, p = 0.001 | 0.02 vs. 0.06, p = 0.469 | 20.2 vs. 21.3, p = 0.034 | 0.14 vs. 0.37, p = 0.920 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, A.; Ferreira Almeida, M.; MacDonald, A.; Cristina Ramos, P.; Rocha, S.; Guimas, A.; Ribeiro, R.; Martins, E.; Bandeira, A.; Jackson, R.; et al. Over Restriction of Dietary Protein Allowance: The Importance of Ongoing Reassessment of Natural Protein Tolerance in Phenylketonuria. Nutrients 2019, 11, 995. https://doi.org/10.3390/nu11050995
Pinto A, Ferreira Almeida M, MacDonald A, Cristina Ramos P, Rocha S, Guimas A, Ribeiro R, Martins E, Bandeira A, Jackson R, et al. Over Restriction of Dietary Protein Allowance: The Importance of Ongoing Reassessment of Natural Protein Tolerance in Phenylketonuria. Nutrients. 2019; 11(5):995. https://doi.org/10.3390/nu11050995
Chicago/Turabian StylePinto, Alex, Manuela Ferreira Almeida, Anita MacDonald, Paula Cristina Ramos, Sara Rocha, Arlindo Guimas, Rosa Ribeiro, Esmeralda Martins, Anabela Bandeira, Richard Jackson, and et al. 2019. "Over Restriction of Dietary Protein Allowance: The Importance of Ongoing Reassessment of Natural Protein Tolerance in Phenylketonuria" Nutrients 11, no. 5: 995. https://doi.org/10.3390/nu11050995
APA StylePinto, A., Ferreira Almeida, M., MacDonald, A., Cristina Ramos, P., Rocha, S., Guimas, A., Ribeiro, R., Martins, E., Bandeira, A., Jackson, R., van Spronsen, F., Payne, A., & César Rocha, J. (2019). Over Restriction of Dietary Protein Allowance: The Importance of Ongoing Reassessment of Natural Protein Tolerance in Phenylketonuria. Nutrients, 11(5), 995. https://doi.org/10.3390/nu11050995